[level-membership-for-hematology-oncology-and-palliative-medicine-category]
22 Laboratory aspects of blood transfusion
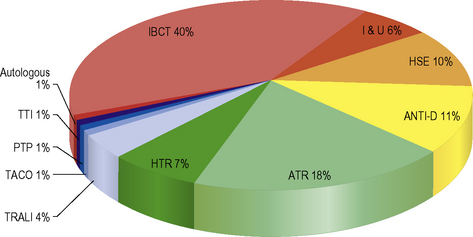
Safe and effective blood transfusion requires the combined efforts of blood transfusion services, biomedical scientists and clinicians to ensure the highest standards are applied to all the systems in a complex process from ‘vein to vein’. This chapter provides a description of the laboratory framework required to provide the right blood components to the right patients at the right time. The increased awareness of what can go wrong with blood transfusion comes from a number of sources including the Serious Hazards of Transfusion UK haemovigilance scheme, which was started in 1996.1,2 This confidential reporting scheme, using detailed root cause analysis of errors, has provided data that have informed both national bodies and local transfusion services of measures to introduce in order to reduce risk (Fig. 22.1). It is clear that multiple errors can contribute to a single adverse event and that many of these are outside the control of the transfusion laboratory.
Within the laboratory setting, the application of strict protocols for sample labelling and testing, robust laboratory procedures, reliable documentation, frequent staff training and competency assessment should be used. Recently, the UK Transfusion Collaborative, comprising representatives of the professional bodies involved in UK transfusion practice, has produced some recommendations to reduce laboratory errors in transfusion. These address issues such training, competency, staffing levels and automated systems.3
This chapter is concerned with the testing of patient samples prior to the provision of appropriate compatible blood components including identification of red cell antibodies. It also covers compatibility testing and investigation of transfusion reactions and the testing required in other special situations including the antenatal and postnatal settings. National professional bodies such as the British Committee for Standards in Haematology (BCSH)4 and the AABB5 issue guidance to transfusion laboratories and this has been referenced where appropriate.
There is a new regulatory framework governing hospital transfusion laboratory practice which was implemented after the publication of two European Union Directives: 2002/98/EC and 2004/33/EC.6 In the UK these are the Blood Safety and Quality Regulations 2005 (Statutory Instruments 2005/50, 2005/1098 and 2006/2013) and they set standards for quality and safety of human blood and blood components in hospital ‘blood banks’ as well as ‘blood establishments’ (the UK Blood Services).7 ‘Blood banks’ are regulated in their roles of storing, distributing and performing compatibility tests, on blood and blood components for use in hospitals. The implications for hospital transfusion practice are the requirement for ‘vein to vein’ traceability of blood components, the importance of maintaining the ‘cold chain’ for all therapeutic blood components, the need to store transfusion records for 30 years and the requirement for a quality management system.6,7 UK laboratories have to assess themselves and submit an annual compliance report to the competent authority, which is the Medicines and Healthcare Products Regulatory Agency (MHRA).8 The MHRA carries out laboratory inspections to assess compliance with these regulations.
The UK government, through the National Blood Transfusion Committee, continues to promote the safe and effective transfusion of blood components and has published three Health Service Circulars.9 These documents aim to ensure that a team approach to blood transfusion safety is taken via the local clinical governance arrangements and to promote good transfusion practice via Hospital Transfusion Committees and Hospital Transfusion Teams supported by Regional Transfusion Committees.
Technology and automation in blood transfusion laboratories
Column agglutination (CAT) and solid-phase technology can both be used on automated machines and CAT can also be used by manual techniques. In UK hospital transfusion laboratories these technologies have replaced tube techniques and liquid-phase microplates for antibody screening and crossmatching (Figs 22.2, 22.3).10
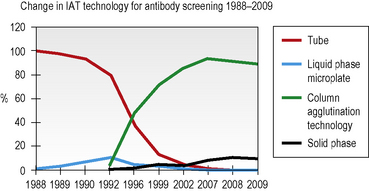
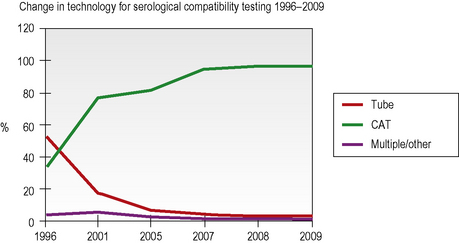
Individual laboratories need to make careful and informed decisions when selecting reagents for pre-transfusion testing. It is vital that any abbreviated testing in an automated or semiautomated system is carefully evaluated for the risks that could ensue if important controls were omitted when using this technology for blood grouping.11
Laboratory information management systems (LIMS) store patient details and results of laboratory tests, allowing timely and accurate access to important information. In the transfusion department, IT systems have a much broader use and the updated BCSH guidelines for the specification and use of Information Technology (IT) systems in blood transfusion practice (2006) reflect this.12 Where possible, using bidirectional or unidirectional interfaces to automated blood grouping analysers, IT systems are used to eliminate errors that can arise when a manual step is employed, including interpretation of test results.
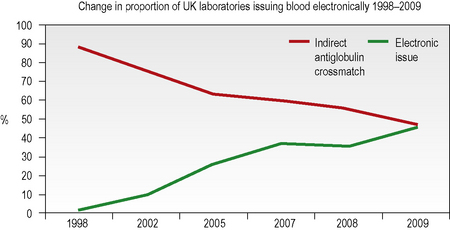
Computer algorithms support the ‘electronic issue’ of blood to patients with a negative antibody screen without the need to perform an antiglobulin crossmatch and, in the UK in 2009, 46% of laboratories were using this system for some or all of their patients (Fig. 22.4). The use of automation for all aspects of compatibility testing is now recommended practice as it is recognized that it is safer.3 Automation brings several or all of the discrete activities of compatibility testing into a single platform process. It provides various levels of increased security over manual testing and may provide justification for abbreviated pre-transfusion testing (e.g. abandoning duplicate D, previously termed ‘RhD’, typing or reverse ABO grouping in the presence of a valid historical group). A risk assessment must be made and documented prior to any abbreviation of an established procedure, with consideration being given to the presence or absence of key functions in the automated equipment. The BCSH guidelines for compatibility procedures12 and guidance from the MHRA13 give a list of factors to be taken into consideration and the reader is advised to consult these prior to implementing automated or semiautomated systems.
Pre-transfusion compatibility systems
Documentation of the Transfusion Process
All stages of the transfusion process must be clearly documented and these records must be kept. In addition to guidance from the UK Royal College of Pathologists on the retention of documents,18 the BSQR 2005 regulations stipulate that the records must be accessible for 30 years.7 This allows any blood component to be traced from the donor to the recipient should information come to light about any potential infective risks to the recipient.
Computer records are easier to search than paper records, but laboratory information systems are likely to become obsolete and be replaced several times within this mandatory 30-year period, so provision must be made to store historical data in an accessible format when procuring a replacement computer system.12 Patient-held records are useful for patients who are treated in more than one institution, particularly if they have red cell antibodies and require phenotyped blood or if they have special requirements because of their underlying disease or its treatment. Credit card-sized records with corresponding patient information leaflets are issued by some transfusion centres to patients with red cell antibodies and similar cards exist for patients who require irradiated cellular blood components.19
Identification and Storage of Blood Samples
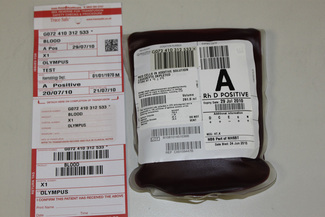
On being received in the laboratory, the details on the request form must be checked against the blood sample. Each blood sample must be labelled with a unique sample number. Barcode labels offer the advantage of positive sample identification and reduce the number of transcription errors. Samples inadequately or inaccurately labelled should NOT be used for pre-transfusion testing.11
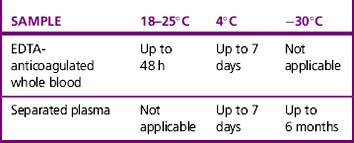
Whole blood samples should be tested as soon as possible because they will deteriorate over time. Problems associated with storage include lysis of the red cells, loss of complement in the serum and decrease in potency of antibodies. The BCSH guidelines11 indicate working limits as outlined in Table 22.1. If separated plasma or serum samples are stored for later serological crossmatch, care must be taken to ensure that the patient has not been transfused in the interim. It has been recommended that samples should be kept for a minimum of 7 days from group and screen, stored at 4°C. Samples should be retained post-transfusion for investigation of acute transfusion reactions and preferably stored for 7 days post-transfusion to enable investigation of delayed transfusion reaction.18
ABO and D grouping
ABO Grouping
This is an excellent built-in check for the ‘forward’ or cell group and has always been considered to be an integral part of ABO grouping, allowing the reading and recording of test results to be split into two discrete tasks. However, with secure, fully automated systems, linked to secure laboratory information management systems that, in combination, have the ability to prevent procedural ABO grouping errors, some laboratories now omit the reverse group when testing samples for which a historical group is available.11 This should only be considered following a careful risk assessment and taking into account that the first sample taken may have been from the wrong patient. This may be in the order of 1:2000 samples.16 Any discrepancy between the forward and reverse groups should be investigated further and any repeat tests should be undertaken using cells taken from the original sample rather than from a prepared cell suspension.
D Grouping
Reagents for D Grouping
DVI is the partial D with the fewest epitopes; therefore of all the D variants, DVI individuals are those most likely to form anti-D and a case of severe haemolytic disease of the fetus and newborn (HDFN) has been described.20 For this reason, anti-D monoclonal reagents that do not detect DVI should be selected for testing patients’ samples.21,22 The use of anti-CDE reagents has led to the misinterpretation of r′ and r″ cells in UK National External Quality Assessment Scheme (NEQAS) exercises and, because they are of no value in routine patient typing, their use is not recommended.10,11 Selection of high-avidity monoclonal anti-D reagents will allow detection of all but the weakest examples of weak D, negating the need to use more sensitive techniques to check the D status of apparent D negatives.
