[level-membership-for-basic-science-category]
CHAPTER 40 Introduction to the Cell Cycle
The cell cycle, is the series of events that leads to the duplication and division of a cell. Research on the molecular events of cell-cycle control revealed that variations of similar mechanisms operate the cell cycles of all eukaryotes from yeasts to humans. Furthermore, the components that regulate cell growth and division also play key roles in the cessation of cell division that accompanies cell differentiation. Control of the cell cycle is of major importance to human health because cancer, which is usually caused by perturbations of cell-cycle regulation, affects 46% of males and 38% of females in the United States.
Although animal cells have a wide variety of specialized cell cycles, the cells in the stratified epithelium that forms skin illustrate the most common lifestyles (Fig. 40-1). The basal layer of the epithelium is composed of stem cells that divide only occasionally. (See Box 41-1.) They can activate the cell cycle on demand and then return to a nondividing state. When specific signals induce stem cells to proliferate, one daughter cell usually remains a stem cell and the other enters a pool of rapidly dividing cells. These dividing cells populate the upper layers of the epithelium, stop dividing, and gradually differentiate into the specialized cells that cover the surface.
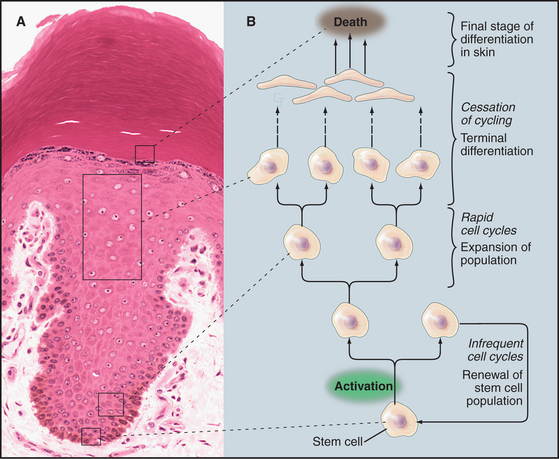
Similarly, the nervous system contains a few stem cells and a few dividing cells, but most neurons, once differentiated, can live for more than 100 years without dividing again. Like stem cells, fibroblasts of the connective tissue (see Fig. 28-2) are typically nondividing, but they can be stimulated to enter the cell cycle following wounding or other stimuli (see Fig. 32-11).
Principles of Cell-Cycle Regulation
The goal of the cell cycle in most cases is to produce two daughter cells that are accurate copies of the parent (Fig. 40-2). The cell cycle integrates a continuous growth cycle (the increase in cell mass) with a discontinuous division or chromosome cycle (the replication and partitioning of the genome into two daughter cells). The chromosome cycle is driven by a sequence of enzymatic cascades that produce a sequence of discrete biochemical “states” of the cytoplasm. Each state arises by destruction or inactivation of key enzymatic activities characteristic of the preceding state and expression or activation of a new cohort of activities. Later sections of this chapter explain these mechanisms.
Phases of the Cell Cycle
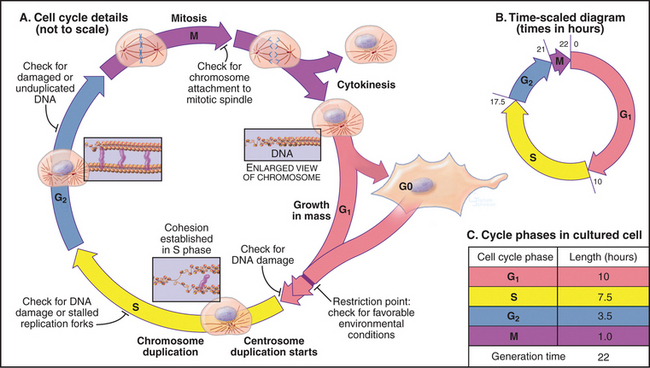
In thinking about the cell cycle, it is convenient to divide the process into a series of phases. Recognition of cell-cycle phases began in 1882, when Flemming named the process of nuclear division mitosis (from the Greek mito, or “thread”) after the appearance of the condensed chromosomes. It initially appeared that cells were active only during mitosis, so the rest of the cell cycle was called interphase (or resting stage) (Box 40-1).
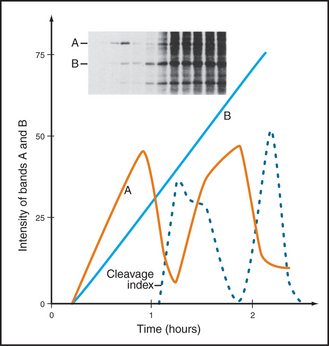
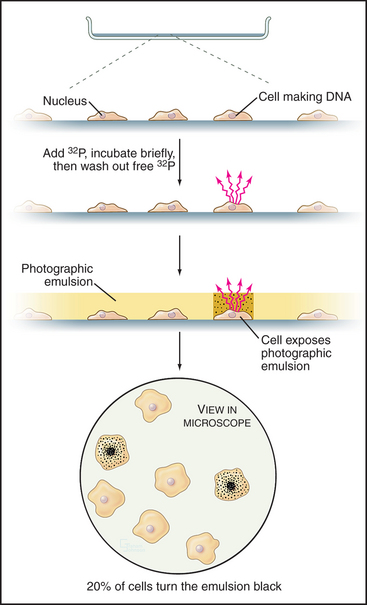
Once DNA was recognized as the agent of heredity in the 1940s, it was deduced that DNA must be duplicated at some time during interphase so that daughter cells can each receive a full complement of genetic material. A key experiment identified the relationship between the timing of DNA synthesis and the mitotic cycle (Fig. 40-3) and defined the four cell-cycle phases as they are known today (Fig. 40-2).
