Lipid disorders
1. What are the major lipids in the bloodstream?
Cholesterol and triglycerides (TGs) are the major circulating lipids. Cholesterol is used by all cells for the synthesis and repair of membranes and intracellular organelles and by the adrenal glands and gonads as a substrate to synthesize adrenal and gonadal steroid hormones. TGs are an energy source that can be stored as fat in adipose tissue or used as fuel by muscle and other tissues.
Cholesterol and TGs are not water soluble and thus cannot be transported through the circulation as individual molecules. Lipoproteins are large, spherical particles that package these lipids into a core surrounded by a shell of water-soluble proteins and phospholipids. Lipoproteins serve as vehicles that transport cholesterol and TGs from one part of the body to another.
3. What are the major lipoproteins in the bloodstream?
Chylomicrons, very-low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs) are the major circulating lipoproteins. Their functions are as follows:
| Chylomicrons | Transport exogenous TGs from the gut to adipose tissue and muscle |
| VLDLs | Transport endogenous TGs from the liver to adipose tissue and muscle |
| LDLs | Transport cholesterol from the liver to peripheral tissues |
| HDLs | Transport cholesterol from peripheral tissues to the liver |
4. What are the apolipoproteins?
Apolipoproteins are located on the surfaces of the lipoproteins. They function as ligands for binding to lipoprotein receptors and as cofactors for metabolic enzymes. Their functions are as follows:
| Apolipoprotein A | Ligand for peripheral HDL receptors |
| Apolipoprotein B | Ligand for peripheral LDL receptors |
| Apolipoprotein E | Ligand for hepatic receptors for remnant particles |
| Apolipoprotein C-II | Cofactor for lipoprotein lipase (LPL) |
5. Name other enzymes and transport proteins that are important in lipoprotein metabolism.
See Table 6-1 and Figure 6-1.
TABLE 6-1.
ENZYMES AND TRANSPORT PROTEINS IMPORTANT IN LIPOPROTEIN METABOLISM
| ENZYME/TRANSPORT PROTEIN | FUNCTION |
| Hydroxy-3-methyl-glutaryl-coenzyme A reductase | The rate-limiting enzyme in hepatic cholesterol synthesis |
| Lipoprotein lipase | Removes TGs from chylomicrons and VLDLs in adipose tissue, leaving remnant particles |
| Hepatic lipase | Removes additional TGs from remnant particles in the liver, converting them into LDLs |
| Lecithin cholesterol acyl transferase | Esterifies cholesterol molecules on the surface of HDLs, drawing them into the HDL core |
| Cholesterol ester transfer protein | Shuttles esterified cholesterol back and forth between HDLs and LDLs |
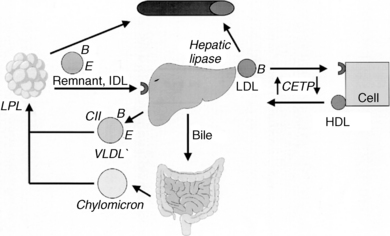
6. Explain the function and metabolism of TGs.
Food and hepatic synthesis are the major sources of TGs. They are transported by chylomicrons (dietary TGs) and VLDLs (endogenous TGs) to adipose tissue and muscle, where lipoprotein lipase and cofactor apolipoprotein C-II (Apo C-II) break down TGs into fatty acids (FAs) and monoglycerides. FAs enter adipose cells to be stored as fat or muscle cells to be used as fuel. The chylomicron and VLDL remnant particles return to the liver, where hepatic lipase converts VLDL remnants into LDL.
7. Describe the function and metabolism of LDL.
LDL transports cholesterol from the liver to peripheral tissues, where surface apolipoprotein B-100 binds to cellular LDL receptors (LDLRs). LDLR clustering in clathrin-coated pits on the cell membrane, promoted by LDLR adaptor protein-1 (LDLRAP1), is necessary for efficient LDL uptake. After LDL is internalized, it is degraded to free cholesterol (FC) for intracellular use. Excess LDL is cleared from the circulation by scavenger macrophages.