Methods
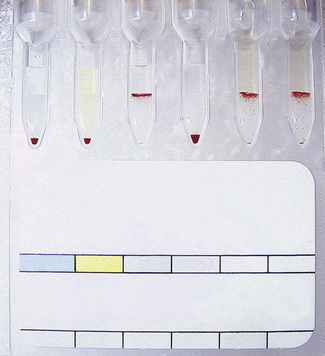
There are several techniques available for routine ABO and D grouping including tube test, slide test, liquid-phase and solid-phase microplates and columns. Other techniques for blood grouping have been described but they are not in routine use. For example, molecular ABO typing is reserved for investigating anomalous ABO groups, in organ transplantation where red cells from the donor are not available, forensic practice and paternity testing.23 Care should be taken to use the appropriate reagent because not all reagents have been validated by the manufacturer for all techniques.
EDTA for diluents
Liquid-phase microplate methods
Liquid-phase microplate technology provides a cheap and secure method for batch testing when semiautomation is utilized for dispensing and reading but it is no longer the grouping technique of choice in the UK (see Fig. 22.2). In 2009, a UK NEQAS survey showed that only 13% of responding laboratories were using microplates for grouping, down from 41% in a similar survey in 2002.10
The plate is usually laid out as 12 × 8 (12 tests and 8 reagents) but may be used in the opposite orientation (8 × 12), depending on the number of reagents and controls required. Particularly if performing this technique manually, the anti-D reagents should be kept away from the anti-A and anti-B reagents because splashing between wells can occur when dispensing reagents and handling the plates during testing if insufficient care is taken. The following method is recommended:24
Column agglutination techniques
CAT techniques (see Chapter 21, p. 502) are now the commonest method for grouping in the UK (80% of laboratories who responded to a UK NEQAS survey in 2009, see Fig. 22.2), especially where automated systems are in place; these should always be performed in accordance with the manufacturer’s instructions. There are several different profiles to choose from and some cards/cassettes include monoclonal antibodies to other blood group antigens (e.g. K) in addition (see Chapter 21, p. 502 and Fig. 22.6). Forward and reverse grouping may be undertaken in separate cards/cassettes.
Controls
Positive and negative controls should be included with every test or batch of manual tests. In fully automated systems, the controls should be set up at least twice in a 24-h period. The timings should coincide with machine startup and changing of reagents, taking account of the length of time that reagents have been kept at room temperature on the machine. The control samples should be loaded in the same way as the test samples. The required controls are shown in Table 22.2. Where controls do not give the expected reactions, investigations should be undertaken to determine the validity of all tests undertaken subsequent to the most recent valid control results.
| Reagent | Positive control | Negative control |
|---|---|---|
| Anti-A | A cells | B cells |
| Anti-B | B cells | A cells |
| Anti-D | D positive cells | D negative cells |
| A1 cells | Anti-A | Anti-B |
| B cells | Anti-B | Anti-A |
Causes of Discrepancies in ABO/D Grouping
T-activation/polyagglutination
Polyagglutination25 describes agglutination of red cells by all or most normal adult sera but not by the patient’s own serum. This is as the result of IgM antibodies reacting with an antigen on the red cells which is usually hidden but can be exposed by enzyme activity. The most common form is T-activation, which occurs when the bacterial enzyme neuraminidase cleaves N-acetyl neuraminic acid from the red cell membrane, exposing the T antigen.
Acquired B
The acquired B antigen is usually caused by a bacterial deacetylase enzyme acting on A1 red cells and producing a B-like substance. Some anti-B reagents react strongly with the acquired B antigen (e.g. those derived from the ES4 clone).26 Such anti-B reagents are rare but should be avoided in routine blood grouping.
Potentiators
Red cells may be coated with IgG as a result of in vivo sensitization. The use of potentiated techniques, such as the antiglobulin test for D typing, or of potentiated reagents for ABO or D typing, may result in a false-positive reaction; the latter would also result in a positive reaction with the diluent control, but UK NEQAS data have shown that some laboratories fail to include an appropriate control or fail to understand the significance of a positive control.27 For this reason, use of potentiated techniques or reagents for blood grouping is not advised.
False-Negative Reactions
D variant phenotypes
Weak D phenotypes are where the entire D antigen is present but there are fewer D antigen sites per cell and most weak D types group as D positive with the currently available high-avidity commercial monoclonal anti-D reagents. Where differing reactions are obtained with two reagents, the patient may be a partial D (i.e. one or more of the epitopes of the D antigen is missing). It was generally thought that patients who are weak D are unable to make anti-D and may be treated as D positive whereas some patients with partial D may be capable of making immune anti-D following sensitization with the missing epitope. This concept has been challenged by reports of weak D patients with anti-D.28
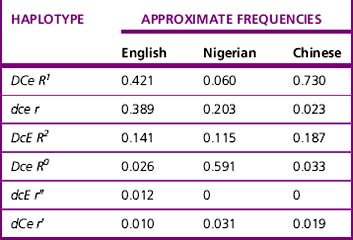
Current advice about choice of D-typing reagents is that because DVI lacks the most epitopes, such individuals are likely to make anti-D when challenged by transfusion or pregnancy. For this reason, anti-D reagents for routine grouping of patients’ samples should not detect DVI.11 There is little evidence to suggest that a DVI donor would elicit an immune response in a recipient who is D negative; however, weak D positive and partial D donors, including DVI donors, should be classified as D positive.29 There are some important ethnic differences in the frequency of different Rh haplotypes, as shown in Table 22.3. Resolution of anomalous D grouping where a partial D is suspected now includes both serological testing and genotypic studies.30
Antibody screening
Red Cell Reagents
In the UK, the following antigens should be expressed as a minimum: C, c, D, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s, M, N and Lea; one cell should be R2R2 and another R1R1 or R1wR1. The following phenotypes should also be represented in the screening set: Jk(a+b−), Jk(a−b+), S+s−, S−s+, Fy(a+b−) and Fy(a−b+) (Table 22.4). These recommendations for homozygosity are based on UK data regarding the incidence of delayed haemolytic transfusion reactions, the need for high sensitivity in the detection of Kidd antibodies and the poorer performance of column agglutination techniques in the detection of some examples of Kidd antibodies using heterozygous cells.11,29 The requirement for the expression of Cw and Kpa antigens on screening cells has been the cause of much debate but in the UK and the USA detection of anti-Cw or anti-Kpa is not a requirement even in the absence of an antiglobulin crossmatch.11,29 This is because these are low-frequency antigens and the antibodies rarely cause delayed haemolytic transfusion reactions or severe haemolytic disease of the newborn.
Table 22.4 Expression of red cell antigens on screening cells11
| Blood group system | Antigen | Homozygous cells recommended |
|---|---|---|
| Rh | C | Yes (R1R1 or R1wR1) |
| c | Yes (R2R2) | |
| D | Yes (R1R1 and R2R2) | |
| E | Yes (R2R2) | |
| e | Yes (R1R1 or R1wR1) | |
| Kell | K | Noa |
| k | No | |
| Duffy | Fya | Yes |
| Fyb | Yes | |
| Kidd | Jka | Yes |
| Jkb | Yes | |
| MNSs | M | No |
| N | No | |
| S | Yes | |
| s | Yes | |
| Lewis | Lea | No |
a Although desirable, KK cells are unlikely to be available.
Methods
Antibody screening should always be carried out by an indirect antiglobulin test as the primary method. Additional methods (e.g. two-stage enzyme or Polybrene) may also be used but are inferior for the detection of some clinically significant antibodies and should not be used alone. A large retrospective study showed that the vast majority of antibodies reactive only by enzyme technique are of no clinical significance.31 In Issitt’s study of 10 000 recently transfused patients, only one anti-c, initially unreactive by indirect antiglobulin test, caused a delayed haemolytic transfusion reaction.32 There has been one SHOT report (1998–1999) of an enzyme-only anti-E that became detectable by indirect antiglobulin test 7 days following the transfusion, causing a delayed haemolytic transfusion reaction and subsequent death of the patient due to renal failure.1
For liquid-phase techniques, BCSH guidelines11 recommend the use of red cells suspended in LISS, rather than in standard normal ionic strength saline (NISS), because LISS increases the speed and sensitivity of detection of many potentially clinically significant antibodies. There are conflicting reports about whether sensitivity can also be improved by adding polyethylene glycol.33
Indirect Antiglobulin Techniques
Column Agglutination
In many countries, including the UK, column agglutination is now more commonly used than traditional tube or liquid-phase microplate techniques (see Figs 22.2 and 22.3) because it has been shown to be at least as sensitive as a standard LISS spin-tube technique,33,34 it is simpler to perform because it requires no washing phase, it uses small volumes of plasma and reagents, it has a more objective reading phase and it is easy to automate. The antihuman globulin (AHG) incorporated in the matrix is available as either a polyspecific or anti-IgG reagent. Red cell concentrations and volumes can be critical and it is important to follow manufacturers’ instructions at all times.
Column agglutination crossmatching tests have been shown to be less sensitive than standard tube techniques in detection of weak ABO antibodies such as anti-A with A2B cells and Kidd antibodies with heterozygous red cells.27,35–37 It has been suggested that these failures may be the result of shear forces occurring during centrifugation, which cause weak agglutinates to be disrupted, especially when the antigen site density is low.38
Solid-Phase Systems
Solid-phase techniques (e.g. Immucor Capture-R and Bio-Rad Solidscreen II) are also becoming more popular because they have been shown to have a high level of sensitivity and they also lend themselves to full automation. In 2009 10% of UK laboratories were routinely using solid phase for antibody screening (see Fig. 22.3).
