CHAPTER 72 Liver Physiology and Energy Metabolism
LIVER CELL TYPES AND ORGANIZATION
Liver cells can be classified into three groups: parenchymal cells include hepatocytes and bile duct epithelia; sinusoidal cells are composed of hepatic sinusoidal endothelial and Kupffer cells (hepatic macrophages); and perisinusoidal cells consist of hepatic stellate cells and pit cells. Hepatocytes comprise 60% of the adult liver cell population, representing ~78% of the tissue volume (see Chapter 71).1
PARENCHYMAL CELLS
Hepatocytes
Hepatocytes are large polyhedral cells approximately 20 to 30 µm in diameter.2 Consistent with their high synthetic and metabolic activity, hepatocytes are enriched in organelles. About 30% of human hepatocytes are binucleate. Hepatocytes are polarized epithelial cells. Their plasma membranes have three distinct domains—(1) the sinusoidal surface (~37% of the cell surface) that comes in direct contact with plasma through the fenestrae of the specialized hepatic sinusoidal endothelial cells; (2) the canalicular surface (~13% of the cell surface) that encloses the bile canaliculus; and (3) contiguous surfaces. By analogy with glandular epithelia, the sinusoidal, canalicular, and contiguous plasma membrane domains are also termed basolateral, apical, and lateral surfaces, respectively.3 The sinusoidal and canalicular surfaces contain microvilli, which greatly extend the surface area of these domains.
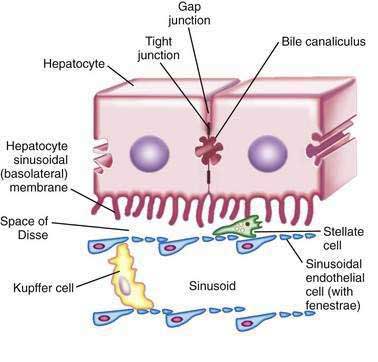
The space between the endothelia and the sinusoidal villi is termed the space of Disse. There is a bidirectional exchange of liquids and solutes between the plasma and hepatocytes at the sinusoidal surface. In many cases, the molecular transfer is augmented by proteins that promote facilitated diffusion or energy-consuming active transport. The canalicular domains of two adjacent hepatocytes are sealed at the periphery by tight junctions (desmosomes), thereby delimiting the bile canaliculus, which is the beginning of the biliary drainage system (see Chapter 62). In contrast to the bidirectional flow at the sinusoidal surface, flow from hepatocytes into the bile canaliculi is predominantly unidirectional.
Plasma Membranes
The plasma membranes consist of lipid bilayers composed of glycerophospholipids, cholesterol, and sphingolipids that provide barrier to water and most polar substances.3,4 The inner and outer leaflets of the plasma membrane differ in lipid, protein, and carbohydrate composition, reflecting their functional differences. Protein molecules within the leaflets mediate transport of specific molecules and serve as a link with cytoskeletal structures and the extracellular matrix. Hepatocyte plasma membranes consist of 36% lipid, 54% protein, and 10% carbohydrate by dry weight. Outer leaflets of hepatocyte plasma membranes are enriched in carbohydrates.
Lipid rafts are microdomains (∼50 nm diameter) of the outer leaflets of the plasma membrane that are highly enriched in cholesterol and sphingolipids.5 These are coupled to cholesterol-rich microdomains in the inner leaflet by an unknown mechanism. Raft lipids and associated proteins diffuse together laterally on the membrane surface. Some surface receptors become associated with the rafts on ligand binding, or they can lead to “clustering” of smaller rafts into larger ones. Lipid rafts are important in signal transduction, apoptosis, cell adhesion and migration, cytoskeletal organization, and protein sorting during both exocytosis and endocytosis (see later). Certain viruses enter cells via the lipid rafts.
Membrane proteins perform receptor, enzyme, and transport functions.6 Integral membrane proteins traverse the lipid bilayer once or multiple times or are buried in the lipid. Additional “extrinsic” protein molecules are associated with plasma membrane. Membrane proteins can rotate or diffuse laterally but usually do not flip-flop from one leaflet to another. Concentration of specific membrane proteins is maintained by a balance between their synthesis and degradation by shedding of membrane vesicles, proteolytic digestion within the membrane, or internalization into the cell. Receptor proteins internalized into the cell may be degraded or recycled to the cell surface.
Cell Junctions
Hepatocytes are organized into sheets (seen as chords in two-dimensional sections) by occluding (“tight”), communicating (“gap”), and anchoring junctions (Fig. 72-1). Tight junctions or desmosomes form gasket-like seals around the bile canaliculi, thereby permitting a concentration difference of solutes between the cytoplasm and bile canaliculus. Desmosomes are specialized membrane structures that anchor intermediate filaments to the plasma membrane and link cells together. Gap junctions are subdomains of contiguous membranes of hepatocytes that comprise ~3% of the total surface membrane. They consist of hexagonal particles with hollow cores, termed connexons, made up of six connexin molecules.7 Connexons of one cell are joined to those of an adjacent cell to form a radially symmetrical cylinder that can open or close the central channel. Gap junctions are involved in nutrient exchange, synchronization of cellular activities, and conduction of electrical impulses.
Cytoskeleton
The hepatocyte cytoskeleton supports the organization of subcellular organelles, cell polarity, intracellular movement of vesicles, and molecular transport.8,9 It comprises microfilaments, microtubules, and intermediate filaments, as well as the cytoskeleton-associated proteins.10 Intermediate filaments are polymers of fibrous polypeptides (cytokeratins and lamins) that provide structural support to the cells. In addition, vimentin is expressed by hepatocytes in tissue culture, and neurofilaments appear in injured hepatocytes and form Mallory bodies (also termed Mallory-Denk bodies or Mallory’s hyaline). Hepatocytes express two cytokeratins, CK8 and CK18. Bile duct epithelial cells express these proteins and CK19. Plectin is a giant protein that cross-links intermediate filaments to each other and to the plasma membrane, microtubules, and actin filaments.
Microtubules are hollow tubular structures (with an outer diameter of 24 nm) that consist of polymerized dimers of α and β tubulin, that are involved in intracellular transport and cellular organization.11,12 Microtubules serve as tracks to the movement of cytoplasmic vesicles, mediated by adenosine triphosphatase (ATPase)-powered motor proteins, kinesin, dynein, and dynamin. Depolymerization of the microtubules, for example, by colchicine treatment, inhibits plasma protein secretion without affecting protein synthesis. Microtubules participate in cellular organization by interacting with the Golgi apparatus, intermediate filaments, and F-actin.13 They also maintain the integrity of the surface membrane during canalicular contraction.14
Microfilaments are composed of double-helical F-actin strands, which are polymers of G-actin. A large number of actin-associated proteins control the polymerization, depolymerization, and splicing of F-actin. Together with myosins, actins maintain the integrity of the cell matrix, facilitate bile canalicular contraction, and control tight junction permeability. Microfilaments are also important in receptor-mediated endocytosis and various transport processes. Collapse of the cellular structure of hepatocytes during apoptosis and formation of apoptotic bodies may be related to remodeling of the actin cytoskeleton of hepatocytes.15
Nucleus
Nuclei of hepatocytes are relatively large and have prominent nucleoli. The two concentric nuclear membranes are stabilized by networks of intermediate filaments, one inside the inner membrane and one outside the outer membrane.16 The outer nuclear membrane is in direct continuity with the ER membranes. The perinuclear space between the two nuclear membranes surrounds the nucleus and is continuous with the endoplasmic reticulum (ER) lumen. The nuclear membrane contains pores through which molecules are selectively transported to and from the cytoplasm. The ribonuclear protein (RNP) network and the perinucleolar chromatin radiate from the nucleolus.
Transport between the Nucleus and the Cytoplasm
Pores of the nuclear envelope are associated with a large number of proteins, organized in an octagonal symmetry.17 The nuclear pore complex (NPC) is a large macromolecular assembly that protrudes into both the cytoplasm and the nucleoplasm. Bidirectional nucleocytoplasmic transport occurs through the central aqueous channel in NPCs.18 Histones, DNA- and RNA-polymerases, transcription factors, and RNA-processing proteins are selectively transported into the nucleus from the cytoplasm, where they are synthesized, whereas tRNAs and mRNAs are synthesized in the nucleus and exported to the cytoplasm through the NPCs.
Endoplasmic Reticulum
The ER is the largest intracellular membrane compartment, consisting of membranous tubules or flattened sacs (cysternae) that enclose a continuous lumen or space and extend throughout the cytoplasm.19 The domain of ER in which active protein synthesis occurs has attached ribosomes and is termed the rough ER. The other domain, termed smooth ER, is devoid of ribosomes and is the site of lipid biosynthesis, detoxification, and calcium regulation. The nuclear envelope is a specialized domain of the ER.20
Golgi Complex
The Golgi complex consists of a stack of flat sac-like membranes (cysternae) that are dilated at the margins.21 Many proteins synthesized in the rough ER are transported to the Golgi apparatus in protein-filled transition vesicles. The aspect of the Golgi complex facing the ER is the cis face; the opposite side is termed the trans face. Glycoproteins are thought to be transported between the Golgi sacs via shuttle vesicles. The highly mannosylated glycosyl moiety of proteins that are N-glycosylated in the ER are processed in the Golgi sacs into mature forms. Some other proteins are O-glycosylated in the Golgi complex. These proteins are then sorted for transport to appropriate cellular organelles (see later discussion of exocytosis and endocytosis).22
Lysosomes
Lysosomes consist of a system of membrane-bound sacs and tubules that contain hydrolytic enzymes that are active at pH 4.5 to 5.23,24 The ATPase-powered proton pump maintains the acid pH by importing hydrogen ions into the lysosomal lumen.23 Lysosomal enzymes are glycoproteins with N-linked oligosaccharides. Following synthesis in the ER, the carbohydrate moieties are modified in the Golgi apparatus, where their mannose residues are phosphorylated. Recognition of these mannose 6-phosphate (M6P) groups by the M6P receptor in trans-Golgi stacks25 results in their segregation and translocation into late endosomes, which transform into lysosomes.26,27
Mitochondria
Mitochondria constitute about 20% of the cytoplasmic volume of hepatocytes and are responsible for cellular respiration.28–30 They contain the enzymes of the tricarboxylic acid cycle, fatty acid oxidation, and oxidative phosphorylation.30,31 Mitochondria conserve the energy generated by oxidation of substrates as high-energy phosphate bonds of ATP. In addition, parts of the urea cycle, gluconeogenesis, fatty acid synthesis, regulation of intracellular calcium concentration, and heme synthesis take place in the mitochondria. Mitochondria play a key role in programmed cell death, or apoptosis (see later).32
Glycolysis and fatty acid oxidation in the mitochondria generate chemical intermediates that feed into the citric acid cycle of energy-yielding reactions.33,34 The citric acid cycle breaks down acetyl coenzyme A (acetyl CoA) into three molecules of nicotinamide adenine dinucleotide (NADH), one molecule of flavin adenine dinucleotide (FADH2), and two molecules of carbon dioxide. Electrons derived from NADH and FADH2 drive an electron transport pathway in the inner mitochondrial membrane, leading to ATP production. Passage of electrons across the inner mitochondrial membrane to the space between the inner and outer membrane generates a proton gradient that drives ATP synthesis.35
Peroxisomes
Peroxisomes are spherical-appearing structures that enclose a matrix that contains a lattice or crystalline core.36 Peroxisomes are abundant in hepatocytes and are thought to be essential for life. Several oxidative catabolic reactions, as well as anabolic reactions, take place in peroxisomes, which provide important links between the metabolism of carbohydrates, lipids, proteins, fats, and nucleic acids.
Exocytosis and Endocytosis
Exocytosis and endocytosis are pathways involved in exporting, importing, and intracellular trafficking of molecules. Addition of new proteins and lipids to the plasma membrane by exocytosis and removal of membrane components into cytoplasmic compartments by endocytosis keep the cell surface in a state of dynamic polarization. During exocytosis, secreted proteins, synthesized in the ER, pass sequentially through the cis-, medial-, and trans-Golgi stacks and the trans-Golgi network and finally appear at the cell surface.37,38 This vectorial transport through the Golgi stacks occurs via vesicles that are coated by proteins termed coatamers or COP (COPI and COPII), which are distinct from clathrin (see later).39,40 Guanosine triphosphate-guanosine diphosphate (GTP-GDP) exchange factors and GTP-activating proteins that are specific for each type of vesicle stimulate membrane binding and catalytic activation of small guanosine triphosphatases (GTPases).