8. What is the function of HDL?
HDL removes excess cholesterol from cells by two mechanisms. Nascent pre-βHDL is made in the liver and intestine. Surface Apo A1 on pre-βHDL acquires FC through the adenosine triphosphate (ATP)–binding cassette (ABC) transporter-A1 (ABCA1) on arterial wall macrophages. Plasma lecithin cholesterol acyl transferase (LCAT) then esterifies the FC to cholesterol ester (CE). In addition, HDL accepts additional FC from arterial macrophages through the ABCG1 transporter and the scavenger receptor, class B, type 1 (SR-B1) receptor. Cholesterol ester transfer protein (CETP) transfers some CE back to LDL particles, and the mature HDL transports the remaining CE to the liver, where transfer occurs through hepatic SR-B1 receptors. In addition to performing reverse cholesterol transport, HDL reduces LDL oxidation, inhibits vascular inflammation, and improves endothelial function. All of these functions make HDL a potent antiatherogenic lipoprotein.
9. Describe the pathogenesis of the atherosclerotic plaque and arterial thrombosis.
LDL can be modified by oxidation. Scavenger macrophages located beneath the intimal surface of arteries engulf oxidized LDL, becoming lipid-laden foam cells, which secrete growth factors that stimulate smooth muscle cell proliferation. These developing plaques also secrete cytokines that attract inflammatory cells, which secrete proteolytic enzymes that erode the fibromuscular plaque cap, making it prone to rupture. When rupture occurs, platelets aggregate and release chemicals that promote vasoconstriction and initiate thrombus formation, which may ultimately occlude the artery.
10. Are elevated serum TG values harmful?
Increased serum TG levels are associated with atherosclerosis and increased coronary disease. The American Heart Association states that triglycerides are not directly atherogenic but represent an important biomarker of cardiovascular risk because of their association with an atherogenic lipid profile (low HDL cholesterol levels and small, dense LDL particles), as well as obesity, insulin resistance, and the metabolic syndrome. It has not yet been shown that decreasing TG levels will reduce coronary disease risk. TG values greater than 1000 mg/dL significantly increase the risk of acute pancreatitis.
11. What is metabolic syndrome?
Metabolic syndrome (MS) is a condition that is diagnosed when a patient has any three of the following findings: elevated fasting blood glucose (≥ 110 mg/dL), high TGs (≥ 150 mg/dL), low HDL (< 40 mg/dL for men, < 50 mg/dL for women), hypertension (≥ 130 mm Hg systolic/85 mm Hg diastolic), and abdominal obesity (waist > 40 inches in men, > 35 inches in women). The common thread among the disorders that constitute MS appears to be insulin resistance. MS carries a high risk for atherosclerotic vascular disease.
12. What is lipoprotein(a) [Lp(a)]?
Lipoprotein(a) [Lp(a)] has approximately 85% amino-acid sequence homology with plasminogen. When an Lp(a) molecule attaches to apoprotein B on the surface of an LDL particle, the new particle is referred to as Lp(a). Excessive Lp(a) promotes atherosclerosis, possibly because it is easily oxidized and engulfed by macrophages, because it inhibits thrombolysis, or both.
13. What are the primary dyslipidemias?
Primary dyslipidemias are inherited disorders of lipoprotein metabolism. The major primary dyslipidemias and their lipid phenotypes are as follows:
| PRIMARY DYSLIPIDEMIA | PHENOTYPE |
| Familial hypercholesterolemia (FH) | ↑↑Cholesterol |
| Polygenic hypercholesterolemia | ↑Cholesterol |
| Familial combined hyperlipidemia (FCH) | ↑Cholesterol and ↑TGs |
| Familial dysbetalipoproteinemia (FDL) | ↑Cholesterol and ↑TGs |
| Familial hypertriglyceridemia (FHT) | ↑TGs |
| Familial hyperchylomicronemia (FHC) | ↑↑TGs |
14. What is familial hypercholesterolemia?
FH is an inherited disease characterized by extreme elevations of serum cholesterol but normal serum TG levels. The disorder has a population frequency of 1:500 for heterozygotes, who generally have serum cholesterol levels of 300 to 800 mg/dL, and 1:1,000,000 for homozygotes, who have serum cholesterol levels of 600 to 1000 mg/dL. Most patients have genetic mutations resulting in deficient or dysfunctional LDL receptors (LDLRs). Other less common monogenic hypercholesterolemic disorders include apoprotein B mutations that produce a defective apo B that cannot bind to LDLR, proprotein convertase subtilisin–like kexin type 9 (PCSK9) mutations that cause accelerated LDLR degradation, LDLR adaptor protein-1 (LDLRAP1) mutations that prevent normal clustering of LDLR in cell surface clathrin-coated pits, and ATP-binding cassette G5 or G8 (ABCG5/8) mutations that cause abnormal cellular transport of cholesterol and plant sterols (sitosterolemia). These disorders are characterized by premature coronary artery disease (CAD), often before age 20 in homozygous FH, and tendon xanthomas.