Liquid-Phase Techniques – Tubes and Microplates
Liquid-phase microplate technology has never achieved a huge popularity for antibody screening because it is relatively difficult to introduce and standardize and it cannot be automated. Because it is becoming increasingly difficult to obtain AHG reagents standardized for use in microplates, no method is detailed here. However, further information is available in the BCSH guidelines, including a recommended method for screening in V-well plates, using a streaming technique.11,24
Antibody identification
Principles
Each antibody specificity should be taken in turn and its presence should be systematically excluded, by identifying antigen-positive cells that have given negative reactions. Wherever possible a negative reaction should be obtained with a red cell with homozygous expression of the relevant antigen. For example, if a negative reaction has been recorded against a Jk(a+b−) cell, then the presence of anti-Jka can be excluded, but if the only negative reactions are against Jk(a+b+) cells, then anti-Jka cannot immediately be safely excluded. This will leave a list of potential specificities, which should be considered by matching the positive reactions to the antigen-positive cells to determine if any are definitely present. Once this process is complete with the initial screen and identification panel, further cells and techniques may need to be used to complete the exclusion process. For example, where anti-S has been identified as being present, an enzyme-treated panel may be required to exclude the presence of anti-E, where all of the E-positive cells were also S positive or a K+S− cell may need to be selected to exclude the presence of anti-K.39 The more specificities present, the more complicated the process, and where resources are limited, samples may need to be sent to a reference centre to elucidate all specificities. If the reagents are available, phenotyping the patient’s red cells early on in the process will allow exclusion of specificities for which the patient is antigen positive.
Phenotyping
Additional Panels/Techniques
The chances of identifying antibodies, where two specificities are present, are significantly improved by using a two-stage enzyme technique, where at least one of the relevant antigens is affected by enzymes.27 For example, Rh antibodies are enhanced by proteolytic enzymes in routine use, whereas M, N, S, Fya and Fyb antigens are destroyed. Similarly, two different panels of reagent cells provide an increased chance of excluding further specificities where a mixture of two antibodies has been identified.
Reagents
An identification panel should consist of red cells from at least eight group O donors, although 10 is more common in commercial panels and allows easier elucidation of antibody mixtures. To be functional, the panel must permit confident identification of the most commonly encountered, clinically significant antibodies. The UK guidelines11 can be summarized as follows:
Antibody Cards
Delayed haemolytic transfusion reactions can occur when antibodies have not been detected in the current antibody screen or have been incorrectly identified, maybe by a different establishment.1,2 It has been suggested that antibody cards, produced either by the hospital or the reference centre, could be carried by the patient for presentation on admission to hospital. To be effective, such cards have to be accompanied by patient information leaflets, explaining the significance of the antibodies and preferably handed personally to the patient by someone with a clear understanding of blood transfusion practice. There are potential pitfalls with this suggestion, not least that the level of proficiency in identifying antibodies and appreciating clinical significance varies between establishments. A better long-term approach may be to have a national database of patients who have been identified to have clinically significant antibodies.
Selection and transfusion of red cells
Once the blood group of a patient has been established and any antibodies have been identified, a set of procedures is required to select units of red cells that are appropriate for transfusion. These include selecting ABO/D compatible units, which will also need to be negative for antigens to which the recipient has clinically significant red cell alloantibodies (Table 22.5); It is also important that consideration is given to certain clinical criteria dictating special requirements (e.g. the requirement for CMV-negative, irradiated or washed red cells). Selection according to these criteria depends on good clinical information, accurate ABO and D typing and a sensitive antibody screen.
Table 22.5 Recommendations for the selection of blood for a patient with red cell antibodies
| System | Antibody | Recommendation |
|---|---|---|
| ABO | Anti-A1 | IAT crossmatch compatible at 37°C |
| Rh | Anti-D, -C, -c, -E, -e | Antigen negative* |
| Rh | Anti-Cw | IAT crossmatch compatible† |
| Kell | Anti-K, -k | Antigen negative* |
| Kell | Anti-Kpa | IAT crossmatch compatible† |
| Kidd | Anti-Jka, -Jkb | Antigen negative* |
| MNS | Anti-M (active at 37°C) | Antigen negative* |
| MNS | Anti-M (not active at 37°C) | IAT crossmatch compatible at 37°C |
| MNS | Anti-N | IAT crossmatch compatible at 37°C |
| MNS | Anti-S, -s, -U | Antigen negative* |
| Duffy | Anti-Fya, -Fyb | Antigen negative* |
| P | Anti-P1 | IAT crossmatch compatible at 37° |
| Lewis | Anti-Lea, -Leb, Lea+b | IAT crossmatch compatible at 37°C |
| Lutheran | Anti-Lua | IAT crossmatch compatible at 37°C |
| Diego | Anti-Wra (anti-Di3) | IAT crossmatch compatible† |
| H | Anti-HI (in A1 and A1B patients) | IAT crossmatch compatible at 37°C |
| All | Other active by IAT at 37°C | Seek advice from blood centre |
IAT, indirect antiglobulin test.
* Antigen negative and crossmatch compatible.
† These recommendations apply when the antibody is present as a sole specificity. If present in combination, antigen-negative blood may be provided by the blood centre to prevent wastage of phenotyped units.
Some patients are identified as definitely needing transfusion of blood, but others may be undergoing surgery where blood is crossmatched to ‘stand by’. Audits have shown that blood is often crossmatched but not transfused. The transfusion ratio, or crossmatch index, can be used to assess how well blood stocks are managed. One option is to perform a blood group and antibody screen on a patient and then save the sample, only crossmatching blood if certain pre-agreed transfusion triggers are met. Local policy should define a maximum surgical blood ordering schedule, indicating which surgical procedures can be ‘group and screen/group and save’ and how many units of blood need to be crossmatched for procedures with a high likelihood of intraoperative transfusion.41 The decision as to whether blood is crossmatched in advance may relate to local factors including the proximity of the blood transfusion laboratory to the operating theatre (or other location where blood is to be transfused) and the time it takes to provide compatible blood following a request.
Once the appropriate red cells have been selected, compatibility needs to be ensured. This is usually referred to as crossmatching, which may include an indirect antiglobulin test to check for incompatibility as a result of IgG antibodies and ABO antibodies or an immediate spin test to check for ABO incompatibility only. Instead of a serological test, an assessment of ABO compatibility may be based on a ‘computer check,’ usually referred to as ‘electronic issue’. Some transfusion laboratories, where surgery takes place in a different hospital, have extended electronic issue to the selection of compatible units, as dictated by the transfusion laboratory, from a remote issue refrigerator.42
The process of crossmatching blood is to prevent the transfusion of incompatible red cells and a subsequent haemolytic transfusion reaction. The different types of crossmatch are outlined in the following paragraphs. Whichever crossmatch technique is chosen, it should be clear that all patients with known red cell antibodies of potential clinical significance, even if currently undetectable, should have an indirect antiglobulin crossmatch and are not suitable for electronic issue.11,12
Crossmatching
Choice of Test
Indirect Antiglobulin Crossmatch
The arguments for retaining an indirect antiglobulin crossmatch are based on the failure to identify antibodies against low-frequency antigens in the antibody screen. However, there is evidence, summarized by Garratty,31 that the likelihood of missing a clinically significant antibody is approx. 1 in 10 615 crossmatches if the indirect antiglobulin test component is omitted and antibody detection relies on a sensitive antibody screen alone. Moreover, the usual outcome of transfusion if an antibody is present but undetected by the antibody screen is limited to shortened red cell survival.
Immediate Spin Crossmatch
There is evidence of poor standardization of this technique,44 but its sensitivity can be optimized by selecting the appropriate cell suspension, incubation time and serum: cell ratio. The following tube method is recommended:
Incubate at room temperature for 2–5 min to enhance the detection of weak ABO antibodies.
Centrifuge at 100 g for 1 min.
Read the reaction carefully using a ‘tip and roll’ technique.
False-negative results (in immediate spin crossmatch)
Incompatibilities between A2B donor cells and group B patient sera are not consistently detected with this technique.45 Of more concern is the potential failure of agglutination with potent ABO antibodies46,47 on account of rapid complement fixation with bound C1 interfering with agglutination if using serum. Red blood cells must therefore be suspended in saline containing EDTA.
Electronic Issue
BCSH guidelines12 and AABB standards48 require that there be concordant determinations of the patient’s ABO and D type either on two separate samples, at least one of which must be a current sample or on one current sample tested twice, depending on the security present in the system. In addition, there must be no clinically significant antibodies detected and no record of any having been detected previously. The AABB recommends that the group of donor units is rechecked, but the BCSH accepts written verification by the supplying transfusion service of the accuracy of the donor unit label. It is strongly recommended by the BCSH that ABO and D grouping procedures are automated with positive sample identification (e.g. bar-codes) and electronic transfer of results from the analyser to the transfusion laboratory computer, whereas the AABB does not have such a requirement. Ideally, computer algorithms should direct the procedure, only allowing issue if all the criteria are fulfilled. For example, issue of red cells for transfusion will be prevented if only one ABO and D group is on file or if the previous and current groups do not agree. Any manually controlled part of this process increases the risk of error. Electronic issue, based on fully validated systems, has been in place in several countries for some time and has proved to be clinically safe, providing the recommendations are rigidly adhered to.49–51
Emergency blood issue
In clinical emergencies in which immediate red cell support is required, there may not be time for full compatibility testing. Either abbreviated testing is employed and rapid techniques are used or group O red cells are issued. There must be a documented procedure for dealing with emergencies. Local policies on this should be formally assessed for risk and adequate training should be given to staff, particularly those providing the service out of routine laboratory hours. Out-of-hours staff should be included in internal proficiency testing as well as external quality assessment exercises designed to test emergency tests and techniques.3,11
Rapid ABO and D Typing
Selection of Units
Some hospitals provide one or two units of O D negative red cells for use by clinicians pending the availability of ABO and D specific compatible red cells. Because of the relatively short supply of O D negative red cells (only 8% of Caucasian blood donors are O D negative in the UK) the National Blood Transfusion Committee has issued guidance to hospitals about the restrictions on the use of O D negative red cells in patients of another blood group.52
Massive Transfusion
Massive transfusion is defined as more than one blood volume within 24 h and is usually taken to mean 8 or 10 units for an adult.53 After this volume of transfusion, it is no longer necessary to undertake an antiglobulin crossmatch, but immediate spin crossmatch or electronic issue is recommended to check for ABO compatibility. If ABO non-identical blood is given initially, blood of the same group as the patient should be used as soon as possible after the first transfusion. In some situations the recipient’s need for red cell transfusion may necessitate the use of incompatible units, but this is a clinical decision based on the need for blood balanced against the known clinical significance of the red cell antibody detected.