S Phase
Chromosomes of higher eukaryotes are so large that replication of the DNA must be initiated at many different sites, termed origins of replication. In budding yeast, the approximately 400 origins are spaced an average of 30,000 base pairs apart. An average human chromosome contains about 150 ×106 base pairs of DNA, about 10 times the size of the entire budding yeast genome, so many more origins are required. Each region of the chromosome that is replicated from a single origin is referred to as a replicon.
During replication, the duplicated DNA molecules, called sister chromatids, become linked to each other by a protein complex called cohesin (see Fig. 13-19). This pairing of sister chromatids is important for their symmetrical segregation later in mitosis (see Fig. 44-16).
M Phase
Prometaphase begins when the nuclear envelope breaks down (in higher eukaryotes) and chromosomes begin to attach randomly to microtubules emanating from the two poles of the forming mitotic spindle. Chromosomes may also nucleate some spindle microtubules. Once both kinetochores on a pair of sister chromatids are attached to opposite spindle poles, the chromosome slowly moves to a point midway between the poles. When all chromosomes are properly attached, the cell is said to be in metaphase.
Checkpoints
The cell cycle is highly regulated, and checkpoints control transitions between cell-cycle stages. Checkpoints are biochemical circuits that detect external or internal problems and send inhibitory signals to the cell-cycle system. There are four major types of checkpoints. The restriction point in the G1 phase is sensitive to the physiological state of the cell and to its interactions with the surrounding extracellular matrix. Cells that do not receive appropriate growth stimuli from their environment do not progress past this point in the G1 phase and may commit suicide by apoptosis (see Chapter 46).
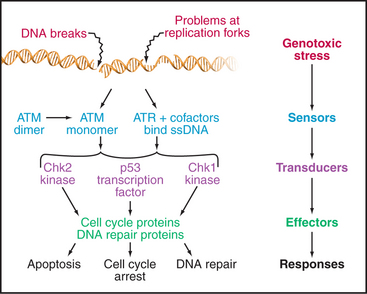
The checkpoints in G1, S, and G2 use common strategies to regulate cell-cycle progression (Fig. 40-4). DNA damage is detected by sensors. These activate transducers, which are often protein kinases but may also be transcriptional activators. The transducers act on effectors, which ultimately block cell-cycle progression and may also fulfill other functions. Two key protein kinases, ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad9 related (ATR), lie at the head of the pathway and may act as sensors of DNA damage. They activate two transducer kinases Chk1 and Chk2 and also stabilize a transcription factor called p53 that induces the expression of a cohort of genes involved in halting cell-cycle progression as well as genes that trigger cell death by apoptosis. Chapters 41 and 43 discuss these proteins in detail. In general, DNA damage checkpoints block cell-cycle progression by inhibiting the cyclin-dependent kinases by a variety of mechanisms.
The Biochemical Basis of Cell-Cycle Transitions
Cyclin-Dependent Kinases
Genetic analysis of the cell cycle in the fission yeast Schizosaccharomyces pombe identified a gene called cell division cycle–2+ (cdc2+) that is essential for cell-cycle progression during both the G1  S and G2
S and G2  M transitions (Box 40-2). The product of this gene, a protein kinase of 34,000D originally called p34cdc2, is the prototype for a family of protein kinases that is crucial for cell-cycle progression in all eukaryotes. This mechanism of cell-cycle control is so well conserved that a human homolog of p34cdc2 can replace the yeast protein, restoring a normal cell cycle to a cdc2 mutant yeast. Boxes 40-3 and 40-4 present a number of the key experiments and experimental systems that led to the identification of the molecules that drive the cell cycle.
M transitions (Box 40-2). The product of this gene, a protein kinase of 34,000D originally called p34cdc2, is the prototype for a family of protein kinases that is crucial for cell-cycle progression in all eukaryotes. This mechanism of cell-cycle control is so well conserved that a human homolog of p34cdc2 can replace the yeast protein, restoring a normal cell cycle to a cdc2 mutant yeast. Boxes 40-3 and 40-4 present a number of the key experiments and experimental systems that led to the identification of the molecules that drive the cell cycle.
BOX 40-2 Use of Genetics to Study the Cell Cycle
Studies of the distantly related budding and fission yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (see Fig. 2-9) have been extremely important for understanding the cell cycle for several reasons. First, the proteins that control the cell cycle are remarkably conserved between yeasts and mammals. Second, both yeast genomes are sequenced and annotated, simplifying characterization of novel gene products. Third, genetic analysis is facilitated, as both yeasts grow as haploids, and both efficiently incorporate cloned DNA into their chromosomes by homologous recombination.
 S transition is particularly amenable to study. In contrast, a fission yeast spends most of its two-hour cell cycle in G2. S phase follows separation of sister chromatids and occurs prior to cytokinesis. Thus, the control of the G2
S transition is particularly amenable to study. In contrast, a fission yeast spends most of its two-hour cell cycle in G2. S phase follows separation of sister chromatids and occurs prior to cytokinesis. Thus, the control of the G2 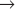 M transition is readily studied in fission yeast. During mitosis, the nuclear envelopes of both yeasts remains intact, so chromosomes segregate on a spindle inside the nucleus.
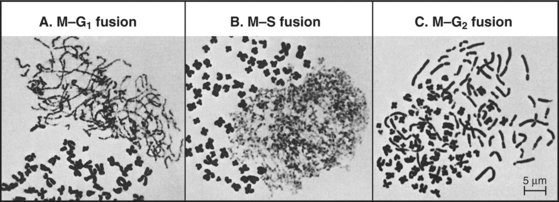
M transition is readily studied in fission yeast. During mitosis, the nuclear envelopes of both yeasts remains intact, so chromosomes segregate on a spindle inside the nucleus.Genetic studies revealed that the yeast cell cycle is a dependent pathway whereby events in the cycle occur normally only after earlier processes are completed. The cell cycle can be modeled as a line of dominoes, each domino corresponding to the action of a gene product that is essential for cell-cycle progression (Fig. 40-5) and the nth domino falling only when knocked down by the (n – 1)th domino. According to the model, mutations in genes that are essential for cell-cycle progression cause an entire culture of yeast to accumulate at a single point in the cell cycle (the point at which the defective gene product first becomes essential). This is referred to as the arrest point. Figure 40-5 shows this by including a “mutant” domino that does not fall over when struck by the upstream domino. Mutants that meet this criterion are called cell division cycle mutants or CDC mutants. (Cdc is used in fission yeast.) Genetic screens for CDC mutants have identified many important genes involved in cell-cycle control.
