Chapter 19 Investigation of a thrombotic tendency
Introduction to thrombophilia
In this chapter, the investigations to detect an acquired thrombotic tendency are presented first, followed by a simplified battery of tests needed to establish the diagnosis of the more important inherited ‘thrombophilias’. Although the number of coagulation factors known to contribute to a thrombotic tendency has increased greatly in the last few years, it remains clear that not all factors have been identified. Hence, the failure to detect one of the traits described does not imply that the individual’s risk of thrombosis is normal. An acquired thrombotic tendency is common and occurs in many conditions but is usually complex, multifactorial and not easily identifiable by a single laboratory test. The large number of traits identified, often with a small associated relative risk, makes their individual utility equally small. Until the interactions of these numerous factors are more completely understood, the clinical history remains a dominant factor in clinical management. The British Committee for Standards in Haematology has published guidelines on the investigation of inherited thrombophilia.1
Tests for the presence of a lupus anticoagulant
The lupus anticoagulant (LAC) is an acquired autoantibody found in various autoimmune disorders and sometimes in otherwise healthy individuals.2 LACs are immunoglobulins that bind to certain proteins when bound to phospholipid. The effective sequestration of phospholipid can then cause prolongation of phospholipid-dependent coagulation tests such as the prothrombin time (PT) or activated partial thromboplastin time (APTT). The name ‘anticoagulant’ is misleading because, despite the in vitro effects, patients do not have a bleeding tendency. Instead, there is a clear association with recurrent venous thromboembolism, cerebrovascular accidents and other arterial events and, in women, with recurrent abortions, fetal loss and other complications of pregnancy.3 Therefore, tests for the presence of the LAC should be carried out in all young individuals with unexplained venous or arterial thrombosis and also in women with recurrent-early or late pregnancy loss.4 Antibodies of this class are members of a larger group called antiphospholipid or anticardiolipin antibodies. (Although not precisely the same these terms are used interchangeably.) Tests for lupus anticoagulant are usually performed in parallel with tests for the presence of antiphospholipid antibodies, usually by ELISA. In general these tests are not performed by haematology laboratories and are therefore not described here. Serological tests for antiphospholipid antibodies are not standardized and agreement between laboratories is poor. A large number of target proteins have been described but the most important, and the only one for which there is evidence of a pathogenic effect, is β2-glycoprotein 1.5 Increasingly, tests specifically for anti-β2-glycoprotein 1 antibodies are performed and possible mechanisms for their prothrombotic activity are being elucidated.6
The presence of a LAC may be detected by the clotting screen and, depending on the reagents and methods used as well as on the potency and avidity of the antibody, either the PT or APTT may be prolonged. However, the sensitivity of both APTT and PT to LAC varies considerably, so that these tests may well be normal and, if clinically suspected, specific tests should always be performed.4 The unmodified test for activated protein C (APC) resistance (see p. 456) is also sensitive to the presence of a LAC.
Patients with a LAC may show other abnormalities, including thrombocytopenia, a positive direct antiglobulin test and a positive antinuclear antibody test. Another frequent target for antiphospholipid antibodies is prothrombin but only rarely are these antibodies sufficient to inhibit or deplete prothrombin activity. Such patients may have a bleeding tendency. A recent international guideline on detection of lupus anticoagulants has been published4 and recommended the following tests:
There are a large number of additional tests which have in the past been successfully used for the detection of LAC, several of which are no longer recommended due to poor reproducibility, technical problems and lack of standardization.4 The kaolin clotting time (KCT) and the dilute thromboplastin inhibition test are retained here because they are still widely used and thought to have some advantages by some authors. Although no single test is sufficiently sensitive to detect all LAC, readers are counselled against performing an excessive (more than two) number of tests because a large number of false positives will be generated. The most recent guidelines for the optimal performance of testing for LAC have been well laid out and are summarized in Box 19.1.4
Box 19.1 Recommendations for the optimal laboratory detection of lupus anticoagulant (LAC)4
(A) Blood collection
(C) Mixing test
(D) Confirmatory test
Dilute Russell’s Viper Venom Time
Principle
Russell’s viper venom (RVV) activates factor X, leading to a fibrin clot in the presence of factor V, prothrombin, phospholipid and calcium ions. A LAC prolongs the clotting time by binding to the phospholipid and preventing the action of RVV. As the following test describes, dilution of the venom and phospholipid makes it particularly sensitive for detecting a LAC.7 Because RVV activates factor X directly, defects of the contact system and factor VIII, IX and XI deficiencies do not influence the test. The DRVVT should be combined with a platelet/phospholipid neutralization procedure to add specificity and this is incorporated into several commercial kits.
Reagents
Platelet Neutralization Test
Reagents for preparation of platelet neutralization reagent
Interpretation
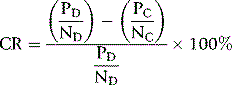
where P is patient’s clotting time and N is the clotting time of normal plasma and D represents the detection procedure and C represents the confirmation (platelet/phospholipid neutralization) procedure.7 A correction of >10% is regarded as positive, but care should be taken to establish a local normal range. Other calculations may also be used such as a simple ratio of PD/PC or percentage correction: [(PD − PC)/PD] × 100%.4
Interpretation of Tests for Lupus Anticoagulant
Detailed instructions for the interpretation of LAC testing have been published.4 No single test detects all lupus-like anticoagulants and, if suspected clinically, then two specific tests should be performed before concluding that a LAC is not present.4,8 Conversely, a single positive test should be repeated 12 weeks later because a transient positive may arise as the result of intercurrent illness or medication. It is crucial to distinguish LAC from specific anti-factor VIII antibodies, which are more typically time dependent but may have some immediate effect as well. Specific factor assays can be useful in discrimination but note that a LAC may result in non-parallelism and spuriously low results in these assays. Similarly, some weak LAC are neutralized by 50:50 mixing with normal plasma and sometimes exhibit a time-dependent effect. Some transient non-specific coagulation inhibitors are not detected by tests for LAC. Tests may be falsely negative while taking warfarin.