Selection of Platelets and Plasma
In patients with hypotensive shock and/or large volume red cell replacement, a coagulopathy develops that requires replacement of other blood components. The use of platelets, fresh frozen plasma and cryoprecipitate may initially be determined by clinical evaluation of the patient’s haemostatic state including observation of diffuse microvascular bleeding. Massive haemorrhage protocols should be developed to enable release of these coagulation factors at the request of the clinicians, in advance of the results of laboratory tests. Ongoing management of the patient can be guided by the results of these initial full blood count and coagulation screen tests, combined with frequent re-evaluation of both the laboratory tests and clinical evaluation of the haemostatic state.53
Potential Errors
Errors leading to transfusion of incorrect blood components are more likely to occur out of routine laboratory hours; therefore it has been recommended that only genuine emergency transfusions should take place out of hours.1,2 There may, however, be circumstances where patients with haemoglobinopathies receiving regular transfusion support are given blood overnight to minimize disruption of their education or employment.
Antenatal serology and haemolytic disease of the newborn
Haemolytic Disease of the Fetus and Newborn
Haemolytic disease of the fetus and newborn is a haemolytic anaemia of the fetus and/or newborn infant that occurs when maternal alloantibody to fetal antigens crosses the placenta and causes haemolysis of fetal red cells or suppression of fetal red cell progenitors, the latter occurring with antibodies within the Kell system.54,55
As IgG is the only immunoglobulin that crosses the placenta, only red cell antibodies of this class are a potential cause of HDFN. Anti-D causes the most severe form of HDFN, but the success of prophylaxis with anti-D immunoglobulin for potentially sensitizing events in pregnancy and after delivery of a D positive baby has reduced the number of cases and routine antenatal anti-D prophylaxis (RAADP) has reduced it even further. The relative proportion of HDFN due to other IgG red cell antibodies has thus increased.56 Although HDFN resulting from anti-D is the most severe form of the disease, anti-c can give rise to significant haemolysis in utero, sufficient in some cases to result in intrauterine death and therefore to warrant intervention in pregnancy. Anti-K has a different mode of action54,55 but can also result in a severely affected fetus. Other IgG antibodies (e.g. anti-E, anti-Ce, anti-Fya and anti -Jka) uncommonly give rise to fetal haemolysis of sufficient severity to merit antenatal intervention. Haemolytic disease of the newborn (HDN) as a result of ABO antibodies can also occur and is described later. For a detailed discussion of the investigation and management of HDFN, the reader is referred to the reviews by Kumar and O’Brien,57 Bowman58 and the textbook Mollison’s Blood Transfusion in Clinical Medicine.30
Antenatal Serology
ABO and D Grouping and Antibody Screening
Maternal ABO and D grouping as well as antibody screening and identification are performed early in pregnancy (i.e. when first seen and ‘booked in’) and then at 28 weeks’ gestation. All pregnant women, whether D positive or D negative, should be screened for red cell antibodies.59 Further testing depends on the specificity of any antibodies detected, whether they are capable of causing HDFN and the obstetric history.
Follow-Up Antibody Screening
Protocols for antenatal screening and follow-up vary from country to country. In the United Kingdom, the following is recommended by the BCSH:59
Prediction of Fetal Blood Group
Partner Testing
The zygosity of the D antigen is usually predicted from the results of tests with anti-c, anti-C, anti-e and anti-E and from the likelihood of the homozygous or heterozygous association with these antigens (Table 22.6, see also Table 22.3). These data have been compiled for different racial groups. It is important, therefore, to tell the specialist laboratory the ethnic origin of the patient. Because the genetic basis for the common D types is now known, DNA typing provides a better alternative for predicting the potential for haemolytic disease of the newborn.61
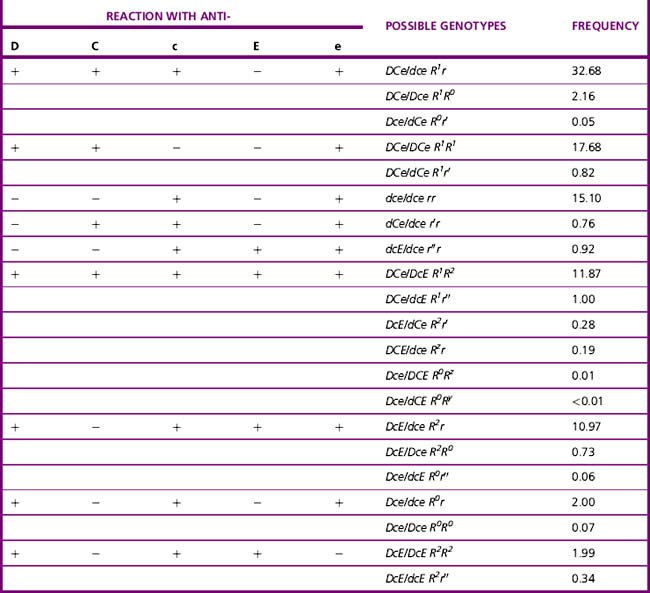
Testing Fetal DNA in the Maternal Circulation
It is now possible to detect fetal DNA in the maternal circulation and, using DNA amplification techniques (see Chapter 8), to obtain D, c, E and K types on these cells. This has proved to be accurate at predicting the D type and, in the UK, it is now offered as a clinical service at the beginning of the 2nd trimester.62 This may replace more invasive tests and supplement partner typing. It can be especially helpful if the father is absent or unknown.
Fetal Blood Sampling
Using ultrasound guidance, it is possible to take a sample of fetal blood for blood grouping, but this carries some risks. Contamination by maternal blood can hinder analysis of the sample obtained, leading to false-negative results. In addition, the procedure itself can lead to fetomaternal haemorrhage (FMH) and hence further sensitization to fetal antigens. There is also a risk of miscarriage.57
Antenatal Assessment of the Severity of Haemolytic Disease of the Fetus and Newborn
Antibody Titrations during Pregnancy
Techniques for antibody titration are described in Chapter 21, but these have variable reproducibility and sensitivity. In many laboratories tube techniques have been replaced by column agglutination technology. Figure 22.7 shows the range of reaction grades used in titrations. The role of the serologist is to carry out serial antibody measurements to determine changes in the titre or concentration of the antibody. It is recommended that the technique chosen for titration should be validated against the National Institute for Biological Standards and Control anti-D standard.59 Hence, laboratories should ensure that titres obtained with the anti-D standard are within one doubling dilution when it is used as an internal control. In addition, antibody titrations performed in pregnancy should be performed in parallel with the previous sample. Increases in titres of more than one doubling dilution should be monitored in conjunction with obstetricians.
Antibody Quantitation
Individual hospital transfusion laboratories should work closely with reference laboratories and obstetricians. Automated quantification is considered to be a more accurate predictor of when to proceed to more active investigation of the fetus but is usually only available for anti-D and anti-c. Results in iu (or μg per ml) are used as part of clinical algorithms to proceed to the next step of fetal investigation.57
Assessment of Fetal Anaemia
This, however, is an indirect measurement, whereas direct fetal blood sampling by ultrasound-guided cordocentesis provides not only direct diagnostic information but also a new approach to fetal therapy by direct fetal intravascular transfusion. However, both of these procedures carry the risk of miscarriage and further fetomaternal haemorrhage.57 It is now common practice for fetal medicine units to offer non-invasive tests to determine fetal anaemia; middle cerebral artery Doppler studies have been very useful in this regard.63 The incidence and severity of HDFN is declining and the increasingly specialized management of severely affected pregnancies has meant that these women are now being referred early in pregnancy to fetal medicine units who specialize in dealing with this condition, thus decreasing the involvement of the routine transfusion laboratory in any but the early stages.
Prevention of Haemolytic Disease of the Fetus and Newborn as a Result of Anti-D
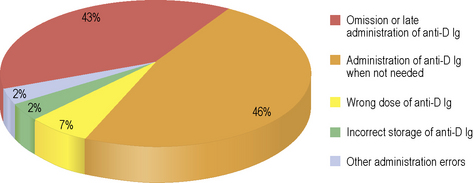
Correct identification of women in early pregnancy who are D negative offers the chance to give intramuscular anti-D immunoglobulin to prevent sensitization to the D antigen at times during the pregnancy when significant fetomaternal haemorrhage is likely to occur, known as ‘potentially sensitizing events’ (PSE).64,65 Accuracy in D grouping is particularly important because women who are D negative, erroneously grouped as D positive, risk not receiving prophylactic anti-D immunoglobulin (or being transfused with D positive cells). Sensitization to the D antigen could then result in severe HDFN in subsequent pregnancies as a result of development of anti-D. Serious Hazards of Transfusion (SHOT) has a reporting category for anti-D errors which includes any adverse event relating to the prescription, administration or omission of anti-D immunoglobulin that has the potential to cause harm to the mother or fetus immediately or in the future (Fig. 22.8).2
Anti-D Prophylaxis
Anti-D immunoglobulin should be given routinely as soon as possible after delivery (but always within 72 h) to women who are D negative who deliver babies that are D positive. It should also be given at times during pregnancy when sensitization could occur, such as during medical or surgical therapeutic termination of pregnancy, chorionic villus sampling, amniocentesis and following any abdominal trauma. It should also be given for episodes of vaginal bleeding where the pregnancy remains viable.64,65 At delivery and for potentially sensitizing events after 20 weeks’ gestation, it is necessary to screen for fetomaternal haemorrhage (FMH) using an acid elution method and estimate the degree of FMH if fetal cells are seen. The BCSH guidelines recommend confirming any FMH >2 ml by flow cytometry so that additional anti-D immunoglobulin can be given if the standard dose in use does not cover the estimated bleed.66
Because of the risk of silent fetomaternal haemorrhage in pregnancy, routine antenatal anti-D prophylaxis (RAADP) is being offered to women in some countries. In the UK, this has been the subject of an appraisal by the National Institute for Health and Clinical Excellence67 which recommends that anti-D immunoglobulin should be given either as two doses at 28 and 34 weeks or a single larger dose at 28 weeks, in addition to the postnatal dose and doses to cover any potentially sensitizing events during pregnancy. Women can decline RAADP, e.g. if they know the father is D negative or if they do not want any further pregnancies. The typing of fetal DNA in the maternal circulation may be used in the future to select women with fetuses that are D positive who would benefit from this additional prophylaxis, but, at the moment, it is not universally offered.68
The administration of prophylactic anti-D results in a positive maternal antibody screen due to passive anti-D. It may be difficult to distinguish between passive anti-D and low-level immune anti-D, particularly in the absence of a history of anti-D administration (this may have taken place at another institution). It is therefore important to take the 28-week sample for blood group and antibody screen before administration of anti-D. Although further antibody screens are not routinely required,59 events in the 3rd trimester may result in a sample being sent to the transfusion laboratory. Laboratories need to have a strategy for dealing with these samples. Some implement screening against rr screening cells, whereas others proceed to full antibody identification on all samples.