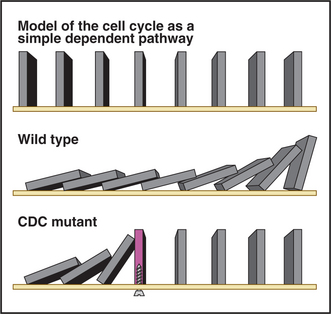
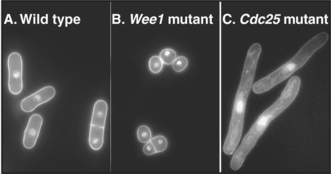
Fission yeasts with CDC mutants affecting the entry into mitosis have distinctive morphologies. Cells that are mutant in Wee1 (a kinase that keeps Cdk1 inactive prior to mitosis) enter mitosis prematurely and are shorter than normal (Fig. 40-6B). In contrast, cells lacking Cdc25 (a phosphatase that counteracts Wee1 and activates Cdk1) are unable to undergo mitosis but continue their growth cycle, therefore becoming greatly elongated (Fig. 40-6C). This simple morphologic assay allowed straightforward classification of yeast CDC genes into those that stimulate progression through mitosis and those that retard entry into mitosis.
BOX 40-3 Studies of the Cell Cycle in Vitro
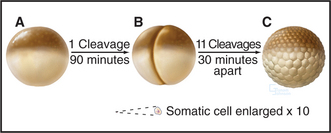
Amphibian oocytes and eggs are storehouses of most components needed for cell-cycle progression. Oocytes are arrested in G2 until a surge of the hormone progesterone causes them to “mature” into eggs, which are then naturally arrested in metaphase of the second meiotic division (see Chapter 45). After fertilization, the embryo of the South African clawed frog (Xenopus laevis) undergoes a rapid burst of cell divisions. An initial cell cycle that is 90 minutes long is followed by a rapid succession of 11 cleavages spaced only 30 minutes apart to produce an embryo of 4096 cells (Fig. 40-7).
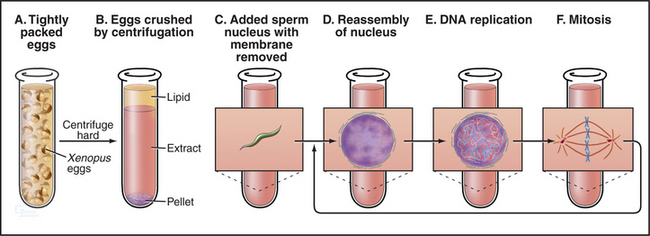
Remarkably, it is possible to make cell-free extracts from Xenopus eggs that progress through the cell cycle in vitro (Fig. 40-8). Nuclei from G1 cells, when added to these extracts, efficiently replicate their DNA and proceed through the cell cycle into mitosis, complete with chromosome condensation, nuclear envelope breakdown, chromosome alignment on a spindle, and anaphase segregation of sister chromatids without any additions to the tube. Because these events occur in a cell-free milieu, they are readily accessible to biochemical manipulation. For example, antibodies and other proteins can be added to the extracts, and their effect on the cell cycle can readily be determined. Thus, the Xenopus extract system offers one of the best tools for testing the role of various proteins in the cell cycle in higher eukaryotes.
BOX 40-4 Discovery of Factors Essential for Cell-Cycle Progression
The best early evidence for the existence of positive inducers of cell-cycle transitions in mammals was obtained in cell fusion experiments. When cultured cells in S phase were fused with cells in G1, the G1 nuclei initiated DNA replication shortly thereafter. In contrast, if S phase cells were fused with G2 cells, the G2 nuclei did not rereplicate their DNA until after passing through mitosis. The most dramatic results were obtained when mitotic cells were fused with interphase cells. This caused the interphase cells to enter into mitosis abruptly (as judged by nuclear envelope breakdown and chromosome condensation). The phenomenon was termed premature chromosome condensation (PCC). The mitotic inducer could work in any cell-cycle phase (Fig. 40-9). If mitotic cells were fused with cells in G1 phase, interphase chromosomes condensed into long, single filaments. If the interphase cell was in G2 phase, the duplicated chromosomes appeared as double filaments. If the interphase cell was in the S phase, the partially replicated chromosomes condensed into a complex pattern of single and double condensed regions separated by regions of decondensed chromatin corresponding to sites where DNA was actively replicating at the time of fusion.
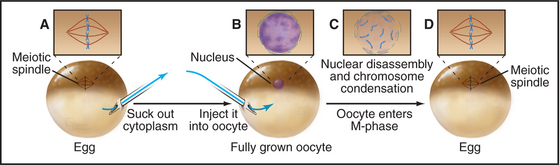
Working independently, developmental biologists who were interested in the control of cell division during early development in frogs also discovered an activity that could cause interphase cells to enter the M phase. They used a micropipette to extract a tiny bit of cytoplasm from a mature egg that was arrested in metaphase of meiosis II and inject it into oocytes (which are in G2 phase). The oocytes rapidly entered M phase, with concomitant chromosome condensation and nuclear envelope disassembly (Fig. 40-10). This stimulation to enter M phase is called maturation, and the unknown factor present in the egg cytoplasm that induced oocyte maturation was termed MPF, or maturation-promoting factor (now often referred to as M phase–promoting factor). It was realized early on that MPF might be related to the inducer of mitosis detected in the PCC experiments. In fact, extracts from mitotic tissue culture cells could induce meiotic maturation when injected into oocytes. Similar extracts from cells in other phases of the cell cycle did not cause the G2/M phase transition in oocytes.