Once bound to the membrane, GTPases induce recruitment of COP proteins. In the ER, the first coat protein to be recruited is COPII, and vesicular/tubular clusters are formed. These clusters are thought to coalesce to form a complex tubular network, termed the ER/Golgi intermediate compartment. Acquisition of COPI proteins by the membranes of this tubular network results in the formation of vesicles that carry out bidirectional protein transport to and from the Golgi stacks. Some vesicles that emerge from the exit side of the Golgi apparatus, termed the trans-Golgi network (TGN), can transport multiple protein molecules simultaneously and release them together into the extracellular medium. Other types of vesicles that carry membrane proteins and enzymes destined for specific intracellular organelles also pass through this secretory pathway. These vesicles are sorted at the TGN, and vesicles carrying specific cargo are delivered to appropriate target organelles.41
Endocytosis is the import of extracellular macromolecules by processes that include pinocytosis, phagocytosis, receptor-mediated endocytosis (RME), and caveolar internalization.42 Pinocytosis refers to nonselective bulk-phase uptake of extracellular fluid via engulfment by plasma membrane invaginations. Phagocytosis is the ingestion of particles as well as regions of the cell surface. In contrast to these nonspecific modes of uptake, RME is a mechanism of uptake of specific molecules (ligands). After the ligands bind to their specific cell surface receptors, the ligand-receptor complexes concentrate in “pits” that are coated on the cytoplasmic surface by three-pronged structures (triskelions) composed of three heavy chains and three light chains of clathrin. The assembled coats consist of a geometric array of 12 pentagons and a variable number of hexagons, depending on the size of the coat. The coated pits pinch off into the underlying cytoplasm as coated vesicles.43 In the next step, the vesicles lose their clathrin coat and are termed endosomes. Endosomal vesicles travel along microtubules and can take three distinct pathways. Some endosomes return to the cell surface, and the contained ligand-receptor complexes are secreted out of the cells by a process termed diacytosis. Transferrin is a prototype ligand for diacytosis. Some other ligands, such as immunoglobulin A (IgA) oligomers, may traverse the cells to be secreted into bile along with the receptor. This process is termed transcytosis.44
The best studied type of RME is the classical endocytotic pathway, in which the interior of the endosome is acidified by the action of a proton pump, thereby leading to ligand-receptor uncoupling.45 By mechanisms that have not been elucidated fully, the dissociated ligands and receptors are sorted into different vesicles. The ligand-containing vesicles proceed to lysosomes, where the ligand is degraded by lysosomal hydrolases. A majority of the ligand-free receptors translocate to the cell surface and replenish the receptor pool. Some receptors, such as the insulin receptor, do not undergo recycling and are rapidly degraded in lysosomes. In addition to the recruitment of clathrin, initiation of the formation of endocytotic vesicles requires adaptor proteins, particularly AP-2, which localizes between the lipid bilayer and clathrin. Non-scaffold proteins, such as the GTPases and dynamin, are also important in the conversion of a coated pit to a coated vesicle. This function of dynamin requires association with a protein termed amphiphysin. Genetic, cell biological, and biochemical studies are identifying additional proteins that are required for clathrin coat and vesicle formation (reviewed by Stockert45). In addition to physiological ligands, many viruses use receptor-mediated endocytosis to enter cells.
Internalization via caveolae is another pathway by which macromolecules can enter cells. Binding of caveolin to the cytoplasmic aspect of cholesterol-rich lipid rafts on the plasma membrane generates 50- to 60-nm flask-shaped invaginations of the plasma membrane. These invaginations bud off into the cytoplasm to form vesicles, termed caveolae or plasmalemmal vesicles. Caveolae perform various functions, including signal transduction, calcium regulation, non-clathrin-dependent internalization, and transcytosis. Glucosyl phosphatidylinositol (GPI)-anchored proteins, the β-adrenergic receptor, and tyrosine kinase are concentrated in caveolae.46
Bile Duct Epithelial Cells
Bile duct epithelial cells, or cholangiocytes, comprise large and small subpopulations of cells, the cell volumes of which correlate roughly with the diameter of the intrahepatic bile ducts (see Chapter 62). The large cholangiocytes have a relatively more developed ER and a lower nucleus-to-cytoplasmic ratio than do the small cholangiocytes.47 The paucity of expression of cytochrome P450–dependent monooxygenase activity imparts a survival advantage to the small cholangiocytes against injury by chemicals. For example, cytochrome P450 2E1–mediated formation of toxic intermediates of carbon tetrachloride leads to the loss of large cholangiocyte function after administration of the pro-toxin, whereas the small cholangiocytes are resistant to the toxic injury.
Bile ducts are not mere passive conduits for biliary drainage but play an active role in the secretion and absorption of biliary components and regulation of the extracellular matrix composition. Cholangiocytes are highly polarized. A sodium-dependent bile salt transporter (ABAT), located at the apical (luminal) surface of cholangiocytes, mediates the uptake of conjugated bile acids by cholangiocytes, whereas an alternatively spliced truncated form of the protein (ASBT), located at the basolateral surface, mediates the efflux of the bile acids in a sodium-independent manner. The sodium-dependent glucose transporter (SGLT1), located at the apical domain, and GLUT1, a facilitative glucose transporter on the basolateral domain, are responsible for glucose reabsorption from bile. Aquaporin-1 at the apical and basolateral surfaces constitute water channels that may mediate hormone-regulated transport of water into bile by cholangiocytes. The purinergic receptor (P2u) stimulates chloride ion efflux. Activation of apical P2u by ATP, which is secreted into the bile by hepatocytes, mobilizes Ca2+ stores, thereby stimulating Cl− efflux from cholangiocytes. The large, but not the small, cholangiocytes express secretin and somatostatin receptors, the chloride/bicarbonate exchanger, and the cystic fibrosis transmembrane regulator, which may enable this population of cholangiocytes to modulate water and electrolyte secretion in response to secretin and somatostatin (see also Chapter 64).48
SINUSOIDAL CELLS
Hepatic Sinusoidal Endothelial Cells
Hepatic sinusoidal endothelial cells (HSECs) account for 20% of total liver cells. These cells are distinguished by the fenestrae (pores) in their flat, thin extensions that form sieve plates. Unlike capillary endothelial cells, HSECs do not form intracellular junctions and simply overlap each other (see Fig. 72-1B). The presence of fenestrae and the absence of a basement membrane permit plasma to enter the space of Disse and come in direct contact with the sinusoidal surface of hepatocytes.49 Diameters of the fenestrae are actively controlled by the actin-containing components of the cytoskeleton in response to changes in the chemical milieu.50 Thus, the specialized endothelial lining of hepatic sinusoids serve as a selective barrier between the blood and the hepatocytes. Hepatic endothelial cells can secrete prostaglandins and a wide variety of proteins, including interleukin (IL)-1 and IL-6, interferon, tumor necrosis factor-α (TNF-α) and endothelin.
Kupffer Cells
Kupffer cells are specialized tissue macrophages that account for 80% to 90% of the total population of fixed macrophages in the body. These cells are derived from bone marrow stem cells or monocytes and are highly active in removing particulate matter and toxic or foreign substances that appear in the portal blood from the intestine.51 Kupffer cells are located in the sinusoidal lumen and are in direct contact with endothelial cells (see Fig. 72-1). They possess bristle-coated micropinocytic vesicles, fuzzy-coated vacuoles, and worm-like structures that are special features of cells that are active in pinocytosis and phagocytosis. An abundance of lysosomes reflects their prominent role in degrading substances taken up from the bloodstream. Kupffer cells secrete a variety of vasoactive toxic mediators, which may be involved in host defense mechanisms and in pathophysiologic processes in some liver diseases. Kupffer cells increase in number and activity in chemical, infectious, or immunologic injury to the liver.52
PERISINUSOIDAL CELLS
Hepatic Stellate Cells
Hepatic stellate cells (HSCs) are also known as Ito cells, vitamin A–storing cells, fat-storing cells, or lipocytes. These cells are a part of the stellate cell system, which includes similar cells in the pancreas, lung, kidney, and intestine. Hepatic stellate cells are located between the endothelial lining and hepatocytes (see Fig. 72-1B). These mesenchymal cells represent 5% to 8% of all liver cells and are important sources of paracrine, autocrine, juxtacrine, and chemoattractant factors that maintain homeostasis in the microenvironment of the hepatic sinusoid. Microfilament and microtubule-enriched flat cytoplasmic extensions of quiescent stellate cells store vitamin A–enriched lipid droplets and spread out parallel to the endothelial lining, contacting several cells.53 HSCs express receptors for retinol-binding protein (RBP), which mediates the endocytosis of RBP-retinol complexes.54
After chronic liver injury, the slender star-shaped HSCs become activated to elongated myofibroblasts. They lose retinoids and up-regulate the synthesis of extracellular matrix components, such as collagen, proteoglycan, and adhesive glycoproteins. Stellate cell activation is the central event in hepatic fibrosis.55 Activation of HSCs is initiated by paracrine stimulation by neighboring sinusoidal endothelial cells, Kupffer cells, endothelial cells, and hepatocytes, as well as platelets and leukocytes. Endothelial cells participate in activation by producing cellular fibronectin and by converting the latent form of TGF-β to its active, profibrogenic form. Binding of TGF-β to its receptor on HSCs plays a critical role in stellate cell activation. Binding of bacterial lipopolysaccharides (LPS) arriving to the liver from the intestine to Toll-like receptor 4 (TLR4) enhances the effect of TGF-β on HSCs by two different mechanisms.56 First, increased chemokine expression by stellate cells results in chemotaxis of Kupffer cells, which secrete TGF-β. Second, LPS binding to TLR4, activates nuclear factor kappa B (NF-κB) via the adapter protein MyD88 (myeloid differentiation response protein), thereby down-regulating the TGF-β pseudoreceptor Bambi (bone morphogenic protein and the activin membrane-bound inhibitor) and thereby sensitizing the HSCs to TGF-β signaling. The three-dimensional structure of the extracellular matrix modulates the shape, proliferation, and function of HSCs, probably by signal transduction via binding to cell surface integrins, followed by changes in cytoskeleton assembly.
Pit Cells
Pit cells, the natural killer (NK) cells of the liver, are located mainly within the sinusoidal lumen, close to Kupffer cells. They have the appearance of large lymphocytes and are adherent to the sinusoidal wall, often anchored with villous extensions (pseudopods).57 In the human liver, pit cells have pronounced polarity, abundant cytoplasm containing dense granules, a conspicuous cytocenter, and a locomotory shape characterized by hyaloplasmic pseudopods and a uropod (a tail-like structure that forms on the trailing end of a moving cell). The cytoplasmic granules appear as pits by microscopy, hence the name pit cells. Pit cells are short-lived and are replenished from extrahepatic sources.
In common with circulating NK cells, the pit cells express OX-8 antigen, and some express asialoganglioside gangliotetrasylceramide (asialo-GMr1). Pit cells do not express the pan–T-cell marker, OX-19, which is expressed by circulating NK cells. Although the source of pit cells remains debated, they are antigenically related to NK cells of other viscera. Pit cells have tumor cell-killing activity in the liver and are also thought to remove virus-infected liver cells. Their per-cell cytolytic activity is greater than that of circulating NK cells. Pit cells may also have a role in controlling the growth and differentiation of liver cells and possibly in liver graft rejection.58
INTEGRATION OF THE FUNCTIONS OF THE DIFFERENT CELL TYPES
Functional integration of the various groups of liver cells occurs through direct cell-to-cell communication (e.g., via gap junctions), paracrine secretion that affects neighboring cells, cell signaling, interaction with the extracellular matrix, and generalized response to endocrine and metabolic fluxes.59 Hepatocytes and hepatic sinusoidal endothelial cells lack a continuous basement membrane, and the spatial relationship of the cells is maintained through interaction with the extracellular matrix. Anchoring to the extracellular matrix is important for the survival of hepatocytes. Anchoring also provides traction for movement and permits liver cells to receive signals from matrix components and matrix-bound growth factors. Hepatic extracellular matrix components are produced during development along the migration path of the hepatocytes and exhibit unique patterns of distribution and organization. Stellate cells, hepatocytes, and, to some extent, endothelial cells are major producers of the extracellular matrix in the liver. Excess deposition of connective tissue causes changes in hemodynamic properties and eventually impairs liver function.55
CELL-MATRIX INTERACTIONS
Cell-matrix interactions in the liver are important in maintaining hepatocyte morphology and proliferation. For example, when plated on a flat layer of collagen, hepatocytes synthesize DNA at a level that is four-fold higher than when they are grown on gels composed of basement membrane proteins. The type of matrix determines the level of expression of albumin and other hepatocyte-specific gene products in cultured hepatocytes.59,60 On the other hand, cell-cell and cell-matrix interactions determine the level of synthesis and deposition of hepatic extracellular matrix proteins by the various types of liver cells. Such interaction also modulates the production of specific enzymes and their inhibitors that mediate remodeling of the extracellular matrix.
Integrin and non-integrin receptors mediate the interaction of liver cells with extracellular matrix. Integrins bind to extracellular matrix proteins at specialized cell attachment sites that often contain the arginine-glycine-aspartate motif, thereby resulting in attachment of the extracellular matrix to the intracellular cytoskeleton network. This attachment results in changes in cell shape, spreading, and migration. Integrins also influence cell proliferation, differentiation, survival, apoptosis, and gene expression via signal transduction.61,62 Non-integrin surface receptors mediate cell attachment by different mechanisms.
