CHAPTER 103 Invasion in Malignant Glioma
There is little question that the infiltrative behavior of malignant gliomas in the brain is one of the primary confounders in modern brain tumor management. A profound paradox exists, however, in that the destructive and infiltrative growth of gliomas in the brain is rarely accompanied by far-flung systemic metastases.1 This is so despite expression of the same cast of proteases, motility factors, and in vitro behavior seen in metastatic tumors from other sources.2 This chapter describes the clinical phenotype of glioma infiltration, its radiologic characteristics, and its significance with regard to clinical outcome. A discussion of the cell biology of glioma cells and their extracellular matrix (ECM)3 and cytoskeletal elements4 follows, as well as a survey of factors associated with invasive behavior5,6 and growth control.7 Finally, these molecules are reviewed as potential therapeutic targets.
Clinical Patterns of Spread
Contiguous Spread
At autopsy, 45% of cases of glioblastoma multiforme (GBM) extend beyond one lobe, 25% involve an entire hemisphere, and 25% to 30% cross over to the opposite hemisphere.8,9 Almost 60% of laterally located supratentorial gliomas spread in an anteroposterior direction, and approximately 20% invade deep supratentorial structures and infratentorial areas by invading along fiber tracts in a vertical direction.9 Frontal lobe gliomas tend to invade the frontal lobe by crossing through the corpus callosum (CC), whereas temporal lobe gliomas are prone to invade the midbrain and pons. These tendencies are observed in high-grade as well as low-grade gliomas. Gliomas arising in regions below the CC are largely confined and tend to invade basal structures, such as the thalamus and peduncles along the corticospinal tracts.10 Bilateral extension occurs through the thalamus, hypothalamus, and anterior commissure, especially in the region of the basal ganglia.8,11 In fact, Scherer found that all thalamic and hypothalamic gliomas extended bilaterally.8 Pontine tumors frequently spread cephalad and invade the midbrain and thalamic areas. Occasionally, caudal spread directly to the upper cervical cord occurs. In untreated lesions, neoplastic cells were observed within 3 cm of the necrotic tissue, whereas in recurrences, the tumor cells extended far beyond the primary tumor and were found in the opposite hemisphere in 80% of cases.12
Distant Recurrence
All treatment strategies for managing high-grade gliomas eventually fail. The pattern of tumor recurrence after treatment has been studied extensively since Kramer advocated whole-brain radiation therapy with a focal boost in 1959.13 The tumor can recur at the same site or close to it (local recurrence), or it can recur at a distant site (distant recurrence), arbitrarily defined as more than 2 cm from the original tumor site. Autopsy studies have shown that radiotherapy somewhat controls local disease (no recurrence at the primary site in 50% of cases), but the incidence of distant recurrence increased from 3% without radiotherapy to 19% to 22% with it.14,15 Clinically, 5% to 7% of high-grade glioma patients may have additional lesions away from the original site when they are seen for the first time.16,17 With respect to the field of radiation, recurrences totally outside the boost field can occur in 2% to 25% of cases, and recurrences partially outside the boost field can occur in 23% to 48% of cases.18,19
Brachytherapy was thought to be more effective than conventional treatment in improving survival, but it also results in an increased incidence of distant recurrence in patients with malignant glioma.20–22 Recurrences 2 to 5 cm away from the original tumor developed in 36% to 50% of patients, and recurrences more than 5 cm from the original tumor developed in 20% to 45% of patients. In another study, the recurrence after brachytherapy was extensive; about 39% of 23 patients experienced multicentric and subependymal spread.23 Other studies, however, do not report such a high incidence of distant metastasis. One analysis of 50 cases of recurrent tumor after brachytherapy found that only 16% of the recurrences extended beyond the 2-cm margin.
The more effectively the disease is controlled locally and the longer the survival, the greater the chance of recurrence at a distant site. Massey and Wallner found that the incidence of distant metastasis is higher in the second recurrence than in the first and observed that once surgery was performed for an initial recurrence, a second, distant recurrence (>4 cm from the original tumor) developed in as many as 25% of cases.24 Choucair and coworkers17 and Sneed and colleagues,25 in contrast, did not find any significant increase in the incidence of distant recurrence; local tumor progression was the predominant pattern of failure after brachytherapy. Late follow-up, though, showed distinctly separate lesions or subependymal or systemic spread in 40% of cases, thus reaffirming that with malignant glioma, increased survival means an increase in the number of distant recurrences. The role of brachytherapy in both newly diagnosed and recurrent glioma remains unclear, however. Reirradiation of recurrent malignant gliomas with GliaSite, in which an aqueous solution of 125I is delivered through an expandable balloon catheter, resulted in only a modest increase in survival.26 Despite initial enthusiasm for the role of 125I brachytherapy, phase III trials have failed to demonstrate a survival benefit.27,28 Since these trials, the use of brachytherapy has decreased greatly.
Radiosurgery has also been evaluated as a mode of delivering local radiation therapy. In a comparison study, distant recurrences were found in approximately 20% of patients treated by radiosurgery or brachytherapy; the rate of distant recurrence was significantly higher, however, in those not treated with either of these modalities.22 Other innovative and aggressive treatments of local glioma have resulted in a similar increase in distant recurrence, with rates as high as 75% after intra-arterial chemotherapy and 57% after radioimmunotherapy.29,30 As with brachytherapy, there was some enthusiasm for the use of radiosurgery to treat glioblastoma after positive results in phase I and II trials, but a randomized prospective trial failed to demonstrate a survival benefit when compared with conventional radiotherapy alone.31
Low-grade gliomas (Kernohan grades I and II) can also recur at distant sites after many years.32 In 20 cases of low-grade recurrence, nearly a fourth were situated outside the radiotherapy field.18
Multifocal Glioma and Gliomatosis Cerebri
Most gliomas occur as single masses. However, wide dissemination of glioma cells may lead to the appearance on magnetic resonance imaging (MRI) of multiple masses separated by intervening brain tissue. These multiple gliomas can be classified as “multifocal” if there is an apparent route of dissemination (i.e., a white matter tract). In contrast, if there is no obvious pathway for spread, it is termed multicentric.33 This is not to be confused with gliomatosis cerebri, which is characterized as a diffuse infiltration of glial tumor cells with preservation of the underlying cytoarchitecture and sparing of neurons. A rare type of neoplasm representing approximately 1% of total glial neoplasms, these tumors involve two or more lobes, often extending bilaterally or to infratentorial structures.34
Surgery, radiotherapy, and other adjuvant treatments, along with increased life expectancy and delayed recurrence, have increased the incidence of multifocal glioma.14,15,17,35,36 The average incidence of multifocal glioma is 1.5% at diagnosis but 7.5% after treatment. In an autopsy series of 209 gliomas, multifocal glioma was found in 27.8% of cases, and the ratio of multiple to multicentric glioma was 10.6:1.37 Tertiary centers may see an unusually high number of multifocal gliomas because of referral bias. In one study, the percentage of multifocal glioma was 30% in 47 malignant gliomas on initial examination and 56% in 25 malignant gliomas on follow-up.38
Multiple lesions occur as a result of tumor invasion from a monoclonal origin. The more time available for glioma cells to migrate, the higher the incidence of multifocal glioma. Anaplastic astrocytoma exhibits more infiltrative growth than GBM does.39,40 Thus, multifocal glioma should occur more frequently in anaplastic astrocytoma than in GBM. In fact, in one study the incidence of multifocal glioma at initial evaluation was higher in moderately anaplastic astrocytoma (“low grade,” 4.3%) than in GBM (0.98%).17 After treatment, only 18 of 405 GBMs (4.4%) recurred with multifocal lesions as compared with 54 of 630 anaplastic gliomas (8.6%) (Fig. 103-1).
Gliomatosis cerebri continues to carry a poor prognosis, although survival is variable. At diagnosis, gliomatosis cerebri is widely migratory and invasive. Despite its often lower grade appearance on histology, a majority of patients with gliomatosis cerebri will have a prognosis similar to that of GBM. Because of the diffusely infiltrative nature of this lesion, resective surgery is often not possible, which leaves radiotherapy and chemotherapy as the mainstays of treatment. Genetic abnormalities are shared with other diffuse gliomas, thus suggesting that gliomatosis may not be a separate entity from gliomas, as has been postulated, but rather a particularly migratory and invasive, but less proliferative subtype of diffuse glioma.41
Computed Tomography and Magnetic Resonance Imaging
Computed tomography (CT) and MRI have aided tremendously in understanding the pathobiology of glioma. Autopsy studies have confirmed the reliability of CT in defining the gross and microscopic extent of the tumor within a 2-cm margin, including the presence of multicentric lesions and recurrences. MRI is more sensitive and accurate than CT in studying gliomas.42–47 The superiority of MRI in assessing the presence of tumor extends to the immediate postoperative period as well. In one study, MRI detected residual tumor postoperatively in 77% of patients, whereas CT detected residual tumor in only 40.5%.45 Contrast-enhanced MRI performed within 72 hours (preferably 24 hours) postoperatively predates the appearance of normal postoperative enhancement and is therefore optimal for evaluating residual macroscopic tumor.48 The persistence of enhancing areas on MRI within 72 hours after tumor resection correlates with a high tendency for tumor recurrence in the early postoperative period.45,49
High-grade gliomas usually consist of a core of macroscopic growth surrounded by tumor cells. These tumor cells infiltrate the parenchyma around the core.12,37,38,44,50 The enhancement seen on MRI and CT correlates with the central mass, which has the highest ratio of abnormal vessels. An area of ring enhancement with surrounding hypodensity is the typical picture of malignant glioma on CT. The solid component of the tumor correlates with the contrast enhancement on CT, the hypodensity within the contrast-enhancing region is usually the central necrotic portion, and the surrounding hypodensity is a mixture of edema and infiltrated glioma cells.12,14,37,51 In contrast to the high-grade variety, low-grade gliomas generally consist of masses of tumor cells that infiltrate functioning parenchyma and frequently appear as hypodensity on CT and as more extensive T2 abnormalities on MRI.38,49
Both autopsy and stereotactic biopsy studies have confirmed the presence of tumor cells in the hypodense areas.30,42,43 In stereotactic biopsy studies by Kelly and associates in 39 patients with low- or high-grade glioma, CT showed tumor cells infiltrating the hypodense regions in 74 of 98 biopsy samples (76%).40 The hypodensity, however, does not delineate tumor-infiltrated brain parenchyma; both stereotactic biopsy and autopsy studies have shown isodense areas beyond the tumor-related hypodensity permeated by tumor cells in as many as 80% of biopsies.12,52
The infiltrating tumor cells in the periphery are better approximated by the hyperintensity on T2-weighted images than by any signal abnormality on T1-weighted MRI or CT with or without contrast enhancement.38,43,49,53,54 It should be emphasized that the T2-weighted abnormality reflects the result of edema, demyelination, and other degenerative changes, not the cellularity or atypia of the tumor cells.44 As with the hypodensity on CT, tumor cells are found infiltrating the brain beyond the hyperintensity signal changes on T2-weighted images when stereotactic biopsies are compared with MRI scans.38,44,49,51 Kelly and associates studied a group of patients with untreated glial tumors, 28% of whom had grade III or IV astrocytoma.40,51 Isolated tumor cell infiltration was found extending at least as far as the T2 weighting–defined abnormality, with tumor cells detected outside this region in approximately half the patients. In high-grade glioma, Watanabe and coworkers found tumor cells in 94% of biopsy specimens within areas of T2-weighted hyperintensity and in 22% of samples outside the areas of T2-weighted hyperintensity.46 Postmortem studies have suggested that T2-weighted hyperintensity may be a sensitive indicator of tumor cell invasion in untreated high-grade glioma patients but may be overrepresented (24%) or underrepresented (28%) in cases of recurrence.49 Heavily T2-weighted fluid attenuation inversion recovery (FLAIR) sequences improve the definition of tumor dissemination. FLAIR may also be a useful imaging modality for assessing response to treatment. Patients treated with bevacizumab, an antiangiogenic agent, have displayed a discordance between enhancement on T1 weighting and FLAIR. This may represent increased tumor dissemination with decreased vascularity.55
Newer MRI techniques have further elucidated the extent of spread in gliomas. Magnetic resonance spectroscopy (MRS) is a technique used to measure metabolites in tissue. Some metabolites commonly detected with MRS include choline, creatine, lactate, lipid, and N-acetylaspartate. Choline is thought to be an indicator of cell membrane turnover, thus being increased in proliferating tissues such as gliomas. In addition, neuronal markers such as N-acetylaspartate and creatine are decreased in glioma tissue. Using these parameters, tumor spread has been found beyond the volume defined by contrast enhancement on T1 imaging or beyond that defined by T2 hyperintensity. In addition, an increase in choline concentration is not necessarily correlated with the area of gadolinium enhancement.56
Diffusion-weighted MRI and diffusion tensor imaging have also been used to define the extent of tumor invasion. Diffusion tensor imaging describes the movement of water in tissue by using two measurements, mean diffusivity and fractional anisotropy, which represent the magnitude and direction of water molecule movement, respectively. Abnormalities in these values are due to a combination of increased water content and tumor infiltration leading to more disorganized diffusion. These alterations in value also extend beyond the border of gadolinium enhancement and T2 hyperintensity, thus suggesting that tumor invasion is more diffuse than was previously believed.57
Mechanism of Glioma Spread
Invasion of tumor cells into peripheral normal tissue is thought to be a multifactorial process requiring the interaction of tumor cells with the ECM and surrounding normal healthy tissue. Unlike other neoplasms, both benign and malignant gliomas infiltrate brain parenchyma early during inception, and they continuously infiltrate and spread from the focus of origin. Scherer defined and categorized the mechanism of glioma spread according to the presence of certain secondary structures that are formed during this process.58 These growths are perineural, surface, perivascular, perifascicular, intrafascicular, interfibrillary, white and gray matter, or a combination of these types. In one analysis, the pattern of spread in malignant glioma was studied by MRI in 47 patients.36,59 Spread along the myelin tract was detected in 34% of invading tumors (28% involving the CC and 6% involving other white matter tracts). T2-weighted studies suggested that a larger proportion of tumors located close to the CC invaded this white matter tract. Among tumors located near the CC (within 2 cm), spread throughout the CC was observed in 16 of 27 patients (59%) at diagnosis and in 14 of 17 patients (82%) at follow-up. Invasion through the adjacent ECM was noted in 38% of patients, along the basement membranes (predominantly subependymal) in 16%, and through cerebrospinal fluid (CSF) in 6%. The track was unclassifiable in 6%. Presumably, the perivascular, perineural, or leptomeningeal growth56 or subependymal spread36 is related to ECM molecules organized as a basement membrane or otherwise. Thus, the most frequent mechanism of spread for gliomas is invasion through the white matter or the ECM.