Kaolin Clotting Time
Principle
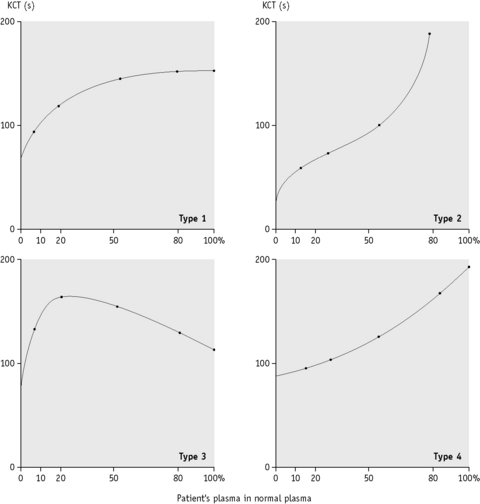
When the APTT is performed in the absence of platelet substitute reagent, it is particularly sensitive to a LAC. If the test is performed on a range of mixtures of normal and patient’s plasma, different patterns of response are obtained, indicating the presence of a LAC, deficiency of one or more of the coagulation factors or the ‘lupus cofactor’ effect.9
Reagents
Results
Plot the clotting times against the proportion of normal to patient’s plasma on linear graph paper as shown in Figure 19.1.
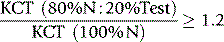
Dilute Thromboplastin Inhibition Test
Principle
When the thromboplastin used for the PT is diluted, the PT becomes prolonged. At a certain point (usually 1:50–1:500 dilution) the concentration of phospholipid is low enough for the test to become sensitive to phospholipid binding antibodies and when a LAC is present the ratio of the test plasma to normal plasma clotting time increases. This test is now considered more useful because some thromboplastin reagents (e.g. Innovin) are more sensitive to LACs. However, it should be noted that diluting thromboplastin makes the system sensitive to low levels of factor VIII as are encountered in mild haemophilia, acquired haemophilia and low levels of factor V or factor VII. Care should be taken that these disorders are not confused. In one study, the test was determined to be positive when the dilute PT ratio (test/mean normal) using Innovin at 1:200 dilution was >1.15.10
Other Acquired Thrombophilic States
There are numerous other disorders that are associated with an increased risk of thrombosis but are not usually diagnosed using coagulation-based tests. Appropriate tests for some of these such as myeloproliferative neoplasms and paroxysmal nocturnal haemoglobinuria are found elsewhere in this book. One of the most important factors precipitating thrombosis is malignancy. However, the value of extensive testing for possible malignancy in patients with thrombosis remains contentious; some studies have shown that a history and examination combined with a few simple tests detect the large majority of malignancy and other systemic disorders. Other studies suggest that more intensive screening including abdominal and pelvic scans are required to achieve this. Large numbers of ‘tumour marker’ analyses result in numerous false positives. Even when tests have been effective in detecting occult malignancy it is not clear there is any improvement in outcome.11,12
Investigation of inherited thrombotic states
Testing for thrombotic syndromes remains frequent, despite doubts about its clinical utility.1 Patients with disorders of pregnancy and those with thrombotic disorders are often referred for investigation. Prior to testing for thrombophilia consideration should be given to the likely benefits including alteration in management that can be achieved. This requires a carefully taken history, noting in particular the circumstances of any previous thrombotic event, a family history of thrombosis and identification of any coexisting disorders. The relevant tests are described below.
Antithrombin (AT)
AT13 (previously known as antithrombin III) is the major physiological inhibitor of thrombin and factors IXa, Xa and XIa. AT deficiency is found in approximately 2% of cases of thrombosis and may be acquired or congenital. Various methods are available for measuring either functional activity or antigenic quantity of AT. The functional methods are based on the reaction with thrombin or factor Xa and can be coagulation based or chromogenic assays. A chromogenic assay is described below.
Antithrombin (AT) Measurement Using a Chromogenic Assay
Interpretation
In an inherited deficiency, the AT concentration is usually <0.7 iu/ml. Most cases are heterozygotes for null mutations (type I deficiency) and have levels of approximately 50% of normal. Be aware that numerous type 2 variants have been described affecting the reactive site, the heparin binding site or having pleiotropic effects, sometimes resulting in assay results that are close to normal. The clinical significance of heterozygous heparin-binding site mutations is probably low. Further tests such as AT antigen, crossed immunoelectrophoresis or mutation analysis may be required to identify variant molecules.14,15 A low level of AT may be acquired as a result of active thrombosis, liver disease, heparin therapy, nephrotic syndrome or asparaginase therapy; very low values are sometimes encountered in fulminant disseminated intravascular coagulation (DIC) or liver failure. Normal newborns have a lower AT concentration (0.60–0.80 iu/ml) than adults. In neonates who are congenitally deficient, very low values (0.30 iu/ml and lower) may be found. It is also important to remember that oral anticoagulant therapy may increase the AT concentration by approximately 0.1 iu/ml in cases of congenital deficiency.
