Left Heart Lesions
Hypoplastic Left Heart Syndrome
Overview: Hypoplastic left heart syndrome (HLHS) is a spectrum of disease characterized by underdevelopment of the left ventricle with obstruction or atresia of ventricular inflow and outflow. HLHS accounts for approximately 2% to 3% of all cases of congenital heart disease, with a slight male predominance.1,2 Chromosomal abnormalities associated with HLHS include Turner syndrome, trisomy 13 and 18, and terminal 11q deletion.1–3
Etiology and Pathophysiology: The structural defects of HLHS include varying degrees of hypoplasia of left heart structures including a hypoplastic ascending aorta and arch, aortic valve atresia or stenosis, hypoplastic left ventricle, mitral atresia or stenosis, and patent ductus arteriosus (PDA) and patent foramen ovale or atrial septal defect (ASD) (Fig. 75-1). In a majority of cases, the ventricular septum is intact, and in severe cases, thickening of the left ventricular endocardium or endocardial fibroelastosis is present.4 Coarctation of the aorta coexists in approximately 80% of patients.5 The right heart structures often are enlarged.5
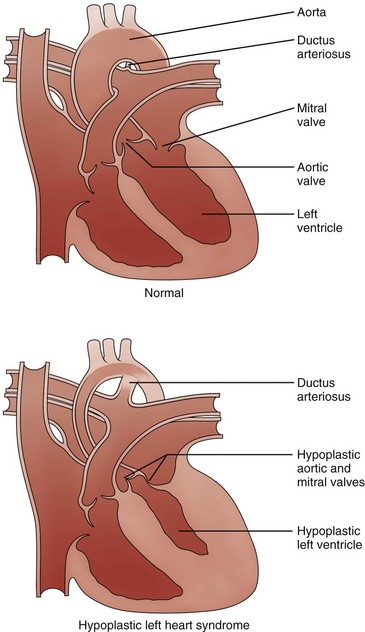
Figure 75-1 Diagram of hypoplastic left heart syndrome.
Compared with the normal heart, the mitral and aortic valves are severely hypoplastic or atretic, the left ventricular cavity and aorta are hypoplastic, and systemic blood flow is supplied by the patent ductus arteriosus. (From American Heart Association. Hypoplastic left heart syndrome. www.americanheart.org. © 2006, American Heart Association, Inc.)
Clinical Presentation: Cyanosis and tachypnea generally are apparent within hours to 2 days after birth. Poor arteriovenous mixing as a result of inadequate interatrial communication can lead to early elevation of left atrial and pulmonary venous pressure, pulmonary edema, and right heart failure. Serious hemodynamic changes occur after the birth of an infant with HLHS when pulmonary vascular resistance begins to drop and the PDA begins to spontaneously close.6 When pulmonary vascular resistance drops, an increase in pulmonary blood flow and a decrease in systemic blood flow occur, leading to a decrease in systemic perfusion. When the PDA constricts, a further decrease in the ductal-dependent systemic and coronary circulations occurs, leading to a decrease in systemic perfusion, myocardial ischemia, shock, and death.
Imaging: HLHS is readily identified in utero. The condition is now discovered in approximately 60% of patients with use of prenatal echocardiography.7,8 Chest radiographic findings are variable. The heart may be normal in size or may be enlarged with a globular configuration, suggesting multichamber enlargement (e-Fig. 75-2). Pulmonary vascularity may be normal in the first hours of life, with a subsequent progressive increase in vascularity if no restriction to blood flow exists at the atrial level. Indistinctness of the pulmonary vessels or pulmonary venous congestion with interstitial lines or pleural fluid may be seen with a restrictive atrial septum.
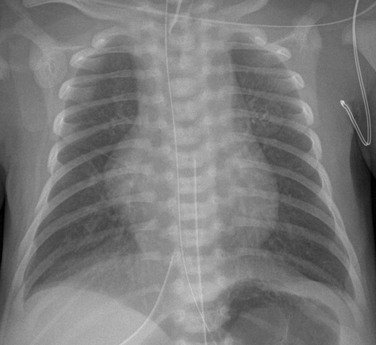
e-Figure 75-2 A newborn with hypoplastic left heart syndrome.
A frontal view of the chest shows a globular cardiac silhouette, consistent with multichamber enlargement. Mild perihilar haziness also is present, indicative of mild venous congestion.
Echocardiography is the imaging method of choice and generally delineates all relevant presurgical anatomy. Cross-sectional imaging can be performed as an adjunct to echocardiography in complex cases.9 Magnetic resonance imaging (MRI) may be especially useful when a marginally hypoplastic left ventricle is present and the possibility exists of a two-ventricle surgical repair (Fig. 75-3 and Video 75-1).4 In these cases, MRI can be used to assess the size of the left atrium, atrial septum, mitral valve orifice, left ventricle, and aortic root to help plan the most appropriate surgical repair. MRI increasingly is being used after the first stage of the palliative single ventricle surgery to reliably quantify the systemic right ventricle systolic function, tricuspid regurgitation, and residual or recurrent coarctation and for pulmonary artery stenosis or hypoplasia before the second-stage bidirectional cavopulmonary anastomosis.10–12
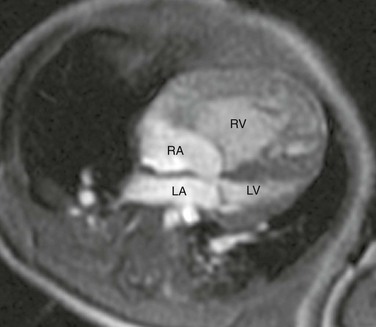
Figure 75-3 Hypoplastic left heart syndrome.
Four-chamber steady-state free-precession (SSFP) magnetic resonance image (MRI) of a 12-week-old with a marginally hypoplastic left ventricle to evaluate for a two-ventricle versus single-ventricle repair. The left atrium (LA) and left ventricle (LV) are small, with the left ventricular volume calculated at 24.2 mL/m2. The study also showed adequate mitral and aortic valve dimensions. Based on the MRI measurements, the patient underwent a successful two-ventricle repair. See Video 75-1 for a four-chamber cine SSFP MRI of the same patient. RA, Right atrium; RV, right ventricle.
Treatment: Initial medical management includes intravenous prostaglandin E1 (PGE1) to maintain ductal patency. Medical therapy, including ventilator adjustments, inhaled agents, and medications, is used to optimize the ratio of pulmonary to systemic blood flow and to minimize the volume load on the single functional ventricle to maintain adequate systemic perfusion.13
Surgical treatment consists of a staged reconstruction procedure (Fig. 75-4). The staged reconstruction involves three surgical procedures with the goal of creating separate pulmonary and systemic circulations supported by the right ventricle and accounts for the high neonatal pulmonary vascular resistance and the subsequent decrease in pulmonary vascular resistance. The first stage of the reconstruction—the Norwood procedure—often is performed in the first week of life.14 This procedure involves transection of the pulmonary trunk proximal to the pulmonary bifurcation and anastomosis of the pulmonary trunk to the aorta. A triangular patch of homograft material is used to augment the hypoplastic ascending aorta, aortic arch, and distal arch. The coronary arteries are then perfused retrograde through the small ascending aorta. The PDA is ligated. A complete atrial septectomy is performed. Blood flow to the lungs is reestablished via a modified Blalock-Taussig shunt (BT shunt) from the subclavian or brachiocephalic artery to the pulmonary artery. (Fig. 75-5). Alternatively, a right ventricle to pulmonary artery (RV-PA) conduit may be placed.15 The potential benefit of the RV-PA conduit is elimination of the diastolic runoff that occurs with a BT shunt. Diastolic runoff can lead to coronary and systemic artery blood flow steal or an increase in the ratio of pulmonary blood flow to systemic blood flow. The potential disadvantages of the RV-PA conduit include right ventricular dysfunction due to the ventriculotomy, the potential for aneurysm formation at the site of the conduit insertion, and right ventricular volume overload from pulmonary regurgitation because there is no valve in the conduit.1,15
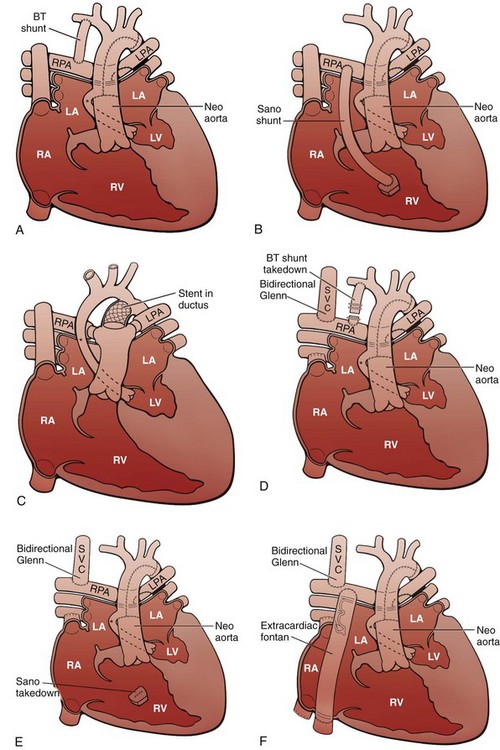
Figure 75-4 Staged reconstruction for hypoplastic left heart syndrome.
