Liver Pathology in Pregnancy
Kamran Badizadegan
Jacqueline L. Wolf
Introduction
Pregnancy is an altered physiologic state designed to support the developing fetus, and gastrointestinal complaints are common during pregnancy. Although symptoms caused by de novo abnormalities of the liver occur infrequently, they require prompt diagnosis and treatment to avoid the potentially high rates of maternal and fetal morbidity and mortality associated with them.
This chapter focuses on the pathophysiology and diagnosis of liver diseases that are unique to pregnancy. In most cases, the panoply of diseases discussed constitutes the differential diagnosis. In practice, some of the most frequent causes of liver disease in pregnancy are common disorders such as viral hepatitis and gallstone disease (Table 53.1), and the differential diagnosis must be broadened to include these abnormalities.
Table 53.1
Hepatobiliary Causes of Abnormal Liver Function Test Results in Pregnancy
| First Trimester* | Second Trimester | Third Trimester |
| Hyperemesis gravidarum | Hyperemesis gravidarum | Gallstones |
| Gallstones | Gallstones | Hepatitis |
| Hepatitis | Hepatitis | Intrahepatic cholestasis of pregnancy |
| Intrahepatic cholestasis of pregnancy | Intrahepatic cholestasis of pregnancy | Preeclampsia or eclampsia |
| Preeclampsia or eclampsia | HELLP syndrome | |
| HELLP syndrome | Acute fatty liver of pregnancy | |
| Hepatic rupture |
* Conditions are ordered by prevalence in each trimester.
HELLP, Hemolysis, elevated liver enzymes, and low platelets.
Laboratory and Histologic Changes of the Liver in Pregnancy
Hepatic histopathology in pregnancy is often nonspecific, and knowledge of the clinical history and physical signs and symptoms is essential for the diagnosis of liver disease. Evaluation of liver disease is further complicated by normal physiologic changes in liver function test results that are a function of gestational age. Despite these well-known physiologic alterations, most clinical laboratories use data from a normal adult population as reference ranges, leaving the correct evaluation of liver function abnormalities in the pregnant patient to the subjective judgment of clinicians and pathologists. Some important changes are summarized in Table 53.2, but the original literature should be consulted for a detailed discussion of the data and controversies regarding physiologic changes that affect liver function test results during normal and uncomplicated pregnancy.1–9
Table 53.2
Physiologic Changes in Common Liver Function Test Results during Pregnancy
| Test | Pattern of Change (max) | Time of Maximum Change |
| Albumin | ↓ (60%) | Second trimester |
| γ-Globulin | No change or ↓ (10%) | First to second trimester |
| Alkaline phosphatase (total) | ↑ (400%) | Third trimester |
| Aminotransferases | No change | NA |
| γ-Glutamyltranspeptidase | No change | NA |
| Bilirubin | No change | NA |
| Prothrombin time | No change | NA |
| Cholesterol | No change or ↑ (200%) | Third trimester |
NA, Not applicable; ↓, decreased; ↑, increased.
Compared with age-matched, nonpregnant women, the total serum protein and albumin levels are significantly lower during all three trimesters, and the concentration of serum albumin can decrease by as much as 60% during the second trimester. The physiologic basis of this change is a subject of debate, although hemodilution during pregnancy is thought to play a role. Among other serum proteins, the levels of coagulation factors VII to X are higher during pregnancy, and fibrinogen levels may increase by as much as 50%. Concentrations of α- and β-globulins are also slightly higher than normal, although γ-globulin levels may decrease.
Serum alkaline phosphatase activity rises during pregnancy and is significantly higher during the third trimester, although most of this activity originates from the placenta. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), γ-glutamyltranspeptidase (GGT), 5′-nucleotidase, bilirubin, and total bile acids and the prothrombin time remain within reference ranges during normal pregnancy. Within the reference range, however, average ALT levels may be slightly higher during the second trimester, whereas average GGT levels appear to be lower during the second and third trimesters compared with nonpregnant controls. Serum 5′-nucleotidase levels are higher during the second and third trimesters compared with the first trimester and compared with nonpregnant control subjects, but average increases are small (10% to 25%), and all values are well within the normal reference range. Total, free, and conjugated bilirubin levels in all trimesters may be lower than in nonpregnant control patients, with the most significant changes (as much as 50%) occurring in total and free bilirubin levels in the second and third trimesters. Serum triglyceride levels are higher in pregnancy, and cholesterol levels may increase by as much as 200% during the third trimester.
The histologic appearance of the liver in uncomplicated pregnancy is essentially normal. Minor nonspecific changes have been described, including mild nuclear pleomorphism, increased glycogen, mild steatosis, mild portal inflammation, and reactive Kupffer cells.10–14
The onset of maternal symptoms in relation to the trimester of pregnancy can help in the differential diagnosis of hepatic pathology (see Table 53.1). Severe nausea and vomiting during the first trimester are key clinical signs of hyperemesis gravidarum, a disease with a relatively benign course and outcome. Later onset of nausea and vomiting suggests preeclampsia when accompanied by headache and peripheral edema or hepatic rupture when accompanied by abdominal pain with or without systemic hypotension. Pruritus in the third trimester, particularly of the palms and soles, is characteristic of intrahepatic cholestasis of pregnancy and typically precedes the clinical manifestation of jaundice. Right upper quadrant and midabdominal pain in the third trimester may indicate acute fatty liver of pregnancy or hepatic rupture, both of which require immediate clinical intervention. Signs and symptoms of acute and chronic viral hepatitis and extrahepatic biliary disease are the same in pregnant women as in nonpregnant women, and their onset is not generally correlated with the gestational age.
Intrahepatic Cholestasis of Pregnancy
With an estimated average incidence of 0.1% to 2%, intrahepatic cholestasis of pregnancy (ICP) is one of the most important causes of jaundice in pregnancy.15–18 Geographic, ethnic, and familial clusterings are prominent in ICP and must be considered in the diagnostic evaluation.
The average incidence of ICP in Europe and the United States varies from 0.1% to 1.5%, whereas the estimated incidence in Chile and Bolivia is higher at 1.5% to 4%. However, the declining incidence of ICP in Chile and Bolivia suggests an environmental contribution to the disease.17,19 For example, the incidence among the Araucanian population in Chile decreased from 27% during 1974 through 1975 to 6.5% during 1988 through 1990.15,17,20
Specific geographic regions have significant ethnic variations. In Chile and Bolivia, the incidence of ICP is significantly lower among white populations than among the native Araucanian (Chile) or Aimara (Bolivia) Indians.21,22 Albeit less striking, similar differences have been described in the United States. The prevalence of ICP in northern California is higher among the Latina population than among whites.23
Studies have suggested that ICP may represent the common clinical manifestation of a pathologically heterogeneous group of liver and biliary disorders.16,24,25 However, no diagnostic criteria have been proposed for specific subclassification of the group of diseases that clinically manifest as ICP.