Protein C (PC)
PC16,17 is a vitamin K-dependent protein. After activation by thrombin, which is accelerated in the presence of thrombomodulin on the vascular endothelium, PC complexes with phospholipids and protein S (PS) to degrade factors Va and VIIIa. Inherited heterozygous PC deficiency is found in 2–4% of first-episode thromboses and 5–7% of all recurrent thromboembolic episodes in young adults.18–20 The importance of the PC–PS system is evidenced by the catastrophic syndrome of purpura fulminans in neonates with homozygous PC or PS deficiency.21 Acquired PC deficiency is found in all conditions associated with vitamin K deficiency or defect, including oral anticoagulant therapy. A low plasma concentration is also found in DIC, sepsis (especially meningococcal septicaemia), in liver disease, sickle cell disease and in the early postoperative period.
PC can be measured using a chromogenic assay, a coagulation assay or an antigenic method.22
Measurement of Functional Protein C by the Protac Method
Reagents
Further Investigation for Protein C Deficiency
If inherited PC deficiency is suspected, an immunological assay may also be carried out with an ELISA-based kit, which will distinguish a type 1 or type 2 deficiency. The amidolytic assay described here does not detect the rare type 2 PC deficiency due to mutations in the Gla domain, although they can be detected by a coagulation-based assay.22 The specificity of the chromogenic substrate is limited and is augmented by the inclusion of substances that inhibit other enzymes capable of cleaving the substrate. In some circumstances, this can fail and spuriously high PC activities can be obtained, which may obscure PC deficiency.23 PC activity and antigen are reduced in patients taking oral vitamin K antagonists, although it is sometimes possible to make a provisional diagnosis of PC deficiency by using a PC:VIIc ratio.24 It is also important to exclude vitamin K deficiency and/or liver disease by assaying other vitamin K-dependent factors. Family studies should be carried out whenever possible.
Clotting-Based Protein C Assay
Principle
Unlike chromogenic PC assays, PC clotting assays are sensitive to functional PC defects such as phospholipid binding (mutations in the Gla domain) and calcium binding. However, they are also sensitive to anticoagulants, Factor V Leiden, LACs and raised factor VIII levels. A functional protein C activity assay can also be performed using the DRVVT which may be less sensitive to these effects.25
Protein S (PS)
PS is also a vitamin K-dependent protein that acts as a cofactor for activated PC. It is similar to the serine proteases of the coagulation system in having a Gla domain and four EGF domains; however, instead of a protease domain it has a large terminal domain closely homologous to sex hormone-binding globulin (SHBG). In plasma, 60% of PS is bound to C4b-binding protein (C4bBP) via the SHBG26 and does not possess any APC cofactor activity; the remaining 40% is free and available to interact with APC. Functional assays of PS are based on the capacity of PS to augment the prolongation of a clotting test time by APC. However, PS has some APC-independent anticoagulant activity that can also be measured in coagulation assays and it can also act as a cofactor for TFPI.27,28 Measurement of the total and free PS antigen is possible using enzyme-linked immuno-assays. All three measurements are considered together here but usually measurement of free PS is adequate.
Enzyme-Linked Immunosorbent Assay of Free and Total Protein S
Principle
The total PS in plasma is detected by a standard ELISA using polyclonal antibodies.29 The analysis is then repeated using plasma in which C4bBP-bound PS has been removed by polyethylene glycol (PEG) precipitation. This gives a measure of free PS.
Reagents
Interpretation of Protein S Functional and Antigenic Assays
PS deficiency has been classified into three subtypes according to the pattern of results obtained in functional and antigenic assays (Table 19.1).
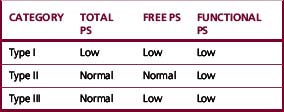
Studies have suggested that the type I and type III patterns are both the result of the same genetic defect and that the difference may be the result of an age-related increase in C4bBP.33,34
Although an estimate of PS functional activity would be ideal for diagnosing PS deficiency, the functional PS assays available are problematic. Like other functional assays they are prone to external influences: factor V Leiden (FVL), LAC and levels of other coagulation factors. Fortunately type II PS defects appear to be extremely rare; many previously diagnosed cases proved to be due to FVL. Thus measurement of free PS is the preferred method for detecting PS deficiency.35,36 Low levels of PS may be an acquired phenomenon during pregnancy and with oral anticoagulation, nephrotic syndrome, use of oral contraceptives, systemic lupus erythematosus, HIV infection and liver disease. Catastrophically low levels have been reported in children after varicella infection owing to autoantibody production.37 It is important to note that the normal range for premenopausal women is significantly lower than in other groups and local normal ranges should be determined to avoid misinterpretation, paying attention to the additional effects of hormonal therapy and artefactual reduction in PS as described earlier.38,39 Although C4bBP is elevated during an acute-phase reaction, the PS-binding β chain does not increase and as a result free PS does not decrease.40
Activated Protein C Resistance
In 1993, Dahlback et al.41 described an inherited tendency to thrombosis characterized by a defective plasma response to activated PC. This became known as activated PC resistance (APCR) and was subsequently shown in >90% of cases to result from a polymorphism encoding the amino acid change Arg506Glu subsequently named factor V Leiden.42 This mutation destroys a cleavage site for APC, which greatly slows APC inactivation of factor Va. It also blocks the conversion by APC of factor V into factor Vi, which acts as a cofactor for APC degradation of factor VIIIa. APCR is found in approximately 20% of patients with a first episode of venous thrombosis.