A, Stage I of the Norwood reconstruction using a modified Blalock-Taussig (BT) shunt. B, Stage I of the Norwood reconstruction using the Sano modification. C, Hybrid procedure. D, Stage II of the Norwood reconstruction. E, Stage II of the Norwood reconstruction using the Sano modification. F, Fontan procedure. Asterisk, Native ascending aorta; LA, left atrium; LPA, left pulmonary artery; LV, left ventricle; RA, right atrium; RPA, right pulmonary artery; RV, right ventricle; SVC, superior vena cava.
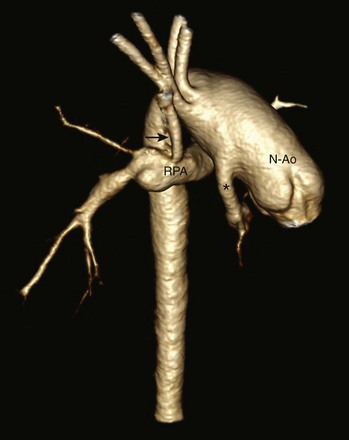
Figure 75-5 A 6-month-old with hypoplastic left heart syndrome after the Norwood I procedure.
Posterior oblique volume-rendered computed tomographic angiography shows the hypoplastic native aorta (asterisk) anastomosed with the native pulmonary artery (now neoaorta) and a Blalock-Taussig shunt (arrow) extending from the right brachiocephalic artery to the right pulmonary artery. N-Ao, Neo-aorta; RPA, right pulmonary artery.
A “hybrid” approach, which combines surgical branch pulmonary artery banding to limit pulmonary blood flow with transcatheter ductal stenting to provide systemic flow, has more recently been advocated to achieve similar physiology to the stage I procedure without requiring cardiopulmonary bypass in the fragile neonate with HLHS (Fig. 75-6).1,16 The comprehensive second-stage procedure then involves cardiopulmonary bypass, removal of the PDA stent and pulmonary artery bands, repair of the aortic arch and pulmonary arteries, division of the diminutive ascending aorta with reimplantation into the pulmonary root, main pulmonary artery to reconstructed aortic anastomosis, atrial septectomy, and a bidirectional Glenn or hemi-Fontan procedure.1,16
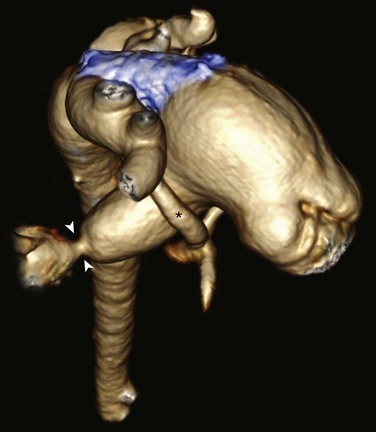
Figure 75-6 A 5-month-old with hypoplastic left heart syndrome after a hybrid procedure.
Posterior oblique volume-rendered computed tomographic angiography shows the hypoplastic native aorta (asterisk). A stent (blue) has been placed to keep the large patent ductus arteriosus open and a pulmonary artery band (arrowheads) has been placed to limit pulmonary blood flow.
The third stage of the reconstruction—the Fontan completion procedure—is generally performed at 18 to 36 months of age. This stage involves directing inferior vena cava blood flow to the pulmonary arteries either via an extracardiac conduit or an intracardiac right atrial baffle (lateral tunnel). If a patch was placed in the right atrium during the second stage of the repair, it is removed. This third stage achieves separation of the systemic and pulmonary circulations. A fenestration may be left in the Fontan circuit so if pressures become high, there can be a pop-off from the Fontan circuit into the heart, which may lead to a more stable postoperative course.17,18 Many fenestrations close spontaneously but also can be closed during a cardiac catheterization procedure.
Survival after the three-stage palliative procedure has improved steadily and now approaches 70%.8 The stage I Norwood procedure carries the highest mortality, ranging from 7% to 19%.15 Cardiac transplantation had been considered an alternative to staged Norwood palliation in the past, but because of limited donor availability and the recent improvement in survival following Norwood palliation, transplantation now generally is reserved for patients for whom staged palliation has failed.1,19
Aortic Stenosis
Overview: Left ventricular outflow tract (LVOT) obstruction can occur at the level of the aortic valve or in the subvalvar or supravalvar regions. This spectrum of disease represents approximately 10% of cases of congenital heart disease.20 Valvar aortic stenosis (AS) is by far the most common form of LVOT obstruction and has a male predominance of nearly 80%.21 Supravalvar AS accounts for 1% to 2% of AS in childhood and occurs spontaneously or may be familial, usually via autosomal dominant transmission. Up to 50% of patients with supravalvar AS have Williams syndrome. Subvalvar AS is slightly more common than supravalvar AS22; it also has a male predominance and may be associated with more complex disease such as double-outlet right ventricle and transposition of the great arteries.
Etiology and Pathophysiology: Congenital aortic valve stenosis results from abnormal valve development rather than the degenerative disease commonly seen in adults. The stenotic valve has variable anatomy, with annular hypoplasia, thickened or tethered leaflets, and/or incompletely developed commissures. Valve morphology may predict clinical severity or associated disease.23,24 Whereas neonatal critical AS often is associated with a unicuspid valve and a small eccentric orifice, bicuspid morphology accounts for up to 95% of cases of congenital valvar AS and is present in up to 30% to 60% of patients with coarctation.25,26 In addition to the abnormality of the bicuspid valve, the aortic root tissue is abnormal in these patients and can lead to significant aortic dilation above a stenotic bicuspid valve. The aortic dilation has been thought to be due to poststenotic dilation, but histologic abnormalities of the ascending aorta can occur without significant valvar stenosis or regurgitation. The histology is similar to the medial disease seen in persons with Marfan syndrome in addition to abnormalities of the smooth muscle, extracellular matrix, elastin, and collagen.27,28
The narrowing in supravalvar AS most commonly is hourglass in shape, occurs immediately above the sinuses of Valsalva at the sinotubular junction, and may be associated with poststenotic dilatation of the aorta, diffuse aortic arch hypoplasia, aortic valve abnormalities, coronary artery ostial stenosis, or left ventricular hypertrophy.29,30 Supravalvar AS often is part of a widespread arteriopathy.