Other cell biologists studying protein synthesis in starfish and sea urchin embryos noticed a curious protein that seemed to accumulate across the cell cycle but was then destroyed during mitosis. They were well aware of the work on MPF, and immediately suspected that their protein, which they called cyclin, might be somehow involved in MPF activity (Fig. 40-11).
In a third line of investigation, geneticists working on yeasts realized that the cell cycle could be dissected through the isolation of cell division cycle (CDC) mutants (Box 40-2). The analysis of the cell cycle with these mutants dominated cell-cycle research to such an extent that many human genes that are important in cell-cycle control bear the CDC name if they are related to well-characterized yeast genes. The best-known genes to emerge from this analysis were Cdc2 (Cdk1) and Cdc25, both of which were determined genetically to encode proteins that actively promote the G2/M transition. Other genes, such as Wee1, were found to encode activities that act as antagonists that inhibit the G2/M transition.
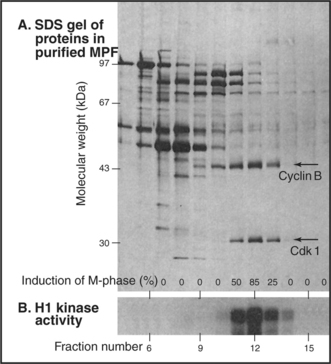
When active MPF was eventually purified from Xenopus eggs (Fig. 40-12), the purified fractions turned out to consist primarily of two polypeptide chains of 45,000 D and 32,000D. The 32,000-D component of MPF is the Xenopus equivalent of the fission yeast Cdc2 (now known as Cdk1) gene product. The 45,000-D component of MPF is a Xenopus B-type cyclin.
Humans have more than 10 distinct protein kinases related to p34cdc2, although only a few are involved in cell-cycle control. To be active, these enzymes must each associate with a regulatory subunit called a cyclin. Thus, they have been termed cyclin-dependent kinases (Cdks). p34cdc2, now termed Cdk1, seems to function primarily in the regulation of the G2  M transition in animal cells. A second family member, Cdk2, is involved in regulation of the G1
M transition in animal cells. A second family member, Cdk2, is involved in regulation of the G1 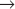 S and G2
S and G2  M transitions, whereas two other family members—Cdk4 and Cdk6—are involved in passage of the restriction point (Appendix 40-1). Cdk7 is important for activation of other Cdks, and also appears to participate in RNA transcription and repair of damaged DNA. Other Cdks participate in diverse processes ranging from transcriptional regulation to neuronal differentiation and may play as-yet-undiscovered roles in cell-cycle regulation. Surprisingly, fibroblasts from mice that lack Cdk2, Cdk4, or Cdk6 are viable; other Cdks must be able to drive the cell cycle if necessary. The mice themselves suffer difficulties because the genes are needed for the differentiation of particular cell types.
M transitions, whereas two other family members—Cdk4 and Cdk6—are involved in passage of the restriction point (Appendix 40-1). Cdk7 is important for activation of other Cdks, and also appears to participate in RNA transcription and repair of damaged DNA. Other Cdks participate in diverse processes ranging from transcriptional regulation to neuronal differentiation and may play as-yet-undiscovered roles in cell-cycle regulation. Surprisingly, fibroblasts from mice that lack Cdk2, Cdk4, or Cdk6 are viable; other Cdks must be able to drive the cell cycle if necessary. The mice themselves suffer difficulties because the genes are needed for the differentiation of particular cell types.
Cyclins
The defining feature of Cdks is that they require binding of cyclins for catalytic activity. Cyclins are a diverse group of proteins ranging in size between 35 kD and 130 kD, all with a similar core structure based on two symmetrical domains of five a-helices (Fig. 40-13). One of these domains, the cyclin box, is highly conserved and is the defining structural feature of these proteins. Cyclins were discovered in rapidly dividing invertebrate embryos as proteins that accumulate gradually during interphase and are abruptly destroyed during mitosis (Fig. 40-11). This process of cyclic accumulation and destruction is the derivation of their name. Subsequently, at least 16 different cyclins have been identified in humans, although only a handful are involved in cell-cycle control. Of those that are, some function during G1 phase, others during G2 phase, and still others during M phase.
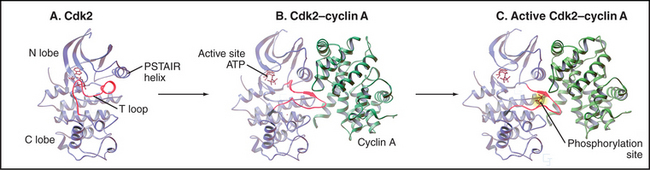
Positive Regulation of Cyclin-Dependent Kinase Structure and Function
The activity of Cdks is regulated with extraordinary care. Like other eukaryotic protein kinases (see Fig. 25-3), Cdks have a bilobed structure with the active site in a deep cleft between a small N-terminal and larger C-terminal domain. However, newly synthesized monomeric Cdks differ from other kinases in that they appear to be incompletely folded: a flexible loop (T loop) blocks the mouth of the catalytic pocket. In addition, misorientation of a short a-helix causes a glutamic acid required for adenosine triphosphate (ATP) hydrolysis to point away from the catalytic cleft. As a result, ATP bound by the monomeric kinase is distorted and cannot transfer its a-phosphate to protein substrates (Fig. 40-13A).