Clinical Features
ICP is characterized by the triad of pruritus, abnormal liver function test results (especially fasting serum bile acid levels greater than 10 µmol/L), and spontaneous resolution of signs and symptoms after delivery or pregnancy termination. ICP occurs most commonly in the third trimester, although it can manifest any time during pregnancy.
Pruritus, which is the typical presenting symptom, tends to be more severe at night and most often affects the palms of hands, the soles of feet, and the trunk. Jaundice occurs in 10% to 25% of cases, making ICP the second leading cause of jaundice in pregnancy (after viral hepatitis). When jaundice occurs, it typically follows the onset of pruritus by 2 to 4 weeks. Other symptoms, such as dark urine, light stools or steatorrhea, nausea, vomiting, and abdominal discomfort, may occur but are less specific.15,20,26 Symptoms of ICP typically persist for the duration of pregnancy, but in most cases, they resolve within 1 to 3 weeks after delivery or pregnancy termination.
Clinical laboratory findings in ICP (Box 53.1) include a mildly elevated serum bilirubin level (mostly direct), mildly elevated levels of serum aminotransferases, and most importantly, markedly elevated levels of fasting serum bile acids.15,27 The concentration of cholic acid that is free or conjugated to taurine or glycine progressively rises from 20 to 40 weeks’ gestation in women with ICP. The ratio of cholic acid to chenodeoxycholic acid is significantly higher in women with ICP (>1.5 : 1) than in women with a normal pregnancy.28 Elevated GGT levels may occur in as many as one half of cases, providing a clue to the cause of the condition.29–31 Dyslipidemia with elevated levels of total cholesterol, low-density lipoprotein cholesterol, and apolipoprotein B-100 has also been observed.32
Except for a mildly elevated risk (<8%) of gallstone disease, the maternal outcome in ICP is generally favorable.15,18,33,34 However, recent cohort studies have found an association of ICP with other liver disorders such as chronic hepatitis C and cirrhosis.25,34a In addition, women with severe ICP may be at increased risk of preterm delivery relative to a healthy pregnancy comparison group (25% versus 6.5%, with adjusted odds ratio of 5.39).34b The fetal complications of ICP are more serious and consist of prematurity (19% to 60%), fetal distress (22% to 33%), meconium staining (9% to 24%), and perinatal death (1% to 2%).34,35
Observations have provided evidence for a direct link between maternal fasting bile acid levels and maternal-fetal complications of ICP. In a prospective cohort study of almost 45,000 pregnancies with 693 cases of ICP, no increase in fetal risk was detected when maternal serum bile acid levels were less than 40 µmol/L.18,36 These observations suggest expectant management of mild ICP (serum bile acids < 40 µmol/L). There are no specific guidelines for the management of more severe ICP, although some studies have shown the effectiveness of ursodeoxycholic acid in treating the pruritus and normalizing liver function test levels.36–40,40a In randomized, clinical trials, ursodeoxycholic acid was more effective than cholestyramine or dexamethasone in providing symptomatic relief for patients with ICP, although the dexamethasone trial was for only 1 week versus 3 weeks for the ursodeoxycholic acid trial.37,38 The benefit of S-adenosyl-l-methionine in treating pruritus, reduction of total bile acid levels, and normalization of liver function test results has been mixed in trials.41,42
Pathologic Features
Because a diagnosis of ICP is typically made according to clinical criteria, liver histopathology in ICP has not been extensively reported. Findings are thought to be subtle and consist primarily of hepatocellular bile and canalicular bile plugs, occurring predominantly in a pericentral distribution with minimal or no hepatocellular necrosis and minimal portal inflammation (Fig. 53.1).43–45 The histologic differential diagnosis of intrahepatic cholestasis is broad and includes other common entities such as drug-induced hepatocellular injury and early extrahepatic biliary obstruction. Nevertheless, the combination of intrahepatic cholestasis with clinical features (i.e., pruritus and elevated levels of serum bile acids) virtually limits the diagnosis to ICP.
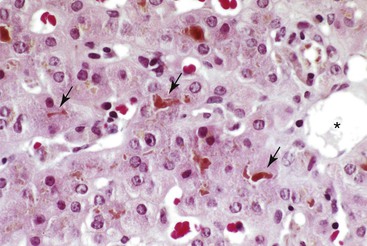
One study showed an association of ICP with abnormalities of the placenta, including syncytial knots, focally thickened amniotic basement membranes, small chorionic villi for gestational age with dense fibrotic stroma, and crowding and congestion of the villi. These abnormalities may increase the incidence of fetal demise.46
Pathogenesis
The pathogenesis of ICP is not fully characterized. Several lines of evidence point to altered metabolism of steroid hormones and bile acids, and various mechanistic roles for disruption of bile acid transport into and out of the hepatocyte by estriol, progesterone, and their intrahepatocellular conjugates have been proposed.17,26,47 Familial clustering and increased incidence in the first-degree relatives of patients with ICP48–51 and linkage to human leukocyte antigens (HLAs)21,49,52 indicate one or more genetic factors. Recently it has been suggested that immune dysregulation resulting in disturbed placental bile acid and serum lipids transportation plays a key role.52a
Environmental factors such as dietary selenium53,54 and seasonal variations in incidence55 suggest one or more exogenous factors in the pathogenesis of ICP. Demonstration of increased intestinal permeability in patients with ICP during and after pregnancy24 provides evidence that the hepatic pathogenesis of ICP may be under the control or influence of extrahepatic or exogenous factors. Collectively, these somewhat disjointed observations and hypotheses about the pathogenesis of ICP and its associated risk factors suggest that ICP may represent the end result of a heterogeneous group of pregnancy-associated hepatic insults rather than a unique pathophysiologic entity.
The most specific data come from genetic studies of a subset of patients with ICP in whom the disease is associated with elevated serum GGT activity. Two pedigrees were initially reported, with ICP in the mothers of children who were born with the autosomal recessive form of progressive familial intrahepatic cholestasis (PFIC) and elevated serum GGT activity, a disease commonly referred to as type 3 PFIC.56,57 The affected children had homozygous mutations in the hepatocellular phospholipid transporter ABCB4 gene (formerly referred to as multidrug resistance 3, or MDR3), whereas ICP developed during pregnancy in mothers who were heterozygous for ABCB4. ABCB4 is a class III multidrug-resistance P-glycoprotein that mediates translocation (i.e., flipping) of phosphatidylcholine (lecithin) across the bile canalicular membrane of hepatocytes.
To better characterize the pathogenic role of ABCB4 in the development of ICP, Dixon and associates investigated eight women with ICP and increased serum GGT activity who had no personal or family history of PFIC.58 DNA sequence analysis revealed a heterozygous missense mutation in ABCB4, resulting in the expression of a nonfunctional protein at the cell surface in one of eight patients. Several subsequent reports on the association between ABCB4 and ICP provide further evidence for a defect in this canalicular transporter protein in the pathogenesis of ICP.59–66 As many as 16% of all patients with ICP are thought to have the disease as a consequence of mutations in the ABCB4 gene, and many different mutations of the gene have been found.30,67,67a,67b
In addition to the compelling data regarding the role of ABCB4 in the pathogenesis of a subset of ICP cases, other cases have been associated with benign, recurrent intrahepatic cholestasis68 (BRIC) (see Fig. 53.1) and with mutations in the same gene.69 BRIC, which is genetically associated with a mutation in the same region of chromosome 18 as type 1 PFIC, is an intrahepatic cholestatic disease in which the affected patients have normal serum GGT activity.68,69 This sharp biochemical contrast with ABCB4-associated cases of ICP points to alternative mechanistic pathways for another subset of women with ICP.