Measurement of Fetomaternal Haemorrhage
The following tests are performed to estimate the quantity of fetal cells in the maternal circulation by the difference between fetal and maternal cells. Most commonly used is acid elution, also known as the Kleihauer test, which depends on the Hb F in fetal cells resisting the acid elution to a greater extent than the Hb A in maternal cells. The calculation of the volume of fetal cells is based on the work by Mollison,30 which assumed that the maternal red cell volume is 1800 ml, fetal red cells are 22% larger than maternal cells and only 92% of fetal cells stain darkly (p. 338). Mollison’s formula for calculating volume of fetomaternal haemorrhage is as follows:
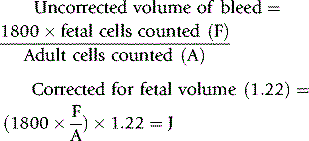
Corrected for staining efficiency (1.09):
Volume of fetomaternal haemorrhage (ml fetal cells) = J × 1.09.
The flow cytometry method uses a fluorochrome-labelled anti-D antibody to measure a minority of D positive cells in the maternal D negative blood and is recommended for confirmation of a positive acid elution test where the estimated FMH exceeds 2 ml. However, flow cytometry may not always be available. Flow cytometric techniques using anti-Hb F have also been developed. The BCSH guidelines66 give full details on performance and use of all these tests.
Recommended Action at Delivery (or Potentially Sensitizing Event)
On the basis of a confirmed FMH result, further anti-D immunoglobulin should be given if the FMH exceeds the volume covered by the standard anti-D immunoglobulin (Fig. 22.9). If the fetomaternal haemorrhage is more than 2 ml, the maternal sample, if possible, should be retested by a second technique such as flow cytometry; alternatively, the test should repeated on the same sample by a different operator.

ABO Haemolytic Disease of the Newborn
Serological Investigation
Compatibility testing in special transfusion situations
Neonates and Infants within First 4 Months of Life
Investigations on the Infant Sample
ABO and D group (cell group only), repeated on the same sample if no historical group
Selection of Blood and Other Components
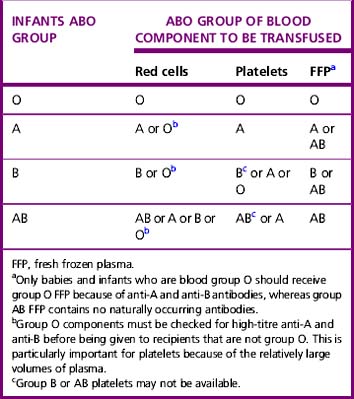
Where possible, the cellular component chosen should be ABO and D identical or an alternative compatible ABO group, taking into account the infant’s blood group and maternal blood group (i.e. presence of maternal anti-A and/or anti-B) (Table 22.7). Care should be taken when giving components containing group O plasma (containing anti-A and anti-B) to infants that are not group O. Despite high-titre anti-A and anti-B donors being excluded from donating components for this group, there have been reports of haemolysis of group A infants’ red cells when given group O platelets.1,2 Current production methods for fresh frozen plasma result in minimal red cell stroma which is not antigenic; therefore plasma components do not need to be D compatible. If the maternal and infant antibody screens are negative (including absent maternal IgG anti-A in the infant), ABO- and D-typed blood can be given with no serological crossmatch, even after repeated small volume transfusions, because formation of red cell antibodies is rare in infants younger than 4 months old. Repeat antibody screen is not necessary if repeat transfusions are required within the first 4 months of life.69 In the case of haemolytic disease of the newborn (non-ABO or ABO) or maternal red cell antibodies, a crossmatch is required against maternal plasma using group O blood (which has been tested and shown to have low titres of anti-A and anti-B or ‘HT negative’) or other group compatible with red cell antibodies in the maternal serum. For neonates and infants, red cells for exchange transfusion should be <5 days old, negative for CMV antibodies, plasma reduced (or washed), Hb S negative, leucodepleted, K negative and irradiated.
Intrauterine (Fetal) Transfusion
Intrauterine (fetal) transfusion is usually carried out in a fetal medicine unit where local protocols should be in place. It is important for maternity units and associated neonatal units to have information about previous intrauterine transfusion because it may influence the selection of appropriate blood components if transfusion is required after delivery (see above). This is particularly important when the clinical care takes place in different institutions which are serviced by different transfusion departments. Shared-care protocols are one way of ensuring effective communication. When providing red cells for intrauterine transfusion, an antiglobulin crossmatch should be performed using maternal plasma and this must be repeated with a fresh maternal sample with every transfusion. Intrauterine transfusion can result in fetomaternal haemorrhage and hence sensitization to new antigens, so antibody identification and quantification or titration must be performed on all maternal samples. Red cells selected should be group O D negative (except where mother has made anti-c, when it will be necessary to give D positive, c-negative blood) and K negative. Further selection of phenotyped blood will depend on the maternal red cell antibody profile. In addition to the stipulations listed earlier for selection of blood for infants and neonates, blood for intrauterine transfusions should also be leucodepleted, K negative and irradiated to prevent transfusion-associated graft-versus-host disease and should be transfused within 24 h of irradiation.69
Patients Receiving Transfusions at Close Intervals
Chronic Transfusion Programmes
Examples of patients in whom a decision has been made that regular transfusions are required include those with β thalassaemia major, some patients with sickle cell anaemia and congenital or acquired bone marrow failure. It is important to establish a treatment plan for each patient with clear triggers for transfusion and regular checks for the adverse effects of transfusion, including iron overload. The risk of developing alloantibodies to red cell antigens on transfused blood influences the timing of blood samples (Table 22.1). If a less rigorous approach is taken for patients in whom repeated transfusions have not led to alloantibody formation, a mutual decision should be made by the clinician and the transfusion department after careful consideration of the risks.
A pre-transfusion Rh phenotype allows matching for D, C, c, E and e. Some ethnic groups commonly have the phenotype cDe (Ro) and D positive blood negative for C and E may be difficult to find, particularly if there are other red cell antibodies. In this situation D negative (cde/cde) blood is selected. Additionally, patients with haemoglobinopathies should have an extended red cell phenotype before they are first transfused to include the antigens K, Jka, Jkb, Fya, Fyb, M, N, S and s.70,71 This may be used to select blood but is also helpful when investigating antibodies formed as a result of repeated transfusions. Although provision of red cells with a matched extended phenotype is undertaken by some units treating haemoglobinopathies,72 the degree of matching depends on local resources and should not impede the delivery of effective transfusion support. There is insufficient evidence to make this recommendation for other patients who have been chronically transfused.
Allogeneic Haemopoietic Stem Cell Transplantation
Prior to the transplant, recipient type red cells, platelets and fresh frozen plasma are given, but following engraftment, the choice of blood components depends on the ABO and D mismatch between the donor/recipient pairs.11 For a recipient who is D positive with a donor who is D negative, D negative components should be used.