Subvalvar AS can be discrete or diffuse. The discrete form, which represents the majority of cases, is a thin fibromuscular diaphragm encircling the LVOT. In the more severe diffuse form, a fibromuscular subaortic band is present along the length of the LVOT, producing a tunnel-like narrowing. The fibrous process often extends to involve the aortic valve cusps or the anterior mitral valve leaflet. The more diffuse form usually is associated with other left heart lesions, including mitral stenosis, supramitral ring, parachute mitral valve, valvar AS, or coarctation of the aorta.31
Clinical Presentation: Severe or critical AS may be diagnosed prenatally or present early in infancy with severe left heart obstruction, congestive heart failure, dyspnea, and poor peripheral circulation. Systemic flow is ductal dependent in cases of critical AS. The clinical manifestations of AS in infancy depend on the degree of valvar obstruction, mitral insufficiency, left atrial hypertension, and left ventricular dysfunction, as well as the amount of shunted atrial and ductal flow and other associated left-sided obstructive lesions. In cases of severe obstruction, the left ventricle may be severely hypoplastic, dilated, or dysfunctional.
Imaging: The chest radiograph in infants with critical AS shows cardiomegaly and pulmonary venous congestion. In older children with mild stenosis, the radiograph generally is normal. When moderate to severe stenosis and left ventricular hypertrophy are present, the cardiac apex is depressed toward the diaphragm and posteriorly to the inferior vena cava. Left atrial enlargement can be seen with severe stenosis. Poststenotic dilation of the ascending aorta is rare in young children (Fig. 75-7).
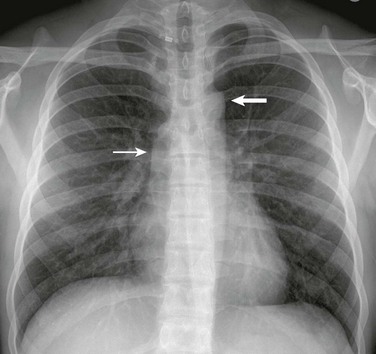
Figure 75-7 Aortic valve stenosis.
A frontal view of the chest in an 18-year-old shows poststenotic dilation of the ascending aorta (thin arrow) and aortic arch (thick arrow).
Echocardiography is the imaging procedure of choice for the evaluation of valvar AS. Cardiac MRI can be used to complement echocardiography in cases of poor acoustic windows or if larger field of view imaging is indicated. Goals of imaging include demonstration of the degree and location of obstruction, valve morphology, leaflet mobility and effective valve orifice area. Systolic valve area calculated by MRI planimetry correlates well with transesophageal echocardiography and cardiac catheterization measurements (Fig. 75-8 and Video 75-2).32 The valve typically appears thickened and doming, with asymmetric or restricted leaflet excursion. The gradient across the aortic valve in millimeters of mercury is calculated using the modified Bernoulli equation (4V2, where V is peak Doppler velocity beyond the aortic valve in meters per second). Aortic valvular regurgitant fraction is calculated with phase-contrast MRI (e-Fig. 75-9). Evaluation of left ventricular size, systolic performance, and diastolic dysfunction is necessary. Assessment of aortic root, ascending aorta, and arch size is crucial in infants with critical AS. These patients may have abnormal endocardium, which can indicate the presence of endocardial fibroelastosis (e-Fig. 75-10). Surveillance of the thoracic aorta is indicated to evaluate for the development of aortic root dilation and aortic dissection associated with aortic valve disease (e-Fig. 75-11 and Video 75-3).28
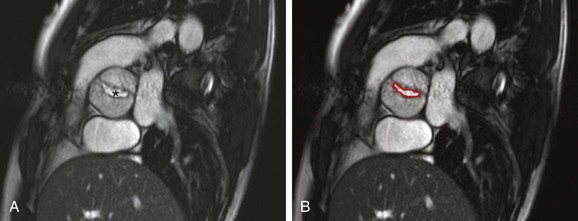
Figure 75-8 Bicuspid aortic valve in a 25-year-old with history of aortic coarctation.
A, A cross-sectional steady-state free-precession (SSFP) systolic image of a severely stenotic bicuspid aortic valve (asterisk) with fusion of the right and noncoronary commissures, giving a “fish-mouth” appearance. See Video 75-2 for a cine SSFP image of the same. B, The aortic valve area (outlined in red) as measured by planimetry is 1.18 cm2.
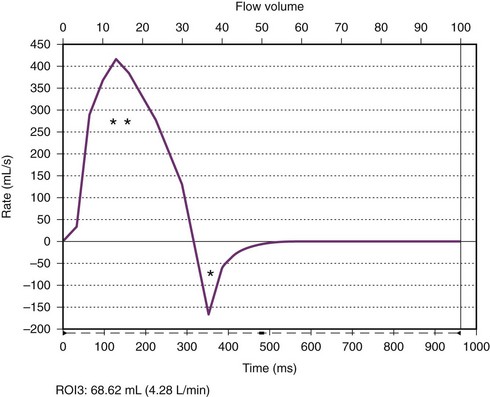
e-Figure 75-9 Aortic valve flow curve.
A flow curve obtained by magnetic resonance phase-contrast imaging acquired just above the aortic valve shows forward (two asterisks) and regurgitant (single asterisk) flow. The regurgitant fraction (regurgitant flow volume/forward flow volume) was calculated to be 18%.
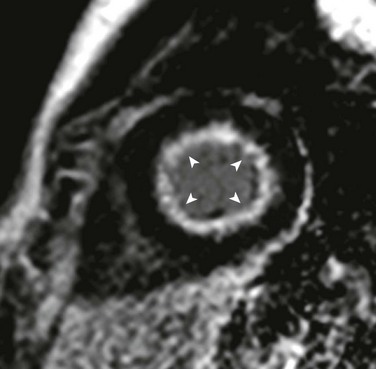
e-Figure 75-10 Endocardial fibroelastosis.
Short-axis delayed enhancement image in a 14-year-old with bicuspid aortic valve after multiple balloon aortic valvuloplasty procedures who, in his teenage years, experienced progressive heart failure symptoms including dyspnea on exertion, increasing fatigue, and decreased exercise tolerance in the setting of normal systolic function. Delayed enhancement inversion recovery images show diffuse subendocardial late enhancement (arrowheads). He underwent a high-risk, but ultimately successful, cardiac transplantation approximately 1 year after he became symptomatic. Pathology of the explanted heart revealed a bicommissural aortic valve, biventricular hypertrophy, hypertrophic subendocardial thebesian veins with prominent intimal thickening, and diffuse endocardial fibroelastosis. (Modified from Robinson JD, Del Nido PJ, Geggel RL, et al. Left ventricular diastolic heart failure in teenagers who underwent balloon aortic valvuloplasty in early infancy. Am J Cardiol. 2010;106[3]:426-429.)
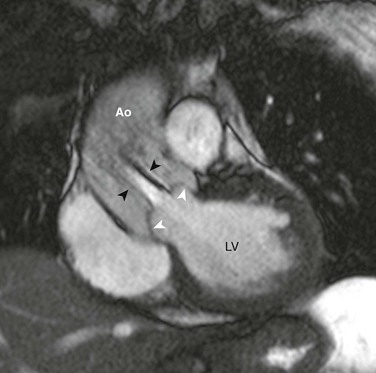
e-Figure 75-11 Bicuspid aortic valve in a 25-year-old with history of aortic coarctation.
Steady-state free-precession (SSFP) magnetic resonance image (MRI) perpendicular to the plane of the bicuspid aortic valve shows upward ballooning of the aortic valve leaflets (white arrowheads) and a turbulent poststenotic jet (black arrowheads) into the dilated ascending aorta (Ao). See Video 75-3 for cine SSFP MRI of the same case with a regurgitant jet visible. LV, Left ventricle.
