[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 10 Investigation of megaloblastic anaemia
cobalamin, folate and metabolite status
Cobalamin absorption and metabolism
Cobalamin in the human diet is a bacterial product ingested and stored by animals and strict vegans are therefore liable to deficiency. The prevalence of cobalamin deficiency, as defined by serum vitamin B12 <200 ng/l and methylmalonic acid >0.27 μmol/l, is 1.6% of subjects over 51 years of age in the 2001–2004 National Health and Nutrition Examination Survey1 in the USA. Of this age group, 3.2% have a low serum B12 <200 ng/l. Ingested cobalamin is released from food proteins by pepsin and acid and bound initially by transcobalamin I or haptocorrin (R binder). This binder also binds other cobinamides in the diet which are metabolically inert. Pancreatic enzymes release the cobalamin from transcobalamin I and permit binding by intrinsic factor. The intrinsic factor–cobalamin complex is attached to cubam, a multiligand receptor, which is a combination of cubilin2 and amnionless and is taken up by endocytosis, into the ileal cell. The cobalamin is then released from the endosome and bound to transcobalamin II (holotranscobalamin) in the ileal cell and exported into the portal circulation. Cobalamin undergoes enterohepatic circulation via the liver and bile ducts with 1.4 μg/day excreted in the bile, of which 1 μg/day is reabsorbed in the ileum. Holotranscobalamin is the active form of cobalamin and is taken up by holotranscobalamin receptors (TCII receptors) on cells throughout the body, particularly liver, kidney and bone marrow cells. At the cellular level in the target tissue the holotranscobalamin undergoes endocytosis via the transmembrane TCII receptor. Holotranscobalamin then undergoes lysosomal degradation, releasing cobalamin for metabolic reactions.
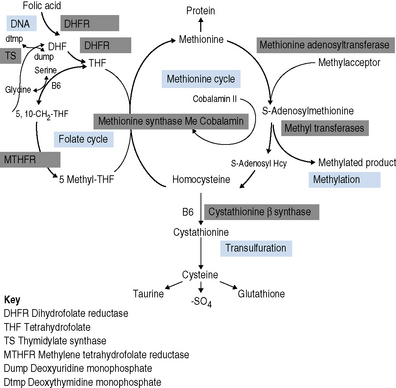
Cobalamin is a cofactor in two important biochemical reactions. In the first, methylcobalamin acts as a cofactor for methionine synthase in the production of methionine from homocysteine. The remethylation of cobalamin requires the donation of the methyl group from methyltetrahydrofolate as it is converted to tetrahydrofolate, thus linking cobalamin to folate and 1-carbon metabolism. The second cobalamin reaction occurs in the mitochondrion. Cobalamin is converted to adenosylcobalamin, a cofactor for the enzyme methylmalonyl-CoA mutase, which converts methylmalonyl-CoA (the product of propionate metabolism) to succinyl-CoA. Methionine produced in the first reaction is converted to adenosylmethionine and is a vital source of methyl groups critical for a series of methylation reactions involving proteins, phospholipids, neurotransmitters, RNA and DNA. In cobalamin deficiency, methylmalonic acid and homocysteine levels are therefore elevated. Reduced methionine synthesis is thought to result in a decrease in methylation of myelin basic protein, resulting in the neuropathies associated with cobalamin deficiency which are irreversible once subacute combined degeneration3 of the cord has occurred.
Folate absorption and metabolism
There is sufficient retention of folate by the renal tubules to prevent urinary vitamin loss; this is achieved by megalin uptake of filtered folate-binding protein4 and bound folate. Cubam,2 which binds intrinsic factor-cobalamin complex, is also important in the uptake of albumen from the renal tubules, which may also contribute to folate retention.
Folates participate in 1-C metabolism and thereby facilitate the essential cellular metabolism of methionine, serine, glycine, choline and histidine in the biosynthesis of purine and deoxythymidine monophosphate (dTMP) in the synthesis of pyrimidines and thus DNA (Fig. 10.1).
Rationale for investigation of cobalamin or folate status
Investigation of the vitamin B12 and folate status of individuals is not restricted to investigation of individuals with classical features of megaloblastic anaemia alone because neuropathy and neuro-psychiatric changes may occur in B12 deficiency in the absence of macrocytosis or anaemia.5–9 The finding that folate supplementation reduced the incidence of neural tube defects10 by 25–46% in the USA and Canada highlights the importance of defining optimum population folate levels. Increased plasma homocysteine and serum methylmalonic acid (MMA) levels have been advocated11–14 as sensitive indicators of folate and cobalamin deficiency that may be subclinical. Introduction of metabolite testing to routine laboratory practice was limited in the past as a result of technical difficulty in measurement but is now becoming more available, though the clinical benefit of this approach has been questioned by some authors.15,16 Elevated homocysteine levels are an independent vascular disease risk factor17 and are also associated with risk of idiopathic venous thrombosis.18 In the USA, dietary supplementation with folate was introduced in 1998 to reduce neural tube defects and by lowering homocysteine levels may achieve a further health gain in reduced myocardial infarction and stroke. Food supplementation with folate remains a controversial step in European countries because of a possible increased risk of cancers19,20 (see Scientific Advisory Committee on Nutrition, at: www.sacn.gov.uk, for update). Increased plasma homocysteine levels occur in both B12 and folate deficiency as a result of reduction in methionine synthesis (Fig. 10.1). Some laboratories have utilized homocysteine as an initial screening test for abnormalities of cobalamin and folate metabolism which may be particularly appropriate for investigation of suspected inherited cobalamin or folate disorders in children.21,22 MMA measurement though not widely available in the UK, requiring gas chromatography-mass spectrometry (GC-MS), is available at a limited number of university departments. In contrast, plasma homocysteine measurement by high-performance liquid chromatography (HPLC) and commercial enzyme immunoassays is widely available. The limitations of total serum B12 measurement have been highlighted by studies that showed poor positive predictive value (i.e. healthy persons with a low level or low cobalamin levels with no evidence of deficiency) and <100% negative predictive value of 95% (i.e. 5% clinically deficient with normal level).15,23 In addition, some publications have highlighted the presence of severe cobalamin deficiency concurrent with a normal cobalamin level using some of the current commercial assays.24,25 The introduction of holotranscobalamin assays,26–28 now available on an Abbott automated immunoassay platform, provides a readily accessible method of assessing the physiologically active form of cobalamin rather than the less relevant total B12. The advent of a commercial holotranscobalamin assay occurred over 20 years after depletion of serum holotranscobalamin II was identified as an early sign of cobalamin deficiency.29 Adoption of new methodology in laboratory medicine in the UK requires careful evaluation in a clinical setting and this research appears inhibited by lack of training in research methodology and financial constraints in service laboratories. Serum methylmalonic acid is increased in renal impairment and 7.9% of subjects over 51 have levels in excess of 0.27 μmol/l.1 MMA must therefore be used in conjunction with serum cobalamin and holotranscobalamin and must be interpreted in the light of renal function. Urinary MMA measurement can be used to compensate for the impact of renal impairment.
Haematological features of megaloblastic anaemia
Megaloblastic anaemia resulting from impaired DNA synthesis is characterized by the presence of megaloblastic red cell precursors in the bone marrow and occasionally also in the blood. Megaloblasts have a characteristic chromatin pattern (Fig. 10.2) and increased cytoplasm as a result of asynchrony of nuclear and cytoplasmic maturation with a relatively immature nucleus for the degree of cytoplasmic haemoglobinization. The delay in nuclear maturation caused by delay in DNA synthesis resulting from lack of vitamin B12 or folate is also seen in all lineages, particularly granulocytic marrow precursors with giant metamyelocytes (Fig. 10.3) and hyperlobated neutrophils with increased lobe size as well as number of nuclear segments (see Chapter 5, Fig. 5.10). In severe pernicious anaemia, a progressive increase in mean red cell volume (MCV) up to 130 fl occurs, with oval macrocytes, poikilocytes and hypersegmentation of neutrophils (>5% with more than five nuclear lobes).31 The neutrophil hypersegmentation index is an equivalent automated parameter on some cell counters, although hypersegmentation does not always respond to a therapeutic trial. The mean platelet volume is decreased and there is increased platelet anisocytosis, as detected by the platelet distribution width (PDW). The MCV falls to 110–120 fl as megaloblastic change advances. Howell–Jolly bodies and basophilic stippling are seen in the red cells.
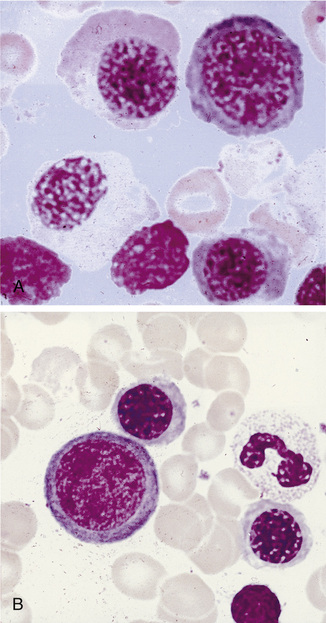
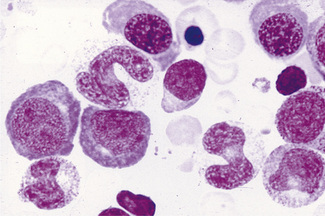
Figure 10.3 Photomicrograph of bone marrow film stained by May–Grünwald–Giemsa showing giant metamyelocytes.
Differential Diagnosis of Macrocytic Anaemia
Macrocytic red cells are also seen in myelodysplastic syndromes, which can be suspected from the presence of hypogranular neutrophils (see Chapter 5, Fig. 5.76) or monocytosis. Excess alcohol consumption results in an increased MCV as a result of round macrocytes, although rarely does it go higher than 110 fl unless coexisting folate deficiency is present. Hypothyroidism, liver disease, aplastic anaemia, rare inherited orotic aciduria or Lesch–Nyhan syndrome also have a high MCV. Automated reticulocyte counts facilitate detection of increased red cell turnover and high MCV as a result of haemolysis or bleeding. Coexisting iron deficiency or thalassaemia trait may mask macrocytic changes, although a high red cell distribution width indicates anisocytosis and the need for blood film review. Congenital dyserythropoietic anaemias types I and III and erythroleukaemia exhibit some features of megaloblastic erythropoiesis that are unrelated to B12 and folate. Drugs interfering with DNA synthesis (e.g. azathioprine, zidovudine or hydroxycarbamide) result in macrocytosis and megaloblastic erythropoiesis. Anticonvulsant therapy interferes with folate metabolism,32 whereas the impact of oral contraceptives on folate absorption and metabolism is controversial.33 Prolonged nitrous oxide anaesthesia destroys methylcobalamin and causes acute megaloblastic change.34 Methotrexate inhibits dihydrofolate reductase and toxicity can be reversed with folinic acid, which is already in the tetrahydrofolate form and effectively reverses the metabolic block, which is not achieved solely with folic acid.