COMPONENTS OF THE EXTRACELLULAR MATRIX
Components of the extracellular matrix include collagens, noncollagenous glycoproteins, and proteoglycans. The liver contains five types of collagen (I, III, IV, V, and VI) and seven classes of noncollagenous glycoproteins (fibronectin, laminin, entactin/nidogen, tenascin, thrombospondin, SPARC [secreted protein, acidic, and rich in cysteine], and undulin). Hepatic extracellular matrix also includes a large number of proteoglycans and glycosaminoglycans, such as membrane-associated syndecan, thrombomodulin, and betaglycan, and extracellular matrix-associated versican, biglycan, decorin, fibromodulin, and perlecan.59,63
REGENERATION AND APOPTOSIS OF LIVER CELLS
REGENERATION
Normal adult hepatocytes divide infrequently, with fewer than 1 in 10,000 hepatocytes undergoing mitosis at any given time—yet the liver possesses a unique capacity to replace tissue mass after liver injury or loss of liver mass. The capacity of the liver to regulate its own growth is evident in liver transplantation, where the size of the transplanted organ increases or decreases as appropriate to the size of the recipient. Such finely regulated hyperplasia of the liver is also seen after successful single-lobe liver transplantation in children.64
Hepatic regeneration has been studied extensively in rodents. Following resection of two thirds of the liver in rats, the residual liver cells proliferate and restore the liver mass within days to weeks. Although generally termed “regeneration,” this process is, in fact, restorative hyperplasia because the total liver mass, rather than the lobulated anatomic configuration, is reconstituted. In the rat, DNA synthesis peaks at 24 hours after partial hepatectomy, when approximately 35% of hepatocytes are in cell cycle. Cell division occurs six to eight hours after DNA synthesis. The time frame of DNA synthesis varies from species to species. For example, in mice, maximum DNA synthesis occurs 36 to 40 hours after hepatic resection. Because 80% to 95% of hepatocytes undergo mitosis, liver mass is restored after one or two cell divisions. All classes of hepatocytes, including diploid, tetraploid, and octaploid cells participate in this quasi-synchronized proliferation, either by mitosis of mononucleated cells or by cytokinesis of binucleated or tetranucleated hepatocytes, after DNA synthesis in all nuclei. Interestingly, adult hepatocytes, rather than liver progenitor cells, contribute to liver regeneration after partial hepatectomy. Only when the proliferation of adult hepatocytes is inhibited because of certain toxic or physical injuries do progenitor cells, often termed oval cells, proliferate. The oval cells are thought to give rise to both hepatocytes and bile duct epithelial cells.65
After liver injury, early signals for hepatocyte replication come from nonparenchymal cells (see Fig. 72-2B).66,67 LPS and intestine-derived cytokines stimulate Kupffer cells and hepatic sinusoidal endothelial cells to produce TNF-α and IL-6. Growth factors, such as hepatic growth factor (HGF), are released from stores in the hepatic matrix and are secreted by HSCs, whereas epidermal growth factor is secreted into portal blood by epithelial cells of the proximal small intestine and salivary glands.66 Hormones, such as triiodothyronine (T3), insulin, and nonrepinephrine, are important cooperative factors in liver regeneration.68 Replication of nonparenchymal cells lag behind that of hepatocytes by 24 to 72 hours. Initially, the newly proliferated hepatocytes form clusters, first in zone 1 and later in other zones of the liver (see Chapter 71). Regenerating endothelial cells invade these clusters and restore the single-cell thick liver plates.
Early as well as late changes occur in the expression of extracellular matrix components and the enzymes that modulate them. The mitotic phase is mostly completed in three days, and the liver mass is restituted in about seven days. Liver cells return to their quiescent state when the liver mass is restored to the original size, give or take ~10%. A balance between mitosis and apoptosis fine-tunes the restoration of hepatic mass. The strictly self-limited nature of hepatocyte replication suggests that strong regulatory pressures are present that favor replicative repression. The ability of the liver to regulate its size is dependent on signals generated outside the liver, such as hormonal or metabolic signals, as well as internal signals generated within the liver.65 Signals for cessation of growth of the regenerating liver are understood less well than those governing hepatocellular replication.
Gene Expression During Hepatic Regeneration
The regenerative process is a cascade of events that move cells from their resting G0 phase through the G1 phase, S (DNA synthesis) phase, G2 phase, and then to M (mitotic cell division) phase (Fig. 72-2A) (see Chapter 3). Expression of a large number of genes is induced or down-regulated after partial hepatectomy at transcriptional or post-transcriptional levels.66,68 The sequence of activation of various genes during liver regeneration has been elucidated by studies using partial hepatectomy and gene knockout mice that lack specific cytokines. These genes include cell cycle genes, metabolic genes, genes coding for extracellular matrix proteins, growth factors, cytokines, and transcription factors. Chronologically, these genes can be grouped into immediate early genes, delayed early genes, and cell cycle–associated genes. Expression of these genes is modulated by signal transduction pathways that receive and transduce stimuli for cell replication and tissue remodeling.
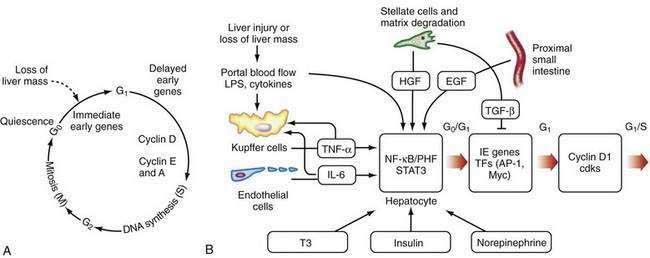
Integration of Cytokine and Growth Factors in Liver Regeneration
The early, reversible phase of liver regeneration, during which hepatocytes can enter the cell cycle by moving from the quiescent G0 state to early G1 phase, is termed priming.66 This phase is initiated by the effect of cytokines, the best studied of which include TNF-α and IL-6. Generation of reactive oxygen species as a consequence of the acute metabolic changes and release of LPS that occur in response to the loss of hepatic functional mass may have a role in triggering the initial cytokine response. During priming, NF-κB and STAT3 are activated, and AP-1 and C/EBP are expressed. Together, these factors lead to the immediate early gene expression response after partial hepatectomy (see earlier). The priming events sensitize hepatocytes to growth factors. In the absence of growth factors, the cells cannot move past a certain “restriction point” in G1.
When the peak level of cyclin D1 expression is reached, cells progress autonomously through the cell cycle, without further need for growth factors. Expression of HGF, TGF-α, and probably epidermal growth factor (EGF) increase after partial hepatectomy. These factors are the direct mitogens for liver regeneration. EGF binds to both the EGF receptor and the TGF-α receptor, and c-met is the receptor for HGF. Growth hormone, thyroid hormones, and parathormone are permissive for liver regeneration, whereas insulin and norepinephrine are considered adjuvant factors.66
HGF and c-met
Major sources of HGF in the liver are Kupffer cells and HSCs. HGF is produced as a single 87- to 90-kd pro-protein by nonparenchymal cells and is cleaved into ~64-kd and ~32-kd peptides that form heterodimers.65,66 HGF mRNA levels are increased 12 to 24 hours after partial hepatectomy in rats. Elevated levels of HGF have been observed in the serum of patients with fulminant hepatic failure, thus suggesting an important role for HGF in regeneration of human liver. C-met, the HGF receptor, is a heterodimer consisting of a 145-kd β-chain and a 45-kd α-chain, linked by disulfide bonds. The two polypeptide chains of c-met are also derived from proteolytic cleavage of a single precursor protein. The β-chain contains the transmembrane region and the intracellular tyrosine kinase domain. HGF binding to the extracellular domain of c-met activates tyrosine kinase, thereby initiating a signal transduction pathway.
PROGRAMMED CELL DEATH
Programmed cell death, or apoptosis, is an integral part of hepatic regeneration. Apoptosis is involved in a fine tuning and remodeling process that results in reconstruction of the hepatic architecture. Apoptosis results in the removal of damaged, senescent, or supernumerary cells, without altering the cellular microenvironment. Loss of function of pro-apoptotic proteins, overexpression of anti-apoptotic proteins, or loss of apoptotic signaling in cells can lead to the survival of DNA-damaged cells, leading in turn to several forms of cancer.69
Apoptotic signals can originate within the cells through mechanisms that sense DNA damage and inappropriate proliferative signals. In other cases, the apoptotic signals come from other cells in at least three ways.70 First, cells recognized as foreign or as pathogens may receive apoptotic signals from immune mediator cells. Second, the nurturing signals of neighboring cells or extracellular matrix may be lost, thus resulting in apoptosis of anchor-dependent cells. Third, some cells undergo apoptosis in response to certain growth factors such as TGF-β1.
In contrast to necrosis, apoptosis is an active process that culminates in cell death. During the latent phase of apoptosis, the cell undergoes molecular and biochemical change but remains morphologically intact. In the execution phase, a series of dramatic structural changes take place that culminate in the fragmentation and condensation of the cell into membrane-enclosed apoptotic bodies. Initially, a variety of stimuli, including DNA damage, growth factor withdrawal, toxins, or radiation, trigger the apoptotic pathway. The signal is transduced by a series of defined protein-protein interactions Finally, cell death is executed by the activation of specific proteases called caspases that cleave multiple substrates, leading to DNA fragmentation, chromatin condensation, cell shrinkage, and membrane blebbing. The apoptotic cell may be phagocytosed or simply lose contact with neighboring cells. Apoptosis does not cause an acute inflammatory reaction. All these morphologic features of apoptosis contrast with those of necrosis, in which the cell swells and releases proinflammatory material into the neighboring space.69
The two major apoptotic pathways include activation of cell surface death receptors70 and mitochondrial permeability transition.71 At least six different cell surface molecules can function as death receptors. One of the best characterized death receptors is Fas (also known as Apo1 or CD95). Fas belongs to the family of TNF receptors. Binding of Fas to Fas ligand leads to an interaction between the cytoplasmic domain of the Fas receptor and the death domain of the adaptor protein, FADD (Fas-associated protein with death domain), which in turn recruits and activates procaspase-8. Once activated, caspase-8 activates downstream caspases such as caspase-3. The second major pathway involves mitochondria and is triggered by various toxic insults. Either Bax or Bak opens channels and thereby releases the electron transport protein cytochrome c and other proteins from the intermembranous space into the cytoplasm. Cytochrome c binds the scaffolding protein Apaf-1. The C-terminal portion of Apaf1 is a negative regulator of apoptosis. The N-terminal region contains a caspase recruitment domain and an ATPase domain. Binding of cytochrome c and deoxyadenosine triphosphate (dATP) removes the negative regulatory influence of the C-terminus of Apaf-1, thereby permitting binding and autoactivation of caspase 9. Activated caspase 9, in turn, activates caspases-3 and -7, thus initiating cell death. In addition, permeabilization of the mitochondrial outer membrane results in the loss of function of the electron transport chain, which is essential for most mitochondrial functions, including ATP generation.
PROTEIN SYNTHESIS AND DEGRADATION IN THE LIVER
HEPATIC GENE EXPRESSION
Compared with most organs, the liver expresses a large number of genes. Over 90% of plasma proteins and about 15% of the total protein mass of the body are produced in the liver.72 As in all mammalian cells, gene expression is initiated by transcription of the gene into an RNA transcript, mediated by RNA polymerase II. The nascent RNA is modified by capping of the 5′-terminus with 7-methylguanosine, excision of the noncoding intervening sequences (introns), splicing together of the coding sequences (exons), and, in most cases, addition of polyadenylate at the 3′-end. The processed mRNA is actively transported out of the nucleus. In the cytoplasm, association of the mRNA with the 40s ribosomal subunit and methionine RNA requires several initiation factors, a cap binding protein, and ATP hydrolysis.
Some genes expressed in hepatocytes, loosely termed “housekeeping genes,” are expressed in many other organs as well. In addition, the expression of many other genes occurs preferentially or uniquely in the liver. Expression of these liver-specific genes permits the liver to perform essential functions of the body, including secretion of plasma proteins, gluconeogenesis, glycogen storage, glucose metabolism, cholesterol homeostasis, bile salt production, and detoxification of endogenous metabolites and exogenous substances. A series of cis-acting elements in specific genes mediate their hepatocyte-preferred expression.73 These cis-acting DNA elements bind different families of HNFs. Although none of these factors is entirely liver-specific, high levels of liver-preferred gene expression occur only in the presence of combinatorial interaction of these transcription factors. Maintenance of hepatocyte-enriched expression of specific transcription factors involves cross-regulation by other unrelated liver-enriched transcription factors. Some of the transcription factors involved in hepatocyte specificity are also important in hepatic tissue specification during embryogenesis. Many of the transcription factors are normally located in the cytoplasm. Binding of hormones or cytokines to their respective cell surface receptors causes conformational changes in the cytoplasmic domain of these receptors, often through phosphorylation. Such conformational changes lead to a series of events that eventually lead to the translocation of specific transcription factors to the nucleus and their binding to the respective cis-acting elements in the regulatory regions of genes. Thus, extracellular signals are transduced to a series of intracellular events, culminating in the induction or repression of gene expression.