In supravalvar AS, imaging is tailored to evaluate for the presence of discrete or diffuse supravalvar narrowing of the ascending aorta, the coronary ostia, the degree of left ventricular hypertrophy, ventricular function, supravalvar pulmonary stenosis, and other sites of vasculopathy as clinically indicated (Fig. 75-12). In patients with subvalvar AS, evaluation for the discrete subaortic membrane or ridge and definition of the long subaortic area of the tunnel-type subaortic lesion is necessary (Fig. 75-13).
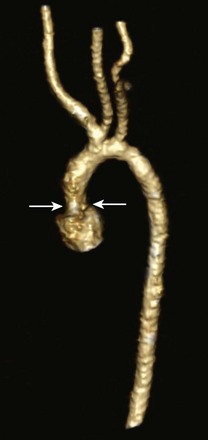
Figure 75-12 Supravalvar aortic stenosis (AS).
An oblique volume-rendered three-dimensional computed tomography angiographic image shows supravalvar AS with an abrupt caliber change in the proximal ascending aorta (arrows).
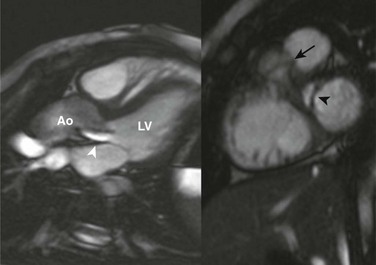
Figure 75-13 Subaortic stenosis.
Steady-state free-precession magnetic resonance imaging long-axis (left) and short-axis (right) images of the left ventricular outflow tract (LVOT) show a subaortic membrane (black arrowhead) causing a jet of flow acceleration across the LVOT (white arrowhead) beginning just below the aortic valve plane (arrow). Ao, Aorta; LV, left ventricle.
Treatment: Infants with severe or critical AS need urgent treatment. Patients are supported with a PGE1 infusion to maintain ductal patency, mechanical ventilation, and inotropic medications. Urgent percutaneous balloon valvuloplasty is the treatment of choice. A neonatal Ross procedure (i.e., replacement of the aortic valve with a pulmonary valve annulus and trunk autograft, coronary reimplantation, and replacement of the pulmonary valve with a homograft conduit) has been advocated by some persons as the treatment of choice in patients with dysplastic valves or small aortic annuli33,34; however, increased mortality and higher rates of autograft deterioration in children compared with adults has resulted in less enthusiasm by others.35,36 During the era of aggressive transcatheter intervention, early mortality for infants with severe or critical AS has fallen from as high as 43% to 4% to 13%.37,38
Balloon valvotomy is considered in older children with a peak systolic ejection gradient of 50 mm Hg or greater at catheterization, for patients with angina, syncope, or congestive heart failure, and for persons who want to play competitive sports or become pregnant.39,40 Routine follow-up is necessary to evaluate for aortic insufficiency and recurrent stenosis. Late surgical valve repair or replacement is necessary in approximately 25% to 35% of patients.38,41 For patients who require valve replacement, a bioprosthetic or mechanical valve may be placed or a Ross procedure may be performed.
Patients with AS are at increased risk for sudden death and endocarditis. The risk of sudden death is thought to significantly increase with symptoms40 but probably has decreased in the era of aggressive and effective transcatheter treatment.42 The incidence of subacute bacterial endocarditis is rare but is approximately 35 times greater than in the general population and also increases with AS severity.43,44
Indications for interventions related to supravalvar AS are less clear, but surgery is performed in symptomatic cases. The high likelihood of progression influences consideration for aortoplasty or LVOT reconstruction. Balloon angioplasty and stenting of associated pulmonary stenoses may be necessary. Complications include aortic aneurysms and infective endocarditis, and surgical mortality is low.45 Given the recurrent and progressive nature of diffuse arteriopathy, long-term follow-up is warranted.
In asymptomatic patients, the timing of surgical intervention for subvalvar AS is controversial and is related to the degree and progression of LVOT obstruction and other associated heart disease.46–48 For persons with discrete subvalvar obstruction, fibromuscular resection with or without septal myectomy is performed, and operative mortality is low. Treatment for the tunnel-like narrowing form of obstruction varies, depending on the size and function of the aortic valve. The Konno procedure (i.e., aortoventriculoplasty, including replacement of the aortic valve) and modifications including valve repair or the Ross procedure (described earlier) may be performed (e-Fig. 75-14). A wide rage in the rates of recurrence and complications such as aortic regurgitation are reported; however, operative mortality, recurrence, and progressive AS are more common after repair of the diffuse type of subvalvar AS.
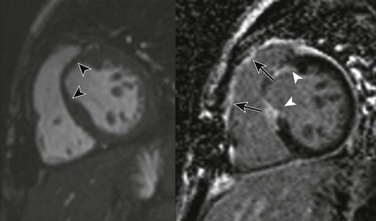
e-Figure 75-14 Ross-Konno procedure for aortic and subaortic stenosis in a 17-year-old.
Magnetic resonance imaging short-axis steady-state free-precession (left) and postcontrast phase-sensitive inversion recovery delayed enhancement (right) views show thinning of the myocardium in the region of the left ventricular outflow tract (black arrowheads) with corresponding late enhancement (white arrowheads) consistent with the Konno ventriculoplasty and Ross procedure. Also, note the postoperative late enhancement in the region of the right ventricular outflow tract related to placement of the pulmonary homograft (arrows).
Coarctation of the Aorta
Overview: Coarctation of the aorta accounts for approximately 7% of cases of congenital heart disease,49 with a male to female ratio of approximately 1.5 : 1.28 Coarctation of the aorta is isolated or present with a PDA in 82% of cases.50 A bicuspid aortic valve is seen in up to 30% to 60% of patients,25,26,51 and ventricular septal defect (VSD) is seen in 11% of patients.50 Other associated lesions include ASD, left-sided obstructive lesions, transposition of the great vessels, double-outlet right ventricle, and atrioventricular septal defect in 7% of patients.50 Eleven percent to 15% of patients with Turner syndrome have aortic coarctation.52,53 Berry aneurysms are seen in up to 10% of patients.54 Coarctation of the aorta is seen in 15% of patients with PHACES syndrome (posterior fossa defects, hemangiomas, arterial anomalies, cardiac defects and coarctation, eye anomalies, and sternal defects or supraumbilical raphe).55
Etiology and Pathophysiology: Coarctation of the aorta is a narrowing of the thoracic aorta that generally occurs adjacent to the insertion site of the ductus arteriosus just distal to the left subclavian artery. The coarctation is almost always juxtaductal and discrete, but varying degrees of tubular hypoplasia of the transverse arch and isthmus may be present and are more commonly seen in infancy.56 The embryology of aortic coarctation is not known, but two theories have been proposed. One theory, called the ductal sling theory, suggests that an abnormal extension of contractile ductal tissue occurs circumferentially around the aortic lumen. Contraction of this tissue with ductal closure leads to a shelflike aortic narrowing in the juxtaductal region.56 The second theory, called the flow theory, postulates that aortic coarctation develops as a result of decreased blood flow through the aortic isthmus as a result of left-sided obstructive lesions. In fetal life the aortic isthmus normally receives a relatively low volume of blood flow. Most of the flow to the descending aorta arises from the right ventricle through the PDA. The left ventricle supplies highly oxygenated blood to the ascending aorta and brachiocephalic vessels, with a small amount of flow going through the aortic valve. With left-sided obstructive lesions, decreased isthmic flow occurs, promoting abnormal isthmic development and leading to aortic coarctation.56,57
The pathophysiology of coarctation of the aorta is related to the severity of the arch obstruction, the presence of collateral vessels, and associated cardiac lesions.50 Initially, the neonate with coarctation will have a PDA, allowing blood flow to bypass the obstruction. With severe obstruction, left ventricular afterload is increased acutely following closure of the PDA, and patients then may present with congestive heart failure and shock. With less severe obstruction, collateral blood vessels develop to bypass the obstruction. The development of adequate collateral vessels may mask the presence of the coarctation until later in childhood or adulthood. Upper extremity hypertension is present in 90% of children and is thought to be secondary to three proposed mechanisms, including mechanical aortic obstruction, abnormal body baroreceptor settings proximal to the obstruction, and hyperactivation of the renin-angiotensin system caused by the underperfused kidneys.50,58,59 Left-sided obstructive lesions may further increase left ventricular afterload, and the presence of a VSD may further increase left ventricular volume load, leading to pulmonary venous and arterial hypertension and heart failure.