Testing strategy for suspected cobalamin or folate deficiency
Microbiological cobalamin and folate assays and competitive radiodilution binding assays for measurement of cobalamin and folate, which were often performed together, have largely been replaced by separate analysis by automated binding assays. The application of a suitable testing strategy for patients suspected of having cobalamin or folate deficiency is shown in Tables 10.1–10.3.
| Symptoms or signs | Possible significance | |
|---|---|---|
| Tiredness, palpitations, pallor | Anaemia | |
| Slight jaundice | Ineffective erythropoiesis | |
| Neurological | ||
| Cognitive impairment, optic atrophy, loss of vibration sense, joint position sense; plantar responses normal or abnormal; tendon reflexes depressed or increased | Cobalamin deficiency, subacute combined degeneration of the spinal cord and sensori/motor peripheral neuropathies | |
| Dietary and gastrointestinal history | ||
| Vegetarian or vegan; poor nutrition (e.g. tea and toast diet in elderly or students); dietary fads | Low iron stores and iron deficiency | |
| Cobalamin deficiency in babies born to mothers who are vegans | ||
| Folate deficiency (often with iron deficiency) | ||
| Weight loss, bloating and steatorrhoea, particularly nocturnal bowel movements | Features of malabsorption and folate deficiency, e.g. due to coeliac disease, tropical sprue | |
| Mouth ulcers, abdominal pain, perianal ulcers, fistulae | Terminal ileal Crohn’s disease – cobalamin deficiency | |
| Glossitis, angular cheilosis and koilonychia | Cobalamin and combined iron deficiency | |
| Alcohol history | Poor diet and interference with folate metabolism | |
| History of autoimmune disease in patient or family | ||
| Hypothyroidism, pernicious anaemia or coeliac disease | Increased likelihood of pernicious anaemia or coeliac disease | |
| Surgery | ||
| Gastrectomy/bowel resection | Cobalamin deficiency usually 2 years post-gastrectomy | |
| Ileal disease resulting in cobalamin deficiency | ||
| Blind loop syndromes | ||
| Physical appearance | ||
| Grey hair, blue eyes, vitiligo | Association with pernicious anaemia | |
| Pregnancy | Increased iron and folate requirements. | |
| Cobalamin levels fall by 30% in the 3rd trimester | ||
| Holotranscobalamin levels are unaltered in late pregnancy | ||
| Malabsorptive syndrome | ||
| Tropical sprue, bacterial overgrowth, fish tape worm in Scandinavian countries | Combined folate and iron deficiency | |
| Cobalamin deficiency | ||
| Drug history | See text | |
| Other haematological disorders | ||
| Myeloproliferative neoplasms, haemolytic anaemias, leukaemias | Increased folate utilization may result in folate deficiency | |
| Myeloma | Paraprotein interference with cobalamin assays resulting in falsely low cobalamin levels, which normalize on treatment of myeloma | |
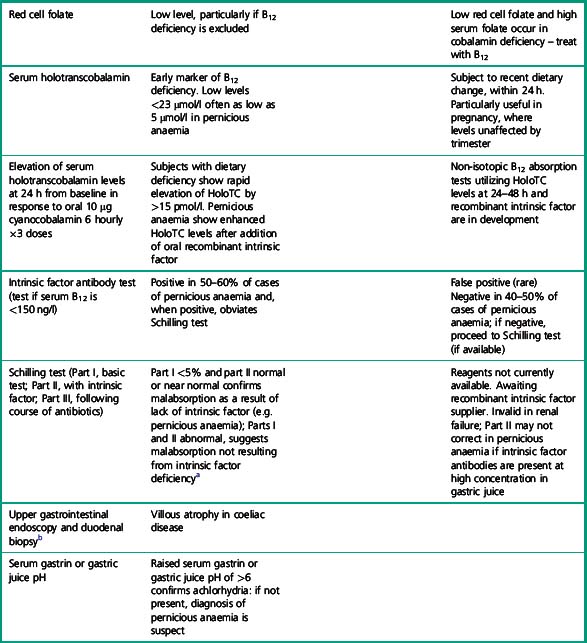

Table 10.3 Clinical and laboratory checklist for diagnosis of pernicious anaemia
| Laboratory criteria | Clinical criteria | |
|---|---|---|
| Minor criteria | Macrocytosis | Parasthesiae, numbness or ataxia |
| Anaemia | Hypothyroidism | |
| Raised plasma homocysteine | Vitiligo | |
| Gastric pH above 6 Raised serum gastrin |
Family history of pernicious anaemia or hypothyroidism | |
| Positive gastric parietal cell antibody | ||
| Major criteria | Low serum B12 (<180 ng/l) or raised serum methylmalonic acid (>0.75 μmol/l) in presence of normal renal function | |
| Megaloblastic anaemia not resulting from folate deficiency | ||
| Positive intrinsic factor antibodies using high-specificity test. | ||
| Holotranscobalamin level <23 μmol/l | ||
| Reference standard criteria | Schilling testa shows malabsorption of oral cyanocobalamin corrected by coadministration of intrinsic factor |
a Reagents for Schilling tests currently unavailable. A non-isotopic B12 absorption test utilizing recombinant intrinsic factor and holotranscobalamin measurement is under development.
Table 10.1 highlights the important clinical details that should be elicited by the clinician and submitted with the request to assist the laboratory in interpretation of the numeric results of cobalamin and folate assays. Ideally, test requests should not be accepted without this information, which could be incorporated into electronic order communication from user to laboratory.
Table 10.2 lists the important laboratory investigations that should be performed – results must not be reported in isolation from other laboratory results and clinical details. If investigations are performed in different laboratories, authorization and release of results requires access to all laboratory data on the individual patient. Laboratory information systems should facilitate this cross-disciplinary access. For example, intrinsic factor antibody results should be available to haematology or clinical chemistry laboratories undertaking cobalamin, MMA or homocysteine assays.
Table 10.3 provides a list of clinical and laboratory features for diagnosis of pernicious anaemia. These criteria avoid undue reliance on a single B12 assay and should help clinicians to make a diagnosis even when some critical tests, e.g. Schilling tests, are not available.
In view of the lack of specificity and sensitivity of serum cobalamin assays and frequent lack of availability of other diagnostic tests, basing the diagnosis of pernicious anaemia, which requires lifelong parenteral B12 therapy, solely on laboratory results, is not straightforward. A checklist of laboratory and clinical diagnostic criteria, as shown in Table 10.2, helps to achieve a greater degree of diagnostic certainty than any single diagnostic test and permits the diagnosis to be made even when a single diagnostic test is anomalous or unavailable. Clinical and other laboratory criteria thus provide additional supportive evidence of an autoimmune aetiology, even if the more demanding diagnostic laboratory criteria are not met.
Limitations of Cobalamin Assays
Sensitivity and Specificity of Cobalamin and Holotranscobalamin Assays
Utility of receiver operator characteristic curves
There has been little data on sensitivity and specificity of current B12 assays, due to the difficulty in defining a truly deficient study population. Some authors have suggested B12 assays and measurement of methylmalonic acid are no better than tossing a coin, to determine the presence or absence of deficiency. The study by Clarke et al.28 provides data which permits calculation of the specificity and sensitivity of a current B12 immunoassay in the detection of cobalamin deficiency in a community study of 1621 subjects over age 65 with normal renal function. Subjects were defined as cobalamin deficient if the methylmalonic acid was elevated above 0.75 μmol/l. Deficiency was found in 4.3% of subjects over 65 years of age with normal renal function. The mean B12 level of these subjects was 151 pmol/l (202 ng/l) by Siemens Centaur assay (range 110–199 pmol/l). Table 10.4 illustrates the calculation to derive specificity and sensitivity for the Siemens Centaur B12 assay using a cut-off point of 200 pmol/l (270 ng/l).
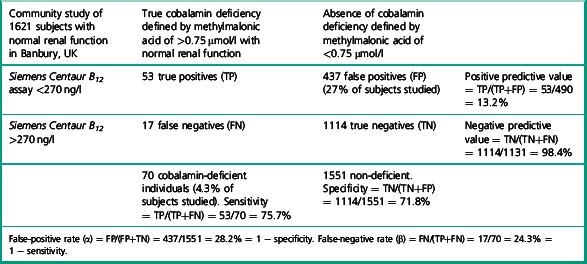
Selection of a cut-off point for cobalamin of below 150 ng/l, will identify subjects with higher probability of presence of cobalamin deficiency. Some authors13 have advocated choosing a cut-off point 25% below the reference range lower limit for any particular assay.
If, for example, a cut-off point of 125 pmol/l (168 ng/l) is chosen, the specificity of a value below this level, i.e. the number of normal individuals who fall below the cut-off point, will be markedly reduced (FP) and therefore the specificity of the test (TN/TN+FP) improves to 95%, although the detection of individuals with true deficiency who lie above the cut-off point, i.e. false negatives will be markedly increased, resulting in a sensitivity (TP/TP+FN) of 35%. The ROC curve therefore allows a laboratory to select a cut-off point that meets the objective of the laboratory – to have either a highly specific but low sensitivity test or to have a test of poor specificity but high sensitivity. Clearly cobalamin assays do not meet the criteria for an ideal test of high sensitivity and high specificity which would lie on the coordinates 0.1 to the left of the graph. The Axis-Shield/Abbott holotranscobalamin assay in this pivotal study is seen to have slightly superior ROC curves (see Fig. 10.4).
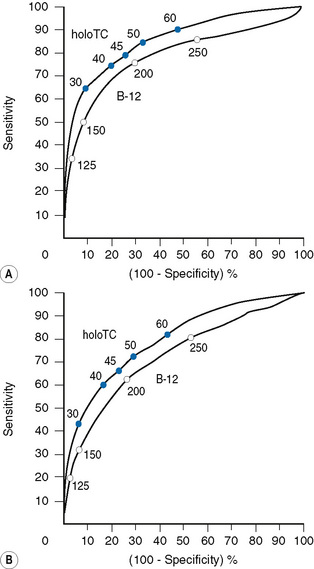
Utility of holotranscobalamin, methylmalonic acid and homocysteine assays
Holotranscobalamin assays gave a greater area under the curve, 0.85 versus 0.76 in the above study, and superior sensitivity and specificity. Holotranscobalamin is the physiologically active fraction of total cobalamin.27–29 An isolated abnormal result of serum B12 should not be the sole criterion on which treatment decisions are based and a repeat assay and other confirmatory and clinical evaluation are necessary prior to a diagnostic conclusion. Additional secondary testing with metabolite levels, or holotranscobalamin and monitoring of treatment response is recommended. High B12 levels have been described in subjects with no myeloproliferative neoplasm and who are not on cobalamin therapy or vitamin supplementation.35 This is thought to be due to immunoglobulin-complexed B12 resulting in assay interference. False normal B12 levels24,25 have been described in subjects with high titre intrinsic factor antibody25 and may also occur due to presence of heterophile antibody interference.
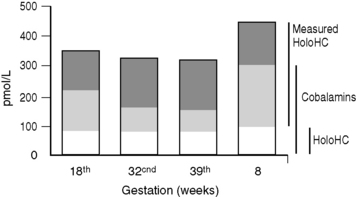
Holotranscobalamin assays27,28 may challenge total B12 assays as a first-line test in cobalamin assessment. Holotranscobalamin has been shown to be unaffected by assay interference from high-titre intrinsic factor antibody levels.36 In addition, holotranscobalamin is not subject to the 30% fall in total B12 levels seen in normal pregnancy, which makes low total B12 levels uninterpretable during pregnancy (Fig. 10.5).30
Access to homocysteine, methylmalonic acid and holotranscobalamin assays facilitates more precise definition of cobalamin and folate status in patients in whom prolonged B12 therapy may have been initiated inappropriately and continued unnecessarily or, conversely, discontinued inappropriately because of lack of confidence in the original assessment. The study by Gorringe et al.16 showed that only 27/49 patients were anaemic or macrocytic with a low total B12 of <170 ng/l and elevated MMA. All of these patients treated with B12 therapy corrected elevated MMA levels, suggesting the presence of a metabolic deficiency. However, only 15/27 showed a haematological response.