Principle43
When activated PC (APC) is added to plasma and an APTT is performed, there is normally a prolongation of the clotting time as a result of factor V and factor VIII degradation. The original detection of this phenomenon was by means of a modified APTT, but it can also be detected using modifications of the PT, DRVVT and factor Xa clotting time. These tests all vary somewhat in their sensitivity and specificity for the FVL mutation, which is generally improved by mixing the test plasma with factor V-deficient plasma. This reduces the effect of other factors such as factor VIII and prothrombin, which can alter estimation of APCR44 and restores the sensitivity of the test in patients who are taking oral anticoagulants. However, the test remains sensitive to interference by LACs. Numerous commercial kits are available for these tests.
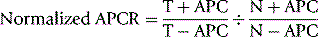
Interpretation
The Leiden thrombophilia survey estimated the relative risk of thrombosis for APCR to be approximately 7.45 Studies using DNA analysis alone have generally found slightly lower relative risks.46 Most testing strategies have been directed toward producing tests that have a high sensitivity and specificity for FVL to avoid the need for DNA analysis. It seems that ‘acquired APCR’ or APCR resulting from other causes represents a prothrombotic state even in the absence of FVL,47 as does the presence of acquired APCR in prothrombotic states such as pregnancy. These are not (except LACs) detected after mixing with factor V-deficient plasma. Some laboratories use a combination of plasma and DNA testing to assess patients’ status but increasingly DNA analysis alone is performed and this can be combined with analysis of the prothrombin gene (below).
Increased Prothrombin, Factor VIII and Other Factors
A later finding from the Leiden thrombophilia survey was that elevated levels of prothrombin were significantly associated with thrombosis.48 Most elevated levels were associated with a mutation in the 3′ untranslated region of the gene (G20210A). The mutation is detected by a simple polymerase chain reaction-based test (see p. 148). Subsequently, other factors, including factor VIII, factor IX and factor XI, have been shown to have an association with thrombosis when elevated.49–51
Heparin Cofactor II
There is no clear evidence that heparin cofactor II (HCII) deficiency is more prevalent in patients with thrombosis than in the normal population; consequently, testing is not recommended as part of thrombophilia investigation.52 (A method for measuring HCII is described in previous editions of this book.)
Fibrinolytic system
Investigation of Suspected Dysfibrinogenaemia
Congenital dysfibrinogenaemia, which may be associated with thrombosis, should be suspected in individuals with a prolonged thrombin time and a slightly or moderately reduced fibrinogen concentration in plasma. The presence of a dysfibrinogen is proved when a significant (usually two-fold) discrepancy is found between the Clauss and clot weight assays. (For details of investigation see p. 412.)
Investigation of the Fibrinolytic System: General Considerations
The investigation of fibrinolysis has an uncertain place in haemostasis. It seems well-established that uncontrolled fibrinolytic capacity as a result of plasmin inhibitor or plasminogen activator inhibitor (PAI-1) deficiency can lead to a haemorrhagic tendency, although these are rare.53,54 Conversely it has been difficult to demonstrate that an impaired fibrinolytic capacity results in a tendency to venous thrombosis. This may be attributed in part to the poor reproducibility of the global tests such as euglobulin clot lysis or fibrin plate lysis but it has not been resolved by use of either specific assays or genetic polymorphic markers.55 More recently a plasma clot lysis time has been developed which has been shown to detect a reduced fibrinolytic potential associated with an increased risk of first and recurrent thrombosis.56,57 Moreover, this defect was associated with levels of thrombin-activatable fibrinolysis inhibitor (TAFI), PAI-1, plasminogen and tissue plasminogen activator (tPA), although for the latter two the association was lost after adjusting for other variables. This test is not yet in routine clinical use. High levels of tPA were shown to be predictive of myocardial infarction in the ECAT (European Concerted Action on Thrombosis and Disabilities) study, but it is possible that this unexpected association can be interpreted as demonstrating an abnormality of endothelial function rather than a problem with fibrinolysis per se.58–61
Fibrinolysis shows considerable diurnal variation as well as interference from plasma lipids and stress. It is therefore generally recommended that these tests be performed in the morning after an overnight fast, after a period of no smoking and after the subject has lain resting for ≥15 min (the plasma half-life of tPA is approximately 5 min). Great care is required in obtaining and handling samples for the assays described later.62 Tests for fibrin and fibrinogen degradation products are described in Chapter 18.
Investigation of ‘Fibrinolytic Potential’
Euglobulin Clot Lysis Time
Reagents
Lysis of Fibrin Plates
Principle
Most commercially available fibrinogen preparations are contaminated with plasminogen. If a standard fibrinogen solution is poured into a Petri dish and clotted with CaCl2 and thrombin, a solid fibrin plate is obtained. If the euglobulin fraction under test is placed on the plate, the plasminogen in the plate is converted into plasmin and a zone of lysis appears around the sample. The area of lysis is proportionate to the concentration of plasminogen activator in the euglobulin fraction.63
Method
Carefully apply 30 μl of the euglobulin fraction, prepared as described in the previous test, to the surface of the plate. There is no need to cut a well. Place in an incubator at 37°C for 24 h. This preparation time can be shortened by the addition of exogenous plasminogen.63
Venous Occlusion Test
Principle
Localized venous occlusion64 of an arm for a standardized period is used as a stimulus for release of tPA from the vessel wall. The original intention was that this would be a better measure of functional defects in fibrinolysis than a resting sample. Preocclusion and postocclusion lysis times, using the previously described euglobulin lysis or the fibrin plate lysis tests, are measured. In normal subjects fibrinolysis is greatly enhanced by occlusion. However, given the problems associated with global assays of fibrinolysis, it seems preferable to perform specific measurements of tPA before and after occlusion.