Clinical Presentation: Infants with critical coarctation of the aorta, especially when it is associated with other cardiac defects, typically present by 7 to 14 days of life as the PDA begins to close and they have dramatic signs of congestive heart failure and poor systemic perfusion. Clinical symptoms include dyspnea, poor feeding, tachycardia, and signs of shock, including oliguria, anuria, and severe acidemia. Femoral pulses are weak or absent, and differential blood pressures generally show a gradient between the upper and lower limbs.50
Coarctation of the aorta also may be diagnosed from infancy to adulthood in asymptomatic or mildly symptomatic patients, with a reported median age of 10 years (range, 1 to 36 years).60 These patients present later because the coarctation is not significant or because adequate collateral circulation has developed. The diagnosis usually is made on the basis of routine physical examination findings such as an incidental murmur, hypertension, or absent or diminished lower extremity pulses.60
Imaging: Infants with severe coarctation present with moderate to marked cardiomegaly and increased pulmonary vascular markings due to venous congestion from obstruction or overcirculation with an associated VSD (e-Fig. 75-15). Congestive heart failure presenting between the first and fourth weeks of life strongly suggests coarctation. Chest radiographs in children older than 1 year and younger than 5 years usually show a normal heart size, but cardiomegaly due to left ventricular hypertrophy can be seen. Rib notching as a result of dilated intercostal collateral arteries forming grooves on the rib undersurfaces is the hallmark of this condition and usually is seen after 5 years of age. The lower borders of the fourth through eighth ribs usually are involved posteriorly.61 The rib notching is bilateral unless an aberrant subclavian artery arising distal to the coarctation is present. Poststenotic dilation of the aorta below the coarctation usually occurs. A “figure three” sign frequently is seen along the left upper mediastinal border, related to the prominent aortic knob and left subclavian artery proximal to the coarctation, the indentation from the coarctation, and the poststenotic aortic segment below the coarctation (Fig. 75-16). The imprint of the aorta on the adjacent barium-filled esophagus, which is known as the “E” sign or “reversed three” sign, is caused by indentation of the esophagus by the dilated aorta proximal and distal to the coarctation.
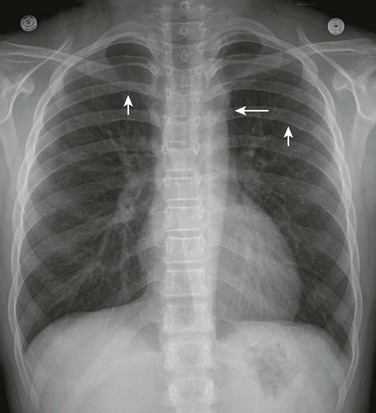
Figure 75-16 Coarctation of the aorta in a 10-year-old.
A frontal view of the chest shows the “figure three” sign (long arrow), which is indicative of aortic coarctation, and bilateral posterior rib notching (short arrows), which is indicative of collateral vessels.
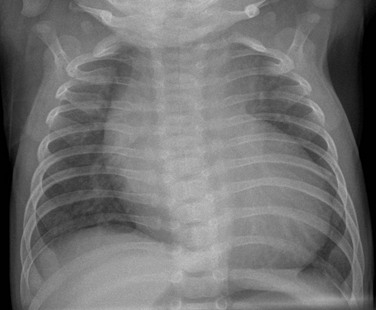
e-Figure 75-15 Critical aortic coarctation in a 2-week-old.
A frontal view of the chest shows cardiomegaly with mild pulmonary vascular congestion.
Echocardiography is the method of choice for imaging coarctation in infancy. Computed tomography and MRI can provide detailed anatomic imaging of the coarctation in older children or adults with limited acoustic windows (Figs. 75-17 and 75-18). MRI also can be used to assess the hemodynamic significance of the coarctation. A complete imaging examination involves imaging for ventricular systolic function, volume and mass; atrioventricular and semilunar valve assessment with particular attention to the aortic valve; and imaging of the aortic root, ascending aorta, arch, coarctation, and descending aorta to the renal arteries. Maximum blood flow velocity, blood flow volume, and flow pattern in the aorta just distal to the coarctation can be assessed using phase-contrast MRI sequences (e-Fig. 75-19 and Video 75-4). The maximal flow velocity obtained across the coarctation can be used in the Bernoulli equation (see the previous aortic stenosis imaging section) to estimate the gradient across the coarctation.62 Aortic flow volume at the diaphragm also can be assessed using phase-contrast sequences. In patients with significant coarctation, the flow volume at the diaphragm may be increased relative to the volume just distal to the coarctation as a result of recruitment of collateral flow through the intercostal arteries and can be an indicator of the significance of the coarctation.63–65
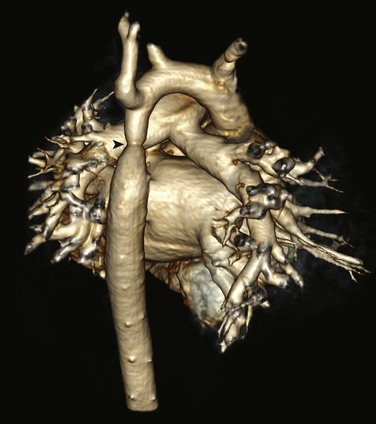
Figure 75-17 Aortic coarctation in a 3-year-old.
Posterior projection from a volume-rendered magnetic resonance angiography image shows a discrete aortic coarctation (arrowhead) distal to the left subclavian artery. No significant collateral vessels are seen.
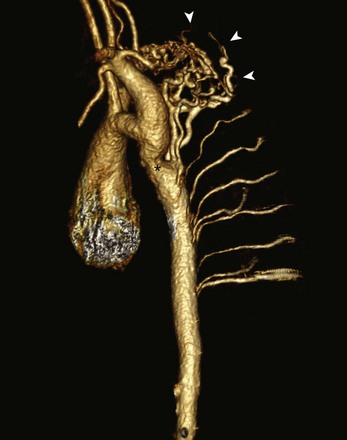
Figure 75-18 Aortic coarctation in a 14-year-old.
Sagittal oblique three-dimensional magnetic resonance angiography reconstruction shows a discrete aortic coarctation distal to the left subclavian artery (asterisk) with multiple dilated tortuous collateral vessels arising proximal to the coarctation (arrowheads) and feeding the descending aorta distal to the coarctation.