Investigation of a transfusion reaction
Adverse events related to transfusion can be acute (within 24 h) or delayed (Table 22.8). Transfusion laboratories should immediately be informed of a suspected transfusion reaction, being ideally placed to coordinate investigation, to communicate with clinicians and transfusion services and to advise about appropriate choice of blood components for subsequent transfusions. Serious adverse events should be reported confidentially to the national haemo-vigilance scheme, the local blood centre and the Hospital Transfusion Team. Acute transfusion reactions are easier to attribute to the transfusion than delayed reactions, although, in patients who are already very ill, they can go undiagnosed. The symptoms and signs of acute transfusion reactions are similar regardless of the cause so treatment and investigation of causes is, by necessity, simultaneous. It is easier to distinguish between the causes of delayed transfusion reactions, but it may be more difficult to recognize their relationship to the transfusion episode because of the delay in onset. The following scheme outlines the role of the laboratory in investigation and management of transfusion reactions and a very useful algorithm can be found in the Handbook of Transfusion Medicine.73
| Acute transfusion reactions | Delayed transfusion reactions |
|---|---|
| Acute haemolytic reaction | Delayed haemolytic reaction |
| Anaphylaxis | Transfusion transmitted infection |
| Bacterial contamination of blood component | Transfusion-associated graft-versus-host disease |
| Transfusion-associated acute lung injury | Post-transfusion purpura |
| Transfusion-associated circulatory overload | Iron overload |
| Allergic reaction | Immunosuppression |
| Febrile non-haemolytic transfusion reaction |
Acute Transfusion Reactions
Acute life-threatening transfusion reactions can result from the following:
Although rare, the onset of an acute transfusion reaction is usually very dramatic and the patient is acutely ill. Treatment is aimed at resuscitating the patient and elucidating the cause to try and prevent any further incidents (Table 22.9). In addition, there are unpleasant but not life-threatening reactions that may occur during transfusion. They include the following:
Table 22.9 Immediate investigations in the case of an acute transfusion reaction
| Check for haemolysis |
| Perform visual examination of patient’s plasma and urine (plasma and urine haemoglobin can be checked but this is not essential). |
| Blood film may show spherocytosis. |
| Bilirubin and lactate dehydrogenase (LDH) levels will be raised. |
| Check for incompatibility |
| Check the documentation and the patient’s identity. |
| Repeat ABO group of patient pre-transfusion and post-transfusion and of the donor unit(s). |
| Screen the patient for red cell antibodies pre-transfusion and post-transfusion. |
| Repeat crossmatch with pre-transfusion and post-transfusion samples. |
| Direct antiglobulin test (DAT) on pre- and post-transfusion samples |
| Eluate from patient’s red cells. |
| Check for disseminated intravascular coagulation |
| Perform blood count and film, coagulation screen and fibrin degradation products (or D-dimers). |
| Check for renal dysfunction |
| Check blood urea, creatinine and electrolytes. |
| Check for bacterial infection |
| Take blood cultures from the patient and donor unit including immediate Gram stain. |
| Immunological investigations |
| Check immunoglobulin A (IgA) levels and anti-IgA antibodies. |
Acute Intravascular Haemolysis
Transfused red cells react with the patient’s own anti-A or anti-B, and the red cells are destroyed in the circulation, causing collapse, renal failure and disseminated intravascular coagulation. Transfusion of ABO-incompatible cells usually results from an identification error. This can occur at point of blood sampling and labelling (wrong blood in tube), laboratory testing (technical error), blood unit labelling (administrative error) and collection from the blood refrigerator or inadequate bedside checking. If red cells are mistakenly transfused to the wrong patient, there is approximately a 1 in 3 chance that ABO incompatibility will occur. The reaction is most severe if group A blood is transfused to a patient who is group O, and only a small volume of red cells is required to cause this reaction. Prompt action in recognizing this acute emergency and stopping the transfusion may lead to a better outcome because the severity depends on the volume of blood transfused. If an acute transfusion reaction is suspected, the laboratory must be informed immediately and the unit of blood and giving set must be returned to the laboratory with blood and urine samples from the patient (Table 22.9).
Delayed Haemolytic Transfusion Reaction
Serological Investigation
1 Serious Hazards of Transfusion (SHOT). Annual Reports, 1996–2009 www.shotuk.org. Available at:
2 Serious Hazards of Transfusion. Annual Report 2009, 2010. Available at: www.shotuk.org July
3 Chaffe B., Jones J., Milkins C., et al. Recommended minimum standards for hospital transfusion laboratories. UK Transfusion Laboratory Collaborative. Transfus Med. 2009;19(4):156-158.
4 British Committee for Standards in Haematology (BCSH). Guidelines. www.bcshguidelines.com. Available at:
5 AABB (previously American Association of Blood Banks). Available at: www.aabb.org
6 . The European Blood Directive 2002 and associated directives. Available at: www.transfusionguidelines.org.uk/docs/pdfs/directive_2002_98_ec.pdf
7 The Blood Safety and Quality Regulations. Available at: www.opsi.gov.uk/si/si2005/20050050.htm, 2005, plus amendments at: www.opsi.gov.uk/si/si2005/20051098 htm, www.opsi.gov.uk/si/si2006/20062013 htm.
8 Medicines and Healthcare Products Regulatory Agency. Available at: www.mhra.gov.uk/Howweregulate/Blood/index.htm
9 Department of Health. Better blood transfusion – safe and appropriate use of blood Health Service Circulars 1998/224, 2002/009 and 2007/001. www.dh.gov.uk/en/Publicationsandstatistics/Lettersandcirculars/Healthservicecirculars/DH_080613. Available at:
10 . Personal communication based on Annual Reports of UK National External Quality Assessment Scheme (Blood Transfusion Laboratory Practice). Contact scheme available via: www.ukneqas.org.uk
11 British Committee for Standards in Haematology. Guidelines for compatibility procedures in blood transfusion laboratories. Transfus Med. 2004;14:59-73.
12 British Committee for Standards in Haematology. The specification and use of Information Technology (IT) systems in Blood Transfusion Practice, 2006 www.bcshguidelines.com/pdf/computer_guide_220506.pdf. Available at:
13 MHRA guidance on electronic issue. Available at: www.mhra.gov.uk/Howweregulate/Blood/index.htm, 2010.
14 British Committee for Standards in Haematology. Guideline on the Administration of Blood Components, 2009 www.bcshguidelines.com/pdf/Admin_blood_components050110.pdf. Available at:
15 Wagar E.A., Stankovic A.K., Raab S., et al. Specimen labeling errors: a Q-probes analysis of 147 clinical laboratories. Arch Pathol Lab Med. 2008;132(10):1617-1622.
16 Dzik W.H., Murphy M.F., Andreu G., et althe Biomedical Excellence for Safer Transfusion (BEST) Working Party of the International Society for Blood Transfusion. An international study of the performance of sample collection from patients. Vox Sang. 2003;85(1):40-47.
17 Murphy M.F., Stearn B.E., Dzik W.H. Current performance of patient sample collection in the UK. Transfus Med. 2004;14:113-121.
18 Guidance from the Royal College of Pathologists and the Institute of Biomedical Science. The retention and storage of pathological records and specimens, 4th ed, 2009. Available at: www.rcpath.org/resources/pdf/g031retentionstorageaugust09.pdf
19 . Information for patients needing irradiated blood components from NHS Blood and Transplant. National Blood Service Hospitals and Science website at: http://hospital.blood.co.uk/library/pdf/INF_PCS_HL_005_01_Irradiated.pdf
20 Cannon M., Pierce R., Taber E.B., et al. Fatal hydrops fetalis caused by anti-D in a mother with partial D. Obstet Gynecol. 2003;102(5):1143-1145.
21 Scott M.L., Voak D. Monoclonal antibodies to RhD: development and uses. Vox Sang. 2000;78(suppl 2):79-82.
22 Jones J., Scott M.L., Voak D. Monoclonal anti-D specificity and Rh D structure: criteria for selection of monoclonal anti-D reagents for routine typing of donors. Transfus Med. 1995;5:171-184.
23 Yip S.P. Sequence variation at the human ABO locus. Ann Hum Genet. 2002;66(1):1-27.
24 BCSH Blood Transfusion Task Force. Guidelines for microplate techniques in liquid-phase blood grouping and antibody screening: a joint publication of the British Society for Haematology and the British Blood Transfusion Society. Clin Lab Haematol. 1990;12:437-460.
25 Horn K.D. The classification, recognition and significance of polyagglutination in transfusion medicine. Blood Rev. 1999;13:36-44.
26 Garratty G., Arndt P., Co A., et al. Fatal haemolytic transfusion reaction resulting from ABO mistyping of a patient with acquired B antigen detectable only by some monoclonal anti-B reagents. Transfusion. 1996;36:351-357.
27 Knowles S.M., Milkins C.E., Chapman J.F., et al. The United Kingdom National External Quality Assessment Scheme (Blood Transfusion Laboratory Practice): trends in proficiency and practice between 1985 and 2000. Transfus Med. 2002;12:11-23.
28 Denomme G.A., Wagner F.F., Fernandes B.J., et al. Partial D, weak D types and novel RHD alleles among 33,864 multiethnic patients: implications for anti-D alloimmunization and prevention. Transfusion. 2005;45(10):1554-1560.
29 Guidelines for the Blood Transfusion Services in the UK. The Red Book, 7th ed, 2005. Available at: www.transfusionguidelines.org.uk/index.aspx?Publication=RB; 2005
30 Klein H.G., Anstee D.J. The Rh blood group system. Mollison’s Blood Transfusion in Clinical Medicine, 11th ed. Blackwell, Oxford, 2005.
31 Garratty G. Screening for RBC antibodies – what should we expect from antibody detection RBCs. Immunohaematology. 2002;18:71-77.
32 Issitt P.D., Combs M.R., Bredehoeft S.J., et al. Lack of clinical significance of ‘enzyme-only’ red cell alloantibodies. Transfusion. 1993;33:284-293.
33 Shirey R.S., Boyd J.S., Ness P.M. Polyethylene glycol versus low-ionic-strength solution in pre-transfusion testing: a blinded comparison study. Transfusion. 1994;34:368-370.
34 Reis K.J., Chachowski R., Cupido A., et al. Column agglutination technology: the antiglobulin test. Transfusion. 1993;33(8):639-643.
35 MDA Evaluation Report MDA/96/14. Six systems for the detection of red cell antibodies. Hannibal House, London: MHRA
36 Bromilow I.M., Eggington J.A., Owen G.A., et al. Red cell antibody screening and identification: a comparison of two column technology methods. Br J Biomed Sci. 1993;50:329-333.
37 Cummins D., Downham B. Failure of DiaMed-ID microtyping system to detect major ABO incompatibility. Lancet. 1994;343:1649-1650.
38 Phillips P., Voak D., Knowles S., et al. An explanation and the clinical significance of the failure of microcolumn tests to detect weak ABO and other antibodies. Transfus Med. 1997;7:47-53.
39 Voak D., Downie D.M., Haigh T., et al. Improved antiglobulin tests to detect difficult antibodies: detection of anti-K by LISS. Med Lab Sci. 1982;39:363-370.
40 Contreras M., Hazlehurst G.R., Armitage S.E. Development of ‘auto-anti-A1 antibodies’ following alloimmunisation in an A2 recipient. Br J Haematol. 1983;55:657-663.
41 British Committee for Standards in Haematology. Guidelines for implementation of a maximum surgical blood order schedule. Clin Lab Haematol. 1990;12:321-327.