Interpretation
Failure to enhance lysis is found in some cases of recurrent venous thrombosis, in people who are obese, after surgery, trauma or severe illness and in Behçet’s syndrome. It may also result from a failure to release the activator because insufficient pressure was applied or the occlusion time was too short. Normal people vary in the degree of response: ‘good’ responders increase the concentration of tPA by three-fold to four-fold, whereas ‘poor’ responders may consistently show only a very slight enhancement of fibrinolysis even with longer occlusion times. When comparing plasma levels of proteins preocclusion and postocclusion, an adjustment for changes in haematocrit may be required.65 The effect of the venous occlusion test and the levels of tPA are very variable over time.66,67
Investigation of Suspected Plasminogen Defect or Deficiency
Inherited plasminogen deficiency or defect may be found in 2–3% of unexplained thromboses in young people.68,69 However, there is no good evidence that deficiency is associated with an increase risk of thrombosis. The only consistent clinical finding appears to be ligneous conjunctivitis.70 The laboratory screening should be carried out using a functional assay based on full transformation of plasminogen into plasmin by activators. Such assays can be caseinolytic, fibrin substrate or chromogenic.
Tissue Plasminogen Activator Amidolytic Assay
Principle
Different amidolytic assays for tPA have been described.71,72 One relies on the activation of purified plasminogen to plasmin in the presence of fibrinogen fragments, which stimulate the tPA activity in the test plasma. The plasmin is measured using a specific chromogenic substrate. In the second method, tPA is captured on specific antibodies bound to a solid-phase matrix such as a microtitre plate; the various plasma inhibitors of tPA and plasmin are washed away, plasminogen is added together with a stimulator of tPA activity and the plasmin produced is measured with chromogenic substrates. Alternatively, chromogenic substrates specific for tPA may be used, but there are specificity problems, especially in the plasma assays.
Plasminogen Activator Inhibitor Activity Assay
Principle
Reagents are available in kit form and the manufacturer’s instructions must be closely followed. The normal range, in particular the lower limit, is not clearly defined and many normal subjects have levels below the assay’s lower limit of detection.73,74 Each laboratory should establish its own range until reliable normal values become available.
Platelet ‘hyperreactivity’ and activation
Platelets may be more reactive than normal as a consequence of in vivo activation by thrombin or non-endothelial surfaces, such as prosthetic valves or Dacron grafts. This can sometimes be detected by a lowered threshold (increased sensitivity) for aggregating agents. Because there is considerable variation in response to aggregating agents in normal people, the attempts to show platelet hyperaggregability are rarely successful and the results are frequently inconsistent. Spontaneous aggregation of platelets in the blood can also be demonstrated.75
Platelets that have formed a part of a platelet thrombus and have been released into the circulation may show a measurable decrease in their ability to aggregate because of a loss of some granular content. The released contents can be measured in plasma; the α-granule proteins, β-thromboglobulin and platelet factor 4 are the constituents most commonly measured. Overall the problems with these tests make them of doubtful utility;59 they are described in previous editions of this book.
Several genetic polymorphisms have been reported to affect the reactivity of platelet glycoproteins and P selectin. Although they may be important in population studies, their clinical significance for individual patients remains unclear.76,77
Platelet Activation: Flow Cytometry
Principle
The activation of platelets is associated with the appearance of new antigenic determinants on the platelet surface. Some of these are molecules present in platelet granules brought to the surface during degranulation (e.g. CD62P, CD63, LAMP-1 and CD40L) and others are new conformations of existing molecules (e.g. the ligand-induced binding site on GpIIbIIIa). These can be detected using fluorescein-conjugated antibodies and the degree of expression can be quantified by flow cytometry. This gives a measure of platelet activation with a much greater degree of sensitivity than platelet factor 4 or β-thromboglobulin estimation and may still be successful in the presence of thrombocytopenia. Samples may need to be collected into inhibitors of platelet activation such as PGE1. Numerous alternative surface molecules are available (Table 19.2). These tests have not yet entered routine laboratory practice but are proving increasingly useful in research.78 An alternative approach is offered by the PFA-100 (see p. 425) in which short closure times may be indicative of platelet hyperreactivity and/or hyperreactive von Willebrand factor species.
Table 19.2 Indicators of platelet activation detectable by flow cytometry
| Name | CD Designation | Comment |
|---|---|---|
| GpIb, IX, V | CD42 | Decreases |
| GpIIbIIIa | CD41 | Increases |
| Phosphatidyl serine | – | Increases Detected by Annexin V binding |
| Lysosomal Integral membrane protein (gp53, granulophysin) | CD63 | Indicates lysosomal degranulation |
| P selectin | CD62P | Indicates α granule release, subsequently cleaved and measurable in plasma |
| Fibrinogen | – | Surface-bound fibrinogen increases |
| IIbIIIa activation | – | Conformation change in IIbIIIa produced by activation, detected by PAC-1 antibody |
Homocysteine
Following the observation that patients with homocystinuria have venous and arterial thromboses with accelerated vascular damage, there has been considerable interest in patients with less marked elevation of plasma homocysteine (hyperhomocysteinaemia). This has been shown to have an association with arterial and venous thrombosis but the assay has little clinical utility and dietary interventions have been ineffective.79,80 Until recently, homocysteine has been measured by high-performance liquid chromatography or mass spectroscopy, but an ELISA-based assay is now available that allows it to fit more easily into coagulation laboratory practice. To standardize study results, homocysteine is measured either while fasting or after a methionine load. Rapid processing of samples is required because homocysteine quickly leaches out of red blood cells.