Homocysteine levels also fell by >25% in 47/49 patients after B12 treatment. Many of these patients had no clinical evidence or symptoms of cobalamin deficiency and may reflect subjects with subclinical deficiency, which could have subtle cognitive impairment, or may just represent a compensated metabolic state of no clinical consequence. Elevation of homocysteine levels is seen in folate deficiency and is therefore less specific than MMA measurement. The causes of cobalamin deficiency are shown in Table 10.5.
| Supportive information/diagnostic tests | ||
|---|---|---|
| Reduced intake | ||
| Strict vegetarian/vegan | Dietary history | |
| Dietary fad that excludes dairy products and meat | Ethnic origin/culture | |
| Breastfed babies of mothers who are vegetarian or cobalamin deficient | ||
| Poor dietary intake in elderly | ||
| Malabsorption as a result of loss or inactivity of intrinsic factor | ||
| Addisonian pernicious anaemia | Diagnostic criteria for pernicious anaemia (see Table 10.3) | |
| Gastrectomy (partial or total) | History of gastric surgery | |
| Bacterial overgrowth or parasitic infestation of small bowel | Radiolabelled lactose breath tests for bacterial overgrowth Repeat Schilling test post-antibiotic therapy |
|
| Pancreatic dysfunction: failure of trypsin release of B12 from R binding proteins | Pancreatic function tests; exocrine pancreatic dysfunction results in abnormal Schilling test but clinical deficiency is rare | |
| Malabsorption as a result of failure of B12-intrinsic factor complex uptake in ileum – ileal resection | Radiological, enteroscopic or capsule camera study of small bowel for Crohn’s disease of terminal ileum or tuberculous ileitis | |
| Congenital Imerslund–Gräsbeck syndrome | Subjects of Scandinavian origin | |
| Tropical sprue | Small bowel biopsy | |
| Zollinger–Ellison syndrome | Multiple gastric and duodenal ulcers | |
| Pancreatic adenoma on imaging | ||
| Food cobalamin malabsorption | ||
| Atrophic gastritis with achlorhydria | Endoscopic and gastric biopsy findings | |
| Gastric surgery | ||
| Abnormal transport proteins | ||
| Transcobalamin II deficiency | Megaloblastic anaemia in presence of normal cobalamin levels; transcobalamin II and holotranscobalamin levels reduced | |
| Transcobalamin I deficiency | No evidence of clinical deficiency but low serum cobalamin levels | |
| Possible fall in holotranscobalamin levels in elderly | ||
| Inborn errors of cobalamin metabolism (see reviews21,22) | Serum and urinary methylmalonic acid and metabolite measurement | |
| Acquired drug effects | ||
| Nitrous oxide: chronic repeated exposure | Drug history | |
| Colchicine: chronic usage impairs B12 uptake resulting from diarrhoea | ||
| Metformin reduces B12 levels in diabetics. | ||
Clinical and Diagnostic Pitfalls of Folate Assays
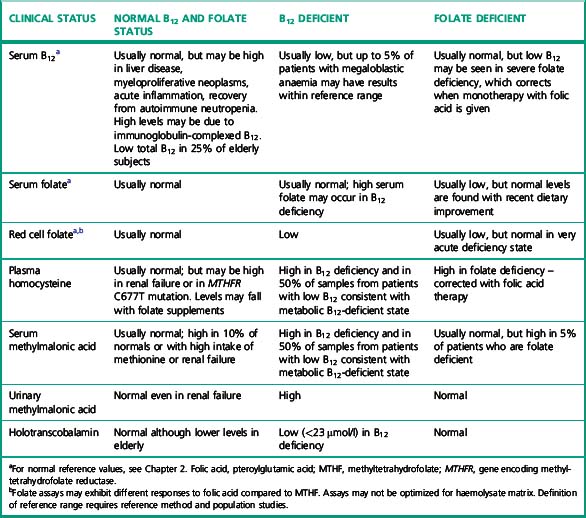
Serum folate is altered by acute dietary change and interruption of enterohepatic recycling; it can therefore be low without significant tissue deficiency. This may be a particular problem in hospital inpatients. Red cell folate was originally advocated as correlating better with megaloblastic change37,38 reflecting the folate status over the lifespan of the red cells (2–3 months), but a subsequent study suggested that little was to be gained by the addition of red cell folate analysis because only 14% of patients with low serum folate also have low red cell folate levels.39 Minor haemolysis in vitro may cause spurious elevation of serum folate levels because the red cell folate may be 10–20 times the serum value. More than half of the patients with severe cobalamin deficiency have a low red cell folate because impaired methionine synthesis results in accumulation of methyltetrahydrofolate (MTHF) monoglutamate, which diffuses out of cells resulting in a high serum folate.40,41 Treatment with cobalamin alone will correct the low red cell folate and high serum folate levels. Concern over the inter-method variability of red cell folate assays, and questions about the additional benefits39 of measurement of both serum and red cell folate, have reduced the use of red cell folate assays. However, satisfactory results are possible if appropriate care is taken with preanalytical sample preparation and analysis. 5-methyl THF is a very labile substance and the addition of sodium ascorbate has reduced the coefficient of variation by half in serum folate assays in external quality assurance surveys.42 The interplay between serum B12, serum folate and red cell folate and plasma homocysteine and MMA is shown in Table 10.7. In view of the limitations of both serum and red cell folate assays, it is prudent to measure both. An international reference method43,44 for serum folate has been recognized by the Joint Committee on Traceability in Laboratory Medicine (JCTLM) and is now used to verify the target values in UK National External Quality Assessment Scheme (NEQAS) Haematinics surveys (www.ukneqas-haematinics.org.uk). This should improve the accuracy of serum folate assays, which have been bedevilled by inter-method differences and variation in recommended reference ranges between manufacturers. Red cell folate assays still suffer from a lack of a standardized method for haemolysate preparation and matrix effects, which result in large inter-method differences.
Clinicians and patients using different laboratories require information about derivation of reference ranges, which will now be included in the standards for laboratory accreditation in the UK. Definition of population reference ranges for both serum and red cell folate have been difficult to achieve due to changing diet and food supplementation and assay variability. Elevated levels of homocysteine have been found in elderly subjects,45,46 indicating possible subclinical folate deficiency in the elderly. Folate reference ranges provided by kit manufacturers also show large inter-method differences. In the USA, some authors have advocated cessation of folate testing since, following dietary supplementation of flour, folate deficiency is very unusual. The causes of clinical deficiency and supportive information or diagnostic tests are shown in Table 10.6 and the interactions between cobalamin and folate are shown in Table 10.7.
| Supportive information/diagnostic tests | ||
|---|---|---|
| Reduced intake | ||
| Poor diet particularly alcoholics (wine and spirits because beer contains folate) | Dietary and alcohol history | |
| Elderly or students ‘tea and toast diet’ | ||
| Dietary fads | ||
| Premature babies | ||
| Unsupplemented parenteral nutrition | ||
| Malabsorption | ||
| Coeliac disease (often with coexisting iron deficiency) Tropical sprue |
Antiendomysial, antigliadin tests, antitissue transglutaminase | |
| Small bowel resection, malabsorption syndromes | Small bowel biopsy | |
| Drug effects | ||
| Sulphasalazine, methotrexate, trimethoprim-sulphamethoxazole, pyrimethamine, phenytoin, sodium valproate, oral contraceptives | Drug history | |
| Bone marrow aspiration | ||
| Hereditary hyperhomocysteinaemia | ||
| Homozygotes for C677T MTHFR have lower folate levels | ||
| Increased folate turnover | ||
| Pregnancy: progressive fall in 3rd trimester | ||
| Increased requirements for breastfeeding | ||
| Skin disease – severe psoriasis or exfoliation | ||
| Haemodialysis | ||
| Haemolysis: haemoglobinopathy, paroxysmal nocturnal haemoglobinuria, autoimmune haemolytic anaemia (see Chapters 11–13) | ||
Standards, Accuracy and Precision of Cobalamin and Folate Assays
There are currently no internationally recognized reference methods for serum cobalamin measurement, but isotope dilution liquid chromatography tandem mass spectrometric methods have recently been accepted as international reference methods for the quantification of folate species in serum.43,44 As a result, international reference materials have been developed (by the World Health Organization, WHO 03/178, and by the National Institute of Standards and Technology, NIST SRM), with values assigned for folate species by the tandem mass spectrometric methods.47 Although reference methods have not been verified for the whole blood matrix as yet, there is a WHO whole blood international standard (95/528) with consensus values for total folate.48
Evaluation of commercial automated binding assays by recovery experiments has shown under-recovery of added 5-methyl THF and over-recovery of pteroylglutamic acid (PGA),49 whereas a suitably calibrated microbiological assay recovers closer to 100%.50 Differential sensitivity of assays to pteroylglutamic acid and genetic variability in the proportion of in vivo formyl folates may be a factor in inter-method variability.
A microbiological assay was the method used to assign a potency value to the British Standard for human serum B12,51 and this was later re-classified as the 1st WHO International Standard (IS) (81/563). The 2nd WHO IS for serum B12, 03/178, was ratified in 2007,47 the values adopted being a consensus of the contemporary B12 protein-binding assays.
Genetic Factors
A number of methylenetetrahydrofolate reductase polymorphisms that alter the proportion of formylfolate in serum have been described52 and this could be a potential source of disparity in the response of some sera to different assays. Individuals homozygous for C677T genetic polymorphism have 25% higher plasma homocysteine levels than controls.53 A genetic–nutrient interactive effect is noted in that the polymorphism confers a greater effect on homocysteine levels in those individuals with low folate levels. Cigarette smoking, age, renal disease, drugs including levodopa and folate supplements all affect homocysteine levels.
Pre-analytical Sample Preparation
The addition of sodium ascorbate 5 mg/ml will stabilize folate in serum, extending sample storage times. Stabilization of serum folate with sodium ascorbate added to the primary blood collection tube would improve the reproducibility of routine serum folate assays as shown in external quality control surveys,42 but would necessitate introduction of separate B12 and folate sample tubes since ascorbate interferes with cobalamin analysis. EDTA plasma is unsuitable and heparinized plasma may result in higher values. Samples must be fibrin free and without bubbles.
Analytical Factors
Methods for cobalamin and folate analysis
Microbiological bioassays and radiodilution assays for serum B12 and folate54 are still used, albeit by a decreasing minority of laboratories and continue to play an important role in the evaluation of new automated methods. They are also used in population studies where they are useful in providing information on the long-term comparability of results. (They are detailed in the 9th edition of this book.)
Serum B12 assays
Binding of B12 to Kit Binder
The binding of B12 to kit binder is the competitive step of the assay. Serum-derived cyanocobalamin competes with labelled cobalamin, which is usually complexed to a chemiluminescent or fluorescent substrate or enzyme, for limited binding sites on porcine intrinsic factor. Specificity for cobalamin is ensured by purification of the intrinsic factor (IF) or by blocking contaminating corrinoid binders (R binders) by addition of excess blocking cobinamide. Specificity of pure and blocked IF can be demonstrated by the addition of cobinamide to sera. There should be no increase in assay value. Some assays use only the alkaline denaturation step to inactivate the endogenous binders. It is important that assays are not be affected by the presence of high-titre anti-intrinsic factor antibody in patient sera. The Siemens Centaur, Beckman Access, Siemens Immulite 2000/2500 and Tosoh assays have all been vulnerable to this false normal B12 effect and carry product literature warnings to this effect. Roche and Abbott assays have not demonstrated this problem with such sera and the holotranscobalamin assay by Axis-Shield is also unaffected.25,26,36
Holotranscobalamin assays
Principle
About 6–20% of B12 is bound to TC II forming the physiologically active complex HoloTC and in this form is taken up by cells. Levels of holotranscobalamin (HoloTC) B12 are 30–160 pmol/l. The remainder of the serum B12 is bound to transcobalamin I (haptocorrin), which is involved in the transport of B12 to the liver and enterohepatic circulation thereof. Haptocorrin also binds B12 in the gastric contents and B12 is released from haptocorrin by pancreatic proteases prior to capture by IF. HoloTC is thought to be the first metabolite to decrease following reduced intake or absorption of B12.28,29 A commercial assay is available from Axis-Shield and Abbott.27 The clinical utility of this assay is being confirmed particularly in pregnancy sera where changes in transcobalamin I make total B12 levels uninterpretable. The relative merits of HoloTC compared with metabolite measurement remain to be clarified. As a sensitive marker of cobalamin malabsorption, holotranscobalamin levels that correct with small oral doses of B12 and the use of recombinant intrinsic factor55–58 could provide the basis for a non-isotopic B12 absorption test to replace the Schilling test, currently unavailable in the UK.