15. What is familial combined hyperlipidemia?
FCH is an inherited disorder characterized by variable elevations of both serum cholesterol and TG. Affected patients have excessive hepatic apolipoprotein B synthesis, with increased numbers of apolipoprotein B–containing VLDL and LDL particles. The genetic basis for the disorder has not yet been determined. These patients are prone to development of premature CAD.
16. What is familial dysbetalipoproteinemia?
Familial dysbetalipoproteinemia (FDL), also known as broad beta disease, is an inherited condition characterized by significant and relatively balanced elevations of both serum cholesterol and TGs. This disorder results from an abnormal apolipoprotein E phenotype (E2/E2), which binds poorly to hepatic receptors, resulting in impaired clearance of circulating VLDL remnants by the liver. Affected patients often have premature CAD. Planar xanthomas in the creases of the palms and soles of the feet are a characteristic finding in patients with FDL.
17. What is polygenic hypercholesterolemia?
Polygenic hypercholesterolemia, which is characterized by mild to moderate elevations of serum cholesterol alone, is the most common type of inherited hypercholesterolemia. This condition generally occurs when one or more mild defects of cholesterol metabolism combine to elevate the serum cholesterol value. Affected patients have an increased risk of CAD.
18. What are familial hypertriglyceridemia and familial hyperchylomicronemia?
Familial hypertriglyceridemia (FHT) is an inherited condition featuring moderate to severe elevations of serum TG levels with normal serum cholesterol levels. Familial hyperchylomicronemia (FHC) is characterized by extremely high serum TG and chylomicron levels. The genetic basis for FHT is unclear, but it may be polygenic or caused by milder forms of the mutations that cause FHC. FHC is due to inactivating mutations in the gene for LPL or Apo CII and mutations of the apolipoprotein AV (APOAV) gene. Severe hypertriglyceridemia with chylomicronemia may predispose to the development of eruptive xanthomas, lipemia retinalis, hepatosplenomegaly, and acute pancreatitis.
19. How do you distinguish between familial combined hyperlipidemia and familial dysbetalipoproteinemia?
Because FCH and FDL are characterized by combined elevations of both cholesterol and TGs, additional tests may be necessary to make the distinction. Patients with FCH have increased serum apolipoprotein B levels, whereas patients with FDL have an E2/E2 apolipoprotein E phenotype and a broad beta-band on lipoprotein electrophoresis. Family studies are also helpful.
20. What causes familial low HDL?
Familial hypoalphalipoproteinemia (familial low HDL), characterized by extremely low serum HDL levels and premature CAD, is caused by inactivating mutations in the genes that encode apolipoprotein A1 (APOA1), ABCA1, or lecithin cholesterol acyl transferase.
21. Name the secondary dyslipidemias.
The secondary dyslipidemias are serum lipid elevations that result from systemic diseases such as diabetes mellitus, hypothyroidism, nephrotic syndrome, renal disease, obstructive liver disease, dysproteinemias, and lipodystrophies. Lipids also may be increased by medications, such as beta-blockers, thiazide diuretics, estrogens, progestins, androgens, retinoids, corticosteroids, cyclosporin A, antipsychotics, and protease inhibitors. These disorders usually improve when the primary condition is treated or the offending drug is discontinued.
 KEY POINTS 1: CAUSES OF LIPID DISORDERS
KEY POINTS 1: CAUSES OF LIPID DISORDERS
1. Elevation of low-density lipoprotein (LDL) cholesterol is a major risk factor for coronary artery disease (CAD).
2. A low level of high-density lipoprotein (HDL) cholesterol is also a significant risk factor for CAD.
3. High levels of serum triglycerides (TGs) are associated with increased CAD, but it is unclear whether lowering TG levels decreases CAD risk.
4. Serum TG levels greater than 1000 mg/dL significantly increase the risk of acute pancreatitis.
5. Inflammation within the atherosclerotic plaque plays a major role in plaque rupture and the occurrence of acute coronary events.