Markers of coagulation activation
Numerous commercial kits are available for measuring molecules produced by coagulation activation.
Principle
An alternative is to measure the concentration of thrombin–antithrombin complexes (TAT), which provides similar information. Plasmin–antiplasmin complexes provide corresponding information about fibrinolysis. These can all be measured using commercially available ELISA kits but are not used routinely and are not required for normal diagnostic work.81 Other tests such as fibrinopeptide A require exceptional care and the use of special anticoagulants to prevent in vitro activation of the sample.
A plasmin cleavage product of crosslinked fibrin, D-dimer (see p. 441) is another measure of activity in the coagulation system. Several studies have shown that elevated levels of D-dimer are an indicator of future risk of thrombosis. It is not yet certain whether they have on their own, or in conjunction with other factors, sufficient predictive value to alter management but they are currently being incorporated into management protocols. The test is usually performed after oral anticoagulants have been discontinued.
Global assays of coagulation
As a response to the failure of reductive approaches to identify assays that reliably predict thrombosis or thrombotic risk, some workers have moved in the opposite direction and devised global assays that assess the overall coagulation potential of a blood or plasma sample. These tests include the endogenous thrombin potential (ETP) and the thromboelastograph. While use of thromboelastography has increased in the management of haemorrhage, their ability to assess thrombotic risk is less well described. There is some evidence that increased ETP can predict patients at increased risk of first or recurrent thrombosis.82 However, neither of these techniques is in routine diagnostic use. Some manufacturers have developed kits for global assessment of the PC–PS pathway.
1 Baglin T., Gray E., Greaves M., et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol. 2010;149(2):209-220.
2 Devreese K., Hoylaerts M.F. Laboratory diagnosis of the antiphospholipid syndrome: a plethora of obstacles to overcome. Eur J Haematol. 2009;83(1):1-16.
3 Palomo I., Segovia F., Ortega C., et al. Antiphospholipid syndrome: a comprehensive review of a complex and multisystemic disease. Clin Exp Rheumatol. 2009;27(4):668-677.
4 Pengo V., Tripodi A., Reber G., et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7(10):1737-1740.
5 de Laat B., Pengo V., Pabinger I., et al. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009;7(11):1767-1773.
6 Agar C., van Os G.M.A., Morgelin M., et al. β2-Glycoprotein I can exist in 2 conformations: implications for our understanding of the antiphospholipid syndrome. Blood. 2010;116(8):1336-1343.
7 Thiagarajan P., Pengo V., Shapiro S.S. The use of the dilute Russell viper venom time for the diagnosis of lupus anticoagulants. Blood. 1986;68(4):869-874.
8 Brandt J.T., Barna L.K., Triplett D.A. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost. 1995;74(6):1597-1603.
9 Exner T., Rickard K.A., Kronenberg H. A sensitive test demonstrating lupus anticoagulant and its behavioural patterns. Br J Haematol. 1978;40(1):143-151.
10 Arnout J., Vanrusselt M., Huybrechts E., et al. Optimization of the dilute prothrombin time for the detection of the lupus anticoagulant by use of a recombinant tissue thromboplastin. Br J Haematol. 1994;87(1):94-99.
11 Monreal M., Lensing A.W., Prins M.H., et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost. 2004;2(6):876-881.
12 Piccioli A., Lensing A.W., Prins M.H., et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost. 2004;2(6):884-889.
13 van Boven H.H., Lane D.A. Antithrombin and its inherited deficiency states. Semin Hematol. 1997;34(3):188-204.
14 Lane D.A., Olds R.J., Conard J., et al. Pleiotropic effects of antithrombin strand 1C substitution mutations. J Clin Invest. 1992;90(6):2422-2433.
15 Kottke-Marchant K., Duncan A. Antithrombin deficiency: issues in laboratory diagnosis. Arch Pathol Lab Med. 2002;126(11):1326-1336.
16 Castellino F.J., Ploplis V.A. The protein C pathway and pathologic processes. J Thromb Haemost. 2009;7(suppl 1):140-145.
17 Bertina R.M. Specificity of protein C and protein S assays. La Ricerca in Clinica e in Laboratorio Journal. 1990;20(2):127-138.
18 Lane D.A., Mannucci P., Bauer K., et al. Inherited thrombophilia: part 1. Thromb Haemost. 1996;76(5):651-662.
19 Lane D.A., Mannucci P., Bauer K., et al. Inherited thrombophilia: part 2. Thromb Haemost. 1996;76(6):824-834.
20 Rosendaal F.R., Reitsma P.H. Genetics of venous thrombosis. J Thromb Haemost. 2009;7(suppl 1):301-304.
21 Marlar R.A., Neumann A. Neonatal purpura fulminans due to homozygous protein C or protein S deficiencies. Semin Thromb Hemost. 1990;16(4):299-309.
22 Khor B., Van Cott E.M. Laboratory tests for protein C deficiency. Am J Hematol. 2010;85(6):440-442.
23 Mackie I.J., Gallimore M., Machin S.J. Contact factor proteases and the complexes formed with alpha 2-macroglobulin can interfere in protein C assays by cleaving amidolytic substrates. Blood Coagul Fibrinolysis. 1992;3(5):589-595.