Spread of Glioma through White Matter
Spread of glioma through white matter was originally observed by Strobe60 and later described in detail by Scherer.58 GBM originates predominantly and spreads diffusely in the subcortical white matter.8,61 Oligodendrogliomas display a similar pattern of growth in white matter.59 Spread of malignant gliomas through all the white matter tracts (i.e., CC, uncinate fasciculus, fasciculus longitudinalis and occipitofrontalis, auditory and visual bundles, and corona radiata) has been documented.11 The CC is the main white matter tract that leads to bilateral growth.8,9 Maxwell’s autopsy study of GBM revealed that in 28 patients with GBM, as many as 75% had interhemispheric extension via the CC visible to the naked eye.10 In MRI studies of living patients, malignant glioma spread across the CC in 34% of cases, and in gliomas that originated within 2 cm of the CC, more than 80% invaded along that pathway.
Spread of Glioma along the Basement Membrane or Extracellular Matrix
Although observed rarely by Scherer,58 subependymal growth was noted in 16% of high-grade gliomas in our series (Fig. 103-2). This disparity might be a reflection of today’s improved survival. Of 48 recurrent cases of GBM studied by Sato and colleagues, 83% extended along the periventricular region.62 The most frequent pathway for the spread of malignant glioma in the study by Rosenblum and coworkers was the adjacent brain parenchyma (38%).38
Cerebrospinal Fluid Dissemination
The spread of malignant glioma through the CSF usually involves the basal leptomeninges and occurs more frequently than is generally realized.59 The reported incidence varies from 6.7% to 21%.35,36,60,63 Because of microscopic seeding that is not apparent clinically or radiographically, CSF dissemination is detected more commonly at autopsy. Children have the highest rate of CSF dissemination at diagnosis; it is either already present or develops during follow-up in a third of children.64,65 Spinal cord seeding can occur in 1.2% to 4.7% of malignant gliomas, and the rate may be higher in those with low-grade glioma.17,18
Molecular Basis of Glioma Invasion
In vivo and in vitro investigations have shown the predilection of human glioma cells or glioma cell lines to invade white matter tracts and along ECM substrates.48,66–72 The CC in particular is a major pathway for the migration of implanted neoplastic cells. Glioma invasion along blood vessels and perivascular spaces, between the ependyma and subependyma, and along the glia limitans can be explained by the migration of glioma cells along the basal lamina. The preference of implanted tumor cells to migrate to perivascular spaces and myelinated fiber tracts has also been demonstrated by Pedersen and associates.69 Prominent invasion of the CC was observed, with tumor cells found in the contralateral hemisphere. Chicoine and Silbergeld found tumor cells in the contralateral hemisphere as well, with cells predominantly in the white matter and lining the CSF pathways after implantation.50 Using cell adhesion and monolayer migration assays, Giese and colleagues demonstrated that myelin and the ECM protein merosin are the most permissive substrates for tumor cell attachment and migration.68 Other investigations have suggested that either fibronectin or laminin produced by mesenchymal cells in the brain (blood vessels, leptomeninges) plays a pivotal role in the non–white matter–dependent infiltration of glioma cells into surrounding parenchyma.67,73
Gliomas have been found to have two discrete populations of cells: a core of proliferating cells surrounded by cells invading the brain parenchyma. The gene expression profile between these two populations has also been found to differ. Several genes currently under investigation, including those for P311,74 death-associated protein 3,75 Fn14,76 and phosphatidylinositol-3′-kinase,77 have been found to have increased expression in invasive-phenotype cells.
Adhesion Molecules in Glioma
ECM molecules, cell surface adhesion molecules (CAMs), and molecules that act as receptors for ECM components or CAMs are collectively called adhesion molecules. They are secreted by cells and accumulate on the cell surface and in the extracellular space around the cells.78–80 It should be noted that these are not distinct classes of molecules. Some ECM molecules may act as CAMs or receptors on the cell surface, and some CAMs may be released and incorporated in the ECM. In vitro, almost all these molecules modify adhesion to the substrates and are therefore called adhesion molecules. Some individual molecules or their fragments may actually have a negative effect on adhesion of cells to the substratum. Besides adhesion, they have a more complex and integral function in modifying the cellular response to external stimuli during development and in the mature state. Such functions include signaling for trophic effect, triggering or suppressing apoptosis, and binding growth factors, proteases, and protease inhibitors; adhesion may or may not be the primary role of these adhesion molecules.
Extracellular Matrix Molecules
The “ground substance” initially recognized by Golgi as a network of fibrillar and amorphous material surrounding the neurons81 was discredited for several decades. This changed in the 1970s and 1980s, when careful examination of the nervous ultrastructure and the use of sensitive immunohistochemistry confirmed the presence of an ECM in the nervous system. The ECM in the brain is scant and unorganized except for the basement membrane around the blood vessels, yet by some estimates about 17% to 20% of total brain volume consists of ECM.82 Some CAMs and certain trophic factors can also be incorporated into the ECM.
The ECM has an important role in the invasion of gliomas into surrounding tissue. Nearly all of the ECM proteins are found in the perivascular space, which is a preferred mode of invasion of glioma cells.83 In addition, various studies have shown that gliomas cause alterations in the composition of ECM and that glioma cells require the presence of ECM proteins to convert to a migratory phenotype.84 Glioma cells grown in isolation in culture do not migrate, whereas exposure to certain ECM components such as laminin, collagen IV, tenascin-C, and vitronectin stimulates radial migration from glioma spheroids (Table 103-1).85
| ECM COMPONENT | CHARACTERISTICS |
|---|---|
| Collagen | Restricted to the mesenchymal component of glioma, except type IV |
| Fibronectin | Expression restricted to glioma blood vessels |
| Laminin | Associates with the mesenchymal component of glioma |
| Tenascin-C |
ECM, extracellular matrix; SPARC, secreted protein, acidic, and rich in cysteine.
Collagen
Collagen is the major glycoprotein in extraneural tissue. At least 19 different forms of collagen and 35 genes that encode for them have been found.77 The mesenchymal tissue (blood vessels and meninges) of normal brain and glioma expresses fibril-forming collagen (types I, III, V, VI, and VII). Collagen type IV, which is classified as a sheet-forming collagen, is extensively expressed in the developing nervous system but is restricted to the synaptic basal lamina in the developed brain. Although glioma cells can deposit different types of collagen in vitro, collagen in situ is restricted to the mesenchymal component of the glioma, with the exception of type IV, which may surround individual glioma cells.77
Fibronectin
Expressed abundantly by fetal neurons and glia, fibronectin is restricted to the mesenchymal structures in normal brain. Fibronectin can be expressed in vitro by several glioma cell lines, whereas in situ expression of fibronectin by glioma cells is scant and somewhat restricted to glioma blood vessels. Fibronectin can also be detected weakly in brain tissue infiltrated by glioma cells.71,86–88
Laminin
Laminin is a cruciform molecule formed by disulfide bonding of three chains. Each chain has a few isoforms that in turn give rise to at least 18 isoforms of laminin. Expression of laminin-1 in the normal human brain and in gliomas is codistributed with collagen type IV; laminin-2, also known as merosin, is the predominant form expressed by reactive astrocytes and occasional glioma cells in situ and by primary astrocytes and glioma cell lines in vitro.71,77,89 Expression of laminin parallels that of fibronectin in low- or high-grade glioma (i.e., it associates with the mesenchymal tissue of the tumor, with only occasional variants of glioma expressing it around individual neoplastic cells).85,86
Tenascin-C
Tenascin-C was initially identified as an antigen that not only is present in the ECM of gliomas but also is expressed in embryonic brain and other fetal tissue.90,91 Its expression generally declines as the brain matures, but in contrast to other ECM proteins such as fibronectin and laminin, it is found in vertebrate brain tissue throughout life.92,93
Each arm has two recurrent motifs: centrifugally, there is a repeat of approximately 13 epidermal growth factor (EGF)-like domains, and centripetally, there is a string of 8 to 15 fibronectin (FN) type III domains.76,78 Besides binding to cell surface integrin receptors through RGD sequences, tenascin also binds fibronectin and chondroitin sulfate proteoglycans such as phosphocan and neurocan in the ECM.94–96
The biologic role of tenascin seems to be more complex than that of any other ECM protein. Tenascin exhibits both adhesive and repulsive properties ascribable to separate molecular domains, mediates neuron-glia interaction, and also creates inhibitory boundaries within the brain and glial scars.90,94 It is upregulated extensively in granulation tissue and astroglial scars, as well as in a variety of mesenchymal tumors and cancers such as glioma, fibrosarcoma, osteosarcoma, melanoma, mammary carcinoma, and squamous cell carcinoma (see Fig. 103-2).86,87 In gliomas, its expression correlates with the degree of anaplasia, although the expression may be more heterogeneous in anaplastic astrocytomas and GBMs.88,92,97
Tenascin-specific antibodies labeled with 131I were used in an attempt to treat gliomas by directing local radiation therapy. Only a slight increase in survival times was noted, although this may have been complicated by study design. In addition, tenascin-C may play a role in angiogenesis. By inhibiting angiogenesis, invasiveness of the tumor may have actually been increased, thus confounding any therapeutic response from local irradiation.98
Thrombospondin
Thrombospondins are a family of at least four related trimeric proteins consisting of three approximately 180-kD subunits and are the product of four related genes. They are expressed in both embryonic and adult brain, and their expression correlates with mitotic and migratory events in certain embryonic nervous tissues.76,77 Thrombospondin-1 and thrombospondin-2 are closely related proteins within the larger group of thrombospondin proteins. Both have been found to have an antiangiogenic effect generated through CD36 receptor signaling.99 In addition, both have been found to have increased expression in glioblastomas.100 This may represent a host antitumor response. Expression levels, however, have not been found to have any prognostic value in patients.101 Given that angiogenesis is found in most glioma cells, it is probable that any antiangiogenic response was overwhelmed by proangiogenic factors.