22. What is the cause of severe elevations of serum TGs?
TG levels above 1000 mg/dL pose a very high risk for the development of acute pancreatitis, a condition with a mortality rate of up to 10%. Most patients with such severe TG elevations have a primary TG disorder, such as FHT, FCH, or FDL, combined with a secondary disorder, most commonly poorly controlled diabetes mellitus, alcohol abuse, estrogen use, or the use of medications for the treatment of human immunodeficiency virus (HIV).
23. Summarize the revised (2004) Coronary Heart Disease (CHD) risk stratification from the Adult Treatment Panel III (ATP III) of the National Cholesterol Education Program (NCEP).
24. What are the revised (2004) LDL cholesterol treatment goals from the ATP III?
| PATIENT RISK | LDL CHOLESTEROL GOAL (MG/DL) |
| High risk | < 100 (optional < 70) |
| Moderately high risk | < 130 (optional < 100) |
| Moderate risk | < 130 |
| Low risk | < 160 |
25. What is therapeutic lifestyle change?
Therapeutic lifestyle change (TLC) should be encouraged for individuals with LDL cholesterol above their risk-stratified goal. Medications should be added. ATP III recommends therapeutic lifestyle change (TLC) for individuals with LDL cholesterol above their risk stratified goal. Medications can be added, as needed, but the TLC should continue. The components of TLC as recommended by ATP III are:
| COMPONENT | GOALS |
| Total fat | 25%-35% of total calories |
| Saturated fat | < 7% of total calories |
| Polyunsaturated fat | < 10% of total calories |
| Monounsaturated fat | < 20% of total calories |
| Carbohydrate | 50%-60% of total calories |
| Protein | Approximately 15% of total calories |
| Total calories | Adjust to achieve and maintain ideal body weight |
| Dietary fiber | 20-30 g/day |
| Physical activity | Expend at least 200 kcal/day |
26. What medications most effectively improve dyslipidemia?
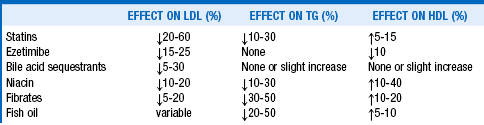
27. How do the currently available statin medications differ?
The statins inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase, the rate-limiting enzyme in cholesterol synthesis. This leads to a decrease in cholesterol synthesis and an increase in LDL receptor–mediated removal of LDL. Some statins are naturally occurring compounds (lovastatin, pravastatin) and others are synthetic. Some are more hydrophilic (pravastatin, rosuvastatin), whereas the others are more lipophilic. The main differences of clinical interest, however, are their LDL-lowering potencies. The most commonly used statins, in order of relative LDL-lowering potencies, are fluvastatin < pravastatin < lovastatin < simvastatin < atorvastatin < rosuvastatin < pitavastatin. The initial statin dose produces the greatest LDL cholesterol reduction. Each subsequent doubling of the statin dose, on average, results only in an additional 6% decrease in serum LDL cholesterol level.
28. How should the statin-intolerant patient be approached?
Myopathy occurs in approximately 10% of patients treated with statins. Myopathy typically manifests as myalgias, with or without elevations in creatine kinase (CK). For CK elevations greater than five times the upper limit of normal or if the patient has moderate to severe symptoms, the statin should be stopped. Once the patient is asymptomatic and the CK level is reduced, reasonable approaches include a trial of low-dose fluvastatin or pravastatin, alternate-daily or weekly dosage of a more potent statin such as rosuvastatin or pitavastatin, or combination of a low-dose statin with a non-statin cholesterol agent (ezetimibe or bile acid sequestrant). Over-the-counter preparations containing natural statin-like agents, such as red yeast rice, can also be tried, although they undergo limited quality control and have low efficacy. In patients with mild symptoms and CK elevations less than five times the upper limit of normal, the statin may be continued. If symptoms worsen, the CK level should be rechecked.
29. How effective and safe are combinations of lipid-lowering medications?
For severe cholesterol elevations, the addition of ezetimibe, niacin, or a bile acid resin to a statin often reduces serum LDL cholesterol by an additional 20%, compared with only 6% when the statin dose is doubled. These combinations are generally safe to use, but side effects can be additive. For elevations of both cholesterol and TGs, adding a fibrate to a statin can lower the serum TG level up to 50%. However, the risk of myositis and frank rhabdomyolysis increases. Fenofibrate appears to be significantly safer than gemfibrozil when combination with a statin is considered necessary.