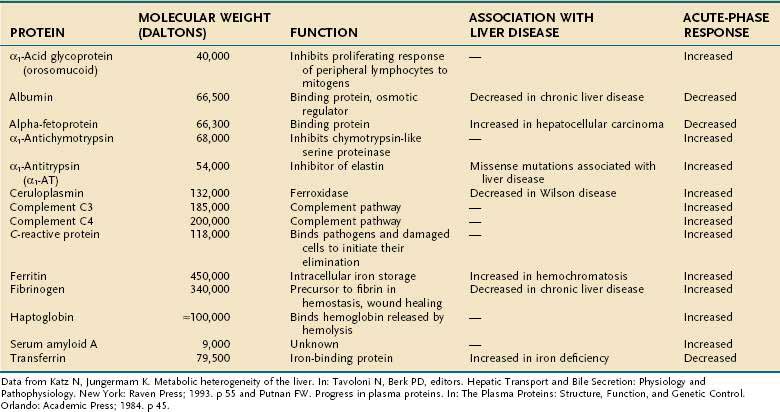
Regulation of gene transcription is the most important, but not the only, mechanism of modulation of gene expression. Stability of the RNA, translational regulation, and post-translational modifications can all affect the steady-state concentration, intracellular or extracellular location, and activity of a given gene product. The major plasma proteins synthesized and secreted by the liver are shown in Table 72-1.
Nuclear Receptors
Modulation of metabolic pathways and detoxicating mechanisms according to the needs of the body often requires coordinated up-regulation or repression of the expression of a set of genes. In many cases, such coordination is mediated by nuclear receptors, such as retinoid X receptor (RXR), liver X-receptor (LXR), farnesoid X-receptor (FXR), constitutive androstane receptor (CAR), peroxisome proliferator activator receptor (PPAR), and thyroid hormone receptor (TR).74 For example, expression of proteins that mediate bilirubin uptake by hepatocytes, intracellular storage of bilirubin, glucuronidation of bilirubin, and bile canalicular excretion of bilirubin glucuronides may all be regulated by CAR. Nuclear receptors mediate induction or repression of genes by small nonprotein molecules. For example, phenobarbital binds to CAR in the cytoplasm, leading to the translocation of CAR to the nucleus and thereby resulting in simultaneous induction of multiple genes that have CAR-binding elements in their cis-regulatory regions. Similarly, bile acids bind to FXR, fibrates bind to PPAR, and thyroid hormones bind to TR. In most cases, nuclear receptors function by forming heterodimers with RXR, although some nuclear receptors can function as homodimers.
PROTEIN FOLDING
Proteins that are destined for export to intracellular membranes or secretion into the plasma are translocated into the ER where folding takes place prior to secretion through the Golgi apparatus.75 The ER contains a number of molecular chaperones and folding catalysts that promote efficient folding. All chaperones enable and promote protein folding and assembly, but their specific functions differ. Many chaperones work in tandem with one other. Some molecular chaperones bind to nascent chains as they emerge from the ribosome and protect aggregation-prone hydrophobic regions. Other chaperones are involved in later stages of folding, particularly for complex proteins that include oligomeric species and multimolecular assemblies.
In addition to promoting proper folding, chaperones play an important role in the “quality control” of proteins, through a complex series of glycosylation and deglycosylation processes and prevention of misfolded proteins from being secreted from the cell.76 The unfolded or misfolded proteins are targeted for degradation through the ubiquitin-proteasome pathway.77 Up to one half of all polypeptide chains fail to satisfy the quality control mechanism in the ER, and for some proteins, such as the cystic fibrosis transmembrane conductance regulator (CFTR), the success rate is even lower. The proportion of molecules that misfold is increased greatly in mutant proteins with amino acid substitutions. Some molecular chaperones are able to rescue misfolded proteins to enable them to have a second chance to fold correctly. Under some circumstances, chaperones can solubilize proteins that have aggregated because of misfolding. In some cases, energy for such active intervention may be derived from ATP hydrolysis. Many molecular chaperones, such as the heat shock protein, are up-regulated in stressful situations, when protein misfolding is more prone to occur.
PROTEIN CATABOLISM
Like protein synthesis, proteolysis, is a major process that contributes to the body protein turnover. The autophagic-lysosomal pathway and the ubiquitin/proteasome pathway are the two major mechanisms of protein degradation. The autophagic-lysosomal mechanism is responsible for bulk degradation of endogenous proteins, as well as degradation of other cellular components such as RNA, carbohydrates, and lipids. This pathway may be seen as a cell restructuring mechanism. The autophagy system is regulated physiologically by plasma levels of the amino acids leucine, glutamine, tyrosine, phenylalanine, proline, methionine, tryptophan, and histidine, probably through binding to cell surface receptors and subsequent intracellular signaling. Protein kinase cascades such as mTOR, Erk, eIF2a, and others may be involved in the regulation of autophagy. Amino acids may exert their effects through these pathways in combination with insulin.78 Chaperone-mediated autophagy (CMA) is a selective mechanism for the degradation of altered cytosolic proteins in lysosomes. Synthesis of a lysosomal receptor that is critical for CMA declines in aged animals, thereby leading to the accumulation of altered proteins and eventually leading to the characteristic functional alterations in the aging liver and other organs. In transgenic mice in which the abundance of the lysosomal receptor for CMA is retained until advanced age, less damaged proteins accumulate intracellularly, and liver functions are maintained at youthful levels.79
The ubiquitin/proteasome pathway is the principal mechanism for turnover of normally short-lived proteins in mammalian cells.80 Ubiquitin is a small protein that can link covalently to itself or to other proteins, either as monomers or as chains of polyubiquitin. Ubiquitin is added to a target protein by ubiquitin-activating, ubiquitin-conjugating, and ubiquitin-ligating enzymes. The first function attributed to ubiquitin was the covalent binding to misfolded proteins, thereby directing proteasome-dependent proteolysis. Now, ubiquitin and ubiquitin-related proteins are also known to direct specific proteins through the endocytotic pathway by modifying cargo proteins, as well as by regulating components of the cytoplasmic protein trafficking machinery. By regulating the turnover of mitotic cyclins, ubiquitination plays an important role in cell cycle regulation.81,82
Although the ubiquitin/proteasome pathway is generally considered to be separate from the lysosomal proteolysis mechanism, ubiquitination is sometimes required for lysosomal proteolysis. A subset of endocytosed proteins must be conjugated to ubiquitin as a trigger for internalization from the plasma membrane.83,84 Thus, ubiquitin conjugation appears to be important in several protein trafficking steps, including endocytosis.
HEPATIC NUTRIENT METABOLISM
The liver is at the hub of numerous metabolic pathways. In this chapter, we will present a brief description of the role of the liver in energy metabolism. The liver provides energy continuously to the entire body through its ability to store and modulate the availability of systemic nutrients.85 In turn, the metabolic function of the liver is regulated by hormones secreted by the pancreas, adrenals, thyroid, and adipose tissue, as well as neuronal inputs. A liver-adipose tissue-brain-pancreas axis,86 as well as a gut-brain-liver axis,87 orchestrates the management of the energy supply to body tissues. In addition to serving as a store for excess energy as lipids, the adipose tissue, particularly visceral fatty tissue that drains into the portal circulation, plays an active role in hepatic energy metabolism by releasing free fatty acids (FFAs) into plasma and releasing a series of adipokines that either increase or decrease insulin sensitivity in the liver and other tissues.86 During nutrient absorption (fed state), the liver regulates nutrient flux as the absorbed nutrients are metabolized, modified for storage in the liver and fatty tissue, or made available to other organs as an energy source. During fasting, the energy supply is maintained from the stored fuel and by synthesis. Starvation induces breakdown of triglycerides in adipose tissues into FFAs and glycerol. FFAs reduce insulin sensitivity, thereby affecting glucose metabolism in muscles and the liver. FFAs bind and activate PPARs in liver and other tissues, thereby affecting their gene expression.88 In the hepatocyte, most of the acetyl CoA produced by oxidation of FFA is used to synthesize ketone bodies (e.g., acetoacetate and β-hydroxybutyrate) that are released into circulation and used as an energy source by many peripheral tissues. The glycerol released by triglyceride hydrolysis is used by the liver for the synthesis of glucose, which is the only source of energy for neurons and red blood cells, or of triglyceride. The triglyceride is packaged into very-low-density lipoproteins (see later) and returns to the adipose tissue.89
The role of a gut-brain-liver axis in glucose homeostasis has also been established.87 In rats, lipids arriving in the intestine give rise to long-chain fatty acyl-coenzyme A by the action of acyl-CoA synthase, which sends an afferent signal to the nucleus of the solitary tract in the hindbrain through the vagus nerve. This signal leads to N-methyl-d-aspartate ion channel-dependent glutamatergic neurotransmission through the efferent vagal fibers that supply the liver, thereby resulting in a reduction in glucose production by the liver that precedes the actual post-absorptive glucose influx from the intestine. Thus, the rapid gut-brain-liver communication helps prevent excessive fluctuation of the blood glucose level. Unfortunately, this mechanism becomes inoperative with continued intake of excessive calories for several days. Detailed reviews of hepatic nutrient metabolism are available elsewhere.90
CARBOHYDRATE METABOLISM
Glucose is the primary energy source for the brain, erythrocytes, muscle, and renal cortex. Maintaining adequate circulating levels of glucose is essential for the central nervous system, which normally uses glucose as its major metabolic fuel. After a person fasts for 24 to 48 hours, the brain can use ketones as a metabolic fuel, thereby reducing its glucose requirement by 50% to 70%.90 The liver is the principal organ that maintains total carbohydrate stores by synthesizing glycogen and generating glucose from precursors.91 Glucose is synthesized from nonoxidative metabolic products of glucose (pyruvate and lactate) that are generated predominantly by red blood cells (RBCs) and from amino acid precursors that are derived predominantly from muscle during prolonged starvation or exercise.
Regulation of Glucose Uptake and Efflux from the Hepatocyte
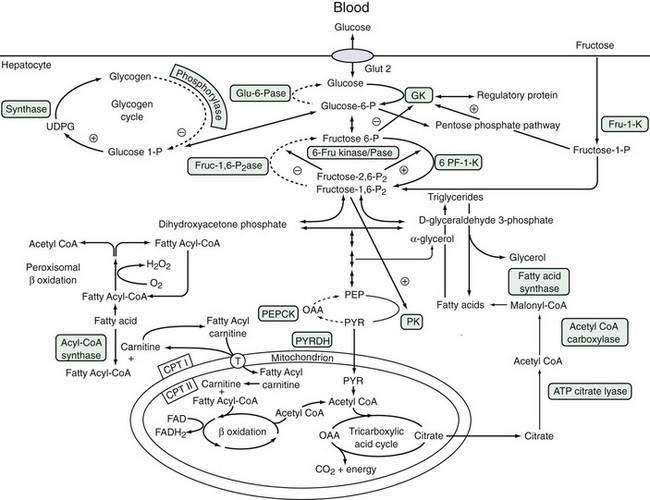
Glucose is a critical molecule in the metabolic pathway because it can be converted to amino acids, fatty acids, or glycogen, the major storage form of glucose. Glucose enters hepatocytes via the glucose transporter-2, which facilitates the diffusion of glucose across the sinusoidal membrane.92 Glucose transporter-2 differs from other members of the glucose transporter family in that it is independent of metabolic conditions or insulin levels. Because of the low-affinity, high-capacity characteristics of glucose transporter-2, intrahepatic glucose concentration is determined by the plasma glucose level, which, in turn, is regulated by glucokinase activity (see “Formation of Glucose-6-Phosphate” later).93 Glucose transporter-1, which is present in the brain, RBCs, and hepatocytes, particularly in zone 3, is a low-capacity, high-affinity glucose transporter that permits glucose uptake by hepatocytes when the circulating glucose concentration is low. Increased expression of glucose transporter-1 during fasting enhances glucose uptake by hepatocytes. Hepatocellular glucose homeostasis is maintained by interlinking pathways that are regulated by multiple signals, which prevent competing pathways from operating at the same time.94 Figure 72-3 illustrates these pathways and the modulating influences that control the metabolic flux of glucose and other sugars, such as fructose.
Formation of Glucose-6-Phosphate
Rapid conversion of glucose to glucose-6-phosphate (glucose-6-P) modulates the glucose concentration within the hepatocyte, thereby regulating influx or efflux of glucose from the hepatocyte.91 Glucose-6-P is a nodal branch point compound that can enter three independent metabolic pathways: (1) synthesis of glycogen, which can be mobilized rapidly during fasting; (2) anaerobic glycolysis via the Embden-Meyerhof pathway, which generates pyruvate or lactate as a substrate for the tricarboxylic acid (Krebs) cycle in mitochondria; or (3) the pentose-phosphate shunt, which generates reducing equivalents necessary for anaerobic glycolysis and fatty acid synthesis. The pentose-phosphate shunt is regulated by the activity of mitochondrial glucose-6-P dehydrogenase.92 Conversion of glucose to glucose-6-P is catalyzed by hexokinase, which accepts several different hexose substrates, and glucokinase (GK, also termed hexokinase type 4 or D), which is expressed predominantly in the liver and pancreas and is specific for glucose.95
A low-affinity, high-capacity system, GK is not inhibited by the reaction product, glucose-6-P. Therefore, the level of GK activity regulates hepatocellular glucose concentration, which determines the net uptake of glucose by hepatocytes from hepatic sinusoidal plasma. GK is activated by insulin and inhibited by glucagon.90 Mutations in the GK gene are associated with some rare cases of maturity-onset diabetes of young adults (MODY).95 Fructose-1-phosphate modulates GK activity by regulating the inhibitory activity of a GK regulatory protein.96 The regulation of GK by fructose is thought to prevent futile cycling between glucose and glucose-6-P that consumes ATP. Starvation decreases GK activity, thereby promoting glucose efflux from the hepatocyte.
