50 Leukaemia
The adjectives ‘myeloid’ and ‘lymphoid’ refer to the predominant cell involved, and the suffix-cytic and -blastic to mature and immature cells, respectively. These characteristics can be determined by a combination of cellular appearances, surface antigen expression and cytogenetic features. The international standard for leukaemia classification is the WHO system (Swerdlow et al., 2008).
Epidemiology
Haematological malignancies account for only 5% of all cancers; of these, CLL is the most common form of leukaemia. UK incidence data are presented in Table 50.1. CLL mainly affects an older age group: 90% of patients are over the age of 50 and nearly two-thirds are over 60 years old at diagnosis. It rarely occurs in young people and is twice as common in men as in women. CML is primarily a disease of middle age with the median onset in the 40–50 year old age group, but it can occur in younger people.
Table 50.1 Incidence of leukaemia in the UK (Leukaemia and Lymphoma Research, 2010)
| New cases/year | Incidence per 100,000 of the population | |
|---|---|---|
| CLL | 2750 | 4.58 |
| CML | 750 | 1.5 |
| ALL | 650 | 1.00 |
| AML | 1950 | 3.25 |
Aetiology
Pathophysiology
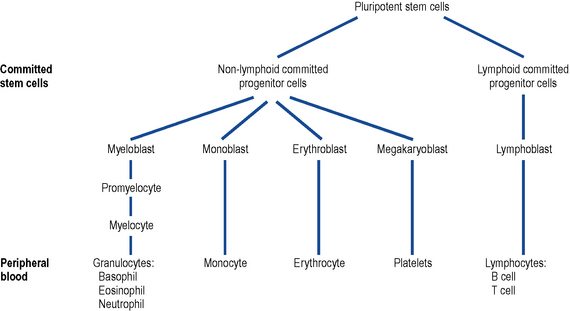
In leukaemia, the normal process of haemopoiesis is altered (Fig. 50.1). Transformation to malignancy appears to occur in a single cell, usually at the pluripotential stem cell level, but it may occur in a committed stem cell with capacity for more limited differentiation. Accumulation of malignant cells leads to progressive impairment of the normal bone marrow function.
Acute leukaemias
Classification of acute myeloblastic leukaemia
AML has traditionally been classified on the basis of morphological features of the disease. Subtypes displaying granulocytic, monocytic, erythroid and megakaryocytic differentiation can be demonstrated. Recently, the World Health Organization (WHO) has updated this system (Table 50.2). AML is now classified using a combination of morphological, genetic and immunological cell marker features (surface antigen expression) in an attempt to define disease groups of greater prognostic significance (Swerdlow et al., 2008).
| Subgroup | Examples |
|---|---|
| AML with recurrent genetic abnormalities | Inversion chromosome 16 (inv 16) |
| t(15;17) | |
| t(8;21) | |
| AML with multilineage dysplasia | |
| Therapy-related AML | |
| AML not otherwise classified | AML without maturation |
| AML with granulocytic maturation | |
| AML with granulocytic and monocytic differentiation | |
| AML with monocytic differentiation | |
| AML with erythroid differentiation | |
| AML with megakaryocytic differentiation |
Classification of acute lymphoblastic leukaemia
As with AML, the WHO classification system takes account of morphological, genetic and immunological features. The disease is, however, mainly classified immunologically, based on the presence or absence of B- or T-cell markers (Table 50.3). Each subtype displays different clinical presentations, response to treatment and, ultimately, prognosis, with pre-B having the best prognosis and B-ALL the worst. It is worth noting that B-ALL (Burkitt’s type), which is associated with translocations of the myc gene normally located on chromosome 8, seems to be a morphologically and biologically distinct form of leukaemia.