Holotranscobalamin ‘Active B12’ Immunoassay
Measurement of the ‘active form of B12’27 is based on a monoclonal antibody to cobalamin bound to TCII known as HoloTC. This ‘active’ form of B12 amounts to 20% of the total serum B12. Reference range values are 19–134 pmol/l; pernicious anaemia values are <5 pmol/l. HoloTC is thought to be more sensitive to early deficiency than the measurement of total B12 and may be a better indicator of vulnerability to impaired cognitive function.28 Levels are elevated by renal failure, but are unaffected by pregnancy.30
HoloTC Radioimmunoassay
The HoloTC radioimmunoassay59 uses magnetic microspheres coated with monoclonal antibody to holotranscobalamin and achieves separation from haptocorrin by magnetic separator. A57Co B12 tracer together with a reducing and a denaturing agent are then added to destroy the HoloTC linkage. When the B12 binder containing IF is added, the free B12 and tracer compete for binding. The unbound tracer is removed by centrifugation and the bound fraction is measured using a gamma counter. The measured radioactivity reflects the competition between tracer and vitamin B12 bound to transcobalamin (i.e. HoloTC). The concentration of vitamin B12 in the sample is calculated from a calibration curve using recombinant human HoloTC. The assay only requires a 0.4 ml sample volume, the coefficient of variation is <10%, the limit of detection is 10 pmol/l and the assay time is 4 h.
Quantitation of Transcobalamin Saturation
Nexo and colleagues60,61 described a method permitting measurement of total TC and HoloTC. The method uses B12 modified by acid treatment and bound to magnetic beads, which can then be used to remove unsaturated TC or apoTC from serum. The remaining HoloTC is then measured by an enzyme-lined immunosorbent assay (ELISA). Thus the total and HoloTC can be measured and the TC saturation (HoloTC/Total TC) can be quantitated.
In a study of 137 healthy blood donors the reference range for HoloTC was 40–150 pmol/l. Some 10% of circulating TC is saturated with a reference range of 5–20%; 15–50% of B12 is bound to TC. In subjects who were B12 deficient, HoloTC was 2–34 pmol/l and the TC saturation was 0.4–3%, well below the reference interval, providing a clear cut-off from normal sera. Nexo’s method combines a sensitive ELISA60 for HoloTC with a simple procedure for removal of the unsaturated TC or apoTC.
Serum folate methods
Binding of Folate to Folate-Binding Protein
β-Lactoglobulin, isolated from cow’s milk, is commonly used as a binding agent in folate assays, i.e. as the folate-binding protein (FBP). Unfortunately, these lactoglobulins are not specific for the attachment of 5-methyl THF and will bind many forms of folate, dependent on pH. These properties have been utilized for the standardization of folate assay. The physiologically active form (5-methyl THF) is very unstable. Pteroylglutamic acid (PGA), present in vitamin supplements, is more stable, binds to FBP and has been used as an alternative standard for the folate assays. Its use as a standard depends on the observation that the binding affinities for 5-methyl THF and PGA are equivalent62 at pH 9.3 ± 0.1. It is therefore essential that the pH is strictly maintained at the binding stage of the procedure. In most of these assays, serum-derived folate competes with chemiluminescent or enzyme-labelled folate for limited sites on FBP. In the Roche Elecsys methods the FBP is labelled with ruthenium and competition exists between endogenous and biotin-labelled folate.
Red cell folate methods
Whereas Lactobacillus casei responds equally to both triglutamates and monoglutamates, the affinity of the FBP varies with the number of glutamate residues. Reproducible protein-binding assays for red cell folate can only be achieved by release and conversion of the protein-bound folate polyglutamates, mainly 5-methyl THF with four or five additional glutamate moieties, to a monoglutamate form. There must be adequate dilution of the red cells in hypotonic solution, a pH between 3.0 and 6.0 (ideally pH 4.5–5.2) for optimal conditions for plasma folate deconjugase (polyglutamate hydrolase) and ascorbic acid to stabilize the reduced forms.63,64
Haemolysate preparations for the newer assay platforms vary widely. The concentration of ascorbic acid varies from 0.09% to 1%, dilution factors from 1:5 to 1:31 and the duration of haemolysate preparation from 40 to 180 min. Some assays require the addition of protein to lysates before analysis and use bovine serum or human serum albumin, whereas others need only aqueous solutions. The pH of lysing diluent varies from 3.0 to 4.0 and that of the deconjugase step from 4.0 to 6.8 (the final pH of the lysate after protein addition varies from 4.4 to 7.5). These various factors may help to explain the large intermethod differences detected in external quality assessment surveys. Inadequate lysis and deconjugation will give falsely low results.65,66
Calculation of Red Blood Cell Folate from Haemolysate Folate Result
Corrected red cell folate (μg/l) =
Red cell folate in μg/l − [serum folate in μg/l × (1 − Hct)/Hct]
e.g. when Hct is 0.45, red cell folate is 180 μg/l and serum folate is 28 μg/l, then
Corrected red cell folate (μg/l) = 180 − [28 × (1 − 0.45)/0.45] = 145.8.
Serum B12 and Folate and Red Cell Folate Assay Calibration
Cobalamin in serum is protein bound and therefore standards for total B12 assays should be gravimetrically prepared cyanocobalamin in either a lyophilized serum or protein adjusted matrix. A WHO lyophilized serum standard with consensus values for serum B12 is available from the National Institute for Biological Standards and Control (NIBSC) (www.nibsc.ac.uk);47 however, the protein binding B12 assays are not usually calibrated with this material.
For serum folate analysis, 5-methyl THF is the physiologically active folate form and therefore should be used as the standard. However, 5-methyl THF is highly unstable and historically PGA has been used as the primary calibrator either gravimetrically added to aqueous standards or used to assign values to secondary serum matrix standards. The principle underlying the use of PGA is dependent on the equimolar binding of 5-methyl THF or PGA at pH 9.3.62 More recent work suggests the pH of equimolar binding may be nearer 8.9 than 9.3.
The recent development of isotope dilution, liquid chromatography, tandem mass spectrometry (ID-LC/MS/MS) reference methods for folate derivatives in serum43,44 has permitted the introduction of higher-order serum and plasma reference materials with accurate values for folates. A lyophilized serum standard (WHO 03/178)47 is available from NIBSC and three frozen plasma preparations (FPP) with assigned values for 5-methyl THF and folic acid (FA) are available from the NIST in the USA (www.nist.gov/index.html).
Whole Blood Folate Standards
An international reference preparation for whole blood folates48 is available from NIBSC although the haemolysate preparations are generally analysed using the serum method protocols and calibration curves.
Methylmalonic acid measurement
Methods
Plasma or serum MMA is extracted, purified and, using tert-butyldimethylsilyl derivatives of MMA, measured by GC-MS. A deuterated stable isotope of MMA is used as an internal standard. The use of dicyclohexyl, another derivative of MMA, is described by Rasmussen.67
A method for screening for inborn errors of metabolism using O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine HCl to derive oxoacids, followed by liquid partition chromatography, was described by Hoffman et al.68 In more recent HPLC or capillary electrophoresis methods,69,70 MMA can be derivatized using the fluorescent compound 1-pyrenyldiazomethane to yield a fluorescent monoester adduct. After separation by HPLC or capillary electrophoresis, short-chain dicarboxylic acids are quantified following laser-induced fluorescence activation.
Reference ranges for serum MMA measured by GC-MS and using mean ± 3SD of log transformed data are 53–376 nmol/l71 and 50–440 nmol/l (Rasmussen et al.72).
Homocysteine measurement
Principle
Homocysteine is a disulphide amino acid present at low concentrations in cells (<1 µmol/l) and in plasma at 5–15 µmol/l.73 Homocysteine has a reactive sulphydryl group that forms disulphide bonds with homocysteine or cysteine or protein sulphydryl groups to form the oxidized form of homocysteine: homocystine, homocysteine-cysteine mixed disulphide and protein-bound homocysteine. The free homocysteine is <2% of plasma homocysteine and 80% is as protein-bound homocysteine; the remainder is homocystine or mixed disulphides. Reducing conditions convert all these species to homocysteine by reduction of disulphides; therefore total homocysteine is measured.
For quantification, plasma homocysteine requires protein precipitation and reduction of disulphide bonds. The S-H group of homocysteine is derivatized using a thiol-specific reagent and the resulting adduct is detected. A variety of methods have evolved73 from ion-exchange amino acid analysers,74 radioenzymatic determination, capillary gas chromatography75 stable isotope dilution combined with capillary GC-MS,76 liquid chromatography electrospray tandem mass spectrometry77 and HPLC methods using fluorochromophore detection.
Immunoassay for Homocysteine Measurement
Automated enzyme immunoassays78 for homocysteine have now been developed by a number of manufacturers and have reduced the dependence of clinical laboratories on highly specialized instrumentation such as gas chromatography, mass spectrometry and HPLC. The general principle of these methods is the release of protein-bound homocysteine by buffered DTT and the reduction of homocystine and mixed disulphides to homocysteine. The enzymatic conversion of free homocysteine to s-adenosyl-L-homocysteine (sAH) is then achieved with s-adenosyl homocysteine hydrolase in the presence of excess adenosine. Competitive reactions are used to quantify total homocysteine and methods are further sub-classified based on differences in the separation and detection of sAH. These include fluorescence polarization immunoassay (FPIA), chemiluminescent immunoassay (CIA), enzyme-linked immunosorbent assay (ELISA) and other enzyme immunoassays (EIA). More recently latex enhanced agglutination immunoassays have also been developed for homocysteine quantitation.
Reference Methods for Homocysteine
There are three procedures quoted on the Joint Committee for Traceability in Laboratory Medicine (JCTLM) database as high-order reference procedures for homocysteine in human serum.79,80 Methods include isotope dilution, gas chromatography, mass spectrometry (ID/GC/MS) and two methods based on isotope dilution, liquid chromatography, tandem mass spectrometry (ID/LC/MS/MS). These methods are used to assign homocysteine (and folate) values to frozen serum reference material (SRM 1955) produced by NIST in the USA. Lyophilized materials with values assigned by the reference procedures are not available yet.
Relative Performance of Homocysteine Methods
Proficiency testing shows that there are small but significant differences in the values produced by the enzymatic methods when compared with HPLC and immunoassays.81–85 While the within-run precision is acceptable for all three types of technology the between-laboratory data reveals a greater imprecision in the HPLC method group.85 When evaluated in the light of intra-individual variability in homocysteine levels, FPIA meets the performance goal suggested by Fraser and Petersen.86
Pre-analytical Variables in Homocysteine Testing
Plasma homocysteine is elevated in both B12 and folate deficiency and is elevated in individuals with a common genetic polymorphism of methylenetetrahydrofolate reductase, C677T polymorphism, the homozygous form of which is present in 10% of Western populations and results in homocysteine levels 25% above the normal upper limit. A raised homocysteine level is an independent risk factor for vascular disease, including myocardial infarction and stroke.18 Homocysteine levels are affected by age, renal function, smoking, excessive coffee consumption and drugs, including levodopa. Correction of elevated homocysteine levels in patients with cobalamin or folate deficiency by administration of one or both vitamins provides proof of a deficiency state, which may be subclinical.