The ABCB11 gene, which is associated with PFIC type 2, may also contribute to ICP. Mutations in the bile salt export pump, which is the product of the ABCB11 gene, have been identified in ICP.29,67a,67b,70,71 Variations in the farnesoid X receptor, a key transcription factor driving the expression of ABCB11, were significantly associated with ICP and markedly reduced the capacity to activate the ABCB11 promoter in vitro.29,72
It is not surprising that different defects in transport across the hepatocyte canalicular membrane can lead to similar clinical phenotypes, which are collectively recognized as ICP. Fundamentally, ICP can result from any disruption in the steady state between the uptake of bile salts into the hepatocytes and transport of bile salts across the canalicular membrane into bile. Estrogens, progesterone, and their conjugates disrupt the steady state by interfering with bile acid transporters at the basolateral membrane and inhibiting efficient bile acid transport across the canalicular membrane. In compromised hosts, such as heterozygous mothers with ABCB4 mutations, the added insult from pregnancy-associated hormones is sufficient to tilt the balance toward a cholestatic disease. As evidenced by the discovery of an association between specific variants of the multidrug-resistance–associated gene (ABCC2) and ICP,70 future research will undoubtedly reveal other molecular pathways and canalicular transporters that can result in ICP through similar or related pathways.
Acute Fatty Liver of Pregnancy
Acute fatty liver of pregnancy (AFLP) is a serious and potentially fatal complication for the pregnant mother and fetus. AFLP occurs in 1 of 4000 to 20,000 pregnancies; a frequency of 1 in 20,000 was found in a population study of pregnant women in the United Kingdom.73–75 The disease is classically associated with first pregnancies, twin gestations, and a male fetus. AFLP usually occurs late in the third trimester (>30 weeks’ gestation) or in the immediate postpartum period,73–79 although rare exceptions manifesting as early as 22 weeks’ gestation have been reported.80–82
Clinical Features
The initial clinical presentation of AFLP is somewhat vague and includes headache, abdominal pain, nausea, vomiting, and a variety of other nonspecific symptoms. Approximately 80% of women have these prodromal symptoms. At least 70% of all patients have right upper quadrant or epigastric pain accompanied by nausea and vomiting.83–86 Prodromal symptoms are typically followed by jaundice as the disease progresses. Progressive and severe hepatic failure accompanied by coagulopathy and encephalopathy occurs rapidly after the onset of jaundice, and it is reported in more than 50% of cases within 1 to 2 weeks of the onset of jaundice. If untreated, patients may rapidly deteriorate, with gastrointestinal bleeding, seizures, coma, and renal failure with acute tubular necrosis.
AFLP may be associated with preeclampsia in 20% to 40% of patients.76,87,88 In these circumstances, the presenting signs and symptoms include those of pregnancy-induced hypertensive disorders (discussed later). A rare association between AFLP and ICP has been reported in which the patient had pruritus at presentation, which is more common for ICP than AFLP.89
Because early diagnosis and prompt treatment of AFLP are essential to maternal and fetal well-being, liver function test parameters must be measured immediately in any pregnant woman past 22 to 24 weeks’ gestation who has any of the aforementioned symptoms or signs. The liver function test results for AFLP typically suggest mild to moderate hepatocellular damage with mild cholestasis (Box 53.2). Serum aminotransferase levels usually are elevated in AFLP, although rarely to the extent observed in acute viral hepatitis. The bilirubin level is normal early in the course, but it rises if the pregnancy is not immediately terminated. Alkaline phosphatase levels are also elevated, but distinguishing hepatic from placental isoenzymes may not be practical or fruitful. The peripheral blood analysis may show leukocytosis and thrombocytopenia,90 and disseminated intravascular coagulation is relatively common.91,92 Blood urea nitrogen and serum creatinine levels may be elevated, but uric acid levels are disproportionately high, making them diagnostically valuable. Blood glucose levels are typically low, and clinically significant hypoglycemia may occur.
Historically, liver biopsy has been the gold standard for the diagnosis of AFLP, but coagulopathy often prevents its application in the acute clinical setting. Fatty infiltration of the liver can be easily assessed by noninvasive imaging methods such as ultrasonography or magnetic resonance imaging (MRI). The Swansea criteria, which use the clinical symptoms and laboratory findings of women with liver disease in pregnancy, constitute a reliable indicator of AFLP, with or without a liver biopsy (Box 53.3).75,93 In one study, application of the Swansea criteria resulted in 100% sensitivity, 57% specificity, an 85% positive predictive value, and a 100% negative predictive value for diffuse or perivenular microvesicular steatosis on liver biopsy.94
Regardless of the cause or type of presentation, the mainstay of therapy for AFLP is immediate delivery and supportive care. Liver transplantation and artificial liver support have been tried as alternative therapies,95–97 but their clinical role remains experimental. Despite all clinical efforts, AFLP continues to be a serious complication of pregnancy with maternal or fetal death occurring in 1% to 20% of all cases.75,98,98a
Pathologic Features
AFLP is commonly diagnosed without a liver biopsy based on clinical presentation and laboratory data. Nevertheless, microvesicular steatosis of hepatocytes is considered the diagnostic hallmark of AFLP. Classically, steatosis involves the pericentral zone and spares the periportal hepatocytes, although periportal involvement may be seen.79,90 In most cases, the fat droplets are large enough and their location well enough preserved to produce readily recognizable vacuolar change on routine sections with hematoxylin and eosin (H&E) staining (Fig. 53.2, A). Occasionally, individual fat droplets may be too small to be resolved by routine light microscopy or too poorly preserved to result in a classic vacuolar pattern on H&E-stained sections (see Fig. 53.2, B and C). In these circumstances, hepatocytes may look essentially normal, be somewhat dilated, or exhibit diffuse cytoplasmic ballooning not readily distinguishable from ballooning degeneration or other forms of acute hepatocellular damage. A portion of the liver biopsy specimen obtained for clinical suspicion of AFLP must be processed as a frozen section for oil red O or Sudan black staining for fat or be submitted for electron microscopy.