| Pre-B ALL | Possessing the common ALL antigen CD 10 |
|---|---|
| B cell type | B-ALL of Burkitt’s type |
| T cell type | T-ALL |
| Null | Non-B, non-T and lacking the common ALL antigen CD 10 |
Chronic leukaemias
Chronic myelocytic leukaemia
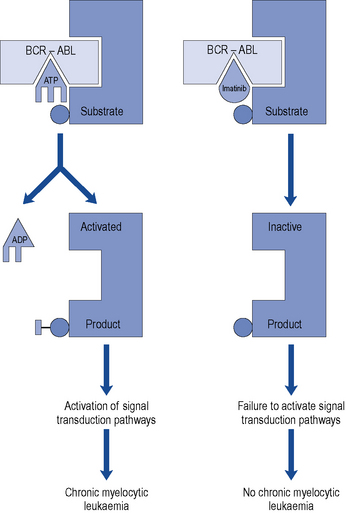
The characteristic feature of CML is the predominance of maturing myeloid cells in blood, bone marrow, liver, spleen and other organs. CML was the first cancer to be associated with a specific chromosomal abnormality: the Philadelphia chromosome translocation (Ph) seen in over 90% of cases. This is a translocation of genetic material between the long arms of chromosome 22 and chromosome 9. This results in the apposition of the BCR gene (chromosome 22) and the ABL gene (chromosome 9). This novel BCR-ABL gene encodes a fusion protein which has tyrosine kinase activity. This genetic event is believed to be crucial in the pathogenesis, or perhaps even to initiate the development, of CML since overactivity of the tyrosine kinase results in the uncontrolled growth characteristic of leukaemic cells (Cilloni and Saglio, 2009).
Clinical manifestations
Investigations
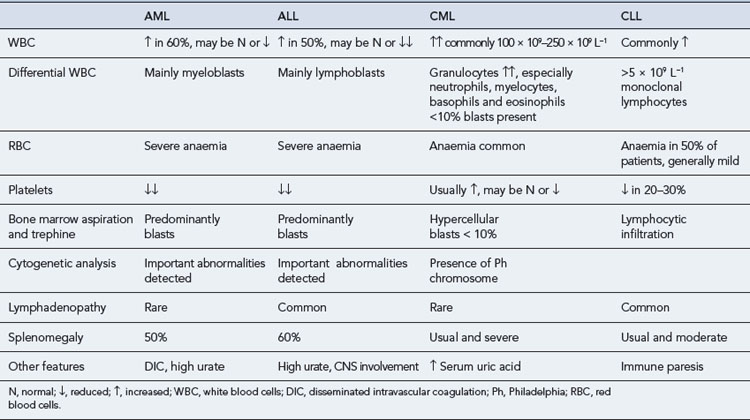
Examinations of peripheral blood and bone marrow are the key laboratory investigations carried out in cases of suspected leukaemia. However, some additional investigations can help in the diagnosis and classification of this group of diseases. Some of the main findings at diagnosis are presented in Table 50.4.
Treatment
Acute leukaemia
Acute lymphoblastic leukaemia
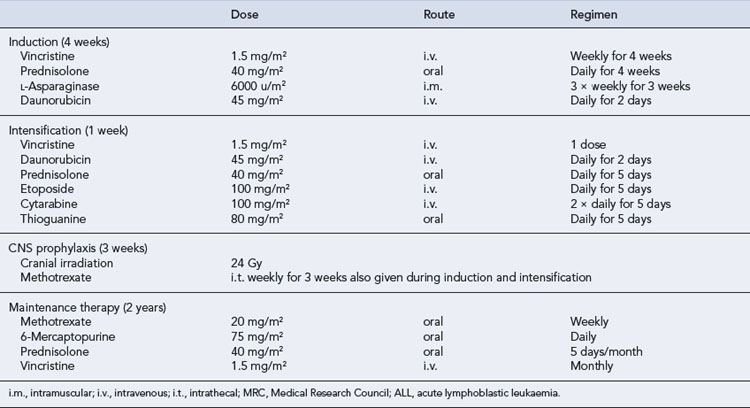
Treatment of ALL in childhood has been one of the success stories of the past 3 decades. Over 80% of children will achieve a remission lasting more than 5 years, and current studies are often focused on trying to identify the 20% of children with poor risk disease and treating them more aggressively (Vrooman and Silverman, 2009). Unfortunately, the results in adults are not so impressive. The combination of vincristine, prednisolone, anthracyclines and asparaginase induces CR in about 90% of children with ALL and 80% of adults, although sadly relapse is far more common in adults (Table 50.5). Other active drugs in the treatment of ALL include methotrexate, 6-mercaptopurine, cyclophosphamide and mitoxantrone.