Factors affecting albumin levels will alter homocysteine because it is protein bound and venepuncture should not be performed after venous stasis or following the subject resting in supine position. Plasma is preferred because cells leak homocysteine, resulting in an increase of 10% during clot formation from uncoagulated samples. Even plasma will show an increase of 10% per hour at room temperature as a result of leakage from red cells.87 Samples should be kept on ice and centrifuged as soon as possible, at least within 1 h.
Dynamic testing of cobalamin folate metabolism
Another dynamic functional test of cobalamin–folate metabolism, utilized primarily in research laboratories, is the deoxyuridine suppression test,88 in which failure of suppression by deoxyuridine of tritiated thymidine 3H-TdR incorporation into DNA in cobalamin or folate deficient is corrected by treatment with the appropriate haematinic. (The method was described in the 9th edition of this book.)
Investigation of the cause of cobalamin deficiency
Once cobalamin deficiency has been confirmed by the finding of an unequivocally low serum B12 result (with or without confirmatory raised metabolite levels and response to therapy), the aetiology of the low cobalamin should be elucidated as in Table 10.5. Gastric parietal cell antibodies are present in 90% of patients with pernicious anaemia, but this is of low specificity, being found in 15% of elderly subjects, and is therefore of little discriminatory use. Achlorhydria as a cause of cobalamin malabsorption may be suspected by the presence of raised gastrin levels.89
Intrinsic factor antibody measurement
Principle
Two types of antibody to IF have been detected in the sera of patients with pernicious anaemia. Type I blocks the binding of B12 to IF, whereas type II prevents the attachment of IF or the IF–B12 complex to ileal receptors. Type II antibodies (precipitating antibodies) may be precipitated by IF–B12 complex and sodium sulphate at pH 8.3 in barbitone buffer. More than 60% of patients with pernicious anaemia are reported to have type I or type II antibodies.90,91 Automated competitive binding assays are now available and have largely superseded radiodilution assays. A number of manufacturers are developing IF antibody assays for their multianalyte immunoassay platforms.
Assay methods have been reviewed90 and one method for detection of types I and II IF antibody based on radiodilution competitive binding was described in the 9th edition of this book.
ELISA Methods for Type I and Type II Intrinsic Factor Antibodies
Serum is incubated in the presence of IF bound to a solid phase in such a way that both the type I and type II binding sites are available for binding IFAb.91 Excess unbound serum is removed and the solid phase is further incubated with conjugate-labelled (e.g. horseradish peroxidase) antihuman IgG. Unbound conjugate is removed and substrate is added to develop the signal, which is proportional to the amount of IFAb in the original serum. The specificity of the IF antibody assay will depend on the purity of the solid-phase IF. Purified porcine intrinsic factor or recombinant intrinsic factor are used in different ELISAs and the UK NEQAS intrinsic factor antibody quality control surveys have shown variable positive rates for different types of ELISA,92 perhaps reflecting different sensitivities and specificity of patient sera.
Investigation of absorption of B12
Development of Non-Isotopic B12 Absorption Tests Using Holotranscobalamin Levels and Recombinant Intrinsic Factor
Holotranscobalamin levels increase 3–4 h after oral cobalamin intake and, after 3 doses of 9 μg cyanocobalamin given 6-hourly on day 1, peak serum HoloTC was detected at 24 h in healthy adults maintained on a normal diet and given bread and juice to aid absorption of each cobalamin dose. Cellular uptake of HoloTC will be greater in deficient individuals and therefore the time course of a rise in HoloTC and the dose of cyanocobalamin may vary. Further work to optimize this approach to develop a standardized B12 absorption test is ongoing.55–57 Bhat et al.58 gave 10 μg of cyanocobalamin at 6-hourly intervals ×3 and defined a normal response as a rise in plasma HoloTC of >15% and >15 pmol above baseline measurement; the majority of Asian women studied corrected low holotranscobalamin levels, suggesting dietary deficiency as the cause.
Non-Isotopic B12 Absorption Test Utilizing Recombinant Intrinsic Factor in Combination with Holotranscobalamin Levels
Native (human) or bovine intrinsic factor is no longer commercially available. Hvas et al.57 have therefore utilized recombinant intrinsic factor from 1 g of the plant leaves of transgenic Arabidopsis thaliana (Pharma Skan, Skanderborg, Denmark) to show enhanced cobalamin absorption and holotranscobalamin levels in subjects with B12 deficiency. Three patterns were observed: one group showed a large increase in HoloTC (mean rise 13 pmol/l) from baseline levels with cyanocobalamin, indicating dietary deficiency; clinically deficient individuals showed only a small rise (mean 5 pmol/l) in HoloTC after cyanocobalamin, which could be enhanced (>10 pmol by day 3) by addition of intrinsic factor, indicating a lack of intrinsic factor (e.g. gastrectomy or autoimmune pernicious anaemia); and a third group with no increase in HoloTC, with or without intrinsic factor, a finding consistent with malabsorption.
Urinary Excretion of Radiolabelled B12 With and Without Intrinsic Factor (Schilling Test)
Radiolabelled cyanocobalamin and bovine intrinsic factor reagents are no longer available. (Details of the principles of the Schilling tests can be found from the references93,94 and in the 10th edition of this book.)
B12 binding capacity of serum or plasma: transcobalamin measurement
Principle
Transcobalamin I (TCI) binds 80% or more of the total serum B12 and the B12–TCI complex is known as holohaptocorrin. TCII is the minor but important transport protein that delivers B12 to the tissues. The B12–TCII complex is known as holotranscobalamin (HoloTC) and 6–25% of total serum B12 is carried in this complex. HoloTC is believed to be the first B12 metabolite that decreases on inadequate B12 absorption. TCII and III are normally virtually unsaturated unless an individual is undergoing B12 treatment. TCII and III should therefore be measured prior to B12 treatment. TCI and III (R binders) are glycosylated proteins differing in their sugar moiety. Chronic myeloid leukaemia, myelofibrosis and other myeloproliferative neoplasms are characterized by increased levels of TCI and therefore total serum B12. Primary liver cancer (fibrolamellar hepatoma) is also associated with synthesis of large quantities of an abnormal form of TCI. It has been suggested that some low B12 levels without evidence of B12 deficiency may result from a decrease in R binder concentration.95 Congenital absence of R binders TCI and III results in very low serum B12 but no evidence of B12 deficiency.96 In congenital absence of TCII,21,22 which results in fulminating pancytopenia and megaloblastosis within 2 months of birth, the serum B12 is normal, unsaturated B12 binding capacity is decreased as a result of absent TCII and B12 absorption is reduced; the deoxyuridine suppression test is abnormal and corrected by B12.
Acknowledgements
The description of metabolic pathways for cobalamin, folate and homocysteine were assisted by Hematology Basic Principles and Practice99 and Homocysteine in Health and Disease.100 We wish to thank Annie Lee, Julie Bonser, staff at UK NEQAS Haematinics Department of Haematology and Good Hope NHS Trust for collaboration on production of international reference standards and experimental work on folate and B12 assays, Dr Christine Pfeiffer at Centers for Disease Control and Prevention (CDC) Atlanta for collaborative work on the international reference method for serum folate and inter-method comparisons and Dr Susan Thorpe at NIBSC for collaborative work on international reference standards. Thanks also to family, friends and colleagues for ever-present support and encouragement.
1 Centers for Disease Control and Prevention. Available at: www.cdc.gov/ncbddd/B12/table4.html.
2 Fyle J.C., Madsen M., Hojrup P., et al. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood. 2004;103:1573-1579.
3 Pearce J. Subacute combined degeneration of the cord. Putnam–Dana Syndrome European Neurology. 2008;60:53-56.
4 Birn H. The kidney in vitamin B12 and folate homeostasis: characterization of receptors for tubular uptake of vitamins and carrier proteins. Am J Physiol Renal Physiol. 2006;291:F22-F36.
5 Waters A.H., Mollin D.L. Observations on the metabolism of folic acid in pernicious anaemia. Br J Haematol. 1963;9:319-327.
6 Jewsbury E.C. Subacute combined degeneration of the cord and achlorhydric peripheral neuropathies without anaemia. Lancet. 1954;3:307-312.
7 Healton E.H., Savage D.G., Brust J.C.M., et al. Neurologic aspects of cobalamin deficiency. Medicine. 1991;70:229-245.
8 Lindenbaum J., Healton E.B., Savage D.G., et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anaemia or macrocytosis. N Engl J Med. 1988;318:1720-1728.
9 Smith D., Refsum H. Vitamin B12 and cognition in the elderly. Am J Clin Nutr. 2009;89:707S-711S.
10 Rush D. Folate supplements prevent recurrence of neural tube defects. FDA Dietary Supplement Task Force. Nutr Rev. 1992;50:22-28.
11 Savage D.G., Lindenbaum J., Stabler S.P., et al. Sensitivity of serum methylmalonic acid and total homocysteine for diagnosing cobalamin and folate deficiencies. Am J Med. 1994;96:239-246.
12 Holleland G., Schneede J., Ueland P.M., et al. Cobalamin deficiency in general practice. Assessment of the diagnostic utility and cost benefit analysis of methylmalonic acid determination in relation to current diagnostic strategies. Clin Chem. 1999;45:189-198.
13 Hvas A.M., Nexo E. Diagnosis and treatment of vitamin B12 deficiency – an update. Haematologica. 2006;91:1506-1512.
14 Klee G. Cobalamin and folate evaluation: Measurement of methylmalonic acid and homocysteine vs vitamin B12 and folate. Clin Chem. 2000;46:1277-1283.
15 Solomon L.R. Cobalamin responsive disorders in the ambulatory care setting: unreliability of cobalamin, methylmalonic acid and homocysteine testing. Blood. 2005;105:978-985.
16 Gorringe A., Ellis R., McDowell I., et al. The limited value of methylmalonic acid, homocysteine and holotranscobalamin in the diagnosis of early B12 deficiency. Haematologica. 2006;91:231-234.
17 Nygard O., Vollset S.E., Refsum H., et al. Total homocysteine and cardiovascular disease. J Intern Med. 1999;246:425-454.
18 Ridker P.M., Hennekens C.H., Selhub J., et al. Interrelation of hyperhomocystinaemia, factor V Leiden and risk of future venous thromboembolism. Circulation. 1997;95:1777-1782.
19 Smith A.D., Kim Y., Refsum H. Is folic acid good for everyone? Am J Clin Nutr. 2008;87:517-533.
20 Ebbing M., Bonaa K.H., Nygard O., et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. J Am Med Assoc. 2009;302:2119-2126.
21 Briddon A. Homocysteine in the context of cobalamin metabolism and deficiency states. Amino Acids. 2003;24:1-12.
22 Whitehead V.M. Acquired and inherited disorders of cobalamin and folate in children. Br J Haematol. 2006;134:125-136.
23 Stabler S.P., Allen R.H., Savage D.G., et al. Clinical spectrum and diagnosis of cobalamin deficiency. Blood. 1990;76:871-881.
24 Devalia V. Diagnosing vitamin B-12 deficiency on the basis of serum B-12 assay. Br Med J. 2006;333:385-386.
25 Hamilton M.S., Blackmore S., Lee A. Possible cause of false normal B-12 assays. Br Med J. 2006;333:654-655.
26 Ulleland M., Eilertsen I., Quadros E.V., et al. Direct assay for cobalamin bound to Transcobalamin (holotranscobalamin) in serum. Clin Chem. 2002;48:526-532.