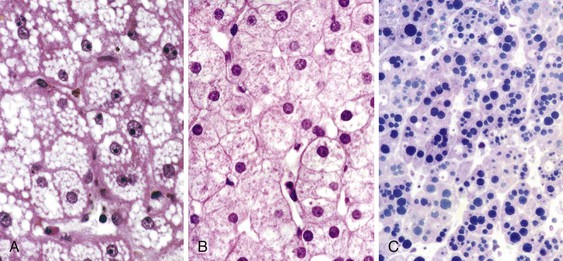
Although fatty change is considered the diagnostic histologic hallmark of AFLP, a host of other microscopic abnormalities were reported in a detailed study of 35 cases by Rolfes and Ishak.79 Hypertrophied Kupffer cells containing lipid or lipofuscin were prominent in areas of fatty change in most cases. Evidence of intrahepatic cholestasis, including bile canalicular plugs and acute cholangiolitis, was seen in two thirds of cases. Significant mononuclear lobular inflammation (comparable with acute viral hepatitis) and inflammation of the central veins were identified in 25% of cases, and 75% showed evidence of extramedullary hematopoiesis with prominent megakaryocytes and cells of erythroid lineage. Most notable by its absence in this large series of cases was sinusoidal fibrin deposition. Despite frequent clinical signs and symptoms of pregnancy-induced hypertensive disorders in patients with AFLP,76,87,88 the absence of fibrin deposition was considered evidence of a lack of histologic overlap between these two entities.79
Although fatty infiltration of the liver is an extremely sensitive diagnostic marker for AFLP, it is nonspecific. The histopathologic differential diagnosis of AFLP is therefore broad and includes essentially all toxic, metabolic, and drug-induced conditions that may lead to microvesicular steatosis of hepatocytes. Definitive diagnosis of AFLP can therefore be made only in conjunction with the appropriate clinical signs and symptoms.
Pathogenesis
Significant progress has been made in understanding the pathogenesis of AFLP. A strong association between fetal fatty acid oxidation (FAO) disorders and maternal AFLP has been established,99–107 suggesting that AFLP is a disorder of mitochondrial fatty acid β-oxidation (see Fig. 53.2, B and C).
Mitochondrial β-oxidation of fatty acids is a critical step in intermediary metabolism in the liver. FAO is a source of energy for the liver under physiologic stress and results in production of various metabolic intermediates that can be used as a source of energy by other vital organs such as the brain. One of the key enzymes in mitochondrial fatty acid β-oxidation is long-chain 3-hydroxyacyl-coenzyme A (CoA) dehydrogenase (LCHAD), which catalyzes the third step in β-oxidation of long-chain fatty acids on the inner mitochondrial membrane. LCHAD activity occurs on the C-terminal portion of the α subunit of the microsomal triglyceride transfer protein (MTTP, also known as trifunctional protein [TFP]). MTTP also contains the active sites of long-chain 2,3-enoyl-CoA hydratase and long-chain 3-ketoacyl-CoA thiolase, catalyzing the second and fourth steps in fatty acid β-oxidation, respectively.
A recessively inherited defect in LCHAD in the infant is responsible for most of the occurrences of AFLP in the mother. Most abnormalities result from a 1528G→C mutation in exon 15 of the α subunit of MTTP, which results in the exchange of glutamic acid for glutamine at amino acid 474. Although MTTP deficiencies have emerged as important causes of metabolic disease in children, only one mutation in the α subunit of MTTP resulting in abnormal LCHAD activity has been associated with maternal liver disease in pregnancy.100–102,105,108–113
In addition to MTTP mutations with LCHAD deficiency, other components of mitochondrial FAO have been associated with maternal disease in pregnancy. Fetal short-chain acyl-CoA dehydrogenase deficiency and fetal carnitine palmitoyltransferase deficiency were each reported in association with maternal AFLP several years ago (Fig. 53.3).114,115 Santos and colleagues provided the first description of a normal fetus with a maternal FAO defect in medium-chain acyl-CoA dehydrogenase resulting in AFLP in the late third trimester.104 In a case-control study comparing fetal oxidation defects with the occurrence of maternal liver disease during pregnancy, two cases of medium chain acyl-CoA dehydrogenase deficiency were associated with AFLP. Remarkably, the maternal liver disease occurring with FAO in the children also included preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets). The risk of liver disease developing in a mother in pregnancy if the child had LCHAD or short- and medium-chain defects was, respectively, 50 times or 12 times more likely than in control patients. Collectively, these results firmly establish a mechanistic association between FAO and AFLP.
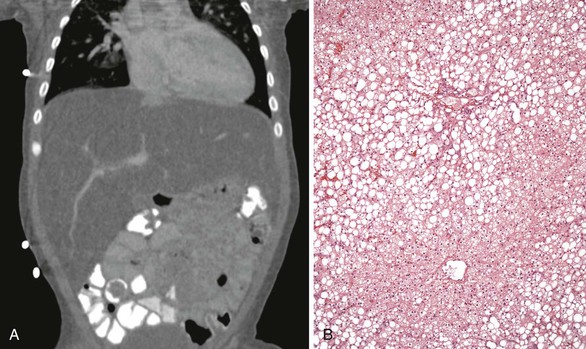
The precise mechanisms through which LCHAD or other FAO deficiencies result in fatty liver or hepatic failure are unknown. It has been postulated that the increasing metabolic demands of the third trimester in a compromised host result in excessive metabolic stress that cannot be handled by the heterozygous mother’s deficient metabolism.105
The placenta may be an important factor in the pathophysiology. The genetic makeup of the placenta is identical to that of the fetus. Natarajan and colleagues found that the placentas at birth in mothers with AFLP showed oxidative stress in the mitochondria and peroxisomes and had compromised mitochondrial function compared with controls.106 The patient’s serum also showed elevation of oxidative and nitrosative stress markers with decreased antioxidant levels. The placentas and sera showed increased levels of arachidonic acid, which caused mitochondrial damage in the Chang liver cell line and increased lipid accumulation as identified by Nile red staining.106 This potential mechanism has many similarities to various forms of ICP in which normally masked deficiencies in bile canalicular transporters or their associated proteins are manifested only during the altered physiologic state of pregnancy.
Preeclampsia and Eclampsia
Preeclampsia is a systemic disorder consisting of hypertension of at least 140 mm Hg systolic or at least 90 mm Hg diastolic on at least two occasions 4 to 6 hours apart after the 20th week of gestation and proteinuria of 0.3 g or more every 24 hours, with or without other associated symptoms. Preeclampsia is characterized by systemic endothelial cell activation and an exaggerated inflammatory response.
Preeclampsia occurs in approximately 2% to 8% of all pregnancies.116–121 When accompanied by new-onset grand mal seizure in a patient without a preexisting brain abnormality, the condition is known as eclampsia.122 Progression to eclampsia is rare, occurring in only 0.1% to 0.2% of all pregnancies, but when it happens, it is associated with significant maternal and fetal morbidity and mortality. In addition to a first pregnancy, which is the most common risk factor for preeclampsia (4.1% for first versus 1.7% for later pregnancies),123 many other clinical and social conditions have been associated with the risk of preeclampsia or eclampsia (Box 53.4).