Acute myeloblastic leukaemia (non-acute promyelocytic leukaemia)
As for ALL, the treatment of AML involves induction and consolidation chemotherapy. In AML therapy, however, the chemotherapy regimens used to achieve remission are much more myelotoxic, and patients require intensive supportive care to survive periods of bone marrow aplasia (Fig. 50.2). The pyrimidine analogue cytarabine has formed the basis of treatment for AML for 20 years. The addition of daunorubicin and oral thioguanine has achieved a CR rate of 75% in patients under the age of 60 years and about 50% in those over 60 years (Dohner et al., 2010). The precise dose and scheduling of these agents is continually being refined in order to improve the response rates. Despite the numbers of patients who achieve CR following induction therapy, the majority relapse, with only about 25% becoming long-term disease-free survivors (Stone et al., 2004). Thus, in common with ALL, additional post-remission therapy is required. Intensive consolidation chemotherapy with high-dose cytarabine and daunorubicin or amsacrine appears to improve survival rates to approximately 50% after 3 years, with even more encouraging results being obtained in patients under 25 years of age (Dohner et al., 2010; Robak and Wierzbowska, 2009). There is generally no role for maintenance therapy in AML. Similarly, CNS prophylaxis is not routinely indicated though patients thought to be at particularly high risk of CNS disease, such as those with testicular or sinus involvement, should receive prophylactic therapy.
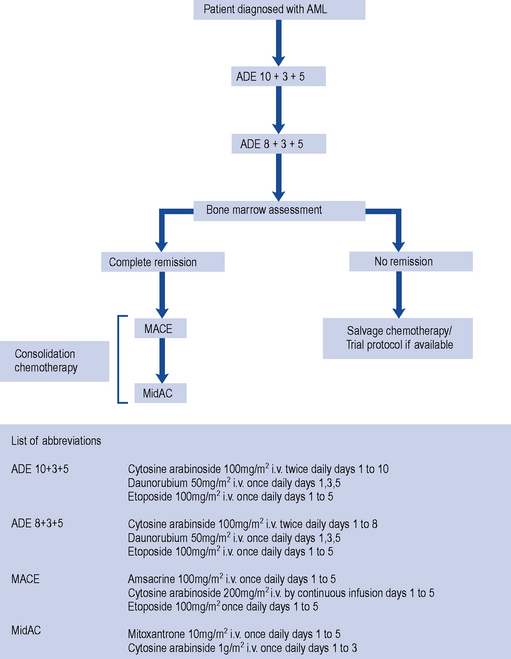
Treatment of AML in relapse is difficult and the prognosis is generally poor. Encouraging results have been seen using a combination of fludarabine, cytosine arabinoside and granulocyte colony-stimulating factor (G-CSF). Novel approaches in AML therapy are often piloted in this group of poor-risk patients. A combination of anti-CD33 antibody, which targets myeloid blasts, with calicheamicin, an anthracycline antibiotic, is a promising and effective approach (Stone et al., 2004), but appears most effective when given in combination with conventional chemotherapy. A newly developed purine analogue, clofarabine, has also been shown to have activity against AML. This drug is a promising agent, particularly in the treatment of older patients, as pilot studies suggest that its toxic effects may be less severe than those associated with other chemotherapy regimens (Robak and Wierzbowska, 2009). 5-Azacytidine is also of interest, especially in older patients and those whose disease has evolved from myelodysplastic syndrome MDS. This agent inhibits DNA methyltransferase resulting in DNA hypomethylation. This process is thought to increase activity of some tumour suppressor genes resulting in anti-tumour effects. The agent has been shown to slow the rate of progression to AML in patients with high-risk myelodysplastic syndrome and is currently the subject of clinical trials in AML.
Acute Promyelocytic Leukaemia
This subtype of AML deserves special consideration as the treatment is quite different from that of other AML variants. APL is associated with the t(15;17) translocation which involves a genetic translocation of material between chromosomes 15 and 17. The disease is clinically characterised by the presence of disseminated intravascular coagulopathy (DIC) at presentation. Since these patients are so prone to life-threatening haemorrhage at diagnosis, the management of a new case of APL is considered a medical emergency. The leukaemic cells are exquisitely sensitive to all-trans retinoic acid (ATRA), which induces blast maturation and can induce remission when used as a single agent (Sanz et al., 2009; Soignet and Maslak, 2004). Using a combination of ATRA and anthracycline chemotherapy, it is now possible to achieve long-term cure in >80% patients. There are some data to suggest that the ongoing use of ATRA in consolidation and maintenance treatment improves outcome further. A number of studies have been published demonstrating the efficacy of arsenic trioxide (ATO) in treating relapsed or refractory APL. Comparison of ATRA and anthracyclines against ATO and ATRA is now the subject of a large UK study of newly diagnosed patients (AML 17). The latter regimen has the potential advantage of avoiding significant myelosuppression.