27 Brady J., Wilson L., McGregor L., et al. Active B12: a rapid automated assay for holotranscobalamin on the Abbott AxSYM analyser. Clin Chem. 2008;54:567-573.
28 Clarke R., Sherliker P., Hin H., et al. Detection of vitamin B12 deficiency in older people by measuring vitamin B12 or the active fraction of vitamin B12 holotranscobalamin. Clin Chem. 2007;53:963-970.
29 Herzlich B., Herbert V. Depletion of serum holotranscobalamin. II. An early sign of negative vitamin B12 balance. Lab Invest. 1988;58:332-337.
30 Morkbak A.L., Hvas A.M., Milman N., et al. Holotranscobalamin remains unchanged during pregnancy. Longitudinal changes of cobalamins and their binding proteins during pregnancy and post partum. Haematologica. 2007;92:1711-1712.
31 Lindenbaum J., Nath B.J. Megaloblastic anaemia and neutrophil hypersegmentation. Br J Haematol. 1980;44:511-513.
32 Rose M., Johnson I. Reinterpretation of the haematological effects of anti-convulsant treatment. Lancet. 1978;1:1349-1350.
33 Shojania A.M., Hornady G.J. Oral contraceptives: effect of folate and B12 metabolism. Can Med Assoc J. 1982;126:244-247.
34 Lassen H.C., Henriksen A., Neukirch F., et al. Treatment of tetanus: severe bone marrow depression after prolonged nitrous oxide anaesthesia. Lancet. 1956;270:527-530.
35 Jeffery J., Millar H., MacKenzie P., et al. An IgG complexed form of vitamin B12 is a common cause of elevated serum concentrations. Clin Biochem. 2010;43:82-88.
36 Hamilton M.S., Blackmore S., Lee A., et al. Ability of holotranscobalamin assay (active B12) to detect severe cobalamin deficiency evidenced by high methylmalonic acid in the presence of high titre intrinsic factor antibody and false normal B12 results. Br J Haematol. 2010;149(supp 1):26. Abs 54
37 Hoffbrand A.V., Newcombe B.F., Mollin D.L. Method of assay of red cell folate activity and the value of the assay as a test for folate deficiency. J Clin Pathol. 1966;19:17-28.
38 Jaffe J.P., Schilling R.E. Erythrocyte folate levels: a clinical study. Am J Hematol. 1991;36:116-121.
39 Phekoo K., Williams Y., Schey S.A., et al. Folate assays serum or red cell? J R Coll Physicians Lond. 1994;31:291-295.
40 Lavoie A., Tripp E., Hoffbrand A.V. The effect of vitamin B12 deficiency on methylfolate metabolism and pteroylpolyglutamate synthesis in human cells. Clin Sci Mol Med. 1974;47:617-630.
41 Perry J., Lumb M., Laundy M., et al. Role of vitamin B12 in folate coenzyme synthesis. Br J Haematol. 1976;32:243-248.
42 Blackmore S., Lee A., Hamilton M.S. The impact of sample stabilization with sodium ascorbate on the analysis of serum folate in the UKNEQAS Haematinics surveys. Clinica Chimica Acta. 2005;355 UKNP 1.3:S459.
43 Pfeiffer C.M., Fazili Z., McCoy L., et al. Determination of folate vitamers in human serum by stable-isotope-dilution tandem mass spectrophotometry and comparison with radioassay and microbiologic assay. Clin Chem. 2004;50:423-432.
44 Nelson B., Pfeiffer C.M., Margolis S., et al. Solid phase extraction-electrospray ionization mass spectrometry for the quantification of folate in human plasma or serum. Anal Biochem. 2004;325:41-51.
45 Selhub J., Jacques P.F., Wilson P.W., et al. Vitamin status and input as primary determinants of homocystinaemia in an elderly population. J Am Med Assoc. 1993;270:2693-2698.
46 Selhub J., Jacques P.F., Rosenbery I.H., et al. Serum total homocysteine concentrations in the third National Health and Nutrition Survey 1991–1994. Population reference ranges and contribution of vitamin status to high serum concentrations. Ann Intern Med. 1999;131:331-339.
47 Thorpe S.J., Heath A., Blackmore S., et al. International standard for serum B12 and serum folate: international collaborative study to evaluate a batch of lyophilised serum for B12 and folate content. Clin Chem Lab Med. 2007;45:380-386.
48 Thorpe S., Sands D., Heath A.B., et al. An international standard for whole blood folate: evaluation of a lyophilised haemolysate in an international collaborative study. Clin Chem Lab Med. 2004;42:533-539.
49 Blackmore S., Pfeiffer C., Hamilton M.S., et al. Recoveries of folate species from serum pools sent to participants of the UKNEQAS Haematinics scheme in February and March 2004. Clinica Chimica Acta. 2005;355 UKNP 1.2:S459.
50 Fazili Z., Pfieffer C.M., Zhang M. Comparison of serum folate species analysed by LC-MS/MS with total folate measured by microbiologic assay and Bio-Rad radioassay. Clin Chem. 2007;53:781-784.
51 Curtis A.D., Mussett M.V., Kennedy D.A. British Standard for human serum vitamin B12. Clin Lab Haematol. 1986;8:135-147.
52 Bagley P.J., Selhub J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc Natl Acad Sci U S A. 1998;95:13217-13220.
53 Jacques P.F., Bostom A.G., Williams R.R., et al. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase and plasma homocysteine concentrations. Circulation. 1996;93:7-9.
54 O’Broin S.D., Kelleher B.P. Microbiological assay on microtitre plates of folate in serum and red cells. J Clin Pathol. 1992;45:344-347.
55 Castel-Roberts K.M., Morkbak A.L., Nexo E., et al. Holotranscobalamin is an indicator of vitamin B12 absorption in healthy adults with adequate vitamin B12 status. American Journal of Nutrition. 2007;85:1057-1061.
56 Hvas A.M., Morkbak A.L., Nexo E. Plasma holotranscobalamin compared with plasma cobalamins for assessment of vitamin B12 absorption: optimization of a non radioactive vitamin B12 absorption test. Clin Chim Acta. 2007;376:150-154.
57 Hvas A.M., Buhl T., Laursen N.B., et al. The effect of recombinant human intrinsic factor on the uptake of vitamin B12 in patients with evident vitamin B12 deficiency. Haematologica. 2006;91:805-807.
58 Bhat D.S., Thuse N.V., Lubree H., et al. Increases in plasma holotranscobalamin can be used to assess vitamin B12 absorption in individuals with low plasma vitamin B12. J Nutr. 2009;139:2119-2123.
59 Ulleland M., Eilertsen I., Quadros E.V., et al. Direct assay for cobalamin bound to transcobalamin (holotranscobalamin) in serum. Clin Chem. 2002;48:526-532.
60 Nexo E., Christensen A., Petersen T.E., et al. Measurement of transcobalamin by ELISA. Clin Chem. 2000;46:1643-1649.
61 Nexo E., Christensen A., Hvas A., et al. Quantification of holotranscobalamin, a marker of vitamin B12 deficiency. Clin Chem. 2002;48:561-562.
62 Givas J., Gutcho S. pH dependence of the binding of folate to milk binder in radioassay of folates. Clin Chem. 1975;21:427-428.
63 Bain B.J., Wickramasinghe S.N., Broom G.W., et al. Assessment of the value of a competitive protein binding radioassay of folic acid in the detection of folic acid deficiency. J Clin Pathol. 1984;37:888-894.
64 Omer A. Factors influencing the release of assayable folate from erythrocytes. J Clin Pathol. 1969;22:217-221.
65 Netteland B., Bakke O.M. Inadequate sample preparation as a source of error in determination of erythrocyte folate by competitive binding radioassay. Clin Chem. 1977;23:1505-1506.
66 Shane B., Tamura T., Stokstad E.L.R. Folate assay: a comparison of radioassay and microbiological methods. Clin Chim Acta. 1980;100:13-19.
67 Rasmussen K. Solid phase sample extraction for rapid determination of methylmalonic acid in serum and urine by a stable isotope dilution method. Clin Chem. 1989;35:260-264.
68 Hoffmann G., Aramaki S., Blum-Hoffmann E., et al. Quantitative analysis for organic acids in biological sample: batch isolation followed by gas chromatographic-mass spectrometric analysis. Clin Chem. 1989;35:587-595.
69 Schneede J., Ulleland P.M. An automated assay for methylmalonic acid in serum and urine based on derivatization with 1-pyrenyldiazomethane, liquid chromatography and fluorescence detection. Clin Chem. 1993;39:392-393.
70 Schneede J., Ulleland P.M. Application of capillary electrophoresis with laser-induced fluorescence detection for routine determination of methylmalonic acid in human serum. Anal Chem. 1995;67:812-819.
71 Allen R.H., Stabler S.P., Savage D.G., et al. Diagnosis of cobalamin deficiency. I. Usefulness of serum methylmalonic acid and total homocysteine concentrations. Am J Hematol. 1990;34:90-98.
72 Rasmussen K., Moller J., Ostergaard K., et al. Methylmalonic acid concentrations in serum of normal subjects: biological variability and effect of oral l-isoleucine loads before and after intramuscular administration of cobalamin. Clin Chem. 1990;36:1295-1299.
73 Ulleland P.M., Refsum H., Stabler S.P., et al. Total homocysteine in plasma or serum: methods and clinical applications. Clin Chem. 1993;39:1764-1779.
74 Rasmussen K., Moller J. Methodologies of testing. In: Carmel R., Jacobsen D.W., editors. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001:9-20.
75 Kataoka H., Takagi K., Makita M. Determination of total plasma homocysteine and related aminothiols by gas chromatography with flame photometric detection. J Chromatogr B Biomed Appl. 1995;664:421-425.
76 Stabler S.P., Marcell P.D., Podell E.R., et al. Quantitation of total homocysteine, total cysteine and methionine in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal Biochem. 1987;162:185-196.
77 Magera M.J., Lacey J.M., Casetta B., et al. Method for the determination of total homocysteine in plasma and urine by stable isotope dilution and electron spray tandem mass spectrometry. Clin Chem. 1999;41:1517-1522.
78 Shipchandler M.T., Moore E.G. Rapid fully automated measurement of plasma homocysteine with the Abbott Imx analyser. Clin Chem. 1995;41:991-994.
79 Satterfield M.B., Sniegoski L.T., Welch M., et al. Comparison of isotope dilution mass spectrometry methods for the determination of total homocysteine in plasma and serum. Anal Chem. 2003;75:4631-4638.
80 Satterfield M.B., Sniegoski L.T., Welch M., et al. Comparison of isotope dilution mass spectrometry methods for the determination of total homocysteine in plasma and serum. [Comparability assessment studies.]. Anal Chem. 2003;75:775-784.
81 Pfeiffer C.M., Twite D., Shih J., et al. Method comparison for total plasma homocysteine between the Abbott Imx analyser and an HPLC assay with internal standardization. Clin Chem. 1999;45:152-153.
82 Pfeiffer CM, Huff DL, Smith SJ, et al. Comparison of plasma total homocysteine measurements in 14 laboratories: an international study. Clin Chem. 45:1261–1288
83 Nexo E., Engbaek F., Ueland P.M., et al. Evaluation of novel assays in clinical chemistry: quantification of plasma total homocysteine. Clin Chem. 2000;46:1150-1156.
84 Huijgen H.J., Tegelaers F.P., Schoenmakers C.H.H., et al. Multicentre analytical evaluation of an enzymatic method for the measurement of plasma homocysteine and comparison with HPLC and immunochemistry. Clin Chem. 2004;50:937-940.
85 Hanson N.Q., Eckfeldt J.H., Schwichtenberg K., et al. Interlaboratory variation of plasma total homocysteine measurements: results of three successive homocysteine proficiency testing surveys. Clin Chem. 2002;48:1539-1545.