Other Glycoproteins
Only a few other ECM glycoproteins have been found to play a significant role in brain tumor biology in that they are upregulated in glioma. Such glycoproteins include vitronectin, osteopontin, and SPARC (secreted protein, acidic, and rich in cysteine). SPARC, also known as osteonectin, is developmentally regulated and plays an important role in cell migration, matrix mineralization, and angiogenesis. SPARC is strongly upregulated in malignant tumors, including malignant glioma. Although its precise mode of action remains unclear, SPARC leads to increased invasiveness in confrontation assays.102 In contrast, increased expression of SPARC is associated with decreased proliferation.103 Thus, SPARC may induce an invasive phenotype in which proliferation is deferred for migration.
Osteopontin, another acidic glycoprotein associated with bones and present at very low levels in normal brain, is upregulated in gliomas in proportion to the degree of malignancy and can be produced by glioma cells themselves.104 Vitronectin, expressed mainly by hepatocytes, is expressed at high levels by mesenchymal tissues in the brain.105 Vitronectin is also expressed in high-grade gliomas but is undetectable in low-grade gliomas. In xenograft models, vitronectin was preferentially detected at invading tumor borders, although it was not clear whether it was expressed by the tumor cells or by the invaded normal brain tissue.106
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are a group of Zn2+-dependent enzymes that are integral for normal ECM turnover. They have also been found to be elevated in glioma cells and may contribute to the invasiveness of these tumors. In particular, MMP-2, MMP-9, and the membrane-type MMPs (MT-MMPs) are thought to have a crucial role in the invasiveness of gliomas. MMP-2, MMP-9, and MT-MMPs can degrade all components of the ECM and are activated in tumor tissues. In addition, activated MMPs have been associated with a poorer prognosis, but small interfering RNA (siRNA) directed toward these enzymes decreases the invasiveness of tumor cells.107
Glycosaminoglycans and Proteoglycans
A proteoglycan consists of a core protein to which glycosaminoglycan chains are attached. Thus far, chondroitin sulfate proteoglycan and heparan sulfate proteoglycan seem to be important for the central nervous system. Some dermatan sulfate proteoglycans such as decorin and biglycan, which may also contain chondroitin sulfate glycosaminoglycans, are expressed in the nervous system and upregulated in neural injury and degenerative processes.76,77 Most of the proteoglycans are present in the ECM, but some may be cell surface proteoglycans and might serve as receptors for growth factors, as well as for ECM components. These proteoglycans include syndecans (which have both heparan sulfate and chondroitin sulfate chains), betaglycan, and NG2.76
Heparan Sulfate Proteoglycans
Most of these proteoglycans, which include syndecans, glypicans, and betaglycan, are cell surface proteoglycans, whereas perlecan is associated with basal lamina.78,79 Even the cell surface proteoglycans are intimately bound and related to the ECM. Expressed in embryonic brain, they are downregulated in adult brain, where they may persist in specialized regions. Heparan sulfate proteoglycan expression is increased in glioma and correlates with the degree of malignancy.108
Chondroitin Sulfate Proteoglycans
The chondroitin sulfate proteoglycans expressed in the nervous system are versican, neurocan, phosphocan, brevican, aggrecan, and related molecules.76,77 Neurocan and brevican are interesting because their expression is restricted to the nervous system. Many chondroitin sulfate proteoglycans such as neurocan and phosphocan have a high-affinity binding site for tenascin, F11/contactin, and integrin receptors.80 Chondroitin sulfate proteoglycans modulate cell interaction and play a significant role in the developing nervous system; some are associated with synapses, and some form part of the astroglial axon barrier. Experimental findings suggest that chondroitin sulfate proteoglycans may act as repulsive molecules and thus facilitate cell division and migration. The overall expression of chondroitin sulfate proteoglycan is not increased in glioma as compared with normal glial cells106 and may actually show a progressive reduction as anaplasia increases in gliomas.109 However, there are a few exceptions. V2, a splice variant of versican, is decreased in glioma ECM but increased in the tumor vessels.77 Brain-enriched hyaluronan binding (BEHAB), which is a truncated form of brevican, is developmentally regulated and absent from adult brain but is expressed in gliomas. NG2, an integral membrane chondroitin sulfate proteoglycan, is downregulated in adult brain but reappears in vascular proliferations of glioma.77,110
Hyaluronic Acid
Hyaluronic acid, also known as hyaluronan or hyaluronate, is different from other glycosaminoglycans in that it has a relatively simple but much larger molecular structure and the absence of any covalent bond to a protein.111 Hyaluronic acid is a ubiquitous and organizing component of the ECM. It organizes large aggregates of proteoglycans by binding different proteoglycans to itself with the help of link proteins. These aggregates are the main component of ECM in the neurophil, form the pericellular network, and play an important role in cell migration and proliferation in the developing central nervous system. In glioma, hyaluronic acid is expressed at the leading edge. The overall increase in expression of hyaluronic acid in glioma correlates with the degree of anaplasia.77,80,106
Cell Surface Adhesion Molecules
Based on their molecular structure and physiologic role, most of the CAMs can be grouped into six classes.112 Four CAMs that involve protein-protein interaction are the immunoglobulin superfamily, cadherins, integrins, and receptor protein tyrosine phosphatases. The other two, which involve binding to carbohydrates, are hyaluronate receptors and selectins. This classification, which is based on molecular homology, is not very distinct. Immunoglobulin domains may occur in other adhesion molecules such as perlecan (i.e., proteoglycan) and some immunoglobulin superfamily adhesion molecules, and certain integrins may contain FN type III repeats.77 Furthermore, certain ECM molecules may act as CAMs and vice versa. For example, certain chondroitin sulfate proteoglycans may act as cell surface receptors, and a form of neural cell adhesion molecule (NCAM) is released and incorporated into the ECM. The classification of CAMs into six groups is not comprehensive. For example, CD24, a glycoprotein that does not traverse the membrane but is covalently linked to the membrane lipid bilayer, is expressed transiently by the developing brain and is upregulated in malignant glioma.113,114
Immunoglobulin Superfamily
NCAM is the prototype of this class of CAMs. Other molecules in this class include contactin, intercellular adhesion molecule (ICAM), and vascular cell adhesion molecule (VCAM). They have multiple immunoglobulin domains—five in NCAM. Some members of this group may be important in tumor biology in that increasing the expression of NCAM in rat glioma cells by transfection decreases their invasiveness.115 L1 (neuron glial CAM [NgCAM]), which may act as a receptor for chondroitin sulfate proteoglycans and bind to other CAMs in the nervous system, is upregulated in glioma.116
Cadherins
The cadherins are a group of calcium-dependent adhesion molecules that consist mainly of E-cadherin, P-cadherin, and N-cadherin. They mediate cell-cell adhesion and have an important role in the organization and construction of multicellular organisms. Cadherins have been found to play a role in various types of cancer. In gliomas, although antibodies directed at N-cadherin were found to decrease invasiveness in U87MG and U373MG glioma cells,117 no correlation was found between cellular motility and N-cadherin expression levels.118 All gliomas lack E- and T-cadherin expression, thus leading to the supposition that breakdown of cadherin-mediated junctions and cellular disorganization lead to increased glioma invasion.
Integrins
Integrins are a large family of heterodimeric glycoproteins composed of α and β subunits. Integrins bind to RGD sequences in most cases. This sequence is found and repeated in several adhesion molecules. Currently, at least 16 α and 8 β subunits are known. Both α and β subunits are transmembrane glycoproteins with a large extracellular domain. Only limited combinations of these subunits exist as functional receptors. Of the α and β subunits constituting the integrin heterodimer, β subunits are used for the subclassification of integrins. Although β1 and β3 subfamilies, which are involved in cell-matrix interaction, are expressed on most cell types, the β2 subfamily, which is involved in cell-cell interaction, is somewhat restricted to leukocytes.110 α3β1 and αvβ3 integrins are upregulated in astrocytomas and GBMs, and occasionally, α2, α5, α6, and β4 subunits can be found upregulated in gliomas.77,119 Antibodies directed toward β1 and αvβ5 integrins have resulted in decreased motility in glioma cells in vitro.120 In addition, C6 glioma cells implanted in nude mice with overexpression of β1 integrin showed diffuse invasion.121 Integrin-mediated effects have also been investigated recently as therapeutic targets.
Hyaluronate Receptors
Although several ECM proteins bind to hyaluronate, two proteins act as major cell surface receptors for it: CD44 and RHAMM (receptor for hyaluronic acid–mediated motility), both of which are expressed by normal glia, as well as by glioma.77 CD44 has more affinity for hyaluronate and is the major hyaluronate receptor in mammalian cells, but RHAMM may play a larger role in cell motility. CD44 exists as a family of more than 20 glycoproteins generated by alternative splicing of 10 variant exons, as well as by posttranslational glycosylation.77 The smallest form without the variant exons is the standard isoform (CD44s); the remainder are larger and can be identified numerically (CD44v). CD44s is expressed by normal astrocytes, as well as by all gliomas. By using polyclonal antisera that reacted to most of the variant epitopes, the variant forms (CD44v) were found to be expressed by most gliomas (and all GBMs), and their expression correlated with the degree of malignancy.122,123
Experimental Models of Brain Tumor Invasion
Various models of glioma invasion have been developed from established cell lines and primary tumor material, and several correlate closely with clinical tumor behavior.124–128 The most commonly used in vitro assays are listed in Table 103-2. Perhaps the most widely used measure of tumor invasiveness is the barrier migration assay,129–131 in which a reconstituted basement membrane (Matrigel) is used as a mechanical barrier through which tumor cells migrate in response to a chemoattractant stimulus and as a result of proteolytic activity.132 This assay is limited, however, in that the dissociated cells are removed from their tissue microenvironment and the barrier substrate, Matrigel, which consists primarily of laminin, does not closely resemble the makeup of the brain ECM.133 Laminin does occur in the central nervous system ECM but is limited to the basement membrane surrounding penetrating blood vessels and the glia limitans externa. Interestingly, recent work has suggested that laminin is upregulated in the region of tumor infiltration and is expressed by normal astrocytes in response to tumor.64,75,134–136
| Assay Type | Substrates | Description |
|---|---|---|
| Monolayer migration assay—scratch assay |
ECM, extracellular matrix.