Chronic leukaemia
Chronic myelocytic leukaemia
Interferon can control symptoms of CML but was also the first agent shown to modify the disease process. It promotes the expression of suppressed normal haemopoiesis at the expense of the malignant clone. Studies have shown that interferon-α therapy prolongs the chronic phase and improves the median survival of patients with CML, and its effects seem to be enhanced by the addition of low-dose cytarabine (Sawyers, 1999).
Therapeutic options for patients with CML have changed dramatically in the past 10 years due to the development of imatinib mesylate. This drug was specifically designed to target the abnormal tyrosine kinase product of the BCR-ABL fusion gene (Fig. 50.3). In a large randomised controlled trial, patients were randomised to receive imatinib or a combination of interferon-α and cytarabine. Many patients were intolerant of the interferon and cytarabine combination and crossed over to receive imatinib after trial commencement. Despite this problem, progression-free survival at 1 year was 97% in the imatinib arm and 80% in the interferon and cytarabine arm in an intention-to-treat analysis (O’Brien et al., 2003). The Philadelphia chromosome became undetectable in 68% of imatinib recipients compared to 7% of those in the alternative arm (Hughes et al., 2003). However, more sensitive testing methods, such as real-time polymerase chain reaction (PCR), have now shown that many patients who are Philadelphia chromosome negative still possess very low levels of the abnormal BCR-ABL gene produced by the Philadelphia translocation.
Chronic lymphocytic leukaemia
Formerly, the alkylating agent chlorambucil was the most common agent used in the treatment of CLL. Corticosteroids can reduce the lymphocyte count without contributing to myelosuppression and are used to treat autoimmune phenomena such as haemolytic anaemia and immune thrombocytopenia. The use of purine analogues, particularly fludarabine, marked the beginning of an exciting phase in the treatment of CLL. Although CRs were unusual, good responses were seen even in patients whose leukaemia was resistant to alkylating agents. With regard to initial therapy of CLL, fludarabine-treated patients show a higher response rate than patients treated with chlorambucil. However, no survival advantage for the use of fludarabine has been demonstrated (Rai et al., 2000). More recently, a large randomised study comparing fludarabine and cyclophosphamide with and without the anti-CD20 antibody, rituximab, has been published (Hallek et al., 2009). This study demonstrated an overall response rate of 86% and 73%, respectively. The median progression-free survival with rituximab is the best published in a large randomised controlled trial to date, at 40 months. This combination of fludarabine, cyclophosphamide and rituximab has been adopted, in the UK, as the regimen of choice for first-line treatment of CLL as long as patients are considered fit enough. Chlorambucil remains an excellent choice for patients with significant co-morbidities as the treatment is less immunosuppressive.
Splenic complications may necessitate splenectomy or splenic irradiation. Radiotherapy can also be used to control localised painful lymphadenopathy. Combination chemotherapy, such as CHOP (see Chapter 51) used in lymphoma, may be beneficial in advanced disease. Campath-1H is a humanised monoclonal anti-CD52 antibody. CD52 is present on most lymphocytes including malignant lymphocytes in CLL. Binding of this antibody induces both antibody-mediated and complement-mediated T-cell cytotoxicity against malignant B cells. In relapsed patients, the duration of response to Campath-1H is relatively short. However, the agent is currently being studied, both in combination with other drugs and as a possible means of purging the bone marrow of residual cells prior to autologous stem cell harvest and transplantation.
Stem cell transplantation
The basic principle
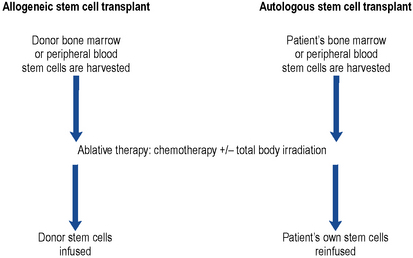
This technique provides a means of rescuing the patient from the potentially lethal effects on the bone marrow of ablative therapy given in an attempt to eradicate all traces of disease (Fig. 50.4). The conditioning regimen most commonly used is a combination of high-dose cyclophosphamide and total body irradiation. Other conditioning regimens include high-dose melphalan, etoposide, busulphan or cytarabine.