Conversion of Glucose-6-Phosphate to Glucose
Conversion of glucose-6-P to glucose is catalyzed by glucose-6-phosphatase (glu-6-Pase), a multisubunit enzyme with its active site located within the ER lumen.97 Thus, glucose-6-P needs to traverse the ER membrane to be dephosphorylated. Inherited deficiency of glu-6-Pase causes glycogen storage disease type Ia (see Chapter 76).97 Glucose-6-P transport is mediated by a microsomal transport protein, which, when defective, causes type Ib glycogen storage disease. As expected, glu-6-Pase activity is increased by starvation, resulting in an increase in hepatocellular glucose concentration and consequent efflux of glucose into the sinusoidal space by the bidirectional glucose transporter-2.
The relative production of fructose 6-P and fructose-1,6-P2 is regulated by the opposing action of 6-phosphofructo-1-phosphokinase (6-PK-1-K) and fructose-1,6-bisphosphatase (fruc-1,62Pase).91 Within this cycle is a unique enzyme: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (6-fru kinase/Pase). This enzyme, which combines the properties of both a 6-PF-2K and its corresponding phosphorylase enzyme activity, produces the regulatory product fructose-2,6-P2. Fructose-2,6-P2 is a potent activator of 6-PF-1-K and inhibitor of fruc-1,62Pase. Moreover, it favors the formation of the fructose-1,6-P2 product. The enzyme is regulated by both hormonal and nutrient regulations and serves as another modulator of glucose metabolism. During starvation, when fructose-2,6-P2 levels are low, gluconeogenesis is enhanced. On the other hand, high levels of 6-fru kinase/Pase found during refeeding and insulin administration promote glycolysis and fatty acid synthesis. The phosphorylation status of the 6-fru kinase/Pase is regulated by the cAMP-dependent kinase site and phosphatase 2A activity.
From fructose-1,6-P2, a sequence of four biochemical reactions leads to the formation of PEP with generation of eight molecules of ATP.85 PEP can then be metabolized into PYR as part of the third regulatory cycle in glucose metabolism. Pyruvate kinase, which transforms PEP to PYR, generates two ATP molecules. PYR is another nodal branch point in the metabolic pathway, from which it can undergo further metabolism in mitochondria to form acetyl-CoA. Thereafter, it can undergo aerobic metabolism by the tricarboxylic acid cycle. In this pathway, PYR may be metabolized ultimately to water and carbon dioxide, with the production of 15 molecules of ATP per molecule of PYR. Other products of the tricarboxylic acid cycle are also precursors for fatty acid (citrate) or amino acids by means of oxaloacetate formation. Fructose-1,6-P2 is also an inducer of PK.98 In the reverse reaction, PYR is metabolized to oxaloacetate, which is a precursor to the amino acid l-aspartate. Oxaloacetate is converted by the energy-dependent activity of phosphoenolpyruvate carboxykinase (PEPCK), an important regulator of gluconeogenesis. PEPCK expression is inhibited by insulin at the transcriptional level90,98 and is up-regulated during fasting and in diabetes mellitus.
Hepatic Metabolism of Galactose and Fructose
Lactose, a major disaccharide present in human and cow milk, is split into glucose and galactose. Galactose can be converted to glucose-6-P, after which it can be used for glycogen synthesis; or it can be oxidized further to form PYR or acetyl-CoA for additional energy generation or fatty acid synthesis.85 Galactose is initially phosphorylated by galactokinase to form galactose-1-phosphate (galactose-1-P). In the presence of uridine diphosphoglucose (UDPG), further metabolism by uridyltransferase forms glucose-1-phosphate (glucose-1-P) and UDP-galactose. UDP-galactose can be epimerized by UDP-glucose-4-epimerase to form UDP-glucose, which is a precursor to glucose-1-P. Glucose-1-P can be converted to glucose-6-P. Thus, like glucose, galactose can participate in the glycolytic pathway.
Glycogen Formation
Glycogen stored in the liver is the main source of rapidly available glucose for the glucose-dependent tissues, such as RBCs, retina, renal medulla, and brain.99 Hepatic glycogen stores contain up to a two-day supply of glucose before gluconeogenesis occurs, mainly from lactate, a three-carbon end-product of anaerobic glucose metabolism.85,100 Hepatic gluconeogenesis produces up to 240 mg of glucose a day, which is approximately twice the metabolic need of the RBCs, retina, and brain. The three-carbon precursors generated by anaerobic metabolism from muscle, intestine, liver, or RBCs may account for up to 50% of the glycogen pool formed during nonabsorptive states. Alanine, another major glucose precursor, is generated by the catabolism of muscle proteins, which is a major cause of muscle wasting during prolonged fasting. Glycogen stored in muscle is used locally and cannot be exported out of the cell because muscles lack glu-6-Pase. The relative contribution of each of the precursors to glycogen synthesis depends on the nutritional status, amount, and route of glucose administration (oral vs. intravenous) and on hormonal regulation.
Rapid switching between glycogen synthesis and breakdown is mediated by a cascade of enzymes that are regulated by local nutrients and hormones.85 Glycogen phosphorylase, which is activated by phosphorylation, catalyzes the breakdown of glycogen subunits, and glycogen synthase, which is activated by dephosphorylation, catalyzes the addition of UDP-glucose to the expanding glycogen chain. In addition, glucose and glucose-6-P are allosteric activators of the synthase enzyme, whereas glucose binding inactivates the phosphorylase.
Regulation of Glycolytic-Gluconeogenic Pathways
The glycolytic-gluconeogenic pathways are regulated by hormonal signals and the relative availability of nutrients. Insulin up-regulates the expression of genes that encode the glycolytic enzymes and represses the expression of metabolic enzymes responsible for gluconeogenesis. Glucagon, catecholamines, corticosteroids, and growth hormone increase cellular cAMP levels, thereby augmenting the gluconeogenic pathway. In many cases, post-transcriptional mRNA stabilization or degradation, post-translational phosphorylation or end-product inhibition, or allosteric modulation contributes to the relative abundance or activity of specific enzymes.91,98 Glucose and fructose modulate the enzyme activities by direct inhibition or by allosteric modulation of the enzymes. In the fed state, high activity of GK, 6-PF-1-K, and PK induced by insulin favors formation of PYR, with low activity of PEPCK and other gluconeogenic enzymes. During fasting, the fall in plasma insulin levels removes the inhibition of the gluconeogenic enzymes PEPCK and fruc-1,6-P2ase. Simultaneously, an increase in glucagon and β-adrenergic agonists raises intracellular cAMP levels, leading to inhibition of 6-PK-2 kinase activity and stimulation of fruc-2,6-Pase, thereby reducing fructose-2,6-P2 concentration and activation of fruc-1,62Pase, with a net increase in gluconeogenesis. After a prolonged fast, gluconeogenesis is further stimulated by an increase in the supply of substrate and alterations in the concentration of various enzymes.
Carbohydrate Metabolism in Cirrhosis
Patients with cirrhosis have an increased frequency of hyperglycemia and relative hyperinsulinemia.101 The hyperglycemia may be explained by decreased glucose uptake by muscle and reduced glycogen storage in liver and muscle. These changes lead to insulin resistance, which causes an increase in plasma insulin levels. Other causes of relative insulin resistance include increased serum FFA levels that can inhibit glucose uptake by muscle, altered second messenger activity after insulin binds to its receptor, and increased serum concentrations of cytokines that result from elevated serum levels of LPS. Increased levels of glucagon and catecholamines may be contributing factors. The net result is impaired nonoxidative use of glucose with decreased storage of glycogen and impaired uptake of glucose by muscle, thereby causing a relative insulin-resistant state similar to that found in patients with diabetes mellitus and obesity.
LIPID METABOLISM
Fatty acids are an important energy source for the liver and serve as an efficient fuel store within and outside the liver because oxidation of fatty acids yields the highest ATP production of any metabolic fuel.85 In addition, most organs are capable of using fatty acids as a fuel.102 The liver plays a central role in regulating the body’s total fatty acid needs. Excess glucose can be converted to fatty acid for future use and stored at distal sites such as adipose tissue and delivered by lipoproteins (see “Lipid Transport” later). Triglycerides are stored in the cytoplasm of hepatocytes, where they are enclosed in a monolayer of phospholipids to form lipid droplets, which are important in the energy balance of the cell and the whole organism. Under conditions of excess lipid accumulation in the hepatocyte, for example, in overnutrition, the risk of acquiring insulin resistance increases. Lipid droplet formation may require a family of PPARα-induced ER proteins, termed fat-inducing transcripts 1 and 2 (FIT-1 and FIT-2).103 Beta oxidation of fatty acids in mitochondria and peroxisomes has different physiologic consequences.104 Furthermore, fatty acids are structural components of cell membranes and are important in cellular function and cell anchoring. The regulation of fatty acid synthesis and transport of fatty acids to other organs in association with lipoproteins constitutes another critical role of the liver in managing the metabolic needs of the entire body.
Fatty Acid Synthesis
Fatty acid synthesis occurs in the cytosol and is regulated closely by the availability of acetyl-CoA, which forms the basic subunit of the developing fatty acid carbon chain.85 Acetyl-CoA is synthesized predominately in mitochondria and is derived mainly from carbohydrate metabolism, with a small fraction coming from amino acids.4,12,13 Acetyl-CoA is condensed with oxaloacetate to form citrate, which is exported from the mitochondria and is then cleaved by the cytosolic ATP citrate lyase to produce oxaloacetate and acetyl-CoA. Conversion of acetyl-CoA to malonyl-CoA by the action of acetyl-CoA carboxylase is the first step in fatty acid synthesis. Acetyl-CoA carboxylase is the key enzyme in regulating fatty acid synthesis because it provides the necessary building blocks for elongation of the fatty acid carbon chain.105
Malonyl-CoA is used by a set of enzymatic activities contained within a single peptide chain that comprises the remarkable fatty acid synthase system.85 Malonyl-CoA binds to acyl carrier protein (ACP). Catalytic activity is contained within two distinct domains that catalyze sequential condensation, reduction, dehydrogenation, and reduction, which constitute the fatty acid synthetic cycle. Two NADPH molecules are required for each two-carbon unit that is added to the growing fatty acid chain. After completion of the first cycle, the 4-carbon butyl group is transferred from ACP to a peripheral thiol, thereby allowing it to accept the next malonyl-CoA group to restart the entire cycle. The cycle continues for an additional six or seven rounds until a carbon-16 (palmitate) or carbon-18 (stearate) fatty acid is synthesized. Fatty acid-CoA is then released and used for other metabolic pathways.
Further elongation of the fatty acid chain can occur either in the mitochondrion or within the microsomal membrane.85 In the mitochondrion, the first step is mediated by enoyl-CoA reductase. Microsomal elongation uses malonyl-CoA to increase the size of fatty acyl-CoA in a process that involves four separate enzymatic reactions. The elongation ability of microsomes is tissue dependent and serves the needs of specific organs. The fatty acid chain elongates until an appropriate length has been achieved, and the fatty acid is then esterified with glycerol to form triglycerides. These newly formed triglycerides can be transported by lipoproteins to distal sites for storage and use. In situations of excess carbohydrates, PYR can be converted to acetyl-CoA by the mitochondrial pyruvate dehydrogenase complex to serve as fatty acid precursors, although lipogenesis from carbohydrates consumes about 25% of the energy contained in the carbohydrates.
Beta Oxidation of Fatty Acids
Mitochondrial Beta Oxidation
Fatty acids are translocated across the mitochondrial membranes by first undergoing fatty acyl-CoA formation by the activity of distinct fatty acyl-CoA synthetases that are specific for short-, medium-, or long-chain fatty acids in the mitochondrial outer membrane.85,106 In the inner mitochondrial membrane, conjugation of fatty acyl-CoA with carnitine is catalyzed by carnitine palmitoyltransferase I, with formation of fatty acylcarnitine, which is translocated into the mitochondrion, in exchange for free carnitine, by an integral inner membrane protein, fatty acylcarnitine:carnitine translocase.107 Inside the mitochondrion, a reverse reaction mediated by carnitine palmitoyltransferase II releases fatty acyl-CoA, which is now a substrate for beta oxidation. The first step that is unique to beta oxidation is formation of trans-enol fatty acid, which is generated by acyl-CoA dehydrogenase. Acyl-CoA dehydrogenase transfers two electrons to flavin adenine dinucleotide (FAD), which then transfers them to the electron transport chain in the mitochondrion. 3-Keto fatty acyl-Co then undergoes a series of sequential reactions to acetyl-CoA and fatty acyl-CoA, which undergo another round of beta oxidation. Acetyl-CoA can enter the tricarboxylic acid cycle, thereby generating 12 ATP, or it can enter the 3-hydroxyl methyl glutaryl-CoA cycle to form ketone bodies. Only mitochondria in the liver are capable of forming ketone bodies. Regulation of mitochondrial beta oxidation lies with fatty acylcarnitine formation, which is catalyzed by carnitine palmitoyltransferase I.107 Malonyl-CoA, the basic subunit of fatty acid synthesis, is a potent inhibitor of carnitine palmitoyltransferase I, and thus prevents beta oxidation and fatty acid synthesis from occurring concurrently.