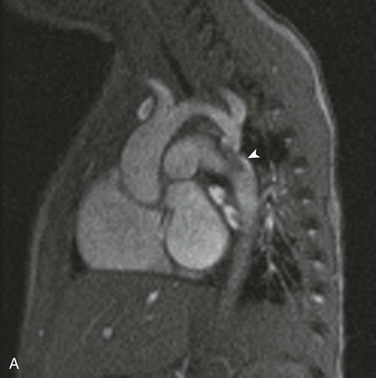
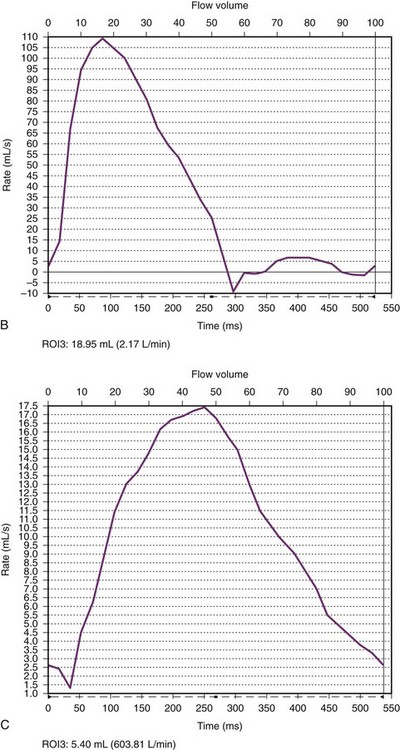
e-Figure 75-19 Aortic coarctation in a 16-month old.
A, An oblique sagittal still image from a cine gradient echo image series shows a systolic flow void distal to the coarctation (arrowhead) indicative of turbulent flow across the coarctation. The transverse aortic arch is mildly hypoplastic. B, A flow curve obtained by phase-contrast imaging in the aortic arch proximal to the coarctation shows a normal arterial flow pattern. C, A flow curve obtained by phase-contrast imaging in the descending aorta distal to the coarctation shows a slow upstroke, lower stroke volume, slow downstroke, and forward flow throughout ventricular diastole. See Video 75-4 for oblique sagittal cine gradient echo images showing systolic flow void beyond the coarctation consistent with turbulent flow across the coarctation.
Treatment: Coarctation of the aorta that presents with congestive heart failure in the neonatal period is managed medically with inotropic drugs and PGE1 to maintain ductal patency and improve blood flow to the descending aorta. After stabilization with medical therapy, urgent surgical repair is the treatment of choice.66,67 Balloon angioplasty can be considered as an initial procedure for neonates at high surgical risk.66
Repair is performed on a more elective basis in patients who present in infancy or childhood with a murmur or upper extremity hypertension compared with critically ill neonates. Conventional therapy for aortic coarctation is surgical. Currently, the surgical procedure used most commonly in infants and children is resection of the coarctation with end-to-end or extended end-to-end anastomosis.50,66,67 Other surgical procedures, including patch aortoplasty and subclavian flap repair, are less commonly performed as the procedure of choice in children because of the risk of aneurysm formation at the patch site with patch aortoplasty and left arm complications and remnant residual ductal tissue with the subclavian flap repair.50,66 The efficacy of balloon angioplasty for native aortic coarctation in infants and children has been a topic of controversy because aneurysm formation and iliofemoral artery injury are reported as being higher68 and the reintervention rate for recurrent stenosis may be higher than for conventional surgical therapy.66 Stent placement can be considered for primary coarctation repair in children who weigh more than 10 to 15 kg.69 The transcatheter approach is accepted therapy for treatment of recoarctation in all age groups.66 Late complications after surgical or endovascular treatment include recurrent coarctation, aneurysm or pseudoaneurysm formation, aortic dissection, and hypertension.50,66,70
Interrupted Aortic Arch
Overview: Interrupted aortic arch (IAA) is rare and accounts for approximately 1.5% of congenital heart anomalies.71 IAA consists of discontinuity between the ascending and descending aorta and is classified according to the location of the arch interruption.72 Type A interruption is the second most common type (seen in 42% of cases), occurs at the isthmus just beyond the origin of the left subclavian artery, and is seen in association with aortopulmonary window with an intact ventricular septum and with transposition of the great arteries and VSD.73 Type B interruption is most common (seen in 58% of cases), occurs between the left common carotid and left subclavian arteries, and usually is associated with conotruncal abnormalities, large posterior malalignment VSDs, and subaortic stenosis.73 Type B interruption is associated with aberrant origin of the right subclavian artery from the descending aorta.74 DiGeorge syndrome is relatively common in patients with type B IAA and often is associated with a right aortic arch.75 Type C interruption is least common (appearing in 4% of cases) and occurs between the right and left common carotid arteries. A PDA is seen in nearly all persons with IAA. Other lesions reported in association with IAA include isolated VSD (73%),71 ASD, bicommissural aortic valve, aortic stenosis, and more complex lesions including truncus arteriosus and the Taussig-Bing type of double-outlet right ventricle (see Chapter 76).76
Etiology, Pathophysiology, and Clinical Presentation: The embryology of IAA depends on the type of interruption. Type A is formed as a result of abnormal distal fourth arch regression during late development, after the ascent of the left subclavian artery. Type B results from an early regression of the fourth arch before migration of the left subclavian artery. Type C likely results from abnormal regression of portions of the left third and fourth arches.
Imaging: IAA may be diagnosed prenatally. Postnatal imaging is directed at defining the arch sidedness and branching pattern, the site of arch interruption, the size of the proximal and distal portions of the arch, the distance of interruption, the size of the aortic annulus, and the presence of a PDA (Fig. 75-20).71 Screening for other cardiovascular abnormalities also is performed. Echocardiography generally is sufficient for preoperative assessment. Cross-sectional imaging is obtained to better define arch branching or other cardiovascular anatomy as needed.
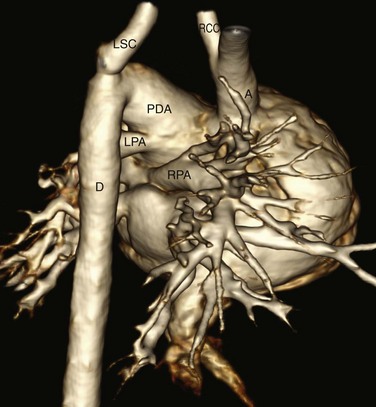
Figure 75-20 Newborn with type B interrupted aortic arch and truncus arteriosus.
A three-dimensional computed tomography reconstruction right posterior view shows interruption of the aortic arch between the right common carotid (RCC) and left subclavian (LSC) arteries. The right (RPA) and left (LPA) pulmonary arteries arise from the pulmonary trunk. A large patent ductus arteriosus (PDA) supplies the descending aorta (D) from which the left subclavian artery arises. The common origin of the aorta and pulmonary artery from a single truncal valve is not shown. A, Ascending aorta.
Treatment: PGE1 infusion is begun upon patient presentation to keep the ductus arteriosus patent. Metabolic abnormalities are treated medically, and surgical repair is undertaken as soon as the infant is clinically stable.71
Complete surgical repair is performed in the neonatal period with generally good outcomes that depend on associated cardiovascular lesions.76 Direct arch anastomosis between the ascending and descending aorta with ductal ligation is the preferred technique. Homograft, pericardial patch, or autologous carotid artery augmentation may be needed if the arch is hypoplastic.76 An intervening conduit generally is not placed unless associated anomalies are present. Surgical technique modifications performed as a result of associated anomalies are based on individual patient anatomy.71
Postoperatively, monitoring for arch obstruction or dilation at the site of anastomosis is necessary. The incidence of postoperative arch obstruction is low, with a reported 74% actuarial freedom from arch obstruction necessitating reintervention at 15 years.76 Assessment for complications related to other cardiovascular anomalies such as recurrent LVOT obstruction also is undertaken. Postoperative imaging generally is performed with echocardiography, with supplementary cross-sectional imaging as needed.
Abbruzzese, PA, Aidala, E. Aortic coarctation: an overview. J Cardiovasc Med (Hagerstown). 2007;8(2):123–128.
Egan, M, Holzer, RJ. Comparing balloon angioplasty, stenting and surgery in the treatment of aortic coarctation. Expert Rev Cardiovasc Ther. 2009;7(11):1401–1412.