Peripheral blood stem cell transplantation
This technique for rescuing bone marrow following ablative conditioning therapy is used to restore haemopoiesis (Russell, 1998). Patients receive the haematopoetic growth factor G-CSF, either alone or following an infusion of high-dose chemotherapy such as high-dose cyclophosphamide. Patients receive G-CSF for a period of about 7 days. This stimulates the release of stem cells into the peripheral circulation. Stem cells are then harvested from the peripheral circulation by a process of cell pheresis. The harvested cells can then be reinfused, fresh, into the patient following conditioning therapy or frozen and stored for later use.
Reduced intensity allografting
Myeloablative allogeneic transplantation, as described earlier, is a very intensive procedure associated with significant morbidity and mortality, hence its restriction to younger patients. Attempts have been made to reduce the intensity of transplant conditioning regimens whilst using increased immunosuppression to facilitate marrow engraftment. Although such an approach reduces the intensity of therapy delivered to any residual tumour, the possible downside of this reduction is offset by an immunological graft-versus-tumour effect and by reduced early post-transplant mortality. This form of transplant is often offered to older patients who are considered unfit to receive a standard allograft. However, the long-term outcomes of this approach are under continual review. Several trials now suggest that the overall survival after myeloablative and reduced intensity transplants may be similar though the causes of death in the two groups are different. More patients die of disease relapse after reduced intensity transplants, and more die of transplant-related complications in the myeloablative group (Pollack et al., 2009).
The place of stem cell transplantation
The place of stem cell transplantation in the management of a particular form of leukaemia depends very much on the prognosis of patients treated with conventional chemotherapy (Table 50.6). For example, the results of intensive chemotherapy in children with ALL are good, and bone marrow transplantation is generally only considered for children who have relapsed and in whom a second remission can be achieved. However, conventional treatment of adults is less successful, and allogeneic bone marrow transplantation may be offered to adults in first remission.
Table 50.6 Indications for allogeneic stem cell transplantation in leukaemia
| AML | First remission with the exception of patients with good risk genetic abnormalities: t(15;17), t(8;21) and inversion of chromosome 16 |
| CML | Chronic phase but only if patient fails to respond to first- and second-line tyrosine kinase inhibitors |
| ALL | First remission in adults |
| Second remission in children | |
| CLL | Not generally appropriate as part of standard therapy but under investigation within several clinical studies |
Patient care
Supportive care
Chapters 51 and 52 examine many of the non-haematological toxicities which result from the use of cytotoxic drugs, and these are clearly pertinent to haematology patients. The major contributors to morbidity and mortality in patients with leukaemia are relapsed disease and infection.
Infection in the immunocompromised patient
A number of intrinsic and extrinsic factors all contribute to the risk of infection in this vulnerable group of patients (Fig. 50.5).
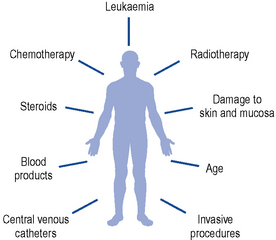
While cross-infection can occur via staff, other patients or contaminated objects, the main sources of infection in this group of patients are endogenous, arising from commensal gut and skin organisms. The normal host defences to infection are broken down; damage to mucous membranes, particularly in the gastro-intestinal tract, occurs with chemotherapy and radiotherapy, allowing infecting organisms to enter the bloodstream. Most infections in neutropenic patients arise from three main sites: the gastro-intestinal and respiratory tracts, and the skin. Table 50.7 lists the main pathogens responsible for infection in this group of patients.
Table 50.7 Pathogens commonly causing infection in neutropenic patients
| Gram-negative bacteria | Pseudomonas spp. |
| Escherichia coli | |
| Klebsiella spp. | |
| Enterobacter spp. | |
| Proteus spp. | |
| Serratia spp. | |
| Legionella pneumophilia | |
| Gram-positive bacteria | Streptococcus spp. |
| Staphylococcus epidermidis | |
| Staphylococcus aureus | |
| Anaerobes | Clostridium difficile |
| Clostridium perfringens | |
| Bacteroides spp. | |
| Fungi | Candida spp. |
| Aspergillus spp. | |
| Viruses | Herpes simplex |
| Herpes zoster | |
| Cytomegalovirus | |
| Hepatitis | |
| Protozoa | Pneumocystis carinii |
Preventive measures
Prophylactic anti-infectives
In general, prophylactic antibiotics are avoided because of the possible development of resistant organisms, but they may have a place in the management of periods of prolonged myelosuppression following chemotherapy and bone marrow transplantation. Prophylactic antifungal agents are often given and patients undergoing bone marrow transplantation and therapy for ALL require prophylaxis against herpes virus and Pneumocystis carinii (Table 50.8).