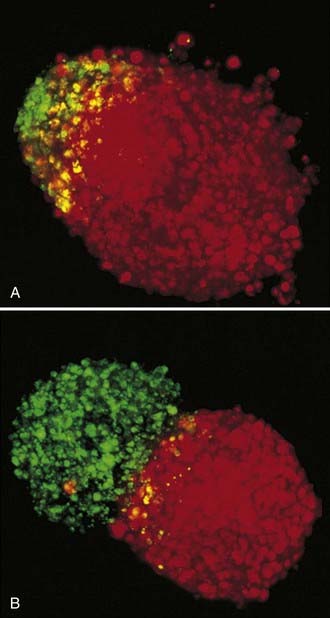
The three-dimensional brain spheroid confrontation model developed by Laerum, Bjerkvig, and others addresses some of the limitations of other glioma models.137–141 It was originally developed to allow in vitro interaction between tumor cells and a normal brain target. Three-dimensional aggregates derived from permanent glioma cell lines or primary tumor explant fragments are maintained in culture and placed in contact with similar aggregates formed by the reaggregation of dissociated fetal rat forebrain cells. These cultures are derived from E18 fetuses and are maintained in culture for several weeks, by which time the cells differentiate. The aggregates may display a laminated cell arrangement, and the cells differentiate into recognizable neurons, glial cells, and ependymal cells. Detailed ultrastructural study has confirmed the differentiated features of the target rat brain spheroid, along with preserved tissue architecture and viable cell-cell interactions.135 Over time, glial cells can be seen to infiltrate and destroy the brain aggregate in the confrontation model. This assay has demonstrated accurate correlation of invasion with clinical tumor grade.137–139 This in vitro culture arrangement maintains access to the invading cells via the culture medium for biochemical characterization and testing of the efficacy of therapeutic agents without having to address pharmacokinetic delivery variables in animals. Other technology has been applied to serial analysis of this assay, such as confocal microscopy,142,143 which appears to be especially useful for quantitation.144
The confrontation method has been used in our laboratory for testing specific protease inhibitors and carboxyamide-triazole (CAI) against human malignant glioma cell lines (see Fig. 103-2).145
Animal Implantation Models
Human xenografts growing in the complex cytoarchitecture of the adult brain closely model human malignant glioma in situ and demonstrate patterns of invasion that recapitulate those seen in clinical material. Mechanical relationships with CSF spaces, vasculature, and the critical anatomic white matter pathways on which tumor cells migrate widely during tumor cell invasion are maintained. The importance of glioma invasion has also been demonstrated in other in vivo models, such as the C6 glioma model of Bernstein and colleagues.146–152 However, as opposed to the spherical growth seen in chemically induced syngeneic tumors such as C6 and 9L, the human glioma xenografts in our model showed a striking propensity to migrate along substrates in the rat brain, which closely resembles the patterns of infiltration in clinical brain tumors. Grafted human glioma cells migrate along the glia limitans, the basement membrane–lined blood vessels and Virchow-Robin spaces, the subpial and subependymal spaces, and the white matter fascicles of anatomic pathways, including the CC, internal capsule, optic tracts, and other parallel and intersecting fiber tracts. The intact vascular basement membrane, however, is not penetrated by glioma cells,148 thus suggesting that the structure of the basement membrane may be critical for guiding the patterns of tumor cell infiltration into the brain and preventing the occurrence of systemic metastases. Although this system is more complex in terms of drug delivery, it closely models the in vivo conditions under which a potential clinical agent would operate. The ability to accurately model the infiltration process with pathologic fidelity lends itself to biologic and therapeutic quantitation and translation to the clinical setting.153 Moreover, techniques of noninvasive imaging make serial and long-term observation of tumor growth in animals possible.154
Barker and coworkers used the 9L rat gliosarcoma to develop an intracerebral tumor model.155,156 This model has been well defined and characterized extensively in neuro-oncologic research. Henderson and associates used it to evaluate radiotherapy in rats implanted with 9L tumor intracerebrally.157 Barker and coworkers investigated combined 1,3-bis-(2-chloroethyl)-1-nitrosourea (BNCU; carmustine) and radiotherapy in the 9L intracerebral tumor model,153 and Kimler and colleagues studied various chemotherapy agents such as BCNU, bleomycin, diaziquone, cis-diamminedichloroplatinum (CDDP; cisplatin), and acivicin applied subcutaneously, intraperitoneally, or intracerebrally.158 Ross and associates evaluated use of the 9L model for detecting a treatment response to chemotherapy152 and gene therapy.159
Other implantation models include a syngeneic intracerebral model with implantation of BT4 An into rat brain160 and metastatic tumors such as Walker 256 tumors implanted into rats.161
C6 murine glioma has been used frequently in experimental neuro-oncology.162 It was derived from the cloned cells of an N-nitrosourea–induced rat glioma.163,164 C6 murine glioma was implanted intracerebrally by Mineura and coworkers, who studied the effect of intracarotid chemotherapy with nimustine on the proliferation characteristics of glioma.165 Glioma cell invasion and migration have been studied with the C6 glioma in rat brain.70 Elaborating on these studies, investigators have introduced human malignant glioma xenografts into the brains of Sprague-Dawley rats and monitored the migration of these tumor cells.68 Grafted astrocytoma cells were found on the glia limitans, in the Virchow-Robin spaces, migrating along the blood vessels, and between subependymal and ependymal layers in the ventricles. The presence of basal lamina and parallel nerve bundles was a common feature in these migration routes. In a further study, these authors implanted human brain tumors (low- and high-grade astrocytomas) in nonimmunosuppressed Sprague-Dawley rats to study brain invasion by these tumors.166 They were able to follow the invasive migration of human tumor cells into rat brain by staining these cells for the human-specific p185c-neu.
Intracerebellar growth of RG-2 (a well-established rat glioma transplacentally induced by N-nitrosourea167 after subdural implantation of multicellular tumor spheroids) has been reported as a model for invasion of normal brain tissue.168 This experimental model is interesting because it does not involve an implantation procedure that disrupts nerve tissue. Molnar and coauthors reported on the effects of dexamethasone on transcapillary transport and blood flow in RG-2 rat gliomas.169 CNS-1,170 another rat glioma, has been ascribed to have invasive characteristics similar to those of human gliomas.
The animal models previously described have several drawbacks. The xenograft models are very reliable in terms of tumor growth rate and survival but do not exactly reproduce the diffusely infiltrative nature of gliomas.171 The mutagenic models, such as C6 and 9L, histologically resemble gliomas, but because the tumors are induced by unknown mutations, the genetic profile of these tumors is unknown, differs among tumors, and is thus unreproducible.172 To overcome these faults, “transgenic” and “knockout” models have been developed. These models have the advantage of producing tumors that closely mimic the behavior of diffuse gliomas while having consistent, measurable genetic profiles. Transgenic mice exhibit expression of oncogenes leading to gliomas. Using a constitutively active Ras driven by a glial fibrillary acidic protein (GFAP) promoter, diffuse astrocytomas were induced.173 Astrocytomas were likewise able to be induced in transgenic mice with v-src coexpressed with GFAP.174 Oligodendrogliomas have also been modeled by transfer of platelet-derived growth factor β (PDGF-β) to brain cells.175,176 A GBM model was developed by using avian leukosis virus–based RCAS (replication-competent avian leukosis virus family splice acceptor) vectors transferred into transgenic mice expressing tv-a, the RCAS receptor. Using this system, GBM-like tumors were produced by transferring constitutively active K-Ras and Akt to nestin-expressing progenitor cells.177
With the speculation that many gliomas may arise as a result of secondary deletions, “knockout” models have been especially useful. Tumors develop in these mice as a result of secondary mutations, which are made permissible by the first deletion. Therefore, these models are useful for identifying factors involved in the initiation or progression of gliomas. Some knockout models were developed through germline deletion of tumor suppressor genes. Other knockout mice had tumor suppressor genes that were conditionally turned off according to tissue or time, thus allowing study of otherwise fatal deletions.178 An astrocytoma model was developed with this method by using T121, a mutant of the SV40 T antigen, to inactivate the Rb family of proteins.179 Oligodendrogliomas have been recapitulated by using a knockout mouse model deficient in p19Arf.180 GBMs have been modeled by crossing two different strains of knockout mice, one deficient in Nf1 and the other in p53. Gliomas of various grades develop in these mice; however, older mice tended to have higher grade tumors. This model may simulate the progression of secondary GBM.181
More recently, studies have shown that a small fraction of glioma cells have the characteristics of primitive neural progenitor cells. The cells also possess tumor-initiating ability and have, as such, been termed tumor stem cells. When implanted into immunodeficient xenograft models, the “neurospheres” formed from these tumor stem cells were highly tumorigenic, even when serially transplanted.182 Furthermore, these tumors closely mimicked GBM in terms of histology and marker expression. These tumors recapitulated the diffusely infiltrative nature of gliomas, thus overcoming some of the shortcomings of previous xenograft models.182,183
Large-Animal Models of Brain Tumors
A rabbit brain tumor model involving VX2 carcinoma has been used to study angiogenesis and evaluate brain edema.184,185
Bayens-Simmonds and colleagues reported on the establishment of 9L tumors in cat brains.186,187 No immunosuppression was required, and an 88% take rate was obtained. The F98 rat glioma clone was xenotransplanted into the internal capsule of cats by Wechsler and associates188 and Hossmann and coworkers,189 who studied the neuropathology and edema formation and protein and water content of edematous regions in the cat brain by MRI. The same research group also reported biochemical studies.190 Kabuto and colleagues used cats to grow the C6 rat glioma intracerebrally.191 The cats were not immunosuppressed and survived an average of 3 weeks after the implantation of 5 × 105 cells. A large-animal human brain tumor xenograft model was developed in immunosuppressed cats by Krushelnycky and associates.192 These authors used the cell line D54MG, derived from a human GBM, and the TE671 human rhabdomyosarcoma cell line. Reproducible results after stereotactic implantation were obtained after pretreating the cats with 120 mg/day of cyclosporine orally for at least 10 days before the implantation of 10 × 106 tumor cells. This animal model could be used for MRI studies. The tumors were reasonably well circumscribed anaplastic gliomas, with some invasion of surrounding normal brain tissue. Although some perivascular lymphocytic cuffing was seen, there was negligible intratumoral lymphocyte infiltration.
Gavin and associates described central nervous system tumors in dogs and other large animals.193 A canine intracerebral gliosarcoma model was used by Whelan and coworkers to study gadolinium-enhanced MRI methodology in brain tumors194 and as a preclinical study of photodynamic therapy for posterior fossa tumors.195 Warnke and colleagues studied the effects of dexamethasone on experimental canine brain tumors.196 Barker and coauthors reported quantitative proton spectroscopy and histology of a canine brain tumor model.197
Berens and coworkers showed the tumorigenic, invasive, karyotypic, and immunologic characteristics of cell lines implanted into fetal beagles in utero.198 Berens’ group also published the detailed development and pathologic outcome of a syngeneic canine glioma model.199 These immunocompetent dogs undergo tolerance induction by subcutaneous injection in utero via a fetoscopic procedure. After birth, the spontaneous canine glioma cell line DG2 is orthotopically implanted into the brain. Pathologically, this tumor exhibits the characteristics of a malignant glioma.