24 Jones D.W., Mackie I.J., Winter M., et al. Detection of protein C deficiency during oral anticoagulant therapy –use of the protein C:factor VII ratio. Blood Coagul Fibrinolysis. 1991;2(3):407-411.
25 Cooper P.C., Cooper S.M., Goodfellow K.J., et al. Evaluation of a new venom-based clotting assay of protein C. International Journal of Laboratory Hematology. 2008;30(5):437-443.
26 Rezende S.M., Simmonds R.E., Lane D.A. Coagulation, inflammation and apoptosis: different roles for protein S and the protein S-C4b binding protein complex. Blood. 2004;103(4):1192-1201.
27 Hackeng T.M., Rosing J. Protein S as cofactor for TFPI. Arterioscler Thromb Vasc Biol. 2009;29(12):2015-2020.
28 Castoldi E., Hackeng T.M. Regulation of coagulation by protein S. Curr Opin Hematol. 2008;15(5):529-536.
29 Comp P.C., Doray D., Patton D., et al. An abnormal plasma distribution of protein S occurs in functional protein S deficiency. Blood. 1986;67(2):504-508.
30 Deffert C., Esteve F., Grimaux M., et al. A direct, automated, immuno-turbidimetric assay of free protein S antigen in plasma. Blood Coagul Fibrinolysis. 2001;12(2):137-141.
31 Serra J., Sales M., Chitolie A., et al. Multicentre evaluation of IL Test Free PS: a fully automated assay to quantify free protein S. Thromb Haemost. 2002;88(6):975-983.
32 Aillaud M.F., Pouymayou K., Brunet D., et al. New direct assay of free protein S antigen applied to diagnosis of protein S deficiency. Thromb Haemost. 1996;75(2):283-285.
33 Simmonds R.E., Zoller B., Ireland H., et al. Genetic and phenotypic analysis of a large (122-member) protein S-deficient kindred provides an explanation for the familial coexistence of type I and type III plasma phenotypes. Blood. 1997;89(12):4364-4370.
34 Zoller B., Garcia d.F.P., Dahlback B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood. 1995;85(12):3524-3531.
35 Lawrie A.S., Lloyd M.E., Mohamed F., et al. Assay of protein S in systemic lupus erythematosus. Blood Coagul Fibrinolysis. 1995;6(4):322-324.
36 Persson K.E., Dahlback B., Hillarp A. Diagnosing protein S deficiency: analytical considerations. Clin Lab. 2003;49(3–4):103-110.
37 Levin M., Eley B.S., Louis J., et al. Postinfectious purpura fulminans caused by an autoantibody directed against protein S. J Pediatr. 1995;127(3):355-363.
38 Faioni E.M., Valsecchi C., Palla A., et al. Free protein S deficiency is a risk factor for venous thrombosis. Thromb Haemost. 1997;78(5):1343-1346.
39 Gari M., Falkon L., Urrutia T., et al. The influence of low protein S plasma levels in young women, on the definition of normal range. Thromb Res. 1994;73(2):149-152.
40 Garcia de Frutos P., Alim R.I., Hardig Y., et al. Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood. 1994;84(3):815-822.
41 Dahlback B., Carlsson M., Svensson P.J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C [see comments]. Proc Natl Acad Sci U S A. 1993;90(3):1004-1008.
42 Bertina R.M., Koeleman B.P., Koster T., et al. Mutation in blood coagulation factor V associated with resistance to activated protein C [see comments]. Nature. 1994;369(6475):64-67.
43 Bertina R.M. Laboratory diagnosis of resistance to activated protein C (APC-resistance). Thromb Haemost. 1997;78(1):478-482.
44 Laffan M.A., Manning R. The influence of factor VIII on measurement of activated protein C resistance. Blood Coagul Fibrinolysis. 1996;7(8):761-765.
45 Koster T., Rosendaal F.R., de R.H., et al. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study [see comments]. Lancet. 1993;342(8886–8887):1503-1506.
46 Ridker P.M., Glynn R.J., Miletich J.P., et al. Age-specific incidence rates of venous thromboembolism among heterozygous carriers of factor V Leiden mutation. Ann Intern Med. 1997;126(7):528-531.
47 de Visser M.C., Rosendaal F.R., Bertina R.M. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood. 1999;93(4):1271-1276.
48 Poort S.R., Rosendaal F.R., Reitsma P.H., et al. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88(10):3698-3703.
49 Koster T., Blann A., Briet E., et al. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep vein thrombosis. Lancet. 1995;345:152-155.
50 Meijers J.C., Tekelenburg W.L., Bouma B.N., et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342(10):696-701.
51 O’Donnell J., Tuddenham E.G., Manning R., et al. High prevalence of elevated factor VIIIc levels in patients referred for thrombophilia screening: role of increased synthesis and relationship to the acute phase reaction. Thromb Haemost. 1997;77:825-828.
52 Tollefsen D.M., van Boven H.H., Lane D.A. Laboratory diagnosis of antithrombin and heparin cofactor II deficiency. Antithrombin and its inherited deficiency states. Semin Thromb Hemost. 1990;16(2):162-168.
53 Fay W.P., Parker A.C., Condrey L.R., Shapiro A.D. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90(1):204-208.
54 Lind B., Thorsen S. A novel missense mutation in the human plasmin inhibitor (alpha2-antiplasmin) gene associated with a bleeding tendency. Br J Haematol. 1999;107(2):317-322.
55 Lane D., Grant P. Role of hemostatic gene polymorphisms in venous and arterial thrombotic disease. Blood. 2000;95(5):1517-1532.