| Gram-negative bacteria | Ciprofloxacin |
| Candidiasis | Nystatin |
| Fluconazole | |
| Itraconazole | |
| Herpes simplex | Aciclovir |
| Pneumocystis carinii | Co-trimoxazole |
Growth factors
An exciting development in the care of patients with leukaemia has been the production of haemopoietic growth factors using recombinant DNA technology. The first of these, G-CSF, given daily by subcutaneous injection or intravenous infusion after completion of chemotherapy, stimulates neutrophil production and may reduce the duration of neutropenia by up to 7 days. The cost of these compounds is a major issue and results of studies investigating the effects of G-CSF on morbidity and mortality, following chemotherapy, have been disappointing (Smith et al., 2006). Newer pegylated growth factors have been introduced. These have a longer half-life than standard agents, and thus fewer injections are required. The impact of these agents on morbidity and mortality post-chemotherapy, however, remains controversial.
Treatment of infection
Commonly, neutropenic patients show no signs of focal infection; they are unable to form pus. The only clinical manifestations of septicaemia might be general malaise, fever or hypotension. A patient’s condition can deteriorate very rapidly, with collapse occurring within hours of the first signs of infection. Treatment should be instigated as soon as infection is suspected. Following a clinically serious febrile episode (temperature: 37.5 °C for more than 1 h or 38 °C or more on a single reading), samples are taken for culture; these may include blood, urine, sputum and stool cultures along with line and throat swabs. Intravenous antibiotics must be started empirically without delay (Sipsas et al., 2005). Standard empirical therapy varies from unit to unit but may involve the combination of an aminoglycoside with an antipseudomonal penicillin such as piperacillin to provide broad-spectrum bactericidal cover. In penicillin-allergic patients, ceftazidime or cefotaxime may be substituted, but local resistance patterns are of paramount importance. Antibiotics with a broad spectrum of activity, such as ciprofloxacin, have been used as single agents.
Although the antifungal activity of the echinocandin, caspofungin, is more limited than that of amphoteracin or voriconazole, it has been shown to be effective for empirical therapy as it has good efficacy against Candida and Aspergillus spp. It has the advantage of reduced toxicity in comparison with other available agents. The most common side effect is hepatotoxicity. Since this agent has a relatively limited spectrum of activity, it is probably best avoided in the setting of presumed fungal sinus or CNS infection, which are often caused by fungi other than Aspergillus or Candida spp. Table 50.9 lists some of the common problems encountered in the treatment of the leukaemias.
| Problem | Cause | Solutions |
|---|---|---|
| Mucositis and oral ulceration | Chemotherapeutic agents directly toxic to mucosal epithelium | Regular mouth toilet including the use of antibacterial mouthwash Prophylactic use of antiviral and antifungal agents for patients in whom myelosuppression is likely to be prolonged |
| Radiotherapy is directly toxic to the mucosa and also reduces saliva production by salivary glands | ||
| Vulnerable mucosa is likely to be attacked by infective agents, for example, herpes simplex, candida | ||
| Fever in neutropenic patients | Infection predominantly caused by bacteria and/or fungi | Broad-spectrum antibiotics must be commenced as soon as blood cultures have been taken A strategy of planned progressive therapy including use of an antifungal agent in non-responsive fever is appropriate |
| Graft-versus-host disease (GVHD) | T lymphocytes from the donor react against host tissues | Use a sibling donor if possible |
| Use the donor most closely HLA matched to the patient | ||
| Consider T-cell depletion of graft (though this may increase the risk of disease relapse) | ||
| Prophylactic therapy with methotrexate and ciclosporin | ||
| Treat GVHD with corticosteroids, ciclosporin, anti-thymocyte, globulin, FK 506, anticytokine monoclonal antibodies | ||
| Irradiate all blood products | ||
| Late complications of treatment | Risks of haemopoietic malignancy and non-haemopoietic malignancy are increased post-chemotherapy | Aim to tailor therapy to the underlying disease, that is, do not overtreat and do not undertreat |
| Late cardiotoxicity secondary to anthracyclines | Do not exceed maximum cumulative doses of anthracyclines | |
| Liposomal anthracyclines may be useful in the future |
The following practice points should be used to control infection in immunocompromised patients.