Nonhuman primates have also been used to develop a brain tumor model. Ausman and colleagues reported on the implantation of choriocarcinoma in the brain of rhesus monkeys,200 as first described by Lewis and associates.201 The effects of dexamethasone on tumor-induced brain edema and its distribution in the brain of monkeys have also been investigated with this model.202 Overall, large-animal models have the advantage of being better suited for imaging and therapeutic studies than models using mice and rats; however, they are much more expensive and labor intensive than small-animal brain tumor models. In addition, nonsyngeneic xenograft models are probably less representative of true glioma activity because of host-graft reaction. Various studies have demonstrated spontaneous regression of tumor in allogeneic xenograft models.203–205
Cytostatics and Anti-Invasion Therapy
Recently, the concept of using cytostatics to restrain tumor progression (rather than inducing cytoreduction as conventional cytotoxic chemotherapies intend) has emerged.206 This concept challenges the current therapeutic model in cancer management, derived from microbiology, in which cancer cells are considered to be different from the host and these differences are exploited therapeutically. Continuing the analogy to infection, conventional wisdom held that unless such cells are killed and totally eliminated, they will overwhelm the host. Research strategies, new drug development, and measures of therapeutic success have been based on this killing paradigm. A regulatory model has been proposed in which cancer is viewed as a dynamic maladaptive process that originates within the host, is constantly in evolution, and is potentially reversible.207 This model is consistent with the molecular genetic understanding of cancer processes such as clonal evolution, as we have demonstrated in gliomas,208 and offers a provocative reinterpretation of long-held clinical and laboratory observations. One of the implications of such a model is that by reimposing biologic control on a cell population, functional control of a tumor may be gained without requiring complete tumor elimination. In fact, not every cancer cell need be killed to achieve control according to this model. Cancer is a process characterized by growth, invasion, and angiogenesis, all of which have their equivalents in normal tissue. Management of these components of the malignant phenotype constitutes a novel avenue for therapeutic research. Conventional antineoplastic approaches such as surgery, radiotherapy, and cytotoxic chemotherapy play roles as debulking modalities in the regulatory model, whereas biologic strategies are intended to induce re-regulation and long-term tumor control. Anti-invasion therapy represents one of these strategies in malignant gliomas and rests on a molecular understanding of the process.
Multiple targets are currently under study as anti-invasive or angio-inhibitory strategies for the treatment of gliomas. Phase II studies of tenascin-specific antibodies labeled with 131I showed slight increases in survival time, although this could have been due to bias in patient selection.97 Other limitations of the study may also have included the fact that the drug was delivered only through diffusion, possibly leaving distantly invasive glioma cells untouched. In addition, inhibiting tenascin-C by decreasing proliferation with the use of antiangiogenesis agents may cause tumor cells to become more invasive by converting cells to an invasion phenotype.
EMD121974 (cilengitide) was designed to block the interaction of αvβ3 and αvβ5 integrin with ECM. This would thus possibly potently inhibit an important process in angiogenesis. In vivo studies demonstrated robust tumor regression,209 and the ensuing clinical phase I studies in which treatment with EMD121974 was combined with radiation therapy have shown encouraging tumor response, as well as clinical tolerance to therapy.210
Thalidomide, a well-known teratogen, has been found to be a potent antiangiogenic factor. D’Amato and colleagues, using the rabbit cornea micropocket assay, showed that orally administered thalidomide inhibited corneal neovascularization induced by basic fibroblast growth factor.211 It requires hepatic metabolism for activation. Because thalidomide was originally developed as a sedative, it is already known to traverse the normal blood-brain barrier. However, phase II studies combining thalidomide with temozolomide, a cytotoxic agent, showed no benefit over temozolomide monotherapy and, moreover, exhibited increased toxicity.212
MMP inhibitors have been shown in vitro to effectively decrease glioma tumor cell invasion.213 Unfortunately, phase III trials of marimastat monotherapy, an orally available MMP inhibitor, showed no survival benefit over placebo. Nonetheless, trials of MMP inhibitors in combination with cytotoxic agents are ongoing.214
Tyrosine kinase inhibitors, which inhibit downstream signaling, have also been investigated as possible therapies. Gefitinib, the best described of the tyrosine kinase inhibitors, was investigated in a phase II trial, but no benefit was found.215 In contrast, it has been discovered that tumors that coexpress PTEN (phosphatase and tensin homologue from chromosome 10) and EGFRvIII were associated with clinical response to erlotinib and gefitinib, thus signaling the importance of biomarkers in tailoring treatment.216
Finally, there is currently much interest in the use of antiangiogenic therapies directed against vascular endothelial growth factor receptors. In particular, sorafenib and bevacizumab have been shown to have clinical activity against a variety of solid tumors.217 Currently, a phase II study is investigating a combination of bevacizumab and irinotecan for recurrent GBMs. The initial results have been promising. Still, given the possibility that antiangiogenic therapies may actually increase invasiveness, these data must be regarded with caution.
Albert FK, Forsting M, Sartor K, et al. Early postoperative magnetic resonance imaging after resection of malignant glioma: objective evaluation of residual tumor and its influence on regrowth and prognosis. Neurosurgery. 1994;34:45-61.
Amar AP, DeArmond SJ, Spencer DR, et al. Development of an in vitro extracellular matrix assay for studies of brain tumor cell invasion. J Neurooncol. 1994;20:1-15.
Batzdorf U, Malamud N. The problem of multicentric gliomas. J Neurosurg. 1963;20:122-136.
Bernstein JJ, Goldberg WJ, Laws ERJr. A model for central nervous system cancer research. J Neurosci Res. 1989;22:134-143.
Chicoine MR, Silbergeld DL. The in vitro motility of human gliomas increases with increasing grade of malignancy. Cancer. 1995;75:2904-2909.
Enam SA, Eisenberg AD, Norman D, et al. Patterns of spread and recurrence of glioma: studies by neuroimaging. In: Mikkelsen T, Bjerkvig R, Laerum OD, et al, editors. Brain Tumor Invasion: Clinical, Biological and Therapeutic Considerations. New York: Wiley-Liss; 1998:133-159.
Giese A, Westphal M. Glioma invasion in the central nervous system. Neurosurgery. 1996;39:235-252.
Kohn EC, Liotta LA. Molecular insights into cancer invasion: strategies for prevention and intervention. Cancer Res. 1995;55:1856-1862.
Kruse CA, Molleston MC, Parks EP, et al. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas. J Neurooncol. 1994;22:191-200.
Larsen LF, Edvardsen K. Cell adhesion molecules in the migration of neural crest cells. In: Mikkelsen T, Bjerkvig R, Laerem OD, et al, editors. Brain Tumor Invasion: Biological, Clinical, and Therapeutic Considerations. New York: Wiley-Liss; 1998:3-12.
Levin VA, Phuphanich S, Yung WK, et al. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol. 2006;78:295-302.
Li X, Lu Y, Pirzkall A, et al. Analysis of the spatial characteristics of metabolic abnormalities in newly diagnosed glioma patients. J Magn Reson Imaging. 2002;16:229-237.
Lund-Johansen M, Engebraaten O, Bjerkvig R, et al. Invasive glioma cells in tissue culture. Anticancer Res. 1990;10:1135-1151.
Mikkelsen T, Edvardsen K. Invasiveness in nervous system tumors. In: Black P, Loeffler JS, editors. Cancer of the Nervous System. Cambridge, MA: Blackwell Scientific, 1995.
Mikkelsen T, Rosenblum ML. Tumor invasiveness. In: Berger M, Mitchell S, Wilson CB, editors. Textbook of Gliomas. Cambridge, MA: Saunders, 1995.
Paulus W. Brain extracellular matrix, adhesion molecules, and glioma invasion. In: Mikkelsen T, Bjerkvig R, Laerem OD, et al, editors. Brain Tumor Invasion: Biological, Clinical, and Therapeutic Considerations. New York: Wiley-Liss; 1998:301-322.
Pilkington GJ. The paradox of neoplastic glial cell invasion of the brain and apparent metastatic failure. Anticancer Res. 1997;17:4103-4105.
Rini BI, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma: a home run or a work in progress? Oncology (Williston Park). 2008;22:388-396.
Schipper H, Goh CR, Wang TL. Shifting the cancer paradigm: must we kill to cure? J Clin Oncol. 1995;13:801-807.
Tonn JC, Wunderlich S, Kerkau S, et al. Invasive behaviour of human gliomas is mediated by interindividually different integrin patterns. Anticancer Res. 1998;18:2599-2605.
Venstrom KA, Reichardt LF. Extracellular matrix 2: role of extracellular matrix molecules and their receptors in the nervous system. FASEB J. 1993;7:996-1003.
Watanabe M, Tanaka R, Takeda N. Magnetic resonance imaging and histopathology of cerebral gliomas. Neuroradiology. 1992;34:463-469.
1 Pilkington GJ. The paradox of neoplastic glial cell invasion of the brain and apparent metastatic failure. Anticancer Res. 1997;17:4103-4105.
2 Silbergeld DL, Chicoine MR. Isolation and characterization of human malignant glioma cells from histologically normal brain. J Neurosurg. 1997;86:525-531.
3 Maidment SL. The cytoskeleton and brain tumour cell migration. Anticancer Res. 1997;17:4145-4149.
4 Chintala SK, Sawaya R, Aggarwal BB, et al. Induction of matrix metalloproteinase-9 requires a polymerized actin cytoskeleton in human malignant glioma cells. J Biol Chem. 1998;273:13545-13551.
5 Westphal M, Rosen EM. Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int J Cancer. 1998;75:19-28.
6 Yamamoto S, Wakimoto H, Aoyagi M, et al. Modulation of motility and proliferation of glioma cells by hepatocyte growth factor. Jpn J Cancer Res. 1997;88:64-77.
7 Cho K-K, Mikkelsen T, Lee YJ, et al. The role of protein kinase Ca in U87 glioma invasion. Int J Dev Neurosci. 1999;17:447-461.
8 Scherer HJ. The forms of growth in gliomas and their practical significance. Brain. 1940;63:1-34.
9 Salazar OM, Rubin P. The spread of glioblastoma multiforme as a determining factor in the radiation treated volume. Int J Radiat Oncol Biol Phys. 1976;1:627-637.
10 Maxwell HP. The incidence of interhemispheric extension of glioblastoma multiforme through the corpus callosum. J Neurosurg. 1946;3:54-57.
11 Matsukado Y, MacCarty CS, Kernohan JW. The growth of glioblastoma multiforme (astrocytomas, grades 3 and 4) in neurosurgical practice. J Neurosurg. 1961;18:636-644.
12 Burger PC, Dubois PJ, Schold SC, et al. Computerized tomographic and pathologic studies of the untreated, quiescent, and recurrent glioblastoma multiforme. J Neurosurg. 1983;58:159-169.
13 Kramer S. Tumor extent as determining factor in radiotherapy of glioblastomas. Acta Radiol. 1959;8:111-117.
14 Hochberg FH, Pruitt A. Assumptions in the radiotherapy of glioblastoma. Neurology. 1980;30:907-911.
15 Wallner KE, Galicich JH, Krol G, et al. Patterns of failure following treatment for glioblastoma multiforme and anaplastic astrocytoma. Int J Radiat Oncol Biol Phys. 1989;16:1405-1409.
16 Bashir R, Hochberg F, Oot R. Regrowth patterns of glioblastoma multiforme related to planning of interstitial brachytherapy radiation fields. Neurosurgery. 1988;23:27-30.
17 Choucair AK, Levin VA, Gutin PH, et al. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J Neurosurg. 1986;65:654-658.
18 Salazar OM, Rubin P, McDonald JV, et al. Patterns of failure in intracranial astrocytomas after irradiation: analysis of dose and field factors. AJR Am J Roentgenol. 1976;126:279-292.
19 Gaspar LE, Fisher BJ, Macdonald DR, et al. Supratentorial malignant glioma: patterns of recurrence and implications for external beam local treatment. Int J Radiat Oncol Biol Phys. 1992;24:55-57.
20 Loeffler JS, Alexander EIII, Hochberg FH, et al. Clinical patterns of failure following stereotactic interstitial irradiation for malignant gliomas. Int J Radiat Oncol Biol Phys. 1990;19:1455-1462.
21 Wen PY, Alexander EIII, Black PM, et al. Long term results of stereotactic brachytherapy used in the initial treatment of patients with glioblastomas. Cancer. 1994;73:3029-3036.
22 Shrieve DC, Alexander EIII, Wen PY, et al. Comparison of stereotactic radiosurgery and brachytherapy in the treatment of recurrent glioblastoma multiforme. Neurosurgery. 1995;36:275-284.