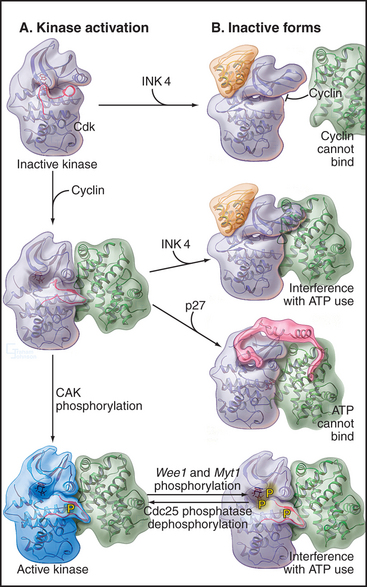
At least four different mechanisms regulate Cdk activity (Fig. 40-14). On one hand, cyclin binding and phosphorylation of the T loop stimulate enzyme activity. On the other, phosphorylation of residues adjacent to the ATP-binding site and binding of inhibitory proteins inhibit Cdks.
Cyclin binding profoundly changes Cdk structure, causing the retraction of the T loop back from the mouth of the catalytic pocket (Fig. 40-13B). In addition, the secondary structure of the N-terminal domain is altered, reorienting the short helix by 90 degrees so that the critical glutamate can interact with the ATP phosphates. This causes the bound ATP to assume a conformation suitable for reaction with substrates. It has been suggested that cyclin binding also causes two residues—threonine14 and tyrosine15 in the roof of the ATP-binding pocket—to reorient so that they become accessible to protein kinases that regulate Cdk1 activity (see later section).
Despite these changes, the Cdk-cyclin complex has only partial catalytic activity. Complete activation of most Cdks requires the action of a kinase called Cdk-activating kinase (CAK), which phosphorylates threonine160 in the T loop of Cdk2-cyclin A (this threonine gives the loop its name). In vertebrates, CAK is composed of Cdk7-cyclin H. Phosphorylated threonine160 fits into a charged pocket on the surface of the enzyme, flattening the T loop back even farther from the mouth of the catalytic pocket (Figs. 40-13C and 40-14A). This stimulates the catalytic activity up to 300-fold, in part because the flattened T loop forms part of the substrate-binding surface. In addition, threonine160 phosphorylation stabilizes the association of Cdk2 with cyclin A.
Negative Regulation of Cyclin-Dependent Kinase Structure and Function
At least two mechanisms slow or stop the cell cycle by inactivating Cdks (Fig. 40-14). During G2 phase, the protein kinases Myt1 and Wee1 hold Cdk1 in check by phosphorylating threonine14 and tyrosine15 in the roof of the ATP-binding site. These phosphates interfere with ATP binding and hydrolysis. Because threonine14 and tyrosine15 are accessible to the regulatory kinases only following cyclin binding, this phosphorylation of Cdks depends, at least in part, on the availability of cyclins.
Three Cdc25 phosphatases (see Fig. 25-5) reverse these inhibitory phosphorylations. Cdc25A is involved in the regulation of both the G1 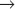 S and G2
S and G2 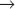 M transitions and is essential for life of the cell. Ccd25B is dispensable for mitosis, but it is essential for the production of gametes in meiosis. Cdc25C is a target of the G2 DNA damage checkpoint that prevents cells from undergoing mitosis with damaged DNA (see Fig. 43-12), but cells can survive without it.
M transitions and is essential for life of the cell. Ccd25B is dispensable for mitosis, but it is essential for the production of gametes in meiosis. Cdc25C is a target of the G2 DNA damage checkpoint that prevents cells from undergoing mitosis with damaged DNA (see Fig. 43-12), but cells can survive without it.
A second strategy for inactivating Cdks involves the binding of small inhibitory subunits of the cyclin-dependent kinase inhibitor (CKI) and inhibitor of Cdk4 (INK4) families (for their names, see Appendix 40-1). CKI molecules inactivate Cdk-cyclin A complexes most efficiently. The CKI p27Kip1 inactivates Cdk2–cyclin A complexes in two ways (Fig. 40-15A). One part of p27Kip1 associates with the cyclin subunit, while another invades the N-terminal domain of the Cdk, profoundly disrupting its structure and competing with ATP for binding to the active site.
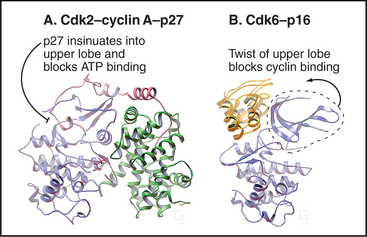
Members of the INK4 family preferentially inactivate Cdk4 and Cdk6. They do this in two ways (Fig. 40-15B). First, interaction with monomeric Cdk opposite the catalytic cleft distorts the orientation of the N- and C-terminal lobes so that cyclin D does not bind. INK4 family inhibitors also inhibit preformed Cdk4/6–cyclin D complexes by binding the Cdk and distorting the ATP-binding site so that the kinase uses ATP much less efficiently.
Cdk inhibitors are important for growth regulation during the G1 and G0 phases of the cell cycle (see Chapter 41). They also play a critical role in the cell-cycle arrest that occurs in response to DNA damage and to antiproliferative signals.
Role of Protein Destruction in Cell-Cycle Control
Ubiquitin-mediated destruction of cyclins involves the action of a series of enzymes (see Fig. 23-8). First, an E1 enzyme (ubiquitin-activating enzyme) activates the small protein ubiquitin by forming a thioester bond between the C-terminus of ubiquitin and a cysteine on the enzyme. Activated ubiquitin is then transferred to another thioester bond on an E2 enzyme (ubiquitin-conjugating enzyme). E2 may either transfer ubiquitin directly to the e-amino group of a lysine of a target protein or combine with a third component (an E3 or ubiquitin-protein ligase) to do so. E3s are particularly important for imparting substrate specificity. Finally, the same enzymes build a chain of ubiquitins by successive conjugations of the C-terminus of a new ubiquitin to a lysine side chain of the previous ubiquitin.
The resulting polyubiquitinated proteins are usually targets for destruction by the cylindrical 26S proteasome (see Fig. 23-5), although depending on the way the ubiquitins are linked together, they may alternatively serve as signaling intermediates. The proteasome is a large multienzyme complex that functions like a cytoplasmic garbage disposal, grinding target proteins down to short peptides and spitting out intact ubiquitin monomers for reuse in further rounds of protein degradation. Its role was originally thought to be the removal of damaged proteins from the cytoplasm; however, it is now recognized as a central factor in cell-cycle control.