Answers
Questions
Answers
Disadvantages of this approach include the need for stimulation of donor haemopoiesis by the G-CSF.
Answers
Answers
Answer
As patients are immunosuppressed, they are also at risk of developing transfusion related graft-versus-host disease. This is a complication of blood product transfusion, caused by engraftment of lymphocytes from the transfused product, which is frequently fatal. This complication can be prevented by irradiation of blood products prior to transfusion. Rituximab is an antibody-based therapy and can be associated with significant reactions which can occasionally be severe and life threatening (see Chapter 51).
Cilloni D., Saglio G. CML: a model for targeted therapy. Balliere’s Best Pract. Res. Clin. Haematol.. 2009;22:285-294.
Dohner H., Estey E.H., Amadori S., et al. Diagnosis and management of acute myeloid leukaemia in adults: recommendations from an international expert panel, on behalf of the European LeukaemiaNet. Blood. 2010;115:453-474.
Hallek M., Fingerle-Rowson G., Fink A.M., et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL Study Group. Blood. 2009;114:535.
Hughes T.P., Kaed J., Branford S., et al. Frequency of major molecular responses to imatinib or interferon-alpha plus cytarabine in newly diagnosed chronic myeloid leukaemia. N. Engl. J. Med.. 2003;349:1423-1432.
Leukaemia and Lymphoma Research. Disease Facts. Available at www.beatbloodcancers.org/leukaemia, 2010.
O’Brien S., Guilhot F., Larson R.A., et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukaemia. N. Engl. J. Med.. 2003;348:994-1004.
Pollack S.M., O’Connor Jr T.P., Hashash J., et al. Non-ablative and reduced intensity conditioning for allogeneic hematopoietic stem cell transplantation: a clinical review. Am. J. Clin. Oncol.. 2009;32:618-628.
Rai K.R., Peterson B.L., Appelbaum F.R., et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukaemia. N. Engl. J. Med.. 2000;343:1750-1757.
Robak T., Wierzbowska A. Current and emerging therapies for acute myeloid leukaemia. Clin. Ther.. 2009;31:2349-2370.
Russell N.H. Developments in allogeneic peripheral blood progenitor cell transplantation. Br. J. Haematol.. 1998;103:594-600.
Sanz M.A., Grimwade D., Tallman M.S., et al. Management of acute promyelocytic leukaemia: recommendations from an expert panel on behalf of European LeukaemiaNet. Blood. 2009;113:1875-1891.
Sawyers C.L. Medical progress: chronic myeloid leukaemia. N. Engl. J. Med.. 1999;340:1330-1340.
Sipsas N.V., Bodey G.P., Kontoyiannis D.P. Perspective for the management of febrile neutropenic patients with cancer in the 21st century. Cancer. 2005;103:1103-1113.
Smith T.J., Khatcheressian J., Lyman G.H., et al. Update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J. Clin. Oncol.. 2006;24:3187-3205.
Soignet S., Maslak P. Therapy of acute promyelocytic leukaemia. Adv. Pharmacol.. 2004;51:35-58.
Stone R.M., O’Donnell M.R., Sekeres M.A. Acute myeloid leukaemia. Hematology. 2004;2004:98-117.
Swerdlow S.H., Campo E., Harris N.L., et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, fourth ed. Lyon: IARC Press; 2008.
Vrooman L.M., Silverman L.B. Childhood ALL: update on prognostic factors. Curr. Opin. Pediatr.. 2009;21:1-8.
Hoffbrand A.V., Pettit J.E., Moss P.A.H. Essential Haematology, fifth ed. Oxford: Blackwell Publishing; 2006.
Howard M.R., Hamilton P.J. Haematology: An Illustrated Colour Text, third ed. Edinburgh: Churchill Livingstone; 2007.
Negrin R.S., Blume K.G. Allogeneic and autologous hematopoietic stem cell transplantation. In Kaushansky K., Lichtman M.A, Beutler E., Kipps T.J., et al, editors: Williams’ Hematology, seventh ed., New York: McGraw-Hill Medical Publishing Division, 2005.
Provan D., Gribben J.G. Molecular Hematology, second ed. Oxford: Blackwell Publishing; 2005.