86 Fraser C.G., Petersen P.H. Analytical performance characteristic should be judged against objective quality specifications. Clin Chem. 1999;45:321-323.
87 Andersson A., Isaksson A., Hultberg B. Homocysteine export from erythrocytes and its implications for plasma sampling. Clin Chem. 1992;38:1311-1315.
88 Wickramasinghe S.N., Matthews J.H. Deoxyuridine suppression: biochemical basis and diagnostic applications. Blood Rev. 1988;2:168-177.
89 Slingerland D.W., Cararelli J.A., Burrows B.A., et al. The utility of serum gastrin levels in assessing the significance of low serum B12 levels. Arch Intern Med. 1984;144:1167-1168.
90 Shackleton P.J., Fish D.I., Dawson D.W. Intrinsic factor antibody tests. J Clin Pathol. 1989;42:210-212.
91 Waters H.M., Smith C., Howarth J.E., et al. A new enzyme immunoassay for the detection of total type I and type II intrinsic factor antibody. J Clin Pathol. 1989;42:307-312.
92 Lee A., Blackmore S., Hamilton M.S. A new external quality assessment (EQA) scheme for intrinsic factor antibody (IFAb) assays: a key diagnostic criterion for pernicious anaemia. Clinica Chimica Acta. 2005;355 UKTP 4.42:S297.
93 Schilling R.F. Intrinsic factor studies. II. The effect of gastric juice on the urinary excretion of radioactivity after the oral administration of radioactive vitamin B12. J Lab Clin Med. 1953;42:860-866.
94 International Committee for Standardization in Haematology. Recommended method for the measurement of vitamin B12 absorption. J Nucl Med. 1981;22:1091-1093.
95 Carmel R. R-binder deficiency: a clinically benign cause of cobalamin deficiency. J Am Med Assoc. 1988;250:1886-1890.
96 Jacob E., Herbert V. Measurement of unsaturated ‘granulocyte-related’ (TCI and TCIII) and ‘liver-related’ (TCII) binders by instant batch separation using a microfine precipitate of silica (QUSO G32). J Lab Clin Med. 1975;88:505-512.
97 Nexo E., Christensen A., Hvas A.B., et al. Quantification of holotranscobalamin, a marker of vitamin B12 deficiency. Clin Chem. 2002;48:561-562.
98 Scott J.M., Bloomfield F.J., Stebbins R., et al. Studies on derivation of transcobalamin III from granulocytes: enhancement by lithium and elimination by fluoride of in vitro increments in vitamin B12-binding capacity. J Clin Invest. 1974;53:228-239.
99 Hoffman R., Furie B., McGlave P., et al, editors. Hematology Basic Principles and Practice, 5th ed., London: Churchill Livingstone Elsevier, 2009. Ch. 39
100 Carmel R., Jacobsen D.W. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 10 Investigation of megaloblastic anaemia
cobalamin, folate and metabolite status
Cobalamin absorption and metabolism
Cobalamin in the human diet is a bacterial product ingested and stored by animals and strict vegans are therefore liable to deficiency. The prevalence of cobalamin deficiency, as defined by serum vitamin B12 <200 ng/l and methylmalonic acid >0.27 μmol/l, is 1.6% of subjects over 51 years of age in the 2001–2004 National Health and Nutrition Examination Survey1 in the USA. Of this age group, 3.2% have a low serum B12 <200 ng/l. Ingested cobalamin is released from food proteins by pepsin and acid and bound initially by transcobalamin I or haptocorrin (R binder). This binder also binds other cobinamides in the diet which are metabolically inert. Pancreatic enzymes release the cobalamin from transcobalamin I and permit binding by intrinsic factor. The intrinsic factor–cobalamin complex is attached to cubam, a multiligand receptor, which is a combination of cubilin2 and amnionless and is taken up by endocytosis, into the ileal cell. The cobalamin is then released from the endosome and bound to transcobalamin II (holotranscobalamin) in the ileal cell and exported into the portal circulation. Cobalamin undergoes enterohepatic circulation via the liver and bile ducts with 1.4 μg/day excreted in the bile, of which 1 μg/day is reabsorbed in the ileum. Holotranscobalamin is the active form of cobalamin and is taken up by holotranscobalamin receptors (TCII receptors) on cells throughout the body, particularly liver, kidney and bone marrow cells. At the cellular level in the target tissue the holotranscobalamin undergoes endocytosis via the transmembrane TCII receptor. Holotranscobalamin then undergoes lysosomal degradation, releasing cobalamin for metabolic reactions.
Cobalamin is a cofactor in two important biochemical reactions. In the first, methylcobalamin acts as a cofactor for methionine synthase in the production of methionine from homocysteine. The remethylation of cobalamin requires the donation of the methyl group from methyltetrahydrofolate as it is converted to tetrahydrofolate, thus linking cobalamin to folate and 1-carbon metabolism. The second cobalamin reaction occurs in the mitochondrion. Cobalamin is converted to adenosylcobalamin, a cofactor for the enzyme methylmalonyl-CoA mutase, which converts methylmalonyl-CoA (the product of propionate metabolism) to succinyl-CoA. Methionine produced in the first reaction is converted to adenosylmethionine and is a vital source of methyl groups critical for a series of methylation reactions involving proteins, phospholipids, neurotransmitters, RNA and DNA. In cobalamin deficiency, methylmalonic acid and homocysteine levels are therefore elevated. Reduced methionine synthesis is thought to result in a decrease in methylation of myelin basic protein, resulting in the neuropathies associated with cobalamin deficiency which are irreversible once subacute combined degeneration3 of the cord has occurred.
Folate absorption and metabolism
There is sufficient retention of folate by the renal tubules to prevent urinary vitamin loss; this is achieved by megalin uptake of filtered folate-binding protein4 and bound folate. Cubam,2 which binds intrinsic factor-cobalamin complex, is also important in the uptake of albumen from the renal tubules, which may also contribute to folate retention.
Folates participate in 1-C metabolism and thereby facilitate the essential cellular metabolism of methionine, serine, glycine, choline and histidine in the biosynthesis of purine and deoxythymidine monophosphate (dTMP) in the synthesis of pyrimidines and thus DNA (Fig. 10.1).
Rationale for investigation of cobalamin or folate status
Investigation of the vitamin B12 and folate status of individuals is not restricted to investigation of individuals with classical features of megaloblastic anaemia alone because neuropathy and neuro-psychiatric changes may occur in B12 deficiency in the absence of macrocytosis or anaemia.5–9 The finding that folate supplementation reduced the incidence of neural tube defects10 by 25–46% in the USA and Canada highlights the importance of defining optimum population folate levels. Increased plasma homocysteine and serum methylmalonic acid (MMA) levels have been advocated11–14 as sensitive indicators of folate and cobalamin deficiency that may be subclinical. Introduction of metabolite testing to routine laboratory practice was limited in the past as a result of technical difficulty in measurement but is now becoming more available, though the clinical benefit of this approach has been questioned by some authors.15,16 Elevated homocysteine levels are an independent vascular disease risk factor17 and are also associated with risk of idiopathic venous thrombosis.18 In the USA, dietary supplementation with folate was introduced in 1998 to reduce neural tube defects and by lowering homocysteine levels may achieve a further health gain in reduced myocardial infarction and stroke. Food supplementation with folate remains a controversial step in European countries because of a possible increased risk of cancers19,20 (see Scientific Advisory Committee on Nutrition, at: www.sacn.gov.uk, for update). Increased plasma homocysteine levels occur in both B12 and folate deficiency as a result of reduction in methionine synthesis (Fig. 10.1). Some laboratories have utilized homocysteine as an initial screening test for abnormalities of cobalamin and folate metabolism which may be particularly appropriate for investigation of suspected inherited cobalamin or folate disorders in children.21,22 MMA measurement though not widely available in the UK, requiring gas chromatography-mass spectrometry (GC-MS), is available at a limited number of university departments. In contrast, plasma homocysteine measurement by high-performance liquid chromatography (HPLC) and commercial enzyme immunoassays is widely available. The limitations of total serum B12 measurement have been highlighted by studies that showed poor positive predictive value (i.e. healthy persons with a low level or low cobalamin levels with no evidence of deficiency) and <100% negative predictive value of 95% (i.e. 5% clinically deficient with normal level).15,23 In addition, some publications have highlighted the presence of severe cobalamin deficiency concurrent with a normal cobalamin level using some of the current commercial assays.24,25 The introduction of holotranscobalamin assays,26–28 now available on an Abbott automated immunoassay platform, provides a readily accessible method of assessing the physiologically active form of cobalamin rather than the less relevant total B12. The advent of a commercial holotranscobalamin assay occurred over 20 years after depletion of serum holotranscobalamin II was identified as an early sign of cobalamin deficiency.29 Adoption of new methodology in laboratory medicine in the UK requires careful evaluation in a clinical setting and this research appears inhibited by lack of training in research methodology and financial constraints in service laboratories. Serum methylmalonic acid is increased in renal impairment and 7.9% of subjects over 51 have levels in excess of 0.27 μmol/l.1 MMA must therefore be used in conjunction with serum cobalamin and holotranscobalamin and must be interpreted in the light of renal function. Urinary MMA measurement can be used to compensate for the impact of renal impairment.
Haematological features of megaloblastic anaemia
Megaloblastic anaemia resulting from impaired DNA synthesis is characterized by the presence of megaloblastic red cell precursors in the bone marrow and occasionally also in the blood. Megaloblasts have a characteristic chromatin pattern (Fig. 10.2) and increased cytoplasm as a result of asynchrony of nuclear and cytoplasmic maturation with a relatively immature nucleus for the degree of cytoplasmic haemoglobinization. The delay in nuclear maturation caused by delay in DNA synthesis resulting from lack of vitamin B12 or folate is also seen in all lineages, particularly granulocytic marrow precursors with giant metamyelocytes (Fig. 10.3) and hyperlobated neutrophils with increased lobe size as well as number of nuclear segments (see Chapter 5, Fig. 5.10). In severe pernicious anaemia, a progressive increase in mean red cell volume (MCV) up to 130 fl occurs, with oval macrocytes, poikilocytes and hypersegmentation of neutrophils (>5% with more than five nuclear lobes).31 The neutrophil hypersegmentation index is an equivalent automated parameter on some cell counters, although hypersegmentation does not always respond to a therapeutic trial. The mean platelet volume is decreased and there is increased platelet anisocytosis, as detected by the platelet distribution width (PDW). The MCV falls to 110–120 fl as megaloblastic change advances. Howell–Jolly bodies and basophilic stippling are seen in the red cells.
Figure 10.3 Photomicrograph of bone marrow film stained by May–Grünwald–Giemsa showing giant metamyelocytes.
Differential Diagnosis of Macrocytic Anaemia
Macrocytic red cells are also seen in myelodysplastic syndromes, which can be suspected from the presence of hypogranular neutrophils (see Chapter 5, Fig. 5.76) or monocytosis. Excess alcohol consumption results in an increased MCV as a result of round macrocytes, although rarely does it go higher than 110 fl unless coexisting folate deficiency is present. Hypothyroidism, liver disease, aplastic anaemia, rare inherited orotic aciduria or Lesch–Nyhan syndrome also have a high MCV. Automated reticulocyte counts facilitate detection of increased red cell turnover and high MCV as a result of haemolysis or bleeding. Coexisting iron deficiency or thalassaemia trait may mask macrocytic changes, although a high red cell distribution width indicates anisocytosis and the need for blood film review. Congenital dyserythropoietic anaemias types I and III and erythroleukaemia exhibit some features of megaloblastic erythropoiesis that are unrelated to B12 and folate. Drugs interfering with DNA synthesis (e.g. azathioprine, zidovudine or hydroxycarbamide) result in macrocytosis and megaloblastic erythropoiesis. Anticonvulsant therapy interferes with folate metabolism,32 whereas the impact of oral contraceptives on folate absorption and metabolism is controversial.33 Prolonged nitrous oxide anaesthesia destroys methylcobalamin and causes acute megaloblastic change.34 Methotrexate inhibits dihydrofolate reductase and toxicity can be reversed with folinic acid, which is already in the tetrahydrofolate form and effectively reverses the metabolic block, which is not achieved solely with folic acid.