42 Staves J., Davies A., Kay J., et al. Electronic remote blood issue: a combination of remote blood issue with a system for end-to-end electronic control of transfusion to provide a ‘total solution’ for a safe and timely hospital blood transfusion service. Transfusion. 2008;48(3):415-424.
43 Ramsey G. Red cell antibodies arising from solid organ transplants. Transfusion. 1991;31(1):76-86.
44 O’Hagan J., White J., Milkins C.E., et al. Direct agglutination crossmatch at room temperature (DRT): the results of a NEQAS (BTLP) questionnaire. Transfus Med. 1999;9(suppl 1):42.
45 Lamberson R.D., Boral L.I., Berry-Dortch S. Limitations of the crossmatch for detection of incompatibility between A2B red blood cells and B patient sera. Am J Clin Pathol. 1986;86:511-513.
46 Shulman I.A., Odono V. The risk of overt acute hemolytic transfusion reaction following the use of an immediate-spin crossmatch. Transfusion. 1994;34:87-88.
47 Berry-Dortch S., Woodside C.H., Boral L.I. Limitations of the immediate spin crossmatch when used for detecting ABO incompatibility. Transfusion. 1985;25:176-178.
48 AABB. American Association of Blood Banks. Guidelines for Implementing the Electronic Crossmatch. Basel: Karger; 2003.
49 Säfwenberg J., Högman C.F., Cassemar B. Computerized delivery control – a useful and safe complement to the type and screen compatibility testing. Vox Sang. 1997;72:162-168.
50 Chapman J.F., Milkins C., Voak D. The computer crossmatch: a safe alternative to the serological crossmatch. Transfus Med. 2000;10:251-256.
51 Judd W.J. Requirements for the electronic crossmatch. Vox Sang. 1998;74(suppl 2):409-417.
52 UK National Blood Transfusion Committee. The appropriate use of group O RhD negative red cells, 2009 www.transfusionguidelines.org.uk/docs/pdfs/nbtc_bbt_o_neg_red_cells_recs_09_04.pdf. Available at:
53 BCSH. BCSH guidelines on the management of massive blood loss. Br J Haematol. 2006;135(5):634-641.
54 Vaughan J.I., Warwick R., Letsky E., et al. Erythropoietic suppression in fetal anemia because of Kell alloimmunization. Am J Obstet Gynecol. 1994;171(1):247-252.
55 Daniels G., Hadley A., Green C.A. Causes of fetal anemia in haemolytic disease due to anti-K. Transfusion. 2003;43:115-116.
56 Koelewijn J.M., Vrijkotte T.G., van der Schoot C.E., et al. Effect of screening for red cell antibodies, other than anti-D, to detect hemolytic disease of the fetus and newborn: a population study in the Netherlands. Transfusion. 2008;48(5):941-952.
57 Kumar S., O’Brien A. Recent developments in fetal medicine. Br Med J. 2004;328:1002-1006.
58 Bowman J. The management of hemolytic disease in the fetus and newborn. Semin Perinatol. 1997;21:39-44.
59 British Committee for Standards in Haematology. Guideline for blood grouping and antibody testing in pregnancy, 2006 www.bcshguidelines.com/pdf/pregnancy_070606.pdf. Available at:
60 Van Dijk B.A., Dooren M.C., Overbeeke M.A. Red cell antibodies in pregnancy: there is no ‘critical titre’. Transfus Med. 1995;5:199-202.
61 Daniels G. Human Blood Groups. Oxford: Blackwell; 2002.
62 Finning K., Martin P., Daniels G. A clinical service in the UK to predict fetal Rh (Rhesus) D blood group using free fetal DNA in maternal plasma. Ann N Y Acad Sci. 2004;1022:119-123.
63 Abdel-Fattah S.A., Soothill P.W., Carroll S.G., et al. Middle cerebral artery Doppler for the prediction of fetal anaemia in cases without hydrops: a practical approach. Br J Radiol. 2002;75:726-730.
64 BCSH. BCSH Guidelines for the use of prophylactic anti-D immunoglobulin, 2006 www.bcshguidelines.com/pdf/Anti-D_070606.pdf. Available at:
65 RCOG. Rh Prophylaxis, Anti-D Immunoglobulin (Green-top 22). Royal College of Obstetricians and Gynaecologists. 2002. Available at: www.rcog.org.uk
66 British Committee for Standards in Haematology. The estimation of fetomaternal haemorrhage, 2009 www.bcshguidelines.com/pdf/BCSH_Fetomaternal_sept2009 pdf. Available at:
67 National Institute for Health and Clinical Excellence. Routine antenatal anti-D prophylaxis for women who are rhesus D negative. Technology Appraisal. 2008:156. Available at: www.nice.org.uk/TA156
68 Finning K., Martin P., Summers J., et al. Effect of high throughput RHD typing of fetal DNA in maternal plasma on use of anti-RhD immunoglobulin in RhD negative pregnant women: prospective feasibility study. Br Med J. 2008;336(7648):816-818.
69 British Committee for Standards in Haematology. Transfusion guidelines for neonates and older children. Br J Haematol. 2004;124:433-453.
70 Sickle Cell Society. Standards for the clinical care of adults with Sickle Cell Disease in the UK, Chapter 6. Blood Transfusion. 2008. Available at: www.sicklecellsociety.org/pdf/CareBook.pdf
71 UK Thalassaemia Society. Standards for the clinical care of children and adults with thalassaemia in the UK, Chapter 6. Blood Transfusion. 2008. Available at: www.ukts.org/pdfs/awareness/ukts-standards-2008.pdf
72 Vichinsky E.P., Luban N.L., Wright E., et al. Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion. 2001;41:1086-1092.
73 McClelland D.B. Handbook of Transfusion Medicine, 4th ed. UK Blood Transfusion Services. The Stationery Office; 2007. Available at: www.transfusionguidelines.org/index.asp?Publication=h1&Section=9&pageid=8
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
22 Laboratory aspects of blood transfusion
Safe and effective blood transfusion requires the combined efforts of blood transfusion services, biomedical scientists and clinicians to ensure the highest standards are applied to all the systems in a complex process from ‘vein to vein’. This chapter provides a description of the laboratory framework required to provide the right blood components to the right patients at the right time. The increased awareness of what can go wrong with blood transfusion comes from a number of sources including the Serious Hazards of Transfusion UK haemovigilance scheme, which was started in 1996.1,2 This confidential reporting scheme, using detailed root cause analysis of errors, has provided data that have informed both national bodies and local transfusion services of measures to introduce in order to reduce risk (Fig. 22.1). It is clear that multiple errors can contribute to a single adverse event and that many of these are outside the control of the transfusion laboratory.
Within the laboratory setting, the application of strict protocols for sample labelling and testing, robust laboratory procedures, reliable documentation, frequent staff training and competency assessment should be used. Recently, the UK Transfusion Collaborative, comprising representatives of the professional bodies involved in UK transfusion practice, has produced some recommendations to reduce laboratory errors in transfusion. These address issues such training, competency, staffing levels and automated systems.3
This chapter is concerned with the testing of patient samples prior to the provision of appropriate compatible blood components including identification of red cell antibodies. It also covers compatibility testing and investigation of transfusion reactions and the testing required in other special situations including the antenatal and postnatal settings. National professional bodies such as the British Committee for Standards in Haematology (BCSH)4 and the AABB5 issue guidance to transfusion laboratories and this has been referenced where appropriate.
There is a new regulatory framework governing hospital transfusion laboratory practice which was implemented after the publication of two European Union Directives: 2002/98/EC and 2004/33/EC.6 In the UK these are the Blood Safety and Quality Regulations 2005 (Statutory Instruments 2005/50, 2005/1098 and 2006/2013) and they set standards for quality and safety of human blood and blood components in hospital ‘blood banks’ as well as ‘blood establishments’ (the UK Blood Services).7 ‘Blood banks’ are regulated in their roles of storing, distributing and performing compatibility tests, on blood and blood components for use in hospitals. The implications for hospital transfusion practice are the requirement for ‘vein to vein’ traceability of blood components, the importance of maintaining the ‘cold chain’ for all therapeutic blood components, the need to store transfusion records for 30 years and the requirement for a quality management system.6,7 UK laboratories have to assess themselves and submit an annual compliance report to the competent authority, which is the Medicines and Healthcare Products Regulatory Agency (MHRA).8 The MHRA carries out laboratory inspections to assess compliance with these regulations.
The UK government, through the National Blood Transfusion Committee, continues to promote the safe and effective transfusion of blood components and has published three Health Service Circulars.9 These documents aim to ensure that a team approach to blood transfusion safety is taken via the local clinical governance arrangements and to promote good transfusion practice via Hospital Transfusion Committees and Hospital Transfusion Teams supported by Regional Transfusion Committees.
Technology and automation in blood transfusion laboratories
Column agglutination (CAT) and solid-phase technology can both be used on automated machines and CAT can also be used by manual techniques. In UK hospital transfusion laboratories these technologies have replaced tube techniques and liquid-phase microplates for antibody screening and crossmatching (Figs 22.2, 22.3).10
Individual laboratories need to make careful and informed decisions when selecting reagents for pre-transfusion testing. It is vital that any abbreviated testing in an automated or semiautomated system is carefully evaluated for the risks that could ensue if important controls were omitted when using this technology for blood grouping.11
Laboratory information management systems (LIMS) store patient details and results of laboratory tests, allowing timely and accurate access to important information. In the transfusion department, IT systems have a much broader use and the updated BCSH guidelines for the specification and use of Information Technology (IT) systems in blood transfusion practice (2006) reflect this.12 Where possible, using bidirectional or unidirectional interfaces to automated blood grouping analysers, IT systems are used to eliminate errors that can arise when a manual step is employed, including interpretation of test results.