The key factor regulating proteolysis of cyclins is a large (20S) complex with E3 activity consisting of 12 to 13 subunits called the anaphase-promoting complex/cyclosome (APC/C) (Fig. 40-16). The APC/C is inactive during the S and G2 phases of the cell cycle. Binding of protein “specificity factors” such as Cdc20 and phosphorylation by Cdk1-cyclin B-Cks1 activate the APC/C in early mitosis. APC/CCdc20 is responsible for triggering the metaphase-anaphase transition.
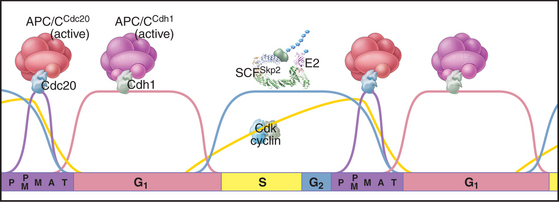
Later in mitosis, the APC/C binds a second specificity factor, Cdh1. Cdk phosphorylation blocks Cdh1 binding to APC/C, so APC/CCdh1 forms only after cyclin levels (and therefore Cdk activity) start to fall late in mitosis. Further repression of Cdk activity and destruction of Cdc20 by APC/CCdh1 are critical both for mitotic exit and during G1 phase in preparing chromatin for the initiation of DNA replication (see Chapter 42). As cells pass from G1 into S phase, a newly synthesized protein, Emi1, binds to APC/CCdh1 and inactivates it. This allows the accumulation of cyclins during S and G2. Remarkably, APC/CCdh1 also has a role in nondividing neurons, where it is involved with regulating the activity of synapses.
A different E3 activity functions after the G1  S transition and throughout the remainder of interphase to regulate the levels of Cdk activity that ultimately results in mitotic entry. This activity is called SCF after three of its four key subunits: Skp1, Cullin, and F-box (Fig. 40-17). SCF is a molecular toolbox built on a bow-shaped scaffold formed by the cullin subunit. The fourth subunit, Rbx1, binds near the C-terminus of cullin and uses a protein motif called a RING finger to dock to an E2 enzyme. Skp1 binds to the other end of cullin, where it provides a docking site for the F-box protein that actually recognizes and binds the substrate. (The F-box got its name because it was first discovered in cyclin F.) Humans have 78 F-box proteins (Caenorhabditis elegans has 350), and this gives SCF an enormous versatility. We discuss two of these: Skp2, which targets the Cdk inhibitor p27, and b-Trcp, which targets Cdc25A, Wee1, and Emi1. SCF is fundamentally different from the APC/C because many F-box proteins bind sub-strates only after they have been phosphorylated, often by Cdks.
S transition and throughout the remainder of interphase to regulate the levels of Cdk activity that ultimately results in mitotic entry. This activity is called SCF after three of its four key subunits: Skp1, Cullin, and F-box (Fig. 40-17). SCF is a molecular toolbox built on a bow-shaped scaffold formed by the cullin subunit. The fourth subunit, Rbx1, binds near the C-terminus of cullin and uses a protein motif called a RING finger to dock to an E2 enzyme. Skp1 binds to the other end of cullin, where it provides a docking site for the F-box protein that actually recognizes and binds the substrate. (The F-box got its name because it was first discovered in cyclin F.) Humans have 78 F-box proteins (Caenorhabditis elegans has 350), and this gives SCF an enormous versatility. We discuss two of these: Skp2, which targets the Cdk inhibitor p27, and b-Trcp, which targets Cdc25A, Wee1, and Emi1. SCF is fundamentally different from the APC/C because many F-box proteins bind sub-strates only after they have been phosphorylated, often by Cdks.
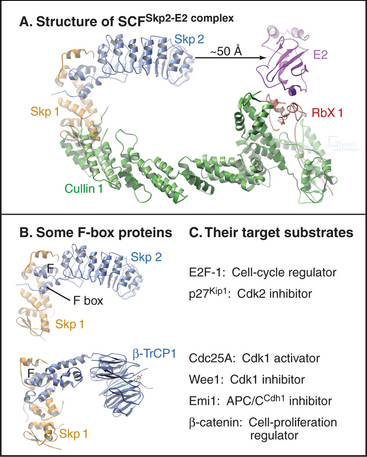
Figure 40-18 shows the activities of two of the major SCF complexes: SCFSkp2, which helps to drive the G1  S transition, and SCFb-Trcp, which functions throughout the cycle, sometimes as an inhibitor and sometimes as a stimulator of cell-cycle progression.
S transition, and SCFb-Trcp, which functions throughout the cycle, sometimes as an inhibitor and sometimes as a stimulator of cell-cycle progression.
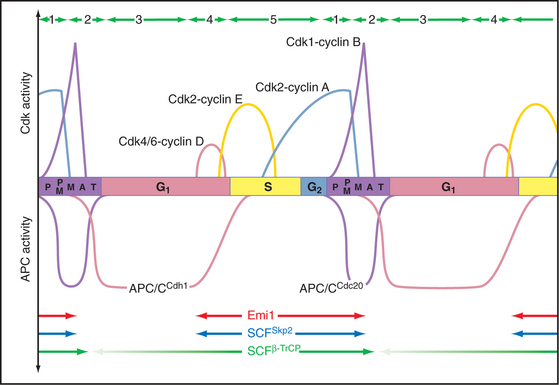
Changing States of the Cytoplasm during the Cell Cycle
The cell cycle is characterized by five discrete physiological states of the cytoplasm, including two phases of mitosis (Fig. 40-18). Transitions between these states are driven by changing levels of Cdk activity, which are sometimes counteracted and sometimes reinforced by targeted proteolysis.
Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science. 1989;246:629-634.
Kastan M, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316-323.