Clinical Features
Preeclampsia usually manifests after 20 weeks’ gestation and is most common near term. Severe cases are characterized by blood pressure of 160/110 mm Hg or higher and proteinuria of greater than 5 g/day, with or without multiorgan involvement such as oliguria of less than 400 mL/day, thrombocytopenia, and severe central nervous system symptoms, such as mental status changes, blurred vision, or headache.124 The liver is a nonspecific target organ, and liver function test results may include increased levels of aminotransferases, which may be accompanied by a mild increase in serum bilirubin, alkaline phosphatase, and uric acid levels (Box 53.5). Serum heat shock protein 70 levels show significant correlation with elevation of AST (not ALT), lactate dehydrogenase, and C-reactive protein levels.124 Clinical and laboratory findings often include moderate thrombocytopenia and disseminated intravascular coagulation with evidence of microangiopathic hemolysis.
Untreated preeclampsia can progress to hypertensive crisis with life-threatening renal failure. Progression to eclampsia is heralded by seizures, and coma eventually ensues. Maternal and fetal morbidity and mortality correlate with the severity of disease, the time of onset of the condition during the pregnancy (before 34 or after 36 weeks’ gestation), multiple gestations, preexisting maternal disease, and clinical management. The overall maternal mortality rate in developed countries is as high as 1.8%, with more than 80% of cases caused by complications of the central nervous system and the remainder resulting from catastrophic hepatic complications such as hepatic rupture and fulminant failure. The highest risk of death is among older women, women without prenatal care, and women with preeclampsia before 28 weeks’ gestation. Patients with severe preeclampsia or eclampsia have similar risks of developing intravascular coagulation (8%), HELLP syndrome (10% to 15%), and liver hematoma (1%).125
Fetal complications are also common and include increased risk of abruptio placentae, prematurity, and intrauterine fetal growth retardation.125 Reduced risk of preeclampsia has been achieved with low-dose aspirin treatment at 16 weeks or earlier but not at a later gestational age.126 Trials of diet, vitamin C, vitamin E, vitamin D, heparin, diuretics, and antihypertensive medications to prevent preeclampsia have been disappointing.120,127 Delivery is the treatment of choice for mild preeclampsia that develops after 34 weeks’ gestation, for severe preeclampsia after 34 weeks’ gestation (controversial before 34 weeks), and for eclampsia.125,128–136
Management of mild preeclampsia remote from term is often expectant management. However, early identification of high-risk mothers is key because aspirin can be used before 16 weeks’ gestation. The long-term sequelae of preeclampsia affect both mother and infant.120 Women with pregnancy complicated by preeclampsia have a subsequent increased risk of preeclampsia in future pregnancies,136a as well as hypertension, type 2 diabetes mellitus, and stroke.129–131 Offspring of women with preeclampsia also have increased blood pressure and double the risk of stroke in later life.120,133 Male children born to mothers with preeclampsia may have alterations in neonatal microvascular adaptation after birth.134 Being born preterm or with intrauterine growth retardation because of preeclampsia or other causes is associated with an increased risk for gestational diabetes and preeclampsia in adulthood.135
Pathologic Features
Hepatic involvement in preeclampsia and eclampsia is primarily characterized by patchy parenchymal and subcapsular hemorrhage. Microscopically, the periportal zone is preferentially affected and may reveal a combination of fibrin deposition, hemorrhage, and hepatocellular necrosis (Fig. 53.4).13,44,137,138 Thrombi and evidence of endothelial damage may be seen in the branches of the hepatic arteries and less commonly in the portal veins. In practice, the diagnosis of preeclampsia or eclampsia is always made clinically, and the classic pathologic changes are seen only in rare instances of maternal death due to severe disease.
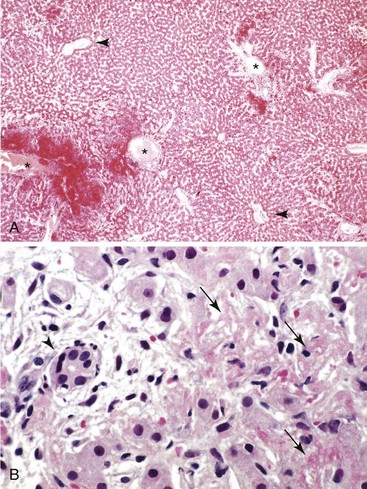
Liver biopsies may be done (but rarely are) for patients with mild or clinically indeterminate liver disease. In these circumstances, the histologic finding may be nonspecific and limited to mild and focal portal or periportal abnormalities. Because of the focal nature of the hepatic involvement, the absence of specific findings on a liver biopsy should not be used to exclude a diagnosis of preeclampsia. These findings are not specific to hypertensive disorders, and an almost identical morphology may be seen in other conditions of altered hemostasis, such as a hypercoagulable state (see Fig. 53.4, B).
Pathogenesis
The placenta plays a key role in the pathogenesis of preeclampsia or eclampsia. Occurrence of the disease in molar pregnancies in which there is no fetal development suggests a required and sufficient role for the placenta in the pathogenesis of preeclampsia, and rapid disappearance of the disease after delivery provides evidence for a placenta-derived factor in the maintenance of maternal symptoms. The clinical manifestations of preeclampsia indicate systemic endothelial dysfunction in the mother.
The pathogenesis of preeclampsia is thought to constitute a two-stage model in which there is an abnormal placentation in early pregnancy followed by events that directly lead to endothelial activation in the third trimester. In the first stage, there is inadequate spiral artery remodeling in the placenta. This condition may result in placental hypoxia or an abnormal flow in the spiral arteries. Because of this hemodynamic abnormality, the placenta is physiologically stressed, resulting in a systemic inflammatory response.139,140 Several genetic predispositions have been suggested to underlie these processes, but a unifying genetic profile has not emerged. It has been argued that one or more placenta-derived circulating factors may be directly responsible for development of preeclampsia.141 Serologic markers of endothelial activation can be seen in women with preeclampsia; the appearance of serologic markers precedes clinically evident disease; and their disappearance accompanies resolution of the disease.141–145
Scientific progress has significantly advanced understanding of the role of placenta-derived factors in the pathophysiology of preeclampsia.142–146 High levels of placenta-derived, soluble FMS-related tyrosine kinase 1 (sFLT1), also known as soluble vascular endothelial growth factor receptor 1 (VEGFR1), herald the onset of clinical symptoms in women with preeclampsia, and circulating levels of sFLT1 correlate with the severity of preeclampsia and proximity to the onset of hypertension or proteinuria.143–146 sFLT1 acts as an antagonist for VEGF and placental growth factor (PGF). The resulting decreased levels of VEGF and PGF produce endothelial dysfunction.120 These conclusions are supported by the fact that overexpression of sFLT1 in pregnant rats results in preeclampsia147 and that antiangiogenic therapy with VEGF in patients with cancer has been associated with preeclampsia-like complications of hypertension and proteinuria.148 The physiological effects of endothelial dysfunction are further magnified by activation of the renin-angiotensin system (RAS) and increased serum levels of angiotensin type II receptor autoantibodies (AT1-AA), resulting in hypertension and proteinuria.148a,148b
The central role of soluble vascular regulatory factors in the pathogenesis of preeclampsia is further advanced by the discovery of the role of endoglin.143,144,146 Endoglin (ENG or CD105) is a cell surface receptor for transforming growth factor-β1 and -β3, and it is highly expressed on the vascular endothelium and placental syncytiotrophoblast. In addition to its central role in the pathogenesis of hereditary hemorrhagic telangiectasia,149 endoglin affects systemic endothelial function through interaction with endothelial nitric oxide synthase and regulation of endothelium-derived nitric oxide.150,151 Nitric oxide is a potent vasorelaxant that plays a key role in the regulation of systemic blood pressure, vascular permeability, and angiogenesis. Placental endoglin is upregulated in preeclampsia, resulting in the release of soluble endoglin (sENG) into the maternal circulation of symptomatic individuals in a dose-dependent fashion.144,146 Elevation of circulating sENG levels begins 2 to 3 months before preeclampsia is clinically apparent.146 sENG interferes with transforming growth factor-β signaling and endothelial nitric oxide synthase activation in maternal endothelial cells.144
Collectively, these findings support the hypothesis that placenta-derived circulating factors that alter maternal endothelial function play a central role in disease development in maternal preeclampsia. Future research will undoubtedly result in translation of these fundamental discoveries into targeted therapies and monitoring strategies for preeclampsia and eclampsia.
