[level-membership-for-pediatrics-category]
Chapter 504 Isolated Glomerular Diseases with Recurrent Gross Hematuria
504.1 Immunoglobulin A Nephropathy (Berger Nephropathy)
Pathology and Pathologic Diagnosis
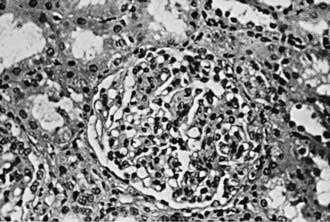
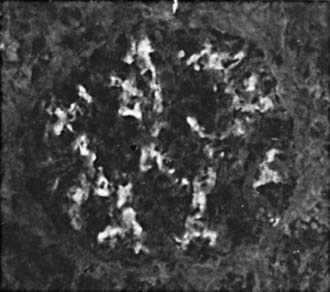
Focal and segmental mesangial proliferation and increased mesangial matrix are seen in the glomerulus (Fig. 504-1). Renal histology demonstrates mesangial proliferation that may be associated with epithelial cell crescent formation and sclerosis. IgA deposits in the mesangium are often accompanied by C3 complement (Fig. 504-2).
Delos Santos NM, Wyatt RJ. Pediatric IgA nephropathies: clinical aspects and therapeutic approaches. Semin Nephrol. 2004;24:269-286.
Dillon JJ. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers for IgA nephropathy. Semin Nephrol. 2004;24:218-224.
Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin Nephrol. 2004;3:225-243.
Gharavi AG, Yan Y, Scolari F, et al. IgA nephropathy, the most common cause of glomerulonephritis is linked to 6q22-23. Nat Genet. 2000;26:354-357.
Naganishi K, Yoshikawa N. Immunoglobulin A nephropathy. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:757-782.
Sanders JT, Wyatt RJ. IgA nephropathy and Henoch-Schönlein purpura nephritis. Curr Opin Pediatr. 2008;20:163-170.
Wang AY, Lai FM, Yu AW, et al. Recurrent IgA nephropathy in renal transplant allografts. Am J Kidney Dis. 2001;38:588-596.
504.2 Alport Syndrome
Pathology
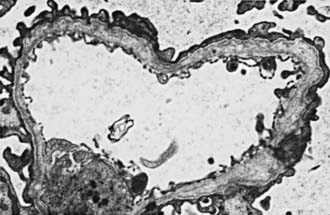
In most patients, electron microscopy reveals diffuse thickening, thinning, splitting, and layering of the glomerular and tubular basement membranes (Fig. 504-3). Ultrastructural analysis of the GBM in all genetic forms of AS may be completely normal, display nonspecific alterations, or demonstrate only uniform thinning.
Prognosis and Treatment
The risk of progressive renal dysfunction leading to end-stage renal disease (ESRD) is highest among hemizygotes and autosomal recessive homozygotes. ESRD occurs before age 30 yr in approximately 75% of hemizygotes with X-linked AS. The risk of ESRD in X-linked heterozygotes is 12% by age 40 yr and 30% by age 60 yr. Risk factors for progression are gross hematuria during childhood, nephrotic syndrome, and prominent GBM thickening. Intrafamilial variation in phenotypic expression results in significant differences in the age of ESRD among family members. No specific therapy is available to treat AS, although angiotensin-converting enzyme inhibitors can slow the rate of progression. Careful management of renal failure complications such as hypertension, anemia, and electrolyte imbalance is critical. Patients with ESRD are treated with dialysis and kidney transplantation (Chapter 529). Approximately 5% of kidney transplant recipients develop anti-GBM nephritis, which occurs primarily in males with X-linked AS who develop ESRD before age 30 yr.
Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009;20:1210-1215.
Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14:2603-2610.
Kashtan CE. Familial hematuria due to type IV collagen mutations: Alport syndrome and thin basement membrane nephropathy. Curr Opin Pediatr. 2004;16:177-181.
Kashtan CE, Gubler MC. Inherited glomerular diseases. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:621-642.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 504 Isolated Glomerular Diseases with Recurrent Gross Hematuria
504.1 Immunoglobulin A Nephropathy (Berger Nephropathy)
Pathology and Pathologic Diagnosis
Focal and segmental mesangial proliferation and increased mesangial matrix are seen in the glomerulus (Fig. 504-1). Renal histology demonstrates mesangial proliferation that may be associated with epithelial cell crescent formation and sclerosis. IgA deposits in the mesangium are often accompanied by C3 complement (Fig. 504-2).