Fernandes, SM, Sanders, SP, Khairy, P, et al. Morphology of bicuspid aortic valve in children and adolescents. J Am Coll Cardiol. 2004;44(8):1648–1651.
Konen, E, Merchant, N, Provost, Y, et al. Coarctation of the aorta before and after correction: the role of cardiovascular MRI. AJR Am J Roentgenol. 2004;182(5):1333–1339.
Ohye, RG, Sleeper, LA, Mahony, L, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362(21):1980–1992.
References
1. Barron, DJ, Kilby, MD, Davies, B. Hypoplastic left heart syndrome. Lancet. 2009;374(9689):551–564.
2. Morris, CD, Outcalt, J, Menashe, VD. Hypoplastic left heart syndrome: natural history in a geographically defined population. Pediatrics. 1990;85(6):977–983.
3. Natowicz, M, Chatten, J, Clancy, R, et al. Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics. 1988;82(5):698–706.
4. Grosse-Wortmann, L, Yun, TJ, Al-Radi, O, et al. Borderline hypoplasia of the left ventricle in neonates: insights for decision-making from functional assessment with magnetic resonance imaging. J Thorac Cardiovasc Surg. 2008;136(6):1429–1436.
5. Hawkins, JA, Doty, DB. Aortic atresia: morphologic characteristics affecting survival and operative palliation. J Thorac Cardiovasc Surg. 1984;88(4):620–626.
6. Abu-Harb, M, Wyllie, J, Hey, E, et al. Presentation of obstructive left heart malformations in infancy. Arch Dis Child Fetal Neonatal Ed. 1994;71(3):F179–F183.
7. Perolo, A, Prandstraller, D, Ghi, T, et al. Diagnosis and management of fetal cardiac anomalies: 10 years of experience at a single institution. Ultrasound Obstet Gynecol. 2001;18(6):615–618.
8. Stumper, O. Hypoplastic left heart syndrome. Postgrad Med J. 2010;86(1013):183–188.
9. Dillman, JR, Yarram, SG, Hernandez, RJ. Imaging of pulmonary venous developmental anomalies. AJR Am J Roentgenol. 2009;192(5):1272–1285.
10. Jones, BO, Ditchfield, MR, Cahoon, GD, et al. Cardiac magnetic resonance imaging prior to bidirectional cavopulmonary connection in hypoplastic left heart syndrome. Heart Lung Circ. 2010;19(9):535–540.
11. Muthurangu, V, Taylor, AM, Hegde, SR, et al. Cardiac magnetic resonance imaging after stage I Norwood operation for hypoplastic left heart syndrome. Circulation. 2005;112(21):3256–3263.
12. Brown, DW, Gauvreau, K, Powell, AJ, et al. Cardiac magnetic resonance versus routine cardiac catheterization before bidirectional glenn anastomosis in infants with functional single ventricle: a prospective randomized trial. Circulation. 2007;116(23):2718–2725.
13. Reddy, VM, Liddicoat, JR, Fineman, JR, et al. Fetal model of single ventricle physiology: hemodynamic effects of oxygen, nitric oxide, carbon dioxide, and hypoxia in the early postnatal period. J Thorac Cardiovasc Surg. 1996;112(2):437–449.
14. Norwood, WI, Lang, P, Hansen, DD. Physiologic repair of aortic atresia-hypoplastic left heart syndrome. N Engl J Med. 1983;308(1):23–26.
15. Ohye, RG, Sleeper, LA, Mahony, L, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362(21):1980–1992.
16. Galantowicz, M, Cheatham, JP. Lessons learned from the development of a new hybrid strategy for the management of hypoplastic left heart syndrome. Pediatr Cardiol. 2005;26(3):190–199.
17. Bridges, ND, Lock, JE, Castaneda, AR. Baffle fenestration with subsequent transcatheter closure. Modification of the Fontan operation for patients at increased risk. Circulation. 1990;82(5):1681–1689.
18. Salazar, JD, Zafar, F, Siddiqui, K, et al. Fenestration during Fontan palliation: now the exception instead of the rule. J Thorac Cardiovasc Surg. 2010;140(1):129–136.
19. Backer, CL, Idriss, FS, Zales, VR, et al. Cardiac transplantation for hypoplastic left heart syndrome: a modified technique [see comment]. Ann Thorac Surg. 1990;50(6):894–898.
20. Samanek, M, Slavik, Z, Zborilova, B, et al. Prevalence, treatment, and outcome of heart disease in live-born children: a prospective analysis of 91,823 live-born children. Pediatr Cardiol. 1989;10(4):205–211.
21. Wagner, HR, Ellison, RC, Keane, JF, et al. Clinical course in aortic stenosis. Circulation. 1977;56(1 suppl):I47–I56.
22. Kitchiner, D, Jackson, M, Malaiya, N, et al. Incidence and prognosis of obstruction of the left ventricular outflow tract in Liverpool (1960-91): a study of 313 patients. Br Heart J. 1994;71(6):588–595.
23. Fernandes, SM, Sanders, SP, Khairy, P, et al. Morphology of bicuspid aortic valve in children and adolescents. J Am Coll Cardiol. 2004;44(8):1648–1651.
24. Roberts, WC, Ko, JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111(7):920–925.
25. Becker, AE, Becker, MJ, Edwards, JE. Anomalies associated with coarctation of aorta: particular reference to infancy. Circulation. 1970;41(6):1067–1075.
26. Teo, LL, Cannell, T, Babu-Narayan, SV, et al. Prevalence of associated cardiovascular abnormalities in 500 patients with aortic coarctation referred for cardiovascular magnetic resonance imaging to a tertiary center. Pediatr Cardiol. 2011;32(8):1120–1127.
27. Biner, S, Rafique, AM, Ray, I, et al. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol. 2009;53(24):2288–2295.
28. Aboulhosn, J, Child, JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation. 2006;114(22):2412–2422.
29. Stamm, C, Li, J, Ho, SY, et al. The aortic root in supravalvular aortic stenosis: the potential surgical relevance of morphologic findings. J Thorac Cardiovasc Surg. 1997;114(1):16–24.
30. Vaideeswar, P, Shankar, V, Deshpande, JR, et al. Pathology of the diffuse variant of supravalvar aortic stenosis. Cardiovasc Pathol. 2001;10(1):33–37.
31. Shone, JD, Sellers, RD, Anderson, RC, et al. The developmental complex of “parachute mitral valve,” supravalvular ring of left atrium, subaortic stenosis, and coarctation of aorta. Am J Cardiol. 1963;11:714–725.
32. John, AS, Dill, T, Brandt, RR, et al. Magnetic resonance to assess the aortic valve area in aortic stenosis: how does it compare to current diagnostic standards? J Am Coll Cardiol. 2003;42(3):519–526.
33. Ohye, RG, Gomez, CA, Ohye, BJ, et al. The Ross/Konno procedure in neonates and infants: intermediate-term survival and autograft function. Ann Thorac Surg. 2001;72(3):823–830.
34. Schoof, PH, Cromme-Dijkhuis, AH, Bogers, JJ, et al. Aortic root replacement with pulmonary autograft in children. J Thorac Cardiovasc Surg. 1994;107(2):367–373.
35. Laudito, A, Brook, MM, Suleman, S, et al. The Ross procedure in children and young adults: a word of caution. J Thorac Cardiovasc Surg. 2001;122(1):147–153.
36. Takkenberg, JJ, Klieverik, LM, Schoof, PH, et al. The Ross procedure: a systematic review and meta-analysis. Circulation. 2009;119(2):222–228.
37. McCrindle, BW. Independent predictors of immediate results of percutaneous balloon aortic valvotomy in children. Valvuloplasty and Angioplasty of Congenital Anomalies (VACA) Registry Investigators. Am J Cardiol. 1996;77(4):286–293.