HELLP Syndrome
The HELLP syndrome is characterized by hemolysis, elevated levels of liver enzymes, and low platelet levels. It is a severe complication that occurs in an estimated 0.5% of all pregnancies (Box 53.6).152 HELLP syndrome is associated with preeclampsia or eclampsia, and in a large study of approximately 12,000 pregnancies complicated by various hypertensive disorders, HELLP syndrome was diagnosed in 20% of patients with severe preeclampsia and 10% of patients with eclampsia.153 This paradoxical inverse relationship between HELLP syndrome and the severity of preeclampsia or eclampsia was not seen in another large population-based study that found a stronger association with eclampsia (35%) than with preeclampsia (12%).154 Lack of standard and quantitative criteria for diagnosis of preeclampsia, eclampsia, and HELLP syndrome confounds these and related studies and may be partly responsible for the observed discrepancies. Although there have been attempts at providing strict laboratory criteria for the diagnosis of HELLP, there appears to be significant overlap between patients who do meet the strict criteria and patients who don’t, questioning the clinical utility of the proposed laboratory criteria.154a
Clinical Features
Despite an apparent association between HELLP syndrome and preeclampsia, these entities have several critical differences. The peak period of clinical onset for HELLP is between 27 and 36 weeks’ gestation, but unlike preeclampsia, as many as one third of cases are diagnosed in the immediate postpartum period.153 The most common presenting symptoms are abdominal pain (40% to 100% of cases), nausea and vomiting (29% to 100%), and headache (29% to 84%), and most patients have peripheral edema (50% to 70%) and proteinuria (85% to 100%).153,155,156
Hypertension may be absent in as many as 12% to 18% of HELLP cases, making a prompt diagnosis difficult.156 This may be further complicated by the fact that the differential diagnosis of thrombocytopenia in pregnancy is often broad and includes gestational thrombocytopenia, immune thrombocytopenia, thrombotic thrombocytopenic purpura, and hemolytic-uremic syndrome.156–159 Gestational thrombocytopenia, which accounts for almost 75% of pregnancy-associated thrombocytopenias, is benign and not associated with significant clinical signs or symptoms. The incidence of immune-mediated thrombocytopenia is increased in pregnancy, and the condition may be difficult to distinguish from HELLP syndrome, although significant liver disease often points to a diagnosis of HELLP.
HELLP syndrome is associated with significant maternal and fetal complications, and the disease may recur in as many as 27% of subsequent pregnancies or be associated with preeclampsia or gestational hypertension in subsequent pregnancies.153,160–164 The maternal mortality rate is 1% or less, and the most serious maternal morbidities are disseminated intravascular coagulation, abruptio placentae, acute renal failure, and pulmonary edema.153,154,156 Infants born to mothers with HELLP syndrome are at significantly increased risk for low birth weight and prematurity.160,161 Infants born at a very early gestational age are at highest risk for perinatal death.156 The mortality rate and the rate of other neonatal complications, however, do not appear to be significantly different from those of matched neonates born to mothers without HELLP syndrome and who were treated in a neonatal intensive care unit.161
Definitive treatment of rapidly progressing thrombocytopenia or fetal distress is delivery. Expectant management and various medical treatment options with steroids and plasma exchange may be considered, but consensus treatment guidelines are lacking.156,165–168 Evidence suggests that corticosteroids administered to women with HELLP or preeclampsia before 34 weeks’ gestation improves perinatal outcome. Treatment may transiently improve maternal thrombocytopenia and liver function test results, but the available data do not show improved maternal mortality rates.156,168,169
Pathologic Features and Pathogenesis
The pathologic features of HELLP syndrome depend on the underlying pathophysiologic processes. In classic forms of HELLP syndrome associated with preeclampsia, the pathologic features are within the spectrum of changes described for preeclampsia or eclampsia.170
The underlying pathophysiology of classic HELLP syndrome is shared with preeclampsia and is fundamentally related to maternal endothelial dysfunction. However, animals treated with sFLT1 develop hypertension and proteinuria but not hemolysis and thrombocytopenia, suggesting that additional cofactors are required for the phenotype of HELLP in the setting of preeclampsia.147 In a subset of patients, this cofactor appears to be sENG, which synergistically works with sFLT1 in a pregnant rat model to induce severe preeclampsia, hemolysis, thrombocytopenia, and elevated liver function test results, similar to the human HELLP syndrome.144 It remains unclear whether sENG is a specific biomarker for HELLP syndrome in pregnant women with preeclampsia.
Despite experimental and clinical evidence for an association between HELLP syndrome and preeclampsia or eclampsia, there is incomplete biologic and clinical overlap between these entities. An intriguing alternative hypothesis for the pathogenesis of HELLP syndrome is its proposed association with AFLP and LCHAD, short-chain acyl-CoA dehydrogenase deficiency (SCAD), and TFP deficiency, as reported in several studies. 99,100,103,107,111,171 In these cases, the liver pathology closely resembles that of AFLP. Although the association would link two divergent pathologic mechanisms (one of presumed endothelial activation and one of mitochondrial FAO), the importance of this mechanism in most cases awaits confirmation, especially in the face of contradictory results.108,172,173 Regardless of the biologic basis, the pathologic distinction between AFLP and vascular abnormalities associated with hypertensive disorders is straightforward and poses no diagnostic challenge. Lastly, the alternative pathway of complement fixation has been proposed to contribute to the pathogenesis of HELLP syndrome, although no specific mechanisms have been proposed.173a,173b
Hepatic Rupture
Clinical Features
Hepatic rupture is a potentially catastrophic complication of pregnancy, with an overall reported incidence of 0.4 to 2.cases per 100,000 deliveries.174,175 Large subcapsular hematomas, the pathologic background in which most cases of hepatic rupture occur, have been reported in 0.9% of pregnant women with HELLP syndrome.153 Although HELLP syndrome is the primary risk factor for this disorder, hepatic rupture has been reported for uncomplicated pregnancies176 and for patients with AFLP,177 connective tissue disorders,178 hepatic abscess,179 liver masses,180,181 history of cocaine use,182 and abdominal trauma.183
Regardless of the underlying cause, hepatic rupture typically manifests with acute abdominal pain associated with nausea and vomiting, followed by abdominal distention and hypovolemic shock. Diagnosis is readily made by abdominal imaging, and emergent surgical and medical treatment is required to prevent maternal and fetal mortality. Maternal mortality rates of 50% to 75% have been reported as a result of massive abdominal hemorrhage in older studies.184 Later series have reported maternal mortality rates of 10% to 30% because of improved therapy.185 Fetal mortality rates are high (60% to 80%) and often related to a combination of maternal hypotension, abruptio placentae, and prematurity.184,185 Recurrence of rupture in subsequent pregnancies is rare.