23 Chun M, McKeough F, Wu A, et al. Interstitial iridium-192 implantation for malignant brain tumours. Part II. Clinical experience. Br J Radiol. 1989;62:158-162.
24 Massey V, Wallner KE. Patterns of second recurrence of malignant astrocytomas. Int J Radiat Oncol Biol Phys. 1990;18:395-398.
25 Sneed PK, Gutin PH, Larson DA, et al. Patterns of recurrence of glioblastoma multiforme after external irradiation followed by implant boost. Int J Radiat Oncol Biol Phys. 1994;29:719-727.
26 Gabayan AJ, Green SB, Sanan A, et al. GliaSite brachytherapy for treatment of recurrent malignant gliomas: a retrospective multi-institutional analysis. Neurosurgery. 2006;58:701-709.
27 Selker RG, Shapiro WR, Burger P, et al. The Brain Tumor Cooperative Group NIH Trial 87-01: a randomized comparison of surgery, external radiotherapy, and carmustine versus surgery, interstitial radiotherapy boost, external radiation therapy, and carmustine. Neurosurgery. 2002;51:343-355.
28 Laperriere NJ, Leung PM, McKenzie S, et al. Randomized study of brachytherapy in the initial management of patients with malignant astrocytoma. Int J Radiat Oncol Biol Phys. 1998;41:1005-1011.
29 Iwadate Y, Namba H, Sueyoshi K. Intra-arterial ACNU and cisplatin chemotherapy for the treatment of glioblastoma multiforme. Neurol Med Chir (Tokyo). 1995;35:598-603.
30 Riva P, Arista A, Franceschi G, et al. Local treatment of malignant gliomas by direct infusion of specific monoclonal antibodies labeled with 131I: comparison of the results obtained in recurrent and newly diagnosed tumors. Cancer Res. 1995;55(Suppl):5952s-5956s.
31 Roberge D, Souhami L. Stereotactic radiosurgery in the management of intracranial gliomas. Technol Cancer Res Treat. 2003;2:117-125.
32 Civitello LA, Packer RJ, Rorke LB, et al. Leptomeningeal dissemination of low-grade gliomas in childhood. Neurology. 1988;38:562-566.
33 Showalter TN, Andrel J, Andrews DW, et al. Multifocal glioblastoma multiforme: prognostic factors and patterns of progression. Int J Radiat Oncol Biol Phys. 2007;69:820-824.
34 Kim DG, Yang HJ, Park IA, et al. Gliomatosis cerebri: clinical features, treatment, and prognosis. Acta Neurochir (Wien). 1998;140:755-762.
35 Salazar OM, Rubin P, McDonald JV, et al. High dose radiation therapy in the treatment of glioblastoma multiforme: a preliminary report. Int J Radiat Oncol Biol Phys. 1976;1:717-727.
36 Liang BC, Thornton AFJr, Sandler HM, et al. Malignant astrocytomas: focal tumor recurrence after focal external beam radiation therapy. J Neurosurg. 1991;75:559-563.
37 Batzdorf U, Malamud N. The problem of multicentric gliomas. J Neurosurg. 1963;20:122-136.
38 Rosenblum ML, Eisenberg AD, Norman D. Brain tumor invasion: clinical patterns of malignant astrocytoma spread [abstract]. J Neurosurg. 1992;76:383A.
39 Daumas-Duport C, Monsaigneon V, Blond S, et al. Serial stereotactic biopsies and CT scan in gliomas: correlative study in 100 astrocytomas, oligo-astrocytomas and oligodendrocytomas. J Neurooncol. 1987;4:317-328.
40 Kelly PG, Daumas-Duport C, Scheithauer BW, et al. Stereotactic histologic correlations of computed tomography– and magnetic resonance imaging–defined abnormalities in patients with glial neoplasms. Mayo Clin Proc. 1987;62:450-459.
41 Cavenee WK, Furnari FB, Nagane M, et al. Diffusely infiltrating astrocytomas. In: Kleihues P, Cavenee WK, editors. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: International Agency for Research on Cancer; 2000:10-21.
42 Bydder GM, Steiner RE, Young IR, et al. Clinical NMR imaging of the brain: 140 cases. AJR Am J Roentgenol. 1982;139:215-236.
43 Brant-Zawadzki M, Badami JP, Mills CM, et al. Primary intracranial tumor imaging: a comparison of magnetic resonance and CT. Radiology. 1984;150:435-440.
44 Le Bas JF, Leviel JL, Decorps M, et al. NMR relaxation times from serial stereotactic biopsies in human brain tumors. J Comput Assist Tomogr. 1984;8:1048-1057.
45 Laster DW, Ball MR, Moody DM, et al. Results of nuclear magnetic resonance with cerebral glioma: comparison with computed tomography. Surg Neurol. 1984;22:113-122.
46 Watanabe M, Tanaka R, Takeda N. Magnetic resonance imaging and histopathology of cerebral gliomas. Neuroradiology. 1992;34:463-469.
47 Forsting M, Albert FK, Kunze S, et al. Extirpation of glioblastomas: MR and CT follow-up of residual tumor and regrowth patterns. AJNR Am J Neuroradiol. 1993;14:77-87.
48 Albert FK, Forsting M, Sartor K, et al. Early postoperative magnetic resonance imaging after resection of malignant glioma: objective evaluation of residual tumor and its influence on regrowth and prognosis. Neurosurgery. 1994;34:45-61.
49 Kyuma Y, Yamashita T, Ishiwata Y, et al. Treatment of recurrent gliomas. No Shinkei Geka. 1984;12:477-483.
50 Chicoine MR, Silbergeld DL. Assessment of brain tumor cell motility in vivo and in vitro. J Neurosurg. 1995;82:615-622.
51 Kelly PJ, Daumas-Duport C, Kispert DB, et al. Imaging-based stereotaxic serial biopsies in untreated intracranial glial neoplasms. J Neurosurg. 1987;66:865-874.
52 Burger PC, Heinz ER, Shibata T, et al. Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J Neurosurg. 1988;68:698-704.
53 Earnest F IV, Kelly PJ, Scheithauer BW, et al. Cerebral astrocytomas: histopathologic correlation of MR and CT contrast enhancement with stereotactic biopsy. Radiology. 1988;166:823-827.
54 Johnson PC, Hunt SJ, Drayer BP. Human cerebral gliomas: correlation of postmortem MR imaging and neuropathologic findings. Radiology. 1989;170:211-217.
55 Zuniga RM, Torcuator R, Jain R, et al. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J Neurooncol. 2009;91:329-336.
56 Li X, Lu Y, Pirzkall A, et al. Analysis of the spatial characteristics of metabolic abnormalities in newly diagnosed glioma patients. J Magn Reson Imaging. 2002;16:229-237.
57 Lu S, Ahn D, Johnson G, et al. Peritumoral diffusion tensor imaging of high-grade gliomas and metastatic brain tumors. AJNR Am J Neuroradiol. 2003;24:937-941.
58 Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34:333-351.
59 Enam SA, Eisenberg AD, Norman D, et al. Patterns of spread and recurrence of glioma: studies by neuroimaging. In: Mikkelsen T, Bjerkvig R, Laerum OD, et al, editors. Brain Tumor Invasion: Clinical, Biological and Therapeutic Considerations. New York: Wiley-Liss; 1998:133-159.
60 Strobe H. Ueber Entstehung und Bau de Gehirngliome. Beitr Pathol Anat. 1895;18:405-411.
61 Russell DS, Rubinstein LJ. Pathology of Tumours of the Nervous System. Baltimore: Williams & Wilkins; 1989.
62 Sato I, Tanaka R, Takeda N, et al. Computed tomographic features of cerebral malignant glioma following treatment and regrowth. Neurol Med Chir (Tokyo). 1988;28:1073-1080.
63 Yamasahi T, Kikuchi H. Three-dimensional analysis of regrowth pattern in recurrent supratentorial glioblastoma and anaplastic astrocytoma with special reference to prognosis. Gon No Rinsho. 1989;35:1261-1271.
64 Grabb PA, Albright AL, Pang D. Dissemination of supratentorial malignant gliomas via the cerebrospinal fluid in children. Neurosurgery. 1992;30:64-71.
65 Packer RJ, Schut L, Siegel KR. Dissemination of primary central nervous system tumors of childhood: incidence and clinical implications. Prog Exp Tumor Res. 1987;30:206-214.
66 Giese A, Rief MD, Loo MA, et al. Determinants of human astrocytoma migration. Cancer Res. 1994;54:3897-3904.
67 Giese A, Loo MA, Rief MD, et al. Substrates for astrocytoma invasion. Neurosurgery. 1995;37:294-302.
68 Giese A, Kluwe L, Laube B, et al. Migration of human glioma cells on myelin. Neurosurgery. 1996;38:755-764.
69 Pedersen P-H, Marienhagen K, Mork S, et al. Migratory pattern of fetal rat brain cells and human glioma cells in the adult rat brain. Cancer Res. 1993;53:5158-5165.
70 Bernstein JJ, Goldberg WJ, Laws ERJr. Human malignant astrocytoma xenografts migrate in rat brain: a model for central nervous system cancer research. J Neurosci Res. 1989;22:134-143.
71 Bernstein JJ, Goldberg WJ, Laws ERJr. Immunohistochemistry of human malignant astrocytoma cells xenografted to rat brain: apolipoprotein E. Neurosurgery. 1989;24:541-546.
72 Bernstein JJ, Goldberg WJ, Laws ERJr, et al. C6 glioma cell invasion and migration of rat brain after neural homografting: ultrastructure. Neurosurgery. 1990;26:622-628.
73 Enam SA, Rosenblum ML, Edvardsen K. Role of extracellular matrix in glioma invasion: migration of glioma cells along fibronectin positive processes of cultured mesenchymal cells. Neurosurgery. 1998;42:599-608.
74 Mariani L, McDonough WS, Hoelzinger DB, et al. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 2001;61:4190-4196.
75 Kissil JL, Deiss LP, Bayewitch M, et al. Isolation of DAP3, a novel mediator of interferon-gamma–induced cell death. J Biol Chem. 1995;270:27932-27936.
76 Tran NL, McDonough WS, Donohue PJ, et al. The human Fn14 receptor gene is upregulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol. 2003;162:1313-1321.
77 Joy AM, Beaudry CE, Tran NL, et al. Migrating glioma cells activate the PI3-K pathway and display decreased susceptibility to apoptosis. J Cell Sci. 1999;116:4409-4417.
78 Venstrom KA, Reichardt LF. Extracellular matrix 2: role of extracellular matrix molecules and their receptors in the nervous system. FASEB J. 1993;7:996-1003.
79 Paulus W. Brain extracellular matrix, adhesion molecules, and glioma invasion. In: Mikkelsen T, Bjerkvig R, Laerem OD, et al, editors. Brain Tumor Invasion: Biological, Clinical, and Therapeutic Considerations. New York: Wiley-Liss; 1998:301-322.
80 Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci. 1991;14:531-570.
81 Golgi C. Intorno alla struttura delle cellule nervose. Communicated to the Societa Medico-chirurgica of Pavia on April 19, 1898. In: Golgi C, editor. Opera Omnia, vol 2. Milan: Hoepli; 1903:643-653.
82 Giese A, Westphal M. Glioma invasion in the central nervous system. Neurosurgery. 1996;39:235-252.
83 Rutka JT, Apodaca G, Stern R, et al. The extracellular matrix of the central and peripheral nervous systems: structure and function. J Neurosurg. 1988;69:155-170.
84 Tysnes BB, Larser LF, Ness GO, et al. Stimulation of glioma-cell migration by laminin and inhibition by anti-α3 and anti-βl integrin antibodies. Int J Cancer. 1996;67:777-784.