Morgan DO. Cyclin-dependent kinases: Engines, clocks and microprocessors. Annu Rev Cell Biol. 1997;13:261-291.
Murray AW. Recycling the cell cycle: Cyclins revisited. Cell. 2004;116:221-234.
Nasmyth K. A prize for proliferation. Cell. 2001;107:689-701.
Nigg EA. Cell division: Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21-32.
Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000;100:71-78.
Peters JM. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol Cell. 2002;9:931-943.
Pines J. Four-dimensional control of the cell cycle. Nat Cell Biol. 1999;1:E73-E79.
Russell P. Checkpoints on the road to mitosis. Trends Biochem Sci. 1998;23:399-402.
APPENDIX 40-1 Inventory of the Enzymes of the Cell Cycle Engine
| Cyclin-Dependent Kinases and Their Cyclin Partners | ||
|---|---|---|
| Kinase | Cyclin (+ Other) Partner | Function |
| Cdk1 (p34cdc2) | A | Mammals: triggers G2 → M transition. Yeasts: triggers G1 → S and G2 → M transitions. Cyclin A is synthesized in S and destroyed starting at prometaphase. Cyclins B are synthesized in S/G2 and destroyed following the completion of chromosome attachment to the spindle. |
| B1, B2 (Xenopus has 5 B-type cyclins) Cdk1–cyclin B binds Cks1. | ||
| Cdk2 | A, E | Triggers G1 → S transition. Can be replaced by other Cdks in mouse. |
| Cdk3 | ? | Poorly understood. May trigger G1 → S transition. |
| Cdk4, Cdk6 | D1–D3 | Phosphorylation of the retinoblastoma susceptibility protein (pRb) in G1. Triggers passage of the restriction point and cyclin E synthesis. Extracellular growth factors control synthesis of D cyclins. Can be replaced by other Cdks in mouse. |
| Cdk5 | p35 (G) | Neuronal differentiation |
| Cdk7 (CAK) | H; also binds assembly factor MAT1. | Cdk activation by phosphorylation of the T loop. Also in TFIIH, important for regulation of RNA polymerase II transcription and DNA repair. |
| Cdk8 | C | Regulation of RNA polymerase II transcription. |
| Cdk9 | T | Regulation of RNA polymerase II transcription. |
| Cyclin Inhibitors | ||
| Inhibitor | Cdk Substrates | Function |
| CKI: p21Cip1/Waf1 | most Cdk-cyclin complexes | Induced by p53 tumor suppresser. Cell cycle arrest after DNA damage. Binds PCNA (see Chapter 42) and inhibits DNA synthesis. Promotes cell cycle arrest in senescence and terminal differentiation. At low levels, may help to assemble active Cdk-cyclin complexes. |
| CKI: p27Kip1 | most Cdk-cyclin complexes | Cell cycle arrest in response to growth suppressers like TGF-β and in contact inhibition and differentiation. |
| CKI: p57Kip2 | most Cdk-cyclin complexes | Important in development of the palate. |
| INK4: p15Ink4b | Cdk4, Cdk6 | Cell cycle arrest in response to TGF-β. Also altered in many cancers. |
| INK4: p16Ink4a | Cdk4, Cdk6 | Cooperates with the retinoblastoma susceptibility protein (pRb) in growth regulation. Cell cycle arrest in senescence. Altered in a high percentage of human cancers. This gene overlaps the gene for p19ARF, an important regulator of the p53 tumor suppresser protein. |
| INK4: p18Ink4c | Cdk4, Cdk6 | Cell cycle arrest in response to growth suppressers. |
| INK4: p19Ink4d | Cdk4, Cdk6 | Cell cycle arrest in response to growth suppressers. |
| Other Components | ||
| Enzyme | Substrates | Functions |
| Wee1 kinase | Cdk1 Y15 | Nuclear kinase. Inhibits Cdk1-cyclinB in G2. |
| Myt1 kinase | Cdk1 T14 + Y15 | Cytoplasmic kinase. Inhibits Cdk1-cyclin B in G2. |
| Cdc25A phosphatase | Cdk1 T14, Y15 | Promotes G1 → S transition and G2 → M transition. Essential for life of the cell. |
| Cdc25B phosphatase | Cdk1 T14, Y15 | Promotes G2 → M transition. Essential in meiosis. |
| Cdc25C phosphatase | Cdk1 T14, Y15 | Promotes G2 → M transition. Dephosphorylates Cdk1 complexed to cyclins A, B at T14 and Y15. Not essential for life. |
| APC/CCDC20 | Cyclin B, many others | E3 ubiquitin ligase active during M. Requires high Cdk activity to function. |
| Destruction of cyclins and other substrates essential for exit from mitosis. | ||
| Contains 12–13 subunits + the CDC20 activator/specificity factor. | ||
| APC/CCdh1 | Cyclins A, B, many others | E3 ubiquitin ligase active during G1. Requires low Cdk activity to function. Keeps Cdk activity low in G1 through cyclin proteolysis. Contains 12–13 subunits + the Cdh1 activator/specificity factor. |
| SCF | Cyclin E, many others | Class of E3 ubiquitin ligases containing Skp1 + cullin + Rbx1 + an F-box protein. C. elegans has over 60 F-box proteins, acting as specificity factors for substrates phosphorylated at specific sites, including cyclin E and Cdk inhibitors. |
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 40 Introduction to the Cell Cycle
The cell cycle, is the series of events that leads to the duplication and division of a cell. Research on the molecular events of cell-cycle control revealed that variations of similar mechanisms operate the cell cycles of all eukaryotes from yeasts to humans. Furthermore, the components that regulate cell growth and division also play key roles in the cessation of cell division that accompanies cell differentiation. Control of the cell cycle is of major importance to human health because cancer, which is usually caused by perturbations of cell-cycle regulation, affects 46% of males and 38% of females in the United States.