Pathologic Features and Pathogenesis
The pathologic processes that lead to hepatic rupture are poorly understood, and they are most likely as varied as the underlying causes of rupture. In hypertensive disorders, the most likely sequence of events is intrahepatic or intracapsular hemorrhage with tissue disruption leading to hematoma formation, followed by distention and rupture of the capsule (Fig. 53.5). The events initiating intrahepatic hemorrhage in hypertensive disorders are unknown, but they are most likely related to parenchymal ischemia that results from fibrin thrombi or reduced blood flow caused by endothelial dysfunction.
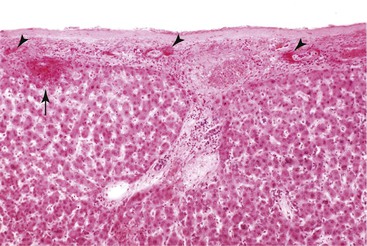
Hepatic rupture is most commonly associated with hematomas that occur on the superior and anterior aspects of the right lobe of the liver. The gross and microscopic pathology is otherwise that of the underlying condition leading to hematoma, tissue disruption, and rupture.
Hyperemesis Gravidarum
Clinical Features
Nausea and vomiting are common during pregnancy, occurring in more than one half of pregnant women.186 Nausea and vomiting most commonly begin at 4 to 7 weeks’ gestation, and they typically regress by the 16th week. Hyperemesis gravidarum (HG) is a severe, intractable form of nausea and vomiting that causes secondary abnormalities such as dehydration and electrolyte imbalance. HG is diagnosed in 0.2% to 3.2% of all pregnancies and may be associated with mildly elevated bilirubin (<5 mg/dL, all unconjugated), aminotransferases (usually less than two times normal), and alkaline phosphatase (less than two times normal) levels.186–191 HG occurs more commonly in young women in their first pregnancy, and the incidence depends on ethnicity and country of birth.190,191
Maternal abnormalities promptly return to normal with supportive treatment, and more aggressive treatment regimens do not appear to improve outcome.192,193 Perinatal outcomes for patients with HG have been the subject of debate. Infants born to mothers with HG may have lower gestational age, lower birth weight, or evidence of mild intrauterine growth retardation, but there is no definitive evidence of significant fetal morbidity or mortality associated with HG.189,191,194–197
Pathologic Features and Pathogenesis
The pathophysiology of HG remains unknown. A causative link between Helicobacter pylori infection and HG has been proposed, but because of the heterogeneity of the studies, small sample sizes, and heavy reliance on serum antibodies to the organism as evidence of active infection, the suggested association cannot be confirmed.198–201 Liver histology in HG is typically normal but may include mild steatosis, mild cholestasis, or occasional necrotic hepatocytes.202,203 Rare cases of severe liver damage have been reported,204 but it is unlikely that such unusual cases represent the pathology of otherwise uncomplicated hyperemesis. No specific mechanisms of liver damage have been described, and the mild hepatic abnormalities are thought to result from dehydration and other systemic changes associated with hyperemesis or pregnancy in general.
Hepatitis E Infection
Clinical Features
Viral hepatitis is the leading cause of jaundice in pregnancy. Hepatitis may manifest any time during pregnancy, and its clinical presentation and pathologic diagnosis are the same as for nongravid patients. With the exception of hepatitis E virus (HEV) infection, the clinical course of viral hepatitis does not seem to be significantly affected by pregnancy.
HEV is uncommon in the United States, but it is responsible for a large number of epidemic and sporadic forms of acute hepatitis in southeast and central Asia, the Middle East, parts of Africa, and Mexico.205–211 HEV is enterically transmitted through contaminated water, food, or blood products, with recently discovered zoonotic spread.
HEV infection manifests in patients after an incubation period of 2 to 10 weeks with a clinical illness resembling other forms of acute viral hepatitis.211,212 The attack rate is the highest among young adults, with a population-based average mortality rate of less than 0.6%.205 In pregnant women, however, the illness can be particularly severe, with average mortality rates as high as 27%.205,208,213,214 HEV is also a leading cause of fulminant hepatic failure in pregnancy. In a study of 100 Indian or Asian women (50 pregnant women and 50 matched nonpregnant patients) with fulminant hepatic failure, HEV seropositivity was found in 76% of the pregnant women and 30% of controls.215 The mortality rate was also significantly higher among pregnant, HEV-positive women (65%) compared with nonpregnant patients (23%). In addition to maternal disease, there is a significant risk of vertical transmission to the infants born to mothers with HEV infection206,216 and an increased risk of poor fetal outcomes in general.217
HEV infection is typically diagnosed by serologic markers and by quantitative measurement of HEV RNA titers. Serologic assays show anti-HEV immunoglobulin M (IgM) antibody early during clinical illness, although the IgM antibody disappears rapidly within a few months. Anti-HEV IgG antibody appears later and persists for at least a few years. Although potentially efficacous anti-HEV vaccines are available,217a none is currently approved for clinical use in the United States.217b
Pathologic Features and Pathogenesis
Histopathology of confirmed HEV infection in pregnancy has been rarely described. It appears to be that of an acute cholestatic hepatitis (Fig. 53.6) with some resemblance to acute hepatitis A infection.218–221 Findings commonly include a dense portal inflammatory infiltrate associated with various forms of bile duct damage, including acute or lymphocytic cholangitis and interface hepatitis. Lobular hepatocellular damage is often prominent and may include canalicular bile stasis, cholestatic hepatocellular rosettes, ballooning degeneration of hepatocytes, acidophilic bodies, and focal or confluent hepatocellular necrosis. The main histologic differential diagnosis in pregnancy is ICP. Markedly elevated levels of aminotransferases may be helpful in distinguishing viral hepatitis from ICP, in which aminotransferase levels are typically only mildly elevated.