30. Does aggressive cholesterol-lowering therapy effectively and safely reduce the risk of coronary artery disease?
Clinical trials have repeatedly demonstrated the efficacy of aggressive cholesterol-lowering with statins in reducing myocardial infarction, strokes, and cardiovascular mortality in patients with a previous history of CAD (secondary prevention—4S (Scandinavian Simvastatin Survival Study), CARE (Cholesterol and Recurrent Events), LIPID (Long-term Intervention with Pravastatin in Ischaemic Disease), HPS (Heart Protection Study), TNT (Treating to New Targets), PROVE IT (Pravastatin or Atorvastatin Evaluation and Infection Therapy), AVERT (Atorvastatin Versus Revascularization Treatment), and ALLIANCE (Aggressive Lipid Lowering to Alleviate New Cardiovascular Endpoints). A meta-analysis from the Cholesterol Treatment Trialists’ Collaboration suggests that each 1-mmol/L reduction in LDL cholesterol results in an approximately 20% reduction in the annual rate of cardiovascular disease.
The role of statins in the setting of primary prevention is less clear. Some trials have demonstrated a benefit—WOSCOPS (West of Scotland Coronary Prevention Study), AFCAPS (Air Force Coronary Atherosclerosis Prevention Study), HPS (Heart Protection Study), ASCOT-LLA, CARDS (Collaborative Atorvastatin Diabetes Study), and JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin). A Cochrane meta-analysis showed that statins can reduce all-cause mortality, cardiovascular mortality, and cardiovascular events. However, another meta-analysis did not find a reduction in all-cause mortality, and the cost effectiveness of statins and effects on quality of life are unclear.
The major safety concerns about statin therapy are hepatotoxicity and myopathy. Both of these problems were relatively rare in the clinical trials but occur more commonly in clinical practice in patients who require higher doses or take them in combination with other medications that may interfere with statin metabolism. In particular, high-dose simvastatin therapy has been associated with an increased risk of myopathy.
31. What is the appropriate role for niacin?
Niacin, which decreases VLDL production, is often used in combination with statins in order to further decrease LDL cholesterol levels, decrease TGs, and increase HDL cholesterol levels. Niacin also decreases lipoprotein(a) [Lp(a)] levels. However, a meta-analysis found no effect of niacin on total mortality or cardiac mortality. The ARBITER 6-HALTS (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6: HDL and LDL Treatment Strategies in Atherosclerosis) study found that among patients taking statins, niacin was superior to ezetimibe on the surrogate end point of regression of carotid intima-media thickness. The later AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL Cholesterol/High Triglyceride and Impact on Global Health Outcomes) study found no benefit to adding niacin to statin therapy. Thus the appropriate role for niacin in the treatment of dyslipidemia is currently unclear.
32. What is the appropriate role for ezetimibe?
Ezetimibe, which inhibits intestinal cholesterol absorption, is commonly used in combination with statins and effectively lowers LDL cholesterol levels. However, ezetimibe in combination with statins has not been conclusively shown to decrease mortality or cardiovascular events. The ENHANCE (Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression) study of individuals with familial hypercholesterolemia did not show any improvement in their surrogate end point of carotid intima-media thickness. The IMPROVE-IT (The Improved Reduction of Outcomes: Vytorin Efficacy International Trial) study, which is currently comparing simvastatin with and without ezetimibe, should provide additional information about the appropriate role for ezetimibe.
33. What is the appropriate role for fibrates?
Fibrates, which decrease VLDL production, are the most effective TG-lowering agents. They also increase HDL cholesterol and modestly decrease LDL cholesterol. A meta-analysis showed that fibrates decreased cardiovascular events but had no effect on stroke, cardiac mortality, or total mortality. The FIELD (Fenofibrate Intervention and Event Lowering in Diabetes) study of fenofibrate in patients with type 2 diabetes reported a nonsignificant 11% reduction in cardiovascular events. The ACCORD (Action to Control Cardiovascular Risk in Diabetes) Lipid Trial in patients with type 2 diabetes did not find any reduction in cardiovascular end points when fenofibrate was added to simvastatin. Given the fact that elevated TGs have not yet been shown to be causally related to cardiovascular risk, the appropriate role for fibrates is still somewhat unclear.
CETP inhibitors interfere with the protein that transfers esterified cholesterol between HDL and LDL, resulting in an increase in HDL of more than 50%. Several CETP inhibitors are currently under development and in clinical trials. Unfortunately, to date these trials have been unable to show an increase in cardiovascular events and mortality, and a trial of torcetrapib was terminated because of an increase in coronary events and mortality in treated patients (see Cannon, 2011, in Bibliography).