Peroxisomal Beta Oxidation
NADH generated in subsequent reactions needs to be removed from the peroxisomes, whereas, in mitochondria, NADH can enter the electron transport cycle and generate additional ATP molecules. Peroxisomal enzymes can metabolize only long-chain fatty acids with a minimal chain length of 10 carbons and a maximal length of 24 carbons. As in mitochondria, beta oxidation in peroxisomes proceeds similarly by 2-carbon acetyl-CoA cleavage until octanoyl-CoA is formed. Octanoyl-CoA is then combined with carnitine to form fatty acyl carnitine, which can be transported by the mitochondrial inner membrane transporter and undergo completion of beta oxidation. Acyl-CoA formed in peroxisomes by beta oxidation of fatty acids can diffuse out of the peroxisomes after formation of acetyl carnitine.107
Increased triglyceride synthesis, reduced synthesis of lipid transport proteins (see later), and a decreased level of beta oxidation can result in the accumulation of fat within hepatocytes (steatosis). A classic example of this process is alcoholic steatosis, which occurs when a large percentage of the total caloric intake is derived from ethanol. Alteration in the redox potential with excess NADH produced by ethanol metabolism results in an increased NADH/NAD ratio, which favors the formation of α-glycerol phosphate, which in turn promotes triglyceride formation. In addition, a decrease in NAD content in mitochondria may reduce fatty acid beta oxidation, thereby contributing to fatty acid accumulation.108
Lipoproteins
Apolipoproteins (apo), which are synthesized by the liver, in combination with triglycerides, phospholipids, cholesterol, and cholesterol esters, constitute circulating lipoproteins, which mediate the transport of lipids from the liver into the plasma and from the plasma into the liver and other tissues. The liver also expresses cell surface receptors for circulating lipoproteins and modulates plasma levels of these important macromolecules. Lipoprotein trafficking has been reviewed elsewhere109 and is summarized in the following section.
Types of Lipoproteins
Lipoproteins were originally classified according to their relative density, which is inversely related to their particle size. Listed in increasing order of density, they are: chylomicrons, very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). Density differences in these particles reflect the type and amount of specific lipids and the proportion of protein present within these lipoprotein fractions.109 Specific apolipoproteins bind lipids to form lipoproteins, which are modified by enzymes in plasma or endothelial cells and act as ligands for specific lipoprotein receptors that mediate their uptake by target tissues.
Tangier’s disease, a rare autosomal recessive disorder characterized by the accumulation of cholesteryl esters in reticuloendothelial cells, including the tonsils, thymus, and lymph nodes, as well as liver, spleen, and gallbladder, in combination with the near absence of serum HDL cholesterol, is now recognized to be caused by mutations in the ATP-binding cassette transporter A1 (ABCA1), a member of the ABC supergene family.110 Affected patients classically present with enlarged, orange-colored tonsils and have a four- to six-fold increased risk of atherosclerotic heart disease. Although the function of the transporter is not completely known, its location at the plasma membrane suggests that it mediates the active transport (“flipping”) of cholesteryl ester from the inner to the outer leaflet of the plasma membrane, from which it can be transferred to apolipoproteins and secreted.110
Apolipoproteins
The major apolipoproteins associated with triglyceride transport are apoB-100, which is synthesized in the liver, and apoB-48, which is synthesized in the intestine.111 Both proteins are translated from the same mRNA. In human intestinal epithelium, the apoB mRNA undergoes post-transcriptional RNA editing, which generates a stop codon by cytidine deamination that results in the translation of a form of apoB that is approximately 48% of the size of the full-length apoB-100 generated in the liver. The carboxy-terminal domain that is absent in apoB-48 is essential for binding to the LDL receptor. Unlike the apoB-100-containing VLDL, chylomicron remnants, which contain apoB-48, are rapidly cleared from plasma and do not give rise to LDL.111
ApoC is synthesized predominately in the liver, with minor expression in the intestine and other organs, and is composed of three different gene products that may inhibit the uptake of chylomicron remnants by the liver. ApoC-1 is a minor component of VLDL, HDL, and IDL and is of unknown function. ApoC-II is present in VLDL, IDL, HDL, and chylomicrons and is an essential activator of lipoprotein lipase (LPL) (see “Intestinal Lipoprotein Metabolism,” later). Inherited deficiency of apoC-II causes hypertriglyceridemia. ApoC-III is present in IDL, HDL, and chylomicrons and may be an inhibitor of LPL activity.112
ApoE is synthesized in the liver and is found on all lipoproteins. ApoE is important for removal of lipoprotein remnants in the serum, can bind to the LDL receptor and other membrane proteins, and is important in targeting lipoproteins to specific receptors on peripheral cells. Three major alleles of the apoE gene exist (ε2, ε3, and ε4), with the ε3 allele being the most abundant and ε2/ε3 genotype being the most frequent. Each allele possesses a different ability to bind to the LDL receptor. Absence of apoE leads to reduced clearance of chylomicron and VLDL remnants, resulting in elevated plasma levels and a consequent increase in the risk of atherosclerosis.113 ApoE is also important in lipid transport in the central nervous system especially after neuronal injury. Inheritance of a single apoε4 allele is associated with a six- to eight-year earlier onset of Alzheimer’s disease than that associated with the ε3/ε3 genotype.114
ApoA-I and -II are synthesized in the liver and intestine. ApoA-I is the major component of HDL lipoproteins. In a lipid-poor state, apoA-I accepts cholesterol from the cell membrane. ApoA-I is a key activator of LCAT, which enhances cholesterol esterification in the plasma, and the absence of a specific, conserved region in apoA-I causes loss of its LCAT activating property. ApoA-II is another component of HDL. ApoA-IV is a minor constituent synthesized in the intestine.115
Lipolytic Enzymes
LPL is synthesized in fat and muscle cells and is located in the luminal surface of the capillary bed of adipose, lung, and muscle tissues.116 LPL catalyzes lipolysis of triglycerides present in VLDL, chylomicrons, or HDL. LPL is stimulated by fasting, fatty acids, hormones, and catecholamines. Patients who are homozygous for LPL deficiency present with severe hypertriglyceridemia in childhood and pancreatitis.
Lipid Transport Proteins
In plasma, lipid exchange between particles is facilitated by the activity of LCAT and cholesteryl ester transfer protein (CETP).116 LCAT is synthesized in the liver, and apoA-I is a cofactor for LCAT activity. CETP is synthesized predominantly in the liver and circulates in association with HDL. CETP mediates the exchange of cholesteryl esters from HDL with triglycerides from chylomicrons or VLDL. The activity of LCAT in combination with the lipid transfer proteins, CETP and phospholipid transfer protein (PLTP), is essential for the transfer of cholesterol from nonhepatic tissue to the liver.116
Intestinal and Hepatic Lipid Transport
The liver functions as the hub for receiving fatty acids and cholesterol from the diet and peripheral tissues, packages them into lipoprotein complexes, and releases the complexes into the circulation (Fig. 72-4). Following absorption by intestinal epithelial cells, fatty acids are formed into triglycerides, and cholesterol is esterified. Both lipids are packaged into nascent chylomicrons composed predominantly of triglycerides (85% to 92%), phospholipids (6% to 12%), cholesteryl ester (1% to 3%), fat-soluble vitamins, and the following apolipoproteins (1% to 3%): apoB-48, apoA-I, apoA-II, and apoA-IV.117 Nascent chylomicrons enter the interstitial space and are carried into the systemic venous circulation via the thoracic duct. In the interstitial space, chylomicrons acquire apoC-II, which activates LPL, thereby promoting triglyceride release. Triglyceride release may be reduced by acquisition of apoC-III, which may inhibit LPL activity. The addition of apoE is critical for targeting the chylomicron remnant, which can then be taken up by hepatocytes through the chylomicron remnant receptor.
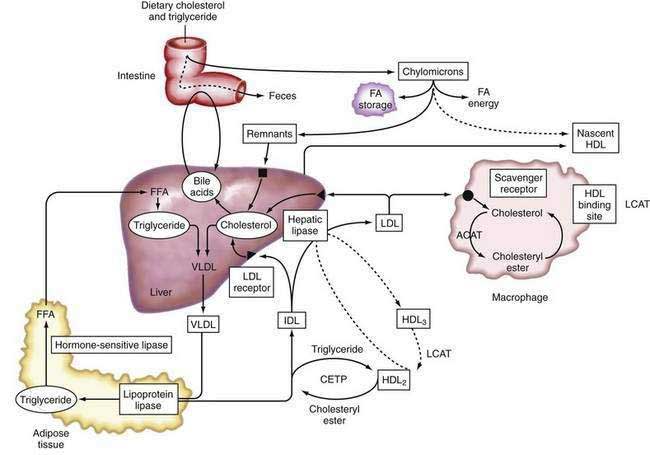
Fatty acids released from adipocytes by the action of intracellular hormone-sensitive lipase are bound to serum albumin and transported to other tissues including the liver, where they are used for synthesis of phospholipids and triglycerides.118 The liver synthesizes cholesterol from low-molecular-weight precursors. Hepatic cholesterol synthesis is regulated by the rate-limiting enzyme 3-hydroxyl-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase). Lipids are exported from the liver as VLDL particles, which are the major carriers of plasma triglycerides during nonabsorptive states.85 Lipids may be stored temporarily in the liver as fat droplets and cholesteryl esters, excreted directly into bile, or metabolized into bile acids. The liver is the major site of sterol excretion from the body and is the site of bile acid synthesis (see Chapter 64).
The coordinated input, synthesis, and excretion of sterols require complex regulation of multiple enzymatic pathways. Bile acids returning to the liver via the enterohepatic circulation modulate these enzyme activities. Bile acids recycle 20 to 30 times per day via the enterohepatic circulation and use specific transmembrane transporters at apical and basolateral domains of hepatocyte plasma membrane, as well as intracellular binding proteins.119 In the terminal ileum, a great majority of the bile acid molecules are reabsorbed via a sodium-dependent bile acid transporter. Bile acids are also important in micellization of fats for intestinal absorption and as coactivators of bile acid–dependent lipase activity. FXR, a member of the sterol nuclear receptor family, binds to and is activated by bile salts. Heterodimers of activated FXR and RXR modulate the coordinated regulation of multiple genes that encode key bile salt transporters, such as the sodium-dependent taurocholate pump (NTCP) at the sinusoidal domain of hepatocytes, bile salt export pump (BSEP) at the canalicular domain, intestinal bile acid transporter (IBAT) in the terminal ileum, and cholesterol-7α-hydroxylase in hepatocytes (see Chapter 64).120
Transport of ApoB-Containing Lipoproteins
In the fasting state, VLDL, which is synthesized in the liver, replaces chylomicrons as the major transporter of triglycerides and cholesterol. In addition to the full-length apoB-100, VLDL contains triglycerides (taken up from plasma or synthesized in the liver), cholesteryl esters (exogenous or endogenous), and phospholipids.121 During fasting, fatty acids in VLDL are derived predominantly from the activity of hormone-sensitive lipase in adipocytes, whereas after a meal, dietary fatty acids are the major source. Fatty acids may be taken up by hepatocytes by passive diffusion or via fatty acid transport proteins in the sinusoidal domain of the cell membrane. In hepatocyte cytosol, fatty acids are stored bound to the abundant 12-kd fatty acid binding protein (FABP) family, which may direct fatty acids to specific subcellular targets, such as the smooth ER for VLDL synthesis or peroxisomes for beta oxidation. FABPs are transcriptionally regulated by peroxisome proliferating agents (e.g., fibrates), suggesting that their role is physiologic in global lipid metabolism.
ApoB-100 is the predominant transport carrier in VLDL; apoC-I, C-II, C-III, and apoE arise from other lipoproteins within the serum. ApoB-100 synthesis and VLDL secretion are regulated by the availability of cotransported lipids and sterols in the smooth ER. ApoB-100 synthesis may change dramatically without alteration in apoB-100 mRNA levels.122 Following synthesis in the smooth ER, apoB-100 interacts with newly synthesized triglycerides and cholesteryl esters that enter the ER via specific membrane transporters. The apoB-lipid complex is translocated into the lumen, transported through the Golgi apparatus, and secreted into the sinusoidal space as VLDL. When the lipid components are not available, apoB-100 undergoes degradation in the ER. During periods of low plasma triglyceride levels, the liver secretes smaller IDL-like particles or even LDL-type particles.