Testing strategy for suspected cobalamin or folate deficiency
Microbiological cobalamin and folate assays and competitive radiodilution binding assays for measurement of cobalamin and folate, which were often performed together, have largely been replaced by separate analysis by automated binding assays. The application of a suitable testing strategy for patients suspected of having cobalamin or folate deficiency is shown in Tables 10.1–10.3.
| Symptoms or signs | Possible significance | |
|---|---|---|
| Tiredness, palpitations, pallor | Anaemia | |
| Slight jaundice | Ineffective erythropoiesis | |
| Neurological | ||
| Cognitive impairment, optic atrophy, loss of vibration sense, joint position sense; plantar responses normal or abnormal; tendon reflexes depressed or increased | Cobalamin deficiency, subacute combined degeneration of the spinal cord and sensori/motor peripheral neuropathies | |
| Dietary and gastrointestinal history | ||
| Vegetarian or vegan; poor nutrition (e.g. tea and toast diet in elderly or students); dietary fads | Low iron stores and iron deficiency | |
| Cobalamin deficiency in babies born to mothers who are vegans | ||
| Folate deficiency (often with iron deficiency) | ||
| Weight loss, bloating and steatorrhoea, particularly nocturnal bowel movements | Features of malabsorption and folate deficiency, e.g. due to coeliac disease, tropical sprue | |
| Mouth ulcers, abdominal pain, perianal ulcers, fistulae | Terminal ileal Crohn’s disease – cobalamin deficiency | |
| Glossitis, angular cheilosis and koilonychia | Cobalamin and combined iron deficiency | |
| Alcohol history | Poor diet and interference with folate metabolism | |
| History of autoimmune disease in patient or family | ||
| Hypothyroidism, pernicious anaemia or coeliac disease | Increased likelihood of pernicious anaemia or coeliac disease | |
| Surgery | ||
| Gastrectomy/bowel resection | Cobalamin deficiency usually 2 years post-gastrectomy | |
| Ileal disease resulting in cobalamin deficiency | ||
| Blind loop syndromes | ||
| Physical appearance | ||
| Grey hair, blue eyes, vitiligo | Association with pernicious anaemia | |
| Pregnancy | Increased iron and folate requirements. | |
| Cobalamin levels fall by 30% in the 3rd trimester | ||
| Holotranscobalamin levels are unaltered in late pregnancy | ||
| Malabsorptive syndrome | ||
| Tropical sprue, bacterial overgrowth, fish tape worm in Scandinavian countries | Combined folate and iron deficiency | |
| Cobalamin deficiency | ||
| Drug history | See text | |
| Other haematological disorders | ||
| Myeloproliferative neoplasms, haemolytic anaemias, leukaemias | Increased folate utilization may result in folate deficiency | |
| Myeloma | Paraprotein interference with cobalamin assays resulting in falsely low cobalamin levels, which normalize on treatment of myeloma | |
Table 10.3 Clinical and laboratory checklist for diagnosis of pernicious anaemia
| Laboratory criteria | Clinical criteria | |
|---|---|---|
| Minor criteria | Macrocytosis | Parasthesiae, numbness or ataxia |
| Anaemia | Hypothyroidism | |
| Raised plasma homocysteine | Vitiligo | |
| Gastric pH above 6 Raised serum gastrin |
Family history of pernicious anaemia or hypothyroidism | |
| Positive gastric parietal cell antibody | ||
| Major criteria | Low serum B12 (<180 ng/l) or raised serum methylmalonic acid (>0.75 μmol/l) in presence of normal renal function | |
| Megaloblastic anaemia not resulting from folate deficiency | ||
| Positive intrinsic factor antibodies using high-specificity test. | ||
| Holotranscobalamin level <23 μmol/l | ||
| Reference standard criteria | Schilling testa shows malabsorption of oral cyanocobalamin corrected by coadministration of intrinsic factor |
a Reagents for Schilling tests currently unavailable. A non-isotopic B12 absorption test utilizing recombinant intrinsic factor and holotranscobalamin measurement is under development.
Table 10.1 highlights the important clinical details that should be elicited by the clinician and submitted with the request to assist the laboratory in interpretation of the numeric results of cobalamin and folate assays. Ideally, test requests should not be accepted without this information, which could be incorporated into electronic order communication from user to laboratory.
Table 10.2 lists the important laboratory investigations that should be performed – results must not be reported in isolation from other laboratory results and clinical details. If investigations are performed in different laboratories, authorization and release of results requires access to all laboratory data on the individual patient. Laboratory information systems should facilitate this cross-disciplinary access. For example, intrinsic factor antibody results should be available to haematology or clinical chemistry laboratories undertaking cobalamin, MMA or homocysteine assays.
Table 10.3 provides a list of clinical and laboratory features for diagnosis of pernicious anaemia. These criteria avoid undue reliance on a single B12 assay and should help clinicians to make a diagnosis even when some critical tests, e.g. Schilling tests, are not available.
In view of the lack of specificity and sensitivity of serum cobalamin assays and frequent lack of availability of other diagnostic tests, basing the diagnosis of pernicious anaemia, which requires lifelong parenteral B12 therapy, solely on laboratory results, is not straightforward. A checklist of laboratory and clinical diagnostic criteria, as shown in Table 10.2, helps to achieve a greater degree of diagnostic certainty than any single diagnostic test and permits the diagnosis to be made even when a single diagnostic test is anomalous or unavailable. Clinical and other laboratory criteria thus provide additional supportive evidence of an autoimmune aetiology, even if the more demanding diagnostic laboratory criteria are not met.
Limitations of Cobalamin Assays
Sensitivity and Specificity of Cobalamin and Holotranscobalamin Assays
Utility of receiver operator characteristic curves
There has been little data on sensitivity and specificity of current B12 assays, due to the difficulty in defining a truly deficient study population. Some authors have suggested B12 assays and measurement of methylmalonic acid are no better than tossing a coin, to determine the presence or absence of deficiency. The study by Clarke et al.28 provides data which permits calculation of the specificity and sensitivity of a current B12 immunoassay in the detection of cobalamin deficiency in a community study of 1621 subjects over age 65 with normal renal function. Subjects were defined as cobalamin deficient if the methylmalonic acid was elevated above 0.75 μmol/l. Deficiency was found in 4.3% of subjects over 65 years of age with normal renal function. The mean B12 level of these subjects was 151 pmol/l (202 ng/l) by Siemens Centaur assay (range 110–199 pmol/l). Table 10.4 illustrates the calculation to derive specificity and sensitivity for the Siemens Centaur B12 assay using a cut-off point of 200 pmol/l (270 ng/l).
Selection of a cut-off point for cobalamin of below 150 ng/l, will identify subjects with higher probability of presence of cobalamin deficiency. Some authors13 have advocated choosing a cut-off point 25% below the reference range lower limit for any particular assay.
If, for example, a cut-off point of 125 pmol/l (168 ng/l) is chosen, the specificity of a value below this level, i.e. the number of normal individuals who fall below the cut-off point, will be markedly reduced (FP) and therefore the specificity of the test (TN/TN+FP) improves to 95%, although the detection of individuals with true deficiency who lie above the cut-off point, i.e. false negatives will be markedly increased, resulting in a sensitivity (TP/TP+FN) of 35%. The ROC curve therefore allows a laboratory to select a cut-off point that meets the objective of the laboratory – to have either a highly specific but low sensitivity test or to have a test of poor specificity but high sensitivity. Clearly cobalamin assays do not meet the criteria for an ideal test of high sensitivity and high specificity which would lie on the coordinates 0.1 to the left of the graph. The Axis-Shield/Abbott holotranscobalamin assay in this pivotal study is seen to have slightly superior ROC curves (see Fig. 10.4).
Utility of holotranscobalamin, methylmalonic acid and homocysteine assays
Holotranscobalamin assays gave a greater area under the curve, 0.85 versus 0.76 in the above study, and superior sensitivity and specificity. Holotranscobalamin is the physiologically active fraction of total cobalamin.27–29 An isolated abnormal result of serum B12 should not be the sole criterion on which treatment decisions are based and a repeat assay and other confirmatory and clinical evaluation are necessary prior to a diagnostic conclusion. Additional secondary testing with metabolite levels, or holotranscobalamin and monitoring of treatment response is recommended. High B12 levels have been described in subjects with no myeloproliferative neoplasm and who are not on cobalamin therapy or vitamin supplementation.35 This is thought to be due to immunoglobulin-complexed B12 resulting in assay interference. False normal B12 levels24,25 have been described in subjects with high titre intrinsic factor antibody25 and may also occur due to presence of heterophile antibody interference.
Holotranscobalamin assays27,28 may challenge total B12 assays as a first-line test in cobalamin assessment. Holotranscobalamin has been shown to be unaffected by assay interference from high-titre intrinsic factor antibody levels.36 In addition, holotranscobalamin is not subject to the 30% fall in total B12 levels seen in normal pregnancy, which makes low total B12 levels uninterpretable during pregnancy (Fig. 10.5).30
Access to homocysteine, methylmalonic acid and holotranscobalamin assays facilitates more precise definition of cobalamin and folate status in patients in whom prolonged B12 therapy may have been initiated inappropriately and continued unnecessarily or, conversely, discontinued inappropriately because of lack of confidence in the original assessment. The study by Gorringe et al.16 showed that only 27/49 patients were anaemic or macrocytic with a low total B12 of <170 ng/l and elevated MMA. All of these patients treated with B12 therapy corrected elevated MMA levels, suggesting the presence of a metabolic deficiency. However, only 15/27 showed a haematological response.
Homocysteine levels also fell by >25% in 47/49 patients after B12 treatment. Many of these patients had no clinical evidence or symptoms of cobalamin deficiency and may reflect subjects with subclinical deficiency, which could have subtle cognitive impairment, or may just represent a compensated metabolic state of no clinical consequence. Elevation of homocysteine levels is seen in folate deficiency and is therefore less specific than MMA measurement. The causes of cobalamin deficiency are shown in Table 10.5.
| Supportive information/diagnostic tests | ||
|---|---|---|
| Reduced intake | ||
| Strict vegetarian/vegan | Dietary history | |
| Dietary fad that excludes dairy products and meat | Ethnic origin/culture | |
| Breastfed babies of mothers who are vegetarian or cobalamin deficient | ||
| Poor dietary intake in elderly | ||
| Malabsorption as a result of loss or inactivity of intrinsic factor | ||
| Addisonian pernicious anaemia | Diagnostic criteria for pernicious anaemia (see Table 10.3) | |
| Gastrectomy (partial or total) | History of gastric surgery | |
| Bacterial overgrowth or parasitic infestation of small bowel | Radiolabelled lactose breath tests for bacterial overgrowth Repeat Schilling test post-antibiotic therapy |
|
| Pancreatic dysfunction: failure of trypsin release of B12 from R binding proteins | Pancreatic function tests; exocrine pancreatic dysfunction results in abnormal Schilling test but clinical deficiency is rare | |
| Malabsorption as a result of failure of B12-intrinsic factor complex uptake in ileum – ileal resection | ||