 KEY POINTS 2: TREATMENT OF LIPID DISORDERS
KEY POINTS 2: TREATMENT OF LIPID DISORDERS
1. Statins are the most effective low-density lipoprotein (LDL) cholesterol–lowering agents and have the strongest evidence base for reducing cardiovascular events.
2. Additional LDL reduction can be achieved by adding ezetimibe, niacin, and bile acid resins.
3. Fibrates are the most effective triglyceride (TG)–lowering agents, but additional reductions can be achieved by adding niacin, fish oils, and high-dose statins.
4. The Adult Treatment Panel III (ATP III) recommends LDL goals of less than 100 mg/dL for patients with coronary heart disease (CHD) or CHD equivalents, less than 130 mg/dL for patients with two or more CHD risk factors, and less than 160 mg/dL for patients with no or one CHD risk factor. An optional LDL goal of less than 70 mg/dL may be more appropriate for the highest risk patients.
5. The ATP III also recommends a non–high-density lipoprotein cholesterol goal of 30 mg/dL above the LDL cholesterol goal in the patient whose serum TG level is greater than 200 mg/dL after the LDL goal has been achieved.
35. Is measurement of inflammatory markers a useful tool in CAD risk assessment?
Inflammation within an atherosclerotic plaque makes the plaque more likely to rupture, precipitating an acute ischemic event. Highly sensitive C-reactive protein (hsCRP), a nonspecific marker of inflammation, appears to predict CAD risk, as do LDL cholesterol levels. The JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin) trial showed a benefit to treatment with rosuvastatin in individuals with LDL cholesterol levels lower than 130 mg/dL and hsCRP levels of 2.0 mg/L or less. Thus information on LDL cholesterol and hsCRP together can be useful to providers making decisions about which patients to treat more aggressively but need not be performed routinely in all patients.
An indirect measure of inflammation is lipoprotein-associated phospholipase A2 (Lp-PLA2), an enzyme produced by inflammatory cells and liver cells that circulates in the plasma primarily bound to LDL particles. It hydrolyzes oxidized phospholipids on LDL particles, producing two inflammatory mediators, lysophosphatidylcholine and oxidized fatty acids, which have been linked to atherosclerotic plaque formation. Measurement of Lp-PLA2 may be considered for selected patients at increased cardiovascular risk as part of the initial clinical assessment. Studies of daraplabid, an Lp-PLA2 inhibitor, are ongoing, but there is no current evidence that lowering Lp-PLA2 will reduce cardiovascular risk.
36. Should we be using measurements of lipoprotein size and number?
Lipoprotein size and number can now be assessed by a variety of commercially available techniques. These analyses provide additional information about the atherogenicity of a lipoprotein profile. The cost-effectiveness of obtaining this additional information has not yet been demonstrated. Decisions regarding the need for treatment and the choice of agents can be made on the basis of the clinical risk factor profile and standard lipid profile in the majority of patients. Therefore these additional tests should be limited to situations in which they are likely to have a clear impact on the choice and aggressiveness of therapy.
37. How should the patient with severe hypertriglyceridemia be managed?
Serum TG levels above 1000 mg/dL must be lowered quickly because of the high risk of precipitating acute pancreatitis. Medications alone are not effective when TG levels are this high. Patients must immediately be started on a very-low-fat (less than 5% fat) diet until the TG level is less than 1000 mg/dL. Such a diet lowers serum TGs approximately 20% each day. Contributing factors, most commonly uncontrolled diabetes mellitus, alcohol abuse, estrogen use, and medications for treatment of human immunodeficiency virus, must simultaneously be addressed. After serum TG levels are less than 1000 mg/dL, the most effective medications to reduce serum TGs further are the fibrates. If these medications do not lower serum TG sufficiently, niacin, fish oils, or a statin may be added to the regimen.
, ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563–1574.
, AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011;365:2255–2267.
Cannon, CP. High-density lipoprotein cholesterol as the Holy Grail. JAMA. 2011;306:2153–2155.
Cannon, CP, Giugliano, RP, Blazing, MA, et al, Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008;156:826–832.
, Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670–1681.
Colhoun, HM, Betteridge, DJ, Durrington, PN, et al, Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS). multicentre randomised placebo-controlled trial. Lancet 2004;364:685–696.