Transport of ApoA-Containing High-Density Lipoprotein
HDL, another major class of lipoproteins secreted by the liver, appears to have a protective role against atherosclerosis. HDL is a heterogeneous population of lipoproteins that can be separated by sophisticated analytic centrifugation techniques. Nascent HDL is formed in the liver and intestine by lipolysis of VLDL and chylomicrons, respectively, with modification by peripheral tissue. The major protein constituents of HDL are apoA-I and apoA-II, with minor amounts of apoA-IV, apoC, apoE, and others.123 In humans, apoA-I is synthesized in the liver and intestine. Nascent apoA-containing lipoprotein complexes that appear as discoid particles can be transformed into HDL particles in the serum by the action of LCAT and the lipid transfer proteins CETP and PLTP.
The HDL3 subclass is particularly important because these cholesterol-poor particles are able to deliver cholesterol extracted from peripheral membranes and provide a substrate for plasma LCAT activity. Cholesteryl esters formed by LCAT are extremely hydrophobic and move into the core of the lipoprotein complex, thereby providing space on the surface of the lipoprotein for extraction of additional cholesterol from cell membranes. This complex enlarges with increasing amounts of cholesteryl esters, which are able to accommodate apoC-II and C-III, thereby resulting in HDL2 formation. CETP removes esterified cholesterol from HDL in exchange for triglycerides, which are eventually hydrolyzed by HTGL, thereby regenerating small HDL. Acquisition of apoC-II also promotes LPL activity, thereby increasing lipolysis.116
Lipoprotein Receptors
The major lipoprotein receptors for LDL, chylomicron remnants, HDL, and the scavenger receptor are members of the larger LDL receptor supergene family.124 These receptors share four major structural features: (1) cysteine-rich complement-type repeats; (2) epidermal growth factor precursor-like repeats; (3) a transmembrane domain; and (4) a cytoplasmic domain.125
LDL Receptor
The LDL receptor exists as an oligomeric surface glycoprotein that plays a pivotal role in LDL clearance and cholesterol homeostasis. It binds ligands at the cell surface, after which the ligand-receptor complex is internalized via the classic endocytotic pathway. The ligand dissociates from the receptor in acidic endosomal vesicles. Subsequently, the ligand is delivered to lysosomes for degradation and the receptor returns to the surface. The LDL receptor is present on all cell types; however, the liver contains approximately 70% of the total body pool of LDL receptors. The LDL receptor recognizes apoE and apoB-100, but not apoB-48. ApoE-containing chylomicron remnants, VLDL, LDL, IDL, and HDL can all be taken up via the LDL receptor. Approximately two thirds of LDL is cleared by this receptor. Homozygous deficiency of the functional LDL receptor occurs in approximately 1 in 1 million persons and is associated with accelerated atherosclerosis manifesting in childhood (familial hypercholesterolemia). LDL receptors are highly conserved among species.124
Very-Low-Density Lipoprotein Receptor
The VLDL receptor has a high-sequence homology with the LDL receptor but is expressed predominantly in extrahepatic tissues such as heart, muscle, and fat. Unlike the LDL receptor, the VLDL receptor does not bind to apoB and may serve specifically to take up triglyceride-rich apoE-containing lipoproteins, such as VLDL or IDL.124,125
Chylomicron Remnant Receptor
The chylomicron remnant receptor accepts apoE as a ligand. Chylomicron remnants are removed from the circulation exclusively by the liver, probably because these large complexes can penetrate the unique sinusoidal vascular space. The multifunctional, α2-macroglobulin/LDL receptor-related protein (LRP) is the chylomicron remnant receptor.126 LRP is present in liver, brain, and muscle. In cultured cells, LRP is able to mediate the endocytosis of apoE-containing chylomicron remnants. Mice that lack LRP in the liver do not have hepatic chylomicron remnant uptake, confirming that LRP is the major chylomicron remnant receptor. Unlike the LDL receptor, LRP is able to bind a number of unrelated ligands, such as lipoprotein, proteinase-inhibitor complex, and protein-lipid complex.
Low-Density Lipoprotein Scavenger Receptor
Ligands for the scavenger receptor A (SR-A) include lipopolysaccharides, polyanionic lipids, and LDL in which some of the free lysine residues have been chemically modified.127 These receptors exist in two forms as trimeric integral membrane glycoproteins in endothelial cells, macrophages, and Kupffer cells. Oxidized LDL is internalized via the scavenger receptors but is metabolized poorly in macrophages, leading to the accumulation of cholesteryl esters within the cell. Monocytes, which migrate into lipid-enriched atherosclerotic lesions, can also be induced to express the scavenger receptor.
High-Density Lipoprotein Receptor
A high-affinity HDL binding protein has been identified in the plasma membrane of hepatocytes, macrophages, adrenal cells, and adipocytes.127 These receptors appear to recognize specifically apoA present in HDL particles. The HDL receptor does not mediate endocytosis but allows only selective delivery of lipids to and from the HDL lipoproteins. By mediating the transfer of cholesterol from the plasma membrane to the HDL lipoprotein, the HDL receptor facilitates reverse cholesterol transport. The HDL receptor is a class B scavenger receptor, referred to as SR-B1.123 This receptor is most abundant in the liver, ovary, and adrenal glands—organs previously shown to be the principal sites of cholesterol uptake from HDL in vivo. HDL is a major source of cholesterol secreted in bile. Overexpression of SR-B1 in mouse liver increases biliary cholesterol secretion and reduces plasma HDL.128 Conversely, deficiency of this receptor results in decreased biliary cholesterol secretion.129
Derangement of Lipid Metabolism in Liver Disease
The most common lipid abnormality in patients with chronic liver disease is hypertriglyceridemia (plasma levels of 250 to 500 mg/dL), which is found in patients with alcoholic or viral liver disease and tends to resolve when the liver disease improves. Excess ethanol ingestion causes predominantly hypertriglyceridemia, due to increased fatty acid synthesis and decreased beta oxidation of fatty acids, resulting from increased NADH production by alcohol metabolism. Moderate alcohol ingestion is associated with increased HDL3 levels, which may reduce the risk of atherosclerosis. LDL, HDL, and total serum cholesterol levels decrease progressively with cirrhosis advancing from Child class A to class C (see Chapters 73 and 90). Serum cholesterol level may be a useful prognostic marker in patients with noncholestatic liver diseases.130
Cholestatic disorders manifest with a distinct pattern of dyslipoproteinemia because of the retention of cholesterol, phospholipids, and bile salts that are normally secreted in bile.131 A prolonged increase in total serum cholesterol and lipid levels, as seen in primary biliary cirrhosis, for example, can be associated with formation of xanthoma. Within the LDL fraction of the serum of cholestatic patients, three distinct lipoproteins can be identified, namely, β2-lipoprotein (triglyceride rich), also known as lipoprotein Y (LP-Y), lipoprotein X (LP-X), and normal LDL. LP-Y appears to be a remnant of a triglyceride-rich lipoprotein that is distinct from IDL. Cholestatic patients with elevated triglyceride levels often have clear (not lipemic) serum because most of the triglycerides are contained in LP-Y and LDL. LP-X is a complex composed of equimolar amounts of excess phospholipid and cholesterol in combination with albumin and certain members of the apoC family. The phospholipid flippase activity of multidrug resistance protein-3 (MDR3), also termed ATP binding cassette protein B4 (ABCB4) (see Chapter 64), is essential for LP-X formation. Mice lacking mdr2 (the murine homolog of MDR3) are unable to form LP-X during cholestasis caused by complete bile duct obstruction.132
Anghel SI, Wahli W. Fat poetry: A kingdom for PPARgamma. Cell Research. 2007;17:486-511. (Ref 86.)
Arias IM. The biology of hepatic endothelial fenestrae. In: Schaffner F, Popper H, editors. Progress in Liver Diseases, vol IX. Philadelphia: WB Saunders; 1990:11-26. (Ref 50.)
Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol. 2003;200:504-15. (Ref 59.)
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37-44. (Ref 42.)
Hui AY, Friedman SL. Molecular basis of hepatic fibrosis. Expert Rev Mol Med. 2003;14:1-23. (Ref 55.)
Karpen SJ. Nuclear receptor regulation of hepatic function. J Hepatol. 2002;36:832-50. (Ref 74.)
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717-21. (Ref 78.)
Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381-8. (Ref 7.)
Nakielny S, Dreyfuss G. Transport of proteins and RNAs in and out of the nucleus. Cell. 1999;99:677-90. (Ref 18.)
Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481-90. (Ref 32.)
Nordlie RC, Foster JD, Lange AJ. Regulation of glucose production by the liver. Annu Rev Nutr. 1999;19:379-406. (Ref 93.)
Miller JP. Liver disease, alcohol and lipoprotein metabolism. In: Zakim D, Boyer TD, editors. Hepatology: A Textbook of Liver Diseases. 4th ed. Philadelphia: WB Saunders; 2003:127. (Ref 109.)
Taub R. Liver regeneration: From myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836-47. (Ref 66.)
Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491-508. (Ref 70.)
Zegers MMP, Hoekstra D. Mechanisms and functional features of polarized membrane traffic in epithelial and hepatic cells. Biochem J. 1998;336:257-69. (Ref 3.)
1. Desmet VJ. Organizational principles. In: Arias IM, Boyer JL, Chisari FV, et al, editors. The Liver: Biology and Pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:3-15.
2. Saxena R, Zucker SD, Crawford JM. Anatomy and physiology of the liver. In: Zakim D, Boyer TD, editors. Hepatology: A Textbook of Liver Diseases. 4th ed. Philadelphia: WB Saunders; 2003:3-30.
3. Zegers MMP, Hoekstra D. Mechanisms and functional features of polarized membrane traffic in epithelial and hepatic cells. Biochem J. 1998;336:257-69.
4. Singer SJ. The molecular organization of membranes. Annu Rev Biochem. 1974;43:805-33.
5. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31-9.
6. Schachter D. The hepatocyte plasma membrane: Organization, differentiation, biogenesis and turnover. In: Arias IM, Boyer JL, Chisari FV, et al, editors. The liver: Biology and Pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:77-81.
7. Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381-8.
8. Novikoff PM, Cammer M, Tao L, et al. Three-dimensional organization of rat hepatocyte cytoskeleton: Relation to the asialoglycoprotein endocytosis pathway. J Cell Sci. 1996;109:21-32.
9. Roma MG, Milkiewicz P, Elias E, Coleman R. Control by signaling modulators of the sorting of canalicular transporters in rat hepatocyte couplets: Role of the cytoskeleton. Hepatology. 2000;32:1342-56.
10. Moll R, Franke WW, Schiller DL, et al. The catalog of human cytokeratins: Patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11-24.
11. Brinkley BR. Microtubule-organizing centers. Annu Rev Cell Biol. 1985;1:145-72.
12. Sobel HJ, Marquet E. Zipper-like structure and centrioles in hepatocytes. Ultrastruct Pathol. 1983;4:115.
13. Satir P. Motor molecules. In: Arias IM, Boyer JL, Chisari FV, et al, editors. The Liver: Biology and Pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:47-58.
14. Tzanakakis ES, Hansen LK, Hu WS. The role of actin filaments and microtubules in hepatocyte spheroid self-assembly. Cell Motil Cytoskeleton. 2001;48:175-89.
15. Blom WM, de Bont HJ, Meijerman I, et al. Remodeling of the actin cytoskeleton of target hepatocytes and NK cells during induction of apoptosis. Cell Motil Cytoskeleton. 2001;49:78-92.
16. Newport J, Forbes DJ. The nucleus: Structure, function and dynamics. Annu Rev Biochem. 1987;56:535-65.
17. Miller M, Park MK, Hanover JA. Nuclear pore complex: Structure, function, and regulation. Physiol Rev. 1991;71:909-49.
18. Nakielny S, Dreyfuss G. Transport of proteins and RNAs in and out of the nucleus. Cell. 1999;99:677-90.
19. DePierre JW, Dallner G. Structural aspects of the membrane of the endoplasmic reticulum. Biochim Biophys Acta. 1975;415:411-72.
20. Sitia R, Meldolesi J. Endoplasmic reticulum: A dynamic patchwork of specialized subregions. Mol Biol Cell. 1993;3:1067-72.
21. Hauri H-P, Schweizer A. Relationship of the ER-Golgi intermediate compartment to endoplasmic reticulum and Golgi apparatus. Curr Opin Cell Biol. 1992;4:600.
22. Altan-Bonnet N, Sougrat R, Lippincott-Schwartz J. Molecular basis for Golgi maintenance and biogenesis. Curr Opin Cell Biol. 2004;16:364-72.
23. deDuve C, Wattiaux R. Functions of lysosomes. Ann Rev Physiol. 1966;28:435-92.
24. Swanson J, Bushnell A, Silverstein SC. Tubular lysosome morphology and distribution within macrophages is dependent on the integrity of cytoplasmic microtubules. Proc Natl Acad. Sci USA. 1987;84:1921-5.
25. Stein M, Zijderhand-Bleekemolen JE, Geuze H, Hasilik A, von Figura K. Mr 46,000 mannose 6-phosphate specific receptor: Its role in targeting of lysosomal enzymes. EMBO J. 1987;6:2677-81.
26. Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;121:1257-69.
27. Dunn KW, Maxfield FR. Delivery of ligands from sorting endosomes to late endosomes occurs by maturation of sorting endosomes. J Cell Biol. 1992;117:301-10.
28. Srere PA. The structure of the mitochondrial inner membrane-matrix compartment. Trends Biochem Sci. 1982;7:375.
29. Ernster L, Kuylenstierna B. Outer membrane of mitochondria. In: Racker E, editor. Membranes of Mitochondria and Chloroplasts. New York: Van Nostrand Reinhold; 1970:172.
30. Ontko JA, Dashti N. Enzymes of mitochondria. In: Altman PI, Katz DD, editors. Cell Biology. Bethesda: Fed Am Soc Exp Biol; 1976:161-179.
31. Hinkle PC, Kumar MA, Resetar A, Harris DL. Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry. 1991;30:3576-82.
32. Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481-90.
33. Racker E, Hortsman LL. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XIII. Structure and function of submitochondrial particles completely resolved with respect to coupling factor. J Biol Chem. 1967;242:2547-51.
34. Walker JE. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. A Rev Biophys. 1992;25:253-324.
35. Trumpower BL. The protonmotive Q cycle-energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J Biol Chem. 1990;265:11409-12.
36. Gould SJ, Raymond GV, Valle D. The peroxisome biogenesis disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:3181-3218.
37. Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135:19-35.
38. Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55-63.
39. Harter C. COP-coated vesicles in intracellular protein transport. FEBS Lett. 1995;369:89-92.
40. Zhao L, Helms JB, Brugger B, et al. Direct and GTP-dependent interaction of ADP ribosylation factor 1 with coatomer subunit beta. Proc Natl Acad. Sci USA. 1997;94:4418-23.
41. Saucan L, Palade GE. Membrane and secretory proteins are transported from the Golgi complex to the sinusoidal plasmalemma of hepatocytes by distinct vesicular carriers. J Cell Biol. 1994;125:733-41.
42. Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37-44.
43. Crowther RA, Pearse BM. Assembly and packing of clathrin into coats. J Cell Biol. 1981;91:790-7.
44. Hemery I, Durand-Schneider AM, Feldmann G., et al. The transcytotic pathway of an apical plasma membrane protein (B10) in hepatocytes is similar to that of IgA and occurs via a tubular pericentriolar compartment. J Cell Sci. 1996;109:1215-27.
45. Stockert RJ. Receptor-mediated endocytosis. In: Arias IM, Boyer JL, Chisari FV, et al, editors. The Liver: Biology and Pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:161-177.
46. Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341-79.
47. Alpini G, McGill JM, Larusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;35:1256-68.
48. Benedetti A, Bassotti C, Rapino K, et al. A morphometric study of the epithelium lining the rat intrahepatic biliary tree. J Hepatol. 1996;24:335-42.
49. Ramadori G, Reider H, Knittel T. Biology and pathobiology of sinusoidal liver cells. In: Tavoloni N, Berk PD, editors. Hepatic Transport and Bile Secretion: Physiology and Pathophysiology. New York: Raven Press; 1993:83-102.
50. Arias IM. The biology of hepatic endothelial fenestrae. In: Schaffner F, Popper H, editors. Progress in Liver Diseases, vol IX. Philadelphia: WB Saunders; 1990:11-26.
51. Wake K, Decker K, Kirn A, et al. Cell biology and kinetics of Kupffer cells in the liver. Int Rev Cytol. 1989;118:173-229.
52. Rogoff TM, Lipsky PE. Role of Kupffer cells in local and systemic immune responses. Gastroenterology. 1981;80:854-60.
53. Sato M, Suzuki S, Senoo H. Hepatic stellate cells: Unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105-12.
54. Senoo H. Structure and function of hepatic stellate cells. Med Electron Microsc. 2004;37:3-15.
55. Hui AY, Friedman SL. Molecular basis of hepatic fibrosis. Expert Rev Mol Med. 2003;14:1-23.
56. Seki E, Minicis SD, Österreicher CH, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med. 2007;13:1324-32.
57. Doherty DG, O’Farrelly C. Innate and adaptive lymphoid cells in the human liver. Immunol Rev. 2000;174:5-20.
58. Wisse E, Luo D, Vermijlen D, et al. On the function of pit cells, the liver-specific natural killer cells. Semin Liver Dis. 1997;17:265-86.
59. Bedossa P, Paradis V. Liver extracellular matrix in health and disease. J Pathol. 2003;200:504-15.
60. Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673-87.
61. Jaeschke H. Cellular adhesion molecules: Regulation and functional significance in the pathogenesis of liver diseases. Am J Physiol. 1997;273:G602-11.
62. Mitaka T. Reconstruction of hepatic organoid by hepatic stem cells. J Hepatobiliary Pancreat Surg. 2002;9:697-703.
63. Greenwel P, Rojkind M. The extracellular matrix of the liver. In: Arias IM, Boyer JL, Chisari FV, et al, editors. The Liver: Biology and Pathobiology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2001:469-473.
64. Steer CJ. Liver regeneration. FASEB J. 1995;9:1396-400.
65. Fausto N. Liver regeneration and repair: Hepatocytes, progenitor cells, and stem cells. Hepatology. 2004;39:1477-87.
66. Taub R. Liver regeneration: From myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836-47.
67. Malik R, Selden C, Hodgson H. The role of non-parenchymal cells in liver growth. Semin Cell Dev Biol. 2002;13:425-31.
68. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60-6.
69. Wyllie AH. Apoptosis and carcinogenesis. Eur J Cell Biol. 1997;73:189-97.
70. Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491-508.
71. Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr Mol Med. 2003;3:527-35.
72. Burt AP, Day CP. Pathophysiology of the liver. In: MacSween RNM, Burt AD, Portmann BC, editors. Pathology of the Liver. 4th ed. London: Churchill Livingstone; 2001:61-106.
73. Cereghini S. Liver-enriched transcription factors and hepatocyte differentiation. FASEB J. 1996;10:267-82.
74. Karpen SJ. Nuclear receptor regulation of hepatic function. J Hepatol. 2002;36:832-50.
75. Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33-45.
76. Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523-9.
77. Bonaficio JS, Lippincott-Schwartz J. Degradation of proteins within the endoplasmic reticulum. Curr Opin Cell Biol. 1991;3:592-600.
78. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717-21.
79. Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:909-10.
80. Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S-237S.
81. Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132-8.
82. Hershko A. The ubiquitin pathway of protein degradation and proteolysis of ubiquitin-protein conjugates. Biochem Soc Trans. 1991;19:726-9.
83. Riezman H, Munn A, Geli MI, Hicke L. Actin-, myosin- and ubiquitin-dependent endocytosis. Experientia. 1996;52:1033-41.
84. Kolling R, Hollenberg CP. The ABC-transporter Ste6 accumulates in the plasma membrane in a ubiquitinated form in endocytosis mutants. EMBO J. 1994;13:3261-71.
85. Seifter S, Englard S. Energy metabolism. In: Arias IM, editor. The Liver: Biology and Pathobiology. 3rd ed. New York: Raven Press; 1994:323-364.
86. Anghel SI, Wahli W. Fat poetry: A kingdom for PPARgamma. Cell Research. 2007;17:486-511.
87. Wang PYT, Caspi L, Lam CKL, et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012-16.
88. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr Rev. 1999;20:649-88.
89. Joseph JW, Koshkin V, Saleh MC, et al. Free fatty acid induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049-56.
90. Felber JP, Golay A. Regulation of nutrient metabolism and energy expenditure. Metabolism. 1995;44:4-9.
91. Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885-909.
92. Pessin JE, Bell GI. Mammalian facilitative glucose transporter family: Structure and molecular regulation. Annu Rev Physiol. 1992;54:911-30.
93. Nordlie RC, Foster JD, Lange AJ. Regulation of glucose production by the liver. Annu Rev Nutr. 1999;19:379-406.
94. Van Schaftingen E. Glycolysis revisited. Diabetologia. 1993;36:581-8.
95. Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647-52.
96. Van Schaftingen E, Detheux M, Veiga da Cunha M. Short-term control of glucokinase activity: Role of a regulatory protein. FASEB J. 1994;8:414-19.
97. Burchell A. Hepatic microsomal glucose transport. Biochem Soc Trans. 1994;22:658-63.
98. Vaulont S, Kahn A. Transcriptional control of metabolic regulation genes by carbohydrates. FASEB J. 1994;8:28-35.
99. McGarry JD, Kuwajima M, Newgard CB, et al. From dietary glucose to liver glycogen: The full circle round. Annu Rev Nutr. 1987;7:51-73.
100. Foster DW. Banting lecture 1984: From glycogen to ketones—and back. Diabetes. 1984;33:1188-99.
101. Nolte W, Hartmann H, Ramadori G. Glucose metabolism and liver cirrhosis. Exp Clin Endocrinol Diabetes. 1995;103:63-74.
102. Coppack SW, Jensen MD, Miles JM. In vivo regulation of lipolysis in humans. J Lipid Res. 1994;35:177-93.
103. Kadereit B, Kumar P, Wang W-J, et al. Evolutionarily conserved gene family important for fat storage. Proc Nat Acad Sci U S A. 2008;105:94-9.
104. Osmundsen H, Bremer J, Pedersen JI. Metabolic aspects of peroxisomal beta-oxidation. Biochim Biophys Acta. 1991;1085:141-58.
105. Kim KH, Tae HJ. Pattern and regulation of acetyl-CoA carboxylase gene expression. J Nutr. 1994;124:1273S.
106. Kerner J, Hoppel D. Fatty acid import into mitochondria. Biochim Biophys Acta. 2001;1486:1-17.
107. Bremer J. The role of carnitine in intracellular metabolism. J Clin Chem Clin Biochem. 1990;28:297-301.
108. Lieber CS. Biochemical factors in alcoholic liver disease. Semin Liver Dis. 1993;13:136-53.
109. Miller JP. Liver disease, alcohol and lipoprotein metabolism. In: Zakim D, Boyer TD, editors. Hepatology: A Textbook of Liver Diseases. 4th ed. Philadelphia: WB Saunders; 2003:127.
110. Oram JF. Tangier disease and ABCA1. Biochim Biophys Acta. 2000;1529:321-30.
111. Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816-19.
112. Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. 1999;19:472-84.
113. Mahley RW, Ji ZS. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1-16.
114. Saunders AM, Trowers MK, Shimkets RA, et al. The role of apolipoprotein E in Alzheimer’s disease: Pharmacogenomic target selection. Biochim Biophys Acta. 2000;1502:85-94.
115. Tall AR. Plasma high-density lipoproteins: Metabolism and relationship to atherogenesis. J Clin Invest. 1990;86:379-84.
116. Bruce C, Chouinard RA, Tall AR. Plasma lipid transfer proteins, high-density lipoproteins, and reverse cholesterol transport. Annu Rev Nutr. 1998;18:297-330.
117. Hussain MM. A proposed model for the assembly of chylomicrons. Atherosclerosis. 2000;148:1-15.
118. Yeaman SJ. Hormone-sensitive lipase—a multipurpose enzyme in lipid metabolism. Biochim Biophys Acta. 1990;1052:128-32.
119. Meier PJ. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. Am J Physiol. 1995;269:G801-12.
120. Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731-44.
121. Yao Z, McLeod RS. Synthesis and secretion of hepatic apolipoprotein B-containing lipoproteins. Biochim Biophys Acta. 1994;1212:152-66.
122. Davis RA. Cell and molecular biology of the assembly and secretion of apolipoprotein B-containing lipoproteins by the liver. Biochim Biophys Acta. 1999;1440:1-31.
123. Krieger M. Charting the fate of the “good cholesterol”: Identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523-58.
124. Hussain MM, Strickland DK, Bakillah A. The mammalian low-density lipoprotein receptor family. Annu Rev Nutr. 1999;19:141-72.
125. Willnow TE. The low-density lipoprotein receptor gene family: Multiple roles in lipid metabolism. J Mol Med. 1999;77:306-15.
126. Rohlmann A, Gotthardt M, Hammer RE, et al. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689-95.
127. Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211-28.
128. Kozarsky KF, Donahee MH, Rigotti A, et al. Overexpression of the HDL receptor SR-B1 alters plasma HDL and bile cholesterol levels. Nature. 1997;387:414-17.
129. Rigotti A, Trigatti BL, Penman M, et al. A targeted mutation in the mutine gene encoding the high density lipoprotein (HDL) metabolism. Proc Natl Acad. Sci USA. 1997;94:12610-15.
130. Cicognani C, Malavolti M, Morselli-Labate AM, et al. Serum lipid and lipoprotein patterns in patients with liver cirrhosis and chronic active hepatitis. Arch Intern Med. 1997;157:792-6.
131. Miller JP. Dyslipoproteinaemia of liver disease. Baillieres Clin Endocrinol Metab. 1990;4:807-32.
132. Oude Elferink RP, Ottenhoff R, van Marle J, et al. Class III P-glycoproteins mediate the formation of lipoprotein X in the mouse. J Clin Invest. 1998;102:1749-57.