Computer algorithms support the ‘electronic issue’ of blood to patients with a negative antibody screen without the need to perform an antiglobulin crossmatch and, in the UK in 2009, 46% of laboratories were using this system for some or all of their patients (Fig. 22.4). The use of automation for all aspects of compatibility testing is now recommended practice as it is recognized that it is safer.3 Automation brings several or all of the discrete activities of compatibility testing into a single platform process. It provides various levels of increased security over manual testing and may provide justification for abbreviated pre-transfusion testing (e.g. abandoning duplicate D, previously termed ‘RhD’, typing or reverse ABO grouping in the presence of a valid historical group). A risk assessment must be made and documented prior to any abbreviation of an established procedure, with consideration being given to the presence or absence of key functions in the automated equipment. The BCSH guidelines for compatibility procedures12 and guidance from the MHRA13 give a list of factors to be taken into consideration and the reader is advised to consult these prior to implementing automated or semiautomated systems.
Pre-transfusion compatibility systems
Documentation of the Transfusion Process
All stages of the transfusion process must be clearly documented and these records must be kept. In addition to guidance from the UK Royal College of Pathologists on the retention of documents,18 the BSQR 2005 regulations stipulate that the records must be accessible for 30 years.7 This allows any blood component to be traced from the donor to the recipient should information come to light about any potential infective risks to the recipient.
Computer records are easier to search than paper records, but laboratory information systems are likely to become obsolete and be replaced several times within this mandatory 30-year period, so provision must be made to store historical data in an accessible format when procuring a replacement computer system.12 Patient-held records are useful for patients who are treated in more than one institution, particularly if they have red cell antibodies and require phenotyped blood or if they have special requirements because of their underlying disease or its treatment. Credit card-sized records with corresponding patient information leaflets are issued by some transfusion centres to patients with red cell antibodies and similar cards exist for patients who require irradiated cellular blood components.19
Identification and Storage of Blood Samples
On being received in the laboratory, the details on the request form must be checked against the blood sample. Each blood sample must be labelled with a unique sample number. Barcode labels offer the advantage of positive sample identification and reduce the number of transcription errors. Samples inadequately or inaccurately labelled should NOT be used for pre-transfusion testing.11
Whole blood samples should be tested as soon as possible because they will deteriorate over time. Problems associated with storage include lysis of the red cells, loss of complement in the serum and decrease in potency of antibodies. The BCSH guidelines11 indicate working limits as outlined in Table 22.1. If separated plasma or serum samples are stored for later serological crossmatch, care must be taken to ensure that the patient has not been transfused in the interim. It has been recommended that samples should be kept for a minimum of 7 days from group and screen, stored at 4°C. Samples should be retained post-transfusion for investigation of acute transfusion reactions and preferably stored for 7 days post-transfusion to enable investigation of delayed transfusion reaction.18
ABO and D grouping
ABO Grouping
This is an excellent built-in check for the ‘forward’ or cell group and has always been considered to be an integral part of ABO grouping, allowing the reading and recording of test results to be split into two discrete tasks. However, with secure, fully automated systems, linked to secure laboratory information management systems that, in combination, have the ability to prevent procedural ABO grouping errors, some laboratories now omit the reverse group when testing samples for which a historical group is available.11 This should only be considered following a careful risk assessment and taking into account that the first sample taken may have been from the wrong patient. This may be in the order of 1:2000 samples.16 Any discrepancy between the forward and reverse groups should be investigated further and any repeat tests should be undertaken using cells taken from the original sample rather than from a prepared cell suspension.
D Grouping
Reagents for D Grouping
DVI is the partial D with the fewest epitopes; therefore of all the D variants, DVI individuals are those most likely to form anti-D and a case of severe haemolytic disease of the fetus and newborn (HDFN) has been described.20 For this reason, anti-D monoclonal reagents that do not detect DVI should be selected for testing patients’ samples.21,22 The use of anti-CDE reagents has led to the misinterpretation of r′ and r″ cells in UK National External Quality Assessment Scheme (NEQAS) exercises and, because they are of no value in routine patient typing, their use is not recommended.10,11 Selection of high-avidity monoclonal anti-D reagents will allow detection of all but the weakest examples of weak D, negating the need to use more sensitive techniques to check the D status of apparent D negatives.
Methods
There are several techniques available for routine ABO and D grouping including tube test, slide test, liquid-phase and solid-phase microplates and columns. Other techniques for blood grouping have been described but they are not in routine use. For example, molecular ABO typing is reserved for investigating anomalous ABO groups, in organ transplantation where red cells from the donor are not available, forensic practice and paternity testing.23 Care should be taken to use the appropriate reagent because not all reagents have been validated by the manufacturer for all techniques.
EDTA for diluents
Liquid-phase microplate methods
Liquid-phase microplate technology provides a cheap and secure method for batch testing when semiautomation is utilized for dispensing and reading but it is no longer the grouping technique of choice in the UK (see Fig. 22.2). In 2009, a UK NEQAS survey showed that only 13% of responding laboratories were using microplates for grouping, down from 41% in a similar survey in 2002.10
The plate is usually laid out as 12 × 8 (12 tests and 8 reagents) but may be used in the opposite orientation (8 × 12), depending on the number of reagents and controls required. Particularly if performing this technique manually, the anti-D reagents should be kept away from the anti-A and anti-B reagents because splashing between wells can occur when dispensing reagents and handling the plates during testing if insufficient care is taken. The following method is recommended:24
Column agglutination techniques
CAT techniques (see Chapter 21, p. 502) are now the commonest method for grouping in the UK (80% of laboratories who responded to a UK NEQAS survey in 2009, see Fig. 22.2), especially where automated systems are in place; these should always be performed in accordance with the manufacturer’s instructions. There are several different profiles to choose from and some cards/cassettes include monoclonal antibodies to other blood group antigens (e.g. K) in addition (see Chapter 21, p. 502 and Fig. 22.6). Forward and reverse grouping may be undertaken in separate cards/cassettes.
Controls
Positive and negative controls should be included with every test or batch of manual tests. In fully automated systems, the controls should be set up at least twice in a 24-h period. The timings should coincide with machine startup and changing of reagents, taking account of the length of time that reagents have been kept at room temperature on the machine. The control samples should be loaded in the same way as the test samples. The required controls are shown in Table 22.2. Where controls do not give the expected reactions, investigations should be undertaken to determine the validity of all tests undertaken subsequent to the most recent valid control results.
| Reagent | Positive control | Negative control |
|---|---|---|
| Anti-A | A cells | B cells |
| Anti-B | B cells | A cells |
| Anti-D | D positive cells | D negative cells |
| A1 cells | Anti-A | Anti-B |
| B cells | Anti-B | Anti-A |
Causes of Discrepancies in ABO/D Grouping
T-activation/polyagglutination
Polyagglutination25 describes agglutination of red cells by all or most normal adult sera but not by the patient’s own serum. This is as the result of IgM antibodies reacting with an antigen on the red cells which is usually hidden but can be exposed by enzyme activity. The most common form is T-activation, which occurs when the bacterial enzyme neuraminidase cleaves N-acetyl neuraminic acid from the red cell membrane, exposing the T antigen.
Acquired B
The acquired B antigen is usually caused by a bacterial deacetylase enzyme acting on A1 red cells and producing a B-like substance. Some anti-B reagents react strongly with the acquired B antigen (e.g. those derived from the ES4 clone).26 Such anti-B reagents are rare but should be avoided in routine blood grouping.
Potentiators
Red cells may be coated with IgG as a result of in vivo sensitization. The use of potentiated techniques, such as the antiglobulin test for D typing, or of potentiated reagents for ABO or D typing, may result in a false-positive reaction; the latter would also result in a positive reaction with the diluent control, but UK NEQAS data have shown that some laboratories fail to include an appropriate control or fail to understand the significance of a positive control.27 For this reason, use of potentiated techniques or reagents for blood grouping is not advised.
False-Negative Reactions
D variant phenotypes
Weak D phenotypes are where the entire D antigen is present but there are fewer D antigen sites per cell and most weak D types group as D positive with the currently available high-avidity commercial monoclonal anti-D reagents. Where differing reactions are obtained with two reagents, the patient may be a partial D (i.e. one or more of the epitopes of the D antigen is missing). It was generally thought that patients who are weak D are unable to make anti-D and may be treated as D positive whereas some patients with partial D may be capable of making immune anti-D following sensitization with the missing epitope. This concept has been challenged by reports of weak D patients with anti-D.28
Current advice about choice of D-typing reagents is that because DVI lacks the most epitopes, such individuals are likely to make anti-D when challenged by transfusion or pregnancy. For this reason, anti-D reagents for routine grouping of patients’ samples should not detect DVI.11 There is little evidence to suggest that a DVI donor would elicit an immune response in a recipient who is D negative; however, weak D positive and partial D donors, including DVI donors, should be classified as D positive.29 There are some important ethnic differences in the frequency of different Rh haplotypes, as shown in Table 22.3. Resolution of anomalous D grouping where a partial D is suspected now includes both serological testing and genotypic studies.30
Antibody screening
Red Cell Reagents
In the UK, the following antigens should be expressed as a minimum: C, c, D, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s, M, N and Lea; one cell should be R2R2 and another R1R1 or R1wR1. The following phenotypes should also be represented in the screening set: Jk(a+b−), Jk(a−b+), S+s−, S−s+, Fy(a+b−) and Fy(a−b+) (Table 22.4). These recommendations for homozygosity are based on UK data regarding the incidence of delayed haemolytic transfusion reactions, the need for high sensitivity in the detection of Kidd antibodies and the poorer performance of column agglutination techniques in the detection of some examples of Kidd antibodies using heterozygous cells.11,29 The requirement for the expression of Cw and Kpa antigens on screening cells has been the cause of much debate but in the UK and the USA detection of anti-Cw or anti-Kpa is not a requirement even in the absence of an antiglobulin crossmatch.11,29 This is because these are low-frequency antigens and the antibodies rarely cause delayed haemolytic transfusion reactions or severe haemolytic disease of the newborn.
Table 22.4 Expression of red cell antigens on screening cells11
| Blood group system | Antigen | Homozygous cells recommended |
|---|---|---|
| Rh | C | Yes (R1R1 or R1wR1) |