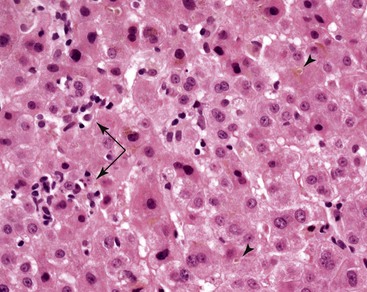
HEV is a nonenveloped, 27- to 34-nm, single-stranded RNA virus that belongs to the genus Hepevirus of the family Hepeviridae.212 The HEV genome has been cloned and the infectious nature of the viral replicons experimentally confirmed. There are four genotypes with specific geographic distribution. Genotype 1 is found in Asia and Africa. Genotype 2 is found in Mexico and Africa. Genotype 3 is found throughout the world, including the United States and Europe. Genotype 4 is found mainly in Asian countries.212
Questions remain about the specific nature of virus interaction with hepatocytes and their pathogenic mechanisms, although evidence points to indirect hepatocyte injury as a result of the immune response to HEV infection.212 For instance, in experimental HEV infection, hepatocellular injury was initially linked to hepatic viral replication, but injury continued despite the appearance of anti-HEV antibodies and the disappearance of the HEV antigen, supporting the hypothesis of a virus-independent mechanism of hepatocellular damage.222
Herpesvirus Infection
Clinical Features
Clinically significant liver disease resulting from nonhepatitic viral infection is uncommon, but it can be devastating in pregnancy. With the exception of herpesviruses, these viral infections are neither more common nor more severe in pregnancy. Among the different serotypes, herpes simplex virus (HSV) infections are the most common, with a seropositivity rate of as much as 30% for HSV type 2 in a large cohort of pregnant women.223
Dissemination of HSV from the primary site of infection is often associated with primary or secondary immunodeficiency states, but in rare occasions, it may occur in otherwise healthy individuals. A disproportionate number of disseminated infections in otherwise healthy individuals appears to be associated with pregnancy, particularly during the third trimester.224–227 Suspicion of the disease and prompt diagnosis with liver biopsy, culture, and serologic tests are important because treatment with antiviral agents may be successful in controlling a disease that otherwise has extremely high maternal and fetal mortality rates.
Clinical presentation is somewhat nonspecific and includes nausea, vomiting, fever, and abdominal pain, and liver function tests often show markedly increased levels of aminotransferases. Timely liver biopsy for tissue diagnosis in clinically suspected cases is important because classic mucocutaneous vesicular lesions may be absent in many pregnancy-associated cases.224,225 Infection with other members of the Hepeviridae family, including cytomegalovirus, Epstein-Barr virus, and varicella-zoster virus, can result in fulminant hepatic disease in pregnancy.
Pathologic Features and Pathogenesis
The underlying factors predisposing some pregnant women to more severe herpesvirus infections are unknown, but altered immunity during the course of pregnancy likely plays a role. Liver pathology in cases of HSV infection is similar to that in the nonpregnant state. It is characterized by irregular areas of hemorrhage and necrosis without significant inflammation but with classic viral inclusions in the surrounding hepatocytes (Fig. 53.7). In a questionable case, immunohistochemical stains and molecular assays for viral markers can provide a definitive diagnosis.
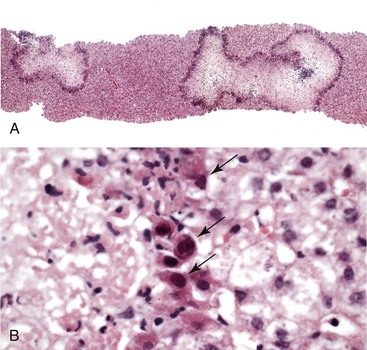
Budd-Chiari Syndrome
Clinical Features
Budd-Chiari syndrome is the clinical manifestation of hepatic venous outflow obstruction and is classically caused by thrombotic occlusion of hepatic vein branches or the inferior vena cava. Pregnancy, including the early postpartum period, accounts for approximately 5% to 15% of documented cases.228,229
Clinical presentation with abdominal pain, hepatomegaly, and ascites is often insidious, although a disproportionate number of pregnancy-related cases have an acute onset and are associated with massive ischemic damage.228,229 Proper diagnosis requires clinical imaging studies, including Doppler ultrasonography and possibly MRI. Treatment with anticoagulants and thrombolytic agents may be sufficient for management of less severe cases, but surgical options such as portacaval shunt or transplantation may be necessary.
Pathologic Features and Pathogenesis
Studies suggest that factor V Leiden deficiency may be responsible for the atypical presentation during pregnancy. In a study of 63 cases by Deltenre and coworkers, Budd-Chiari syndrome with massive ischemic necrosis was encountered only in carriers of factor V Leiden mutation, 3 (75%) of whom were pregnant; the fourth was on oral contraceptives.229 Another rare case of severe acute Budd-Chiari syndrome with factor V Leiden mutation in pregnancy was reported separately.230 Rautou and associates reported that among seven pregnant or postpartum women with Budd-Chiari syndrome, two had factor V Leiden mutation, one of whom had a myeloproliferative disease; two had protein S deficiency, one of whom had a myeloproliferative disease; one had antiphospholipid syndrome and a myeloproliferative disease; one had myeloproliferative disease alone; and one had no identified predisposing factor.231
These observations suggest that a physiologic decrease in the level of protein S during pregnancy may be a predisposing factor.232 The liver histopathology, differential diagnosis, and pathogenesis of Budd-Chiari syndrome in pregnancy are otherwise identical to those in nonpregnant patients (see Chapter 51).
Conclusions
Liver pathobiology is affected by the pathophysiologic alterations and hormonal effects of pregnancy. Although many of the hepatic disorders encountered during pregnancy are similar to those that occur in the nonpregnant population, recognition of liver diseases specific to pregnancy is critical because they have a profound impact on maternal and fetal outcomes. Despite significant advances in understanding the underlying molecular and cellular mechanisms, histopathology of the liver in pregnancy-related disorders is often nonspecific.
The primary role of the pathologist in evaluation of liver disease in pregnancy is to provide confirmatory evidence for a clinically suspected diagnosis or to guide the clinical team toward an alternative diagnosis. For these goals to be accomplished, a clear dialogue must take place between clinicians and pathologists to convey clinically relevant data and to ensure proper handling and triage of biopsy material.
Related posts:
 Diagnostic Cytology of the Biliary Tract and Pancreas
Diagnostic Cytology of the Biliary Tract and Pancreas
 Algorithmic Approach to Diagnosis of Inflammatory Disorders of the Gastrointestinal Tract
Algorithmic Approach to Diagnosis of Inflammatory Disorders of the Gastrointestinal Tract
 Epithelial Neoplasms of the Stomach
Epithelial Neoplasms of the Stomach
 Gallbladder, Extrahepatic Biliary Tract, and Pancreas Tissue Processing Techniques and Normal Histology
Gallbladder, Extrahepatic Biliary Tract, and Pancreas Tissue Processing Techniques and Normal Histology
 Inflammatory and Neoplastic Disorders of the Anal Canal
Inflammatory and Neoplastic Disorders of the Anal Canal
 Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
Enteropathies Associated with Chronic Diarrhea and Malabsorption in Childhood