85 Ohnishi T, Arita N, Hiraga S, et al. Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis. 1997;15:538-546.
86 Higuchi M, Ohnishi T, Arita N, et al. Immunohistochemical localization of fibronectin, laminin, fibronectin receptor in human malignant gliomas: in relation to tumor invasion. Brain Nerve. 1991;43:17-23.
87 Morris CS, Esiri MM. Immunocytochemical study of macrophages and microglial cells and extracellular matrix components in human CNS disease. 1. Gliomas. J Neurol Sci. 1991;101:47-58.
88 Bellon G, Caulet T, Cam Y, et al. Immunohistochemical localization of macromolecules of the basement membrane and extracellular matrix of human gliomas and meningiomas. Acta Neuropathol (Berl). 1985;66:245-252.
89 Chintala SK, Sawaya R, Gokaslan ZL, et al. Immunohistochemical localization of extracellular matrix proteins in human glioma, both in vivo and in vitro. Cancer Lett. 1996;101:107-114.
90 Bourdon MA, Wikstrand CJ, Furthmayr H, et al. Human glioma-mesenchymal extracellular matrix antigen defined by monoclonal antibody. Cancer Res. 1983;43:2796-2805.
91 Chiquet M, Fambrough DM. Chick myotendinous antigen. I. A monoclonal antibody as a marker for tendon and muscle morphogenesis. J Cell Biol. 1984;98:1926-1936.
92 Erickson HP, Bourdon MA. Tenascin: an extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu Rev Cell Biol. 1989;5:71-92.
93 Hoffman S, Crossin KL, Edelman GM. Molecular forms, binding functions, and development expression patterns of cytotactin and cytotactin-binding proteoglycan, an interactive pair of extracellular matrix molecules. J Cell Biol. 1988;106:519-532.
94 Grumet M, Milev P, Sakurai T, et al. Interactions with tenascin and differential effects on cell adhesion of neurocan and phosphocan, two major chondroitin sulfate proteoglycans of nervous tissue. J Biol Chem. 1994;269:12142-12146.
95 Bourdon M, Rouslahti E. Tenascin mediates cell attachment through an RGD-dependent receptor. J Cell Biol. 1989;108:1149-1155.
96 Steindler DA. Glial boundaries in the developing nervous system. Annu Rev Neurosci. 1993;16:445-470.
97 Castellani P, Dorcaratto A, Siri A, et al. Tenascin distribution in human brain tumors. Acta Neurochir (Wien). 1995;136:44-50.
98 Reardon DA, Akabani G, Coleman RE, et al. Phase II trial of murine 131I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20:1389-1397.
99 Guo N, Krutzsch HC, Inman JK, et al. Thrombospondin 1 and type I repeat peptides of thrombospondin I specifically induce apoptosis of endothelial cells. Cancer Res. 1997;57:1735-1742.
100 Pijuan-Thompson V, Grammar JR, Stewart J, et al. Retinoic acid alters the mechanism of attachment of malignant astrocytoma and neuroblastoma cells to thrombospondin-1. Exp Cell Res. 1999;249:86-101.
101 Kazuno M, Tokunaga T, Oshika Y, et al. Thrombospondin-2 (TSP2) expression is inversely correlated with vascularity in glioma. Eur J Cancer. 1999;35:502-506.
102 Porter PL, Sage EH, Lane TF, et al. Distribution of SPARC in normal and neoplastic human tissue. J Histochem Cytochem. 1995;43:791-800.
103 Rempel SA, Golembieski WA, Ge S, et al. SPARC: a signal of astrocytic neoplastic transformation and reactive response in human primary and xenograft gliomas. J Neuropathol Exp Neurol. 1998;57:1112-1121.
104 Saitoh Y, Kuratsu J, Takeshima H, et al. Expression of osteopontin in human glioma: its correlation with the malignancy. Lab Invest. 1995;72:55-63.
105 Seiffert D, Bordin GM, Loskutoff DJ. Evidence that extrahepatic cells express vitronectin mRNA at rates approaching those of hepatocytes. Histochem Cell Biol. 1996;105:195-201.
106 Gladson CL, Wilcox JN, Sanders L, et al. Cerebral microenvironment influences expression of the vitronectin gene in astrocytic tumors. J Cell Sci. 1995;108:947-956.
107 Van Meter TE, Rooprai HK, Kibble MM, et al. The role of matrix metalloproteinase genes in glioma invasion: codependent and interactive proteolysis. J Neurooncol. 2001;53:213-235.
108 Steck PA, Moser RP, Bruner JM, et al. Altered expression and distribution of heparan sulfate proteoglycans in human gliomas. Cancer Res. 1989;49:2096-2103.
109 Bertolotto A, Goia L, Schiffer D. Immunohistochemical study of chondroitin sulfate in human gliomas. Acta Neuropathol (Berl). 1986;72:189-196.
110 Schrappe M, Klier FG, Spiro RC, et al. Correlation of chondroitin sulfate proteoglycan expression on proliferating brain capillary endothelial cells with the malignant phenotype of astroglial cells. Cancer Res. 1991;51:4986-4993.
111 Alberts B, Bray D, Lewis J, et al, editors. Molecular Biology of the Cell. New York: Garland, 1989.
112 Larsen LF, Edvardsen K. Cell adhesion molecules in the migration of neural crest cells. In: Mikkelsen T, Bjerkvig R, Laerem OD, et al, editors. Brain Tumor Invasion: Biological, Clinical, and Therapeutic Considerations. New York: Wiley-Liss; 1998:3-12.
113 Redondo P, Garcia-Foncillas J, Okroujnov I, et al. CD24 expression on human keratinocytes. Exp Dermatol. 1998;7:175-178.
114 Poncet C, Frances V, Gristina R, et al. CD24, a glycosylphosphatidylinositol-anchored molecule, is transiently expressed during the development of human central nervous system and is a marker of human neural cell lineage tumors. Acta Neuropathol (Berl). 1996;91:400-408.
115 Edvardsen K, Pedersen PH, Bjerkvig R, et al. Transfection of glioma cells with neural cell adhesion molecule NCAM: effect on glioma cell invasion and growth in vivo. Int J Cancer. 1994;58:116-122.
116 Tsuzuki T, Izumoto S, Ohnishi T, et al. Neural cell adhesion molecule L1 in gliomas: correlation with TGF-beta and p53. J Clin Pathol. 1998;51:13-17.
117 Kotolevets L, van Hengel J, Bruyneel E, et al. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J Cell Biol. 2001;155:1129-1135.
118 Shinoura N, Paradies NE, Warnick RE, et al. Expression of N-cadherin and alpha-catenin in astrocytomas and glioblastomas. Br J Cancer. 1995;72:629-633.
119 Gladson CL, Cheresh DA. Glioblastoma expression of vitronectin and the αvβ3 integrin. J Clin Invest. 1991;88:1924-1932.
120 Tonn JC, Wunderlich S, Kerkau S, et al. Invasive behaviour of human gliomas is mediated by interindividually different integrin patterns. Anticancer Res. 1998;18:2599-2605.
121 Paulus W, Baur I, Beutler AS, et al. Diffuse brain invasion of glioma cells requires beta 1 integrins. Lab Invest. 1996;75:819-826.
122 Frank S, Rihs HP, Stöcker W, et al. Combined detection of CD44 isoforms by exon-specific RT-PCR and immunohistochemistry in primary brain tumors and brain metastases. Biochem Biophys Res Commun. 1996;222:794-801.
123 Eibl RH, Pietsch T, Moll J, et al. Expression of variant CD44 epitopes in human astrocytic brain tumors. J Neurooncol. 1995;49:199-203.
124 Mikkelsen T, Rosenblum ML. Tumor invasiveness. In: Berger M, Mitchell S, Wilson CB, editors. Textbook of Gliomas. Cambridge, MA: Saunders, 1995.
125 Mikkelsen T, Edvardsen K. Invasiveness in nervous system tumors. In: Black P, Loeffler JS, editors. Cancer of the Nervous System. Cambridge, MA: Blackwell Scientific, 1995.
126 Bjerkvig R, Laerum OD, Rucklidge GJ. Immunocytochemical characterization of extracellular matrix proteins expressed by cultured glioma cells. Cancer Res. 1989;49:5424-5428.
127 Pilkington GJ, Bjerkvig R, DeRidder L, et al. In vitro and in vivo models for the study of brain tumor invasion. Anticancer Res. 1997;17:4107-4109.
128 Chicoine MR, Silbergeld DL. The in vitro motility of human gliomas increases with increasing grade of malignancy. Cancer. 1995;75:2904-2909.
129 Albini A, Iwamoto Y, Kleinman HK, et al. A rapid in vitro assay for quantifying the invasive potential of tumor cells. Cancer Res. 1987;47:3239-3245.
130 Amar AP, DeArmond SJ, Spencer DR, et al. Development of an in vitro extracellular matrix assay for studies of brain tumor cell invasion. J Neurooncol. 1994;20:1-15.
131 Albini A. Tumor and endothelial cell invasion of basement membranes: the Matrigel chemoinvasion assay as a tool for dissecting molecular mechanisms. Pathol Oncol Res. 1998;4:230-241.
132 Deryugina EI, Luo GX, Reisfeld RA, et al. Tumor cell invasion through Matrigel is regulated by activated matrix metalloproteinase-2. Anticancer Res. 1997;17:3201-3210.
133 Rutka JT, Apodaca G, Stern R, et al. The extracellular matrix of the central and peripheral nervous systems: structure and function. J Neurosurg. 1988;69:155-170.
134 Knott JC, Mahesparan R, Garcia-Cabrera I, et al. Stimulation of extracellular matrix components in the normal brain by invading glioma cells. Int J Cancer. 1998;75:864-872.
135 Ohnishi T, Arita N, Hiraga S, et al. Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis. 1997;15:538-546.
136 Ohnishi T, Arita N, Hayakawa T, et al. Motility factor produced by malignant glioma cells: role in tumor invasion. J Neurosurg. 1990;73:881-888.
137 Bjerkvig R, Steinsvag SK, Laerum OD. Reaggregation of fetal rat brain cells in a stationary culture system. I. Methodology and cell identification. In Vitro Cell Dev Biol. 1986;22:180-192.
138 Bjerkvig R, Laerum OD, Mella O. Glioma cell interactions with fetal rat brain aggregates in vitro and with brain tissue in vivo. Cancer Res. 1986;46:4071-4079.
139 Engebraaten O, Bjerkvig R, Lund-Johansen M, et al. Interaction between human brain tumour biopsies and fetal rat brain tissue in vitro. Acta Neuropathol (Berl). 1990;81:130-140.
140 Lund-Johansen M, Engebraaten O, Bjerkvig R, et al. Invasive glioma cells in tissue culture. Anticancer Res. 1990;10:1135-1151.
141 Bjerkvig R, Tonnesen A, Laerum OD, et al. Multicellular tumor spheroids from human gliomas maintained in organ culture. J Neurosurg. 1990;72:463-475.
142 Nygaard SJ, Pedersen PH, Mikkelsen T, et al. Glioma cell invasion visualized by scanning confocal laser microscopy in an in vitro co-culture system. Invasion Metastasis. 1995;15:179-188.
143 Nygaard SJ, Tysnes OB. Quantification of glioma cell invasion by confocal laser scanning microscopy in an in vitro co-culture system. Cancer Lett. 1996;105:45-49.
144 Nygaard SJ, Haugland HK, Laerum OD, et al. Dynamic determination of human glioma invasion in vitro. J Neurosurg. 1998;89:441-447.
145 Jacobs W, Mikkelsen T, Smith R, et al. Inhibitory effects of CAI in glioblastoma growth and invasion. J Neurooncol. 1997;32:93-101.
146 Bernstein JJ, Goldberg WJ, Laws ERJr. A model for central nervous system cancer research. J Neurosci Res. 1989;22:134-143.