56 Lisman T., de Groot P.G., Meijers J.C., et al. Reduced plasma fibrinolytic potential is a risk factor for venous thrombosis. Blood. 2005;105(3):1102-1105.
57 Meltzer M.E., Bol L., Rosendaal F.R., et al. Hypofibrinolysis as a risk factor for recurrent venous thrombosis; results of the LETS follow-up study. J Thromb Haemost. 2010;8(3):605-607.
58 Juhan V.I., Pyke S.D., Alessi M.C., et al. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European Concerted Action on Thrombosis and Disabilities. Circulation. 1996;94(9):2057-2063.
59 Pyke S.D., Thompson S.G., Buchwalsky R., et al. Variability over time of haemostatic and other cardiovascular risk factors in patients suffering from angina pectoris. ECAT Angina Pectoris Study Group. Thromb Haemost. 1993;70(5):743-746.
60 Ridker P.M., Vaughan D.E., Stampfer M.J., et al. Endogenous tissue-type plasminogen activator and risk of myocardial infarction [see comments]. Lancet. 1993;341(8854):1165-1168.
61 Thompson S.G., Kienast J., Pyke S.D., et al. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group [see comments]. N Engl J Med. 1995;332(10):635-641.
62 Kluft C., Meijer P. Update 1996: Blood collection and handling procedures for assessment of plasminogen activators and inhibitors (Leiden fibrinolysis workshop). Fibrinolysis. 1996;10(suppl 2):171-179.
63 Marsh N.A., Gaffney P.J. The rapid fibrin plate – a method for plasminogen activator assay. Thromb Haemost. 1977;38(2):545-551.
64 Juhan V.I., Valadier J., Alessi M.C., et al. Deficient t-PA release and elevated PA inhibitor levels in patients with spontaneous or recurrent deep venous thrombosis. Thromb Haemost. 1987;57(1):67-72.
65 Wieczorek I., Ludlam C.A., MacGregor I. Venous occlusion does not release von Willebrand factor, factor VIII or PAI-1 from endothelial cells – the importance of consensus on the use of correction factors for haemoconcentration [letter] [see comments]. Thromb Haemost. 1993;69(1):91-93.
66 Marckmann P., Sandstrom B., Jespersen J. The variability of and associations between measures of blood coagulation, fibrinolysis and blood lipids. Atherosclerosis. 1992;96(2–3):235-244.
67 Stegnar M., Mavri A. Reproducibility of fibrinolytic response to venous occlusion in healthy subjects. Thromb Haemost. 1995;73(3):453-457.
68 Dolan G., Greaves M., Cooper P., et al. Thrombovascular disease and familial plasminogen deficiency: a report of three kindreds. Br J Haematol. 1988;70(4):417-421.
69 Heijboer H., Brandjes D.P., Buller H.R., et al. Deficiencies of coagulation-inhibiting and fibrinolytic proteins in outpatients with deep-vein thrombosis [see comments]. N Engl J Med. 1990;323(22):1512-1516.
70 Mehta R., Shapiro A.D. Plasminogen deficiency. Haemophilia. 2008;14(6):1261-1268.
71 Holvoet P., Cleemput H., Collen D. Assay of human tissue-type plasminogen activator (t-PA) with an enzyme-linked immunosorbent assay (ELISA) based on three murine monoclonal antibodies to t-PA. Thromb Haemost. 1985;54(3):684-687.
72 Mahmoud M., Gaffney P.J. Bioimmunoassay (BIA) of tissue plasminogen activator (t-PA) and its specific inhibitor (t-PA/INH). Thromb Haemost. 1985;53(3):356-359.
73 Agren A., Wiman B., Stiller V., et al. Evaluation of low PAI-1 activity as a risk factor for hemorrhagic diathesis. J Thromb Haemost. 2006;4(1):201-208.
74 Santamaria A., Borrell M., Mateo J., et al. What is the clinical impact of low plasminogen activator inhibitor-1 (PAI-1) activity? A case report and study of the incidence of low PAI-1 antigen in a healthy population. J Thromb Haemost. 2007;5(7):1565-1566.
75 Wu K.K., Hoak J.C. Spontaneous platelet aggregation in arterial insufficiency: mechanisms and implications. Thromb Haemost. 1976;35(3):702-711.
76 Afshar-Kharghan V., Li C.Q., Khoshnevis-Asl M., Lopez J.A. Kozak sequence polymorphism of the glycoprotein (GP) Ibalpha gene is a major determinant of the plasma membrane levels of the platelet GP Ib-IX-V complex.[see comment]. Blood. 1999;94(1):186-191.
77 Bennett J.S., Catella-Lawson F., Rut A.R., et al. Effect of the Pl(A2) alloantigen on the function of beta(3)-integrins in platelets. Blood. 2001;97(10):3093-3099.
78 Harrison P. Progress in the assessment of platelet function. Br J Haematol. 2000;111(3):733-744.
79 Gatt A., Makris M. Hyperhomocysteinemia and venous thrombosis. Semin Hematol. 2007;44(2):70-76.
80 Undas A., Brozek J., Szczeklik A. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94(5):907-915.
81 Bauer K.A., Rosenberg R.D. Activation markers of coagulation. Baillières Clin Haematol. 1994;7(3):523-540.
82 Besser M., Baglin C., Luddington R., et al. High rate of unprovoked recurrent venous thrombosis is associated with high thrombin-generating potential in a prospective cohort study. J Thromb Haemost. 2008;6(10):1720-1725.