Davidson, MH, Ballantyne, C, Jacobson, TA, et al, Clinical utility of inflammatory markers and advanced lipoprotein testing. advice from an expert panel of lipid specialists. J Clin Lipidology 2011;5:338–367.
Downs, JR, Clearfield, M, Weis, S, et al, Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels. results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1995;279:1615–1622.
Eckel, RH. Approach to the patient who is intolerant of statin therapy. J Clin Endocrinol Metab. 2010;95:2015–2022.
, Emerging Risk Factors Collaboration. Lipid-related markers and cardiovascular disease prediction. JAMA 2012;307:2499–2506.
Executive Summary of the Third Report of the National Education Program (NCEP): 2001 expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III), JAMA 285:2486–2497.
Ford, I, Murray, H, Packard, CJ, et al. Long-term follow-up of the West of Scotland Coronary Prevention Study. N Engl J Med. 2007;357:1477–1486.
Greving, JP, Visseren, FLJ, De Wit GA, et al, Statin treatment for primary prevention of vascular disease. whom to treat? Cost-effectiveness analysis. BMJ 2011;342:1672.
Grundy, SM, Cleeman, JI, Merz, CNB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239.
, Heart Protection Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomized placebo-controlled trial. Lancet 2002;360:7–22.
, Heart Protection Study Collaborative Group. C-reactive protein concentration and the vascular benefits of statin therapy: an analysis of 20,536 patients in the Heart Protection Study. Lancet 2011;377:469–476.
Hou, R, Goldberg, AC, Lowering low-density lipoprotein cholesterol. statins, ezetimibe, bile acid sequestrants, and combinationscomparative efficacy and safety. Endocrinol Metab Clin N Am 2009;38:79–97.
Jun, M, Foote, C, Lv, J, et al, Effects of fibrates on cardiovascular outcomes. a systematic review and meta-analysis. Lancet 2010;375:1875–1884.
Kastelein, JJ, Akdim, F, Stroes, ESG, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443.
Keech, A, Simes, RJ, Barter, P, et al, Effects of long-term fenofibrate therapy on cardiovascular events in 9795. people with type 2 diabetes mellitus (the FIELD Study). randomised controlled trial. Lancet 2005;366:1849–1861.
Koren, MJ, Hunninghake, DB, for the ALLIANCE Investigators. Clinical outcomes in managed care patients with coronary heart disease treated aggressively in lipid lowering disease management clinics. J Am Coll Cardiol 2004;44:1772–1779.
LaRosa, J, Grundy, SM, Waters, DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–1435.
, Lipid Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 1998;339:1349–1357.
Miller, M, Stone, NJ, Ballantyne, C, et al, Triglycerides and cardiovascular disease. a scientific statement from the American Heart Association. Circulation 2011;123:2292–2333.
Pitt, B, Waters, D, Brown, WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med. 1999;341:70–76.
Ray, KK, Seshasai, SRK, Erqou, S, et al, Statins and all-cause mortality in high-risk primary prevention. a meta-analysis of 11 randomized controlled trials. Arch Intern Med 2010;170:1024–1031.
Ridker, PM, Danielson, E, Fonseca, FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207.
Sacks, FM, Pfeffer, MA, Moye, LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–1009.
, Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444. patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S. Lancet 1994;344:1383–1389.
Semenkovich, CF, Goldberg, AC, Goldberg, IJ. Disorders of lipid metabolism. In Melmed S, Polonsky KS, Larsen PR, eds.: Williams Textbook of Endocrinology, 12th edition, Philadelphia: Elsevier Saunders, 2011.
Sever, PS, Dahlof, B, Poulter, NR, et al, Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower than average cholesterol concentrations, in the Anglo-22. Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA). a multicenter randomized controlled trial. Lancet 2003;361:1149–1158.
Shepherd, J, Cobbe, SM, Ford, I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301–1307.
Studer, M, Briel, M, Leimenstoll, B, et al, Effect of different antilipidemic agents and diets on mortality. a systematic review. Arch Inter Med 2005;165:725–730.
, Study of Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomized trial. Lancet 2010;376:1658–1669.
Taylor, AJ, Villines, TC, Stanek, EJ, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–2122.
Taylor, F, Ward, K, Moore, THM, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database of Systematic Reviews. 1, 2011. CD004816
, Third Report of the National Education Program (NCEP). Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation 2002;106:3143–3421.