147 Ji Y, Powers SK, Brown JT, et al. Characterization of the tumor invasion area in the rat intracerebral glioma. J Neurooncol. 1996;30:189-197.
148 Whittle IR, Macarthur DC, Malcolm GP, et al. Can experimental models of rodent implantation glioma be improved? A study of pure and mixed glioma cell line tumours. J Neurooncol. 1998;36:231-242.
149 Bernstein JJ. Local invasion and intraparenchymal metastasis of astrocytomas. Neuropathol Appl Neurobiol. 1996;22:421-424.
150 Bernstein JJ, Woodard CA. Glioblastoma cells do not intravasate into blood vessels. Neurosurgery. 1995;36:124-132.
151 Laws ERJr, Goldberg WJ, Bernstein JJ. Migration of human malignant astrocytoma cells in the mammalian brain: Scherer revisited. Int J Dev Neurosci. 1993;11:691-697.
152 Goldberg WJ, Laws ERJr, Bernstein JJ. Individual C6 glioma cells migrate in adult rat brain after neural homografting. Int J Dev Neurosci. 1991;9:427-437.
153 DeArmond SJ, Stowring L, Amar A, et al. Development of a non-selecting, non-perturbing method to study human brain tumor cell invasion in murine brain. J Neurooncol. 1994;20:27-34.
154 Kim B, Chenevert TL, Ross BD. Growth kinetics and treatment response of the intracerebral rat 9L brain tumor model: a quantitative in vivo study using magnetic resonance imaging. Clin Cancer Res. 1995;1:643-650.
155 Barker M, Hoshino T, Gurcay O, et al. Development of an animal brain tumor model and its response to therapy with 1,3bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1973;33:976-986.
156 Barker M, Deen DF, Baker DG. BCNU and x-ray therapy of intracerebral 9L rat tumors. Int J Radiat Oncol Biol Phys. 1997;5:1581-1583.
157 Henderson SD, Kimler BJ, Morantz RA. Radiation therapy in 9L rat brain tumors. Int J Radiat Oncol Biol Phys. 1981;7:497-502.
158 Kimler BJ, Liu C, Evans RG, et al. Intracerebral chemotherapy in the 9L rat brain tumor model. J Neurooncol. 1992;14:191-200.
159 Ross BD, Kim B, Davidson BL. Assessment of ganciclovir toxicity to experimental intracranial gliomas following recombinant adenoviral-mediated transfer of the herpes simplex virus thymidine kinase gene by magnetic resonance imaging and proton magnetic resonance spectroscopy. Clin Cancer Res. 1995;1:651-657.
160 Mella O, Bjerkvig R, Schem BC, et al. A cerebral glioma model for experimental therapy and in vivo invasion studies in syngeneic BDIX rats. J Neurooncol. 1990;9:93-104.
161 King WA, Black KL, Ikezaki K, et al. Tumor-associated neurological dysfunction prevented by lazaroids in rats. J Neurosurg. 1991;74:112-115.
162 Benda P, Someda K, Messer J, et al. Morphological immunochemical studies of rat glial tumors and clonal strains propagated in culture. J Neurosurg. 1971;34:310-323.
163 Bissell M. Production of glial fibrillary acidic protein in the absence of gliofibrillogenesis. Brain Res. 1974;82:77.
164 Benda P, Lightbody J, Sato G, et al. Differential rat glial cell strain in tissue culture. Science. 1968;161:370-371.
165 Mineura K, Watanabe K, Izumi I, et al. Modulation of BudR labeling index in rat brain tumors following intracarotid ACNU administration. J Neurooncol. 1992;14:201-205.
166 Bernstein JJ, Anagnostopoulos AV, Hattwick EA, et al. Human-specific c-neu proto-oncogene protein overexpression in human malignant astrocytomas before and after xenografting. J Neurosurg. 1993;78:240-251.
167 Ko L, Koestner A, Wechsler W. Morphological characterization of nitrosourea-induced glioma cell lines and clones. Acta Neuropathol (Berl). 1980;51:23-31.
168 Krajewski S, Kiwit JCW, Wechsler W. RG2 glioma growth in rat cerebellum after subdural implantation. J Neurosurg. 1986;65:222-229.
169 Molnar P, Lapin GD, Groothuis DR. The effects of dexamethasone on experimental brain tumors. I. Transcapillary transport and blood flow in RG-2 rat gliomas. J Neurooncol. 1995;25:19-28.
170 Kruse CA, Molleston MC, Parks EP, et al. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas. J Neurooncol. 1994;22:191-200.
171 Finkelstein SD, Black P, Nowak TP, et al. Histological characteristics and expression of acidic and basic fibroblast growth factor genes in intracerebral xenogeneic transplants of human glioma cells. Neurosurgery. 1994;34:136-143.
172 Koestner A, Swenberg JA, Wechsler W. Transplacental production with ethylnitrosourea of neoplasms of the nervous system in Sprague-Dawley rats. Am J Pathol. 1971;63:37-56.
173 Ding H, Roncari L, Shannon P, et al. Astrocyte-specific expression of activated p21-Ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61:3826-3836.
174 Weiss WA, Burns MJ, Hackett C, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003;63:1589-1595.
175 Uhrbom L, Hesselager G, Nistér M, et al. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275-5279.
176 Dai C, Celestino JC, Okada Y, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913-1925.
177 Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55-57.
178 Macleod KF, Jacks T. Insights into cancer from transgenic mouse models. J Pathol. 1999;187:43-60.
179 Xiao A, Wu H, Pandolfi PP, et al. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157-168.
180 Kamijo T, Bodner S, van de Kamp E, et al. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217-2222.
181 Reilly KM, Loisel DA, Bronson RT, et al. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109-113.
182 Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011-7021.
183 Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
184 Weissman DE, Grossman SA. A model for quantitation of peritumoral brain edema. J Neurosci Methods. 1988;23:207-210.
185 Zagzag D, Goldenberg M, Brem S. Angiogenesis and blood-brain barrier breakdown modulate CT contrast enhancement: an experimental study in a rabbit brain-tumor model. Am. J Radiol. 1989;153:141-146.
186 Bayens-Simmonds J, Boisvert DP, Castro ME, et al. A feline model for experimental studies of peritumor brain edema. J Neurooncol. 1988;6:371-378.
187 Bayens-Simmonds J, Boisvert DP, Baker GB. Regional monoamine and metabolite levels in a feline brain tumor model. Mol Chem Neuropathol. 1989;10:63-75.
188 Wechsler W, Szymas J, Bilzer T, et al. Experimental transplantation gliomas in the adult cat brain. 1. Experimental model and neuropathology. Acta Neurochir (Wien). 1989;98:77-89.
189 Linn F, Seo K, Hossmann KA. Experimental transplantation gliomas in the adult cat brain. 3. Regional biochemistry. Acta Neurochir (Wien). 1989;99:85-93.
190 Hossmann KA, Szymas J, Seo K, et al. Experimental transplantation gliomas in the adult cat brain. 2. Pathophysiology and magnetic resonance imaging. Acta Neurochir (Wien). 1989;98:189-200.
191 Kabuto M, Hayashi M, Nakagawa T, et al. Experimental brain tumor in adult mongrel cat. Brain Nerve. 1990;42:339-343.
192 Krushelnycky BW, Farr-Jones MA, Mielke B, et al. Development of a large-animal human brain tumor xenograft model in immunosuppressed cats. Cancer Res.. 1991;51:2430-2437.
193 Gavin PR, Fike JR, Hoopes PJ. Central nervous system tumors. Semin Vet Med Surg (Small Anim). 1995;10:180-189.
194 Whelan HT, Clanton JA, Wilson RE, et al. Comparison of CT and MRI brain tumor imaging using a canine glioma model. Pediatr Neurol. 1988;4:279-283.
195 Whelan HT, Schmidt MH, Segura AD, et al. The role of photodynamic therapy in posterior fossa brain tumors: a preclinical study in a canine glioma model. J Neurosurg. 1993;79:562-568.
196 Warnke PC, Molnar P, Lapin GD, et al. The effects of dexamethasone on experimental brain tumors. II. Canine brain tumors. J Neurooncol. 1995;25:29-38.
197 Barker M, Hoshino T, Gurcay O, et al. Development of an animal brain tumor model and its response to therapy with 1,3bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1973;33:976-986.
198 Berens ME, Bjotvedt G, Levesque DC, et al. Tumorigenic, invasive, karyotypic, and immunocytochemical characteristics of clonal cell lines derived in vitro. Cell Dev Biol Anim. 1993;29A:310-318.
199 Berens ME, Giese A, Shapiro JR, et al. Allogeneic astrocytoma in immune competent dogs. Neoplasia. 1999;1:107-112.
200 Ausman J, Levin VA, Brown WE, et al. Brain tumor chemotherapy: pharmacological principles derived from a monkey brain tumor model. J Neurosurg. 1977;46:155-164.
201 Lewis JLJr, Brown WEJr, Hertz R, et al. Heterotransplantation of human choriocarcinoma in monkeys. Cancer Res. 1968;28:2032-2038.
202 Yamada K, Bremer AM, West CR. Effects of dexamethasone on tumor-induced brain edema and its distribution in the brain of monkeys. J Neurosurg. 1979;50:361-367.
203 Parsa AT, Chakrabarti I, Hurley PT, et al. Limitations of the C6/Wistar rat intracerebral glioma model: implications for evaluating immunotherapy. Neurosurgery. 2004;47:993-1000.
204 Beutler AS, Banck MS, Wedekind D, et al. Tumor gene therapy made easy: allogeneic major histocompatibility complex in the C6 rat glioma model. Hum Gene Ther. 1999;10:95-101.
205 Vince GH, Bendszus M, Schweitzer T, et al. Spontaneous regression of experimental gliomas—an immunohistochemical and MRI study of the C6 glioma spheroid implantation model. Exp Neurol. 2004;190:478-485.
206 Kohn EC, Liotta LA. Molecular insights into cancer invasion: strategies for prevention and intervention. Cancer Res. 1995;55:1856-1862.
207 Schipper H, Goh CR, Wang TL. Shifting the cancer paradigm: must we kill to cure? J Clin Oncol. 1995;13:801-807.
208 Sidransky D, Mikkelsen T, Schwechheimer K, et al. Clonal expansion of p53 mutant cells is associated with brain tumor progression. Nature. 1992;355:846-847.
209 MacDonald TJ, Taga T, Shimada H, et al. Preferential susceptibility of brain tumours to the antiangiogenic effects of an alpha(v) integrin antagonist. Neurosurgery. 2001;48:151-157.
210 Nabors LB, Mikkelsen T, Rosenfeld SS, et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007;25:1637-1638.
211 D’Amato RJ, Loughnan MS, Flynn E, et al. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082-4085.
212 Groves MD, Puducalli VK, Chang SM, et al. A North American Brain Tumor Consortium (NABTC 99-04) phase II trial of temozolomide plus thalidomide for recurrent glioblastoma multiforme. J Neurooncol. 2007;81:271-277.
213 Tonn JC, Kerkau S, Hanke A, et al. Effect of synthetic matrix-metalloproteinase inhibitors on invasive capacity and proliferation of human malignant gliomas in vitro. Int J Cancer. 1999;80:764-772.
214 Levin VA, Phuphanich S, Yung WK, et al. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol. 2006;78:295-302.
215 Rich JN, Sathornsumetee S, Keir ST, et al. ZD6474, a novel tyrosine kinase inhibitor of vascular endothelial growth factor receptor and epidermal growth factor receptor, inhibits tumor growth of multiple nervous system tumors. Clin Cancer Res. 2005;11:8145-8157.
216 Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
217 Rini BI, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma: a home run or a work in progress? Oncology (Williston Park). 2008;22:388-396.