38. McElhinney, DB, Lock, JE, Keane, JF, et al. Left heart growth, function, and reintervention after balloon aortic valvuloplasty for neonatal aortic stenosis. Circulation. 2005;111(4):451–458.
39. Bonow, RO, Carabello, BA, Chatterjee, K, et al. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2008;118(15):e523–e661.
40. Keane, JF, Driscoll, DJ, Gersony, WM, et al. Second natural history study of congenital heart defects. Results of treatment of patients with aortic valvar stenosis. Circulation. 1993;87(2 suppl):I16–I27.
41. Hawkins, JA, Minich, LL, Shaddy, RE, et al. Aortic valve repair and replacement after balloon aortic valvuloplasty in children. Ann Thorac Surg. 1996;61(5):1355–1358.
42. Brown, DW, Dipilato, AE, Chong, EC, et al. Sudden unexpected death after balloon valvuloplasty for congenital aortic stenosis. J Am Coll Cardiol. 2010;56(23):1939–1946.
43. Gersony, WM, Hayes, CJ, Driscoll, DJ, et al. Bacterial endocarditis in patients with aortic stenosis, pulmonary stenosis, or ventricular septal defect. Circulation. 1993;87(2 suppl):I121–I126.
44. Wilson, W, Taubert, KA, Gewitz, M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007;116(15):1736–1754.
45. Stamm, C, Kreutzer, C, Zurakowski, D, et al. Forty-one years of surgical experience with congenital supravalvular aortic stenosis. J Thorac Cardiovasc Surg. 1999;118(5):874–885.
46. Douville, EC, Sade, RM, Crawford, FA, Jr., et al. Subvalvar aortic stenosis: timing of operation. Ann Thorac Surg. 1990;50(1):29–33. [discussion 33-34].
47. Gersony, WM. Natural history of discrete subvalvar aortic stenosis: management implications. J Am Coll Cardiol. 2001;38(3):843–845.
48. Karamlou, T, Gurofsky, R, Bojcevski, A, et al. Prevalence and associated risk factors for intervention in 313 children with subaortic stenosis. Ann Thorac Surg. 2007;84(3):900–906. [discussion 906].
49. Report of the New England Regional Infant Cardiac Program. Pediatrics. 1980;65(2 Pt 2):375–461.
50. Abbruzzese, PA, Aidala, E. Aortic coarctation: an overview. J Cardiovasc Med (Hagerstown). 2007;8(2):123–128.
51. Ward, C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83(1):81–85.
52. Dulac, Y, Pienkowski, C, Abadir, S, et al. Cardiovascular abnormalities in Turner’s syndrome: what prevention? Arch Cardiovasc Dis. 2008;101(7-8):485–490.
53. Kim, HK, Gottliebson, W, Hor, K, et al. Cardiovascular anomalies in Turner syndrome: spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. AJR Am J Roentgenol. 2011;196(2):454–460.
54. Connolly, HM, Huston, J, 3rd., Brown, RD, Jr., et al. Intracranial aneurysms in patients with coarctation of the aorta: a prospective magnetic resonance angiographic study of 100 patients. Mayo Clin Proc. 2003;78(12):1491–1499.
55. Metry, D, Heyer, G, Hess, C, et al. Consensus statement on diagnostic criteria for PHACE syndrome. Pediatrics. 2009;124(5):1447–1456.
56. Allan, LD, Crawford, DC, Tynan, M. Evolution of coarctation of the aorta in intrauterine life. Br Heart J. 1984;52(4):471–473.
57. Rudolph, AM, Heymann, MA, Spitznas, U. Hemodynamic considerations in the development of narrowing of the aorta. Am J Cardiol. 1972;30(5):514–525.
58. de Divitiis, M, Pilla, C, Kattenhorn, M, et al. Vascular dysfunction after repair of coarctation of the aorta: impact of early surgery. Circulation. 2001;104(12 suppl 1):I165–I170.
59. de Divitiis, M, Rubba, P, Calabro, R. Arterial hypertension and cardiovascular prognosis after successful repair of aortic coarctation: a clinical model for the study of vascular function. Nutr Metab Cardiovasc Dis. 2005;15(5):382–394.
60. Strafford, MA, Griffiths, SP, Gersony, WM. Coarctation of the aorta: a study in delayed detection. Pediatrics. 1982;69(2):159–163.
61. Ferguson, EC, Krishnamurthy, R, Oldham, SA. Classic imaging signs of congenital cardiovascular abnormalities. Radiographics. 2007;27(5):1323–1334.
62. Konen, E, Merchant, N, Provost, Y, et al. Coarctation of the aorta before and after correction: the role of cardiovascular MRI. AJR Am J Roentgenol. 2004;182(5):1333–1339.
63. Araoz, PA, Reddy, GP, Tarnoff, H, et al. MR findings of collateral circulation are more accurate measures of hemodynamic significance than arm-leg blood pressure gradient after repair of coarctation of the aorta. J Magn Reson Imaging. 2003;17(2):177–183.
64. Steffens, JC, Bourne, MW, Sakuma, H, et al. Quantification of collateral blood flow in coarctation of the aorta by velocity encoded cine magnetic resonance imaging. Circulation. 1994;90(2):937–943.
65. Varaprasathan, GA, Araoz, PA, Higgins, CB, et al. Quantification of flow dynamics in congenital heart disease: applications of velocity-encoded cine MR imaging. Radiographics. 2002;22(4):895–905. [discussion 905-906].
66. Egan, M, Holzer, RJ. Comparing balloon angioplasty, stenting and surgery in the treatment of aortic coarctation. Expert Rev Cardiovasc Ther. 2009;7(11):1401–1412.
67. Kaushal, S, Backer, CL, Patel, JN, et al. Coarctation of the aorta: midterm outcomes of resection with extended end-to-end anastomosis. Ann Thorac Surg. 2009;88(6):1932–1938.
68. Cowley, CG, Orsmond, GS, Feola, P, et al. Long-term, randomized comparison of balloon angioplasty and surgery for native coarctation of the aorta in childhood. Circulation. 2005;111(25):3453–3456.
69. Holzer, R, Qureshi, S, Ghasemi, A, et al. Stenting of aortic coarctation: acute, intermediate, and long-term results of a prospective multi-institutional registry—Congenital Cardiovascular Interventional Study Consortium (CCISC). Catheter Cardiovasc Interv. 2010;76(4):553–563.
70. Vriend, JW, Mulder, BJ. Late complications in patients after repair of aortic coarctation: implications for management. Int J Cardiol. 2005;101(3):399–406.
71. Jonas, RA. Interrupted aortic arch. In Mavroudis C, Backer CL, eds.: Pediatric cardiac surgery, 3rd ed, Philadelphia: Mosby, 2003.
72. Celoria, GC, Patton, RB. Congenital absence of the aortic arch. Am Heart J. 1959;58:407–413.
73. Van Praagh, R, Bernhard, WF, Rosenthal, A, et al. Interrupted aortic arch: surgical treatment. Am J Cardiol. 1971;27(2):200–211.
74. Reardon, MJ, Hallman, GL, Cooley, DA. Interrupted aortic arch: brief review and summary of an eighteen-year experience. Tex Heart Inst J. 1984;11(3):250–259.
75. Moerman, P, Goddeeris, P, Lauwerijns, J, et al. Cardiovascular malformations in DiGeorge syndrome (congenital absence of hypoplasia of the thymus). Br Heart J. 1980;44(4):452–459.
76. Brown, JW, Ruzmetov, M, Okada, Y, et al. Outcomes in patients with interrupted aortic arch and associated anomalies: a 20-year experience. Eur J Cardiothorac Surg. 2006;29(5):666–673. [discussion 673-664].