Although animal cells have a wide variety of specialized cell cycles, the cells in the stratified epithelium that forms skin illustrate the most common lifestyles (Fig. 40-1). The basal layer of the epithelium is composed of stem cells that divide only occasionally. (See Box 41-1.) They can activate the cell cycle on demand and then return to a nondividing state. When specific signals induce stem cells to proliferate, one daughter cell usually remains a stem cell and the other enters a pool of rapidly dividing cells. These dividing cells populate the upper layers of the epithelium, stop dividing, and gradually differentiate into the specialized cells that cover the surface.
Similarly, the nervous system contains a few stem cells and a few dividing cells, but most neurons, once differentiated, can live for more than 100 years without dividing again. Like stem cells, fibroblasts of the connective tissue (see Fig. 28-2) are typically nondividing, but they can be stimulated to enter the cell cycle following wounding or other stimuli (see Fig. 32-11).
Principles of Cell-Cycle Regulation
The goal of the cell cycle in most cases is to produce two daughter cells that are accurate copies of the parent (Fig. 40-2). The cell cycle integrates a continuous growth cycle (the increase in cell mass) with a discontinuous division or chromosome cycle (the replication and partitioning of the genome into two daughter cells). The chromosome cycle is driven by a sequence of enzymatic cascades that produce a sequence of discrete biochemical “states” of the cytoplasm. Each state arises by destruction or inactivation of key enzymatic activities characteristic of the preceding state and expression or activation of a new cohort of activities. Later sections of this chapter explain these mechanisms.
Phases of the Cell Cycle
In thinking about the cell cycle, it is convenient to divide the process into a series of phases. Recognition of cell-cycle phases began in 1882, when Flemming named the process of nuclear division mitosis (from the Greek mito, or “thread”) after the appearance of the condensed chromosomes. It initially appeared that cells were active only during mitosis, so the rest of the cell cycle was called interphase (or resting stage) (Box 40-1).
Once DNA was recognized as the agent of heredity in the 1940s, it was deduced that DNA must be duplicated at some time during interphase so that daughter cells can each receive a full complement of genetic material. A key experiment identified the relationship between the timing of DNA synthesis and the mitotic cycle (Fig. 40-3) and defined the four cell-cycle phases as they are known today (Fig. 40-2).
S Phase
Chromosomes of higher eukaryotes are so large that replication of the DNA must be initiated at many different sites, termed origins of replication. In budding yeast, the approximately 400 origins are spaced an average of 30,000 base pairs apart. An average human chromosome contains about 150 ×106 base pairs of DNA, about 10 times the size of the entire budding yeast genome, so many more origins are required. Each region of the chromosome that is replicated from a single origin is referred to as a replicon.
During replication, the duplicated DNA molecules, called sister chromatids, become linked to each other by a protein complex called cohesin (see Fig. 13-19). This pairing of sister chromatids is important for their symmetrical segregation later in mitosis (see Fig. 44-16).
M Phase
Prometaphase begins when the nuclear envelope breaks down (in higher eukaryotes) and chromosomes begin to attach randomly to microtubules emanating from the two poles of the forming mitotic spindle. Chromosomes may also nucleate some spindle microtubules. Once both kinetochores on a pair of sister chromatids are attached to opposite spindle poles, the chromosome slowly moves to a point midway between the poles. When all chromosomes are properly attached, the cell is said to be in metaphase.
Checkpoints
The cell cycle is highly regulated, and checkpoints control transitions between cell-cycle stages. Checkpoints are biochemical circuits that detect external or internal problems and send inhibitory signals to the cell-cycle system. There are four major types of checkpoints. The restriction point in the G1 phase is sensitive to the physiological state of the cell and to its interactions with the surrounding extracellular matrix. Cells that do not receive appropriate growth stimuli from their environment do not progress past this point in the G1 phase and may commit suicide by apoptosis (see Chapter 46).
The checkpoints in G1, S, and G2 use common strategies to regulate cell-cycle progression (Fig. 40-4). DNA damage is detected by sensors. These activate transducers, which are often protein kinases but may also be transcriptional activators. The transducers act on effectors, which ultimately block cell-cycle progression and may also fulfill other functions. Two key protein kinases, ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad9 related (ATR), lie at the head of the pathway and may act as sensors of DNA damage. They activate two transducer kinases Chk1 and Chk2 and also stabilize a transcription factor called p53 that induces the expression of a cohort of genes involved in halting cell-cycle progression as well as genes that trigger cell death by apoptosis. Chapters 41 and 43 discuss these proteins in detail. In general, DNA damage checkpoints block cell-cycle progression by inhibiting the cyclin-dependent kinases by a variety of mechanisms.
The Biochemical Basis of Cell-Cycle Transitions
Cyclin-Dependent Kinases
Genetic analysis of the cell cycle in the fission yeast Schizosaccharomyces pombe identified a gene called cell division cycle–2+ (cdc2+) that is essential for cell-cycle progression during both the G1 S and G2 M transitions (Box 40-2). The product of this gene, a protein kinase of 34,000D originally called p34cdc2, is the prototype for a family of protein kinases that is crucial for cell-cycle progression in all eukaryotes. This mechanism of cell-cycle control is so well conserved that a human homolog of p34cdc2 can replace the yeast protein, restoring a normal cell cycle to a cdc2 mutant yeast. Boxes 40-3 and 40-4 present a number of the key experiments and experimental systems that led to the identification of the molecules that drive the cell cycle.
BOX 40-2 Use of Genetics to Study the Cell Cycle
Studies of the distantly related budding and fission yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (see Fig. 2-9) have been extremely important for understanding the cell cycle for several reasons. First, the proteins that control the cell cycle are remarkably conserved between yeasts and mammals. Second, both yeast genomes are sequenced and annotated, simplifying characterization of novel gene products. Third, genetic analysis is facilitated, as both yeasts grow as haploids, and both efficiently incorporate cloned DNA into their chromosomes by homologous recombination.
[/not-level-membership-for-basic-science-category]
