[level-membership-for-basic-science-category]
Introduction to biopharmaceutics
Marianne Ashford
Chapter contents
Key points
• Pharmacodynamics is the study of the biochemical and physiological effects of the drug on the body.
What is biopharmaceutics?
Biopharmaceutics can be defined as the study of how the physicochemical properties of drugs, dosage forms and routes of administration affect the rate and extent of drug absorption.
The relationship between the drug, its dosage form and the route by which it is administered governs how much of the drug enters the systemic circulation and at what rate. For a drug to be effective, enough of it needs to reach its site(s) of action and stay there long enough to be able to exert its pharmacological effect. This is determined by the route of administration, the form in which the drug is administered and the rate at which it is delivered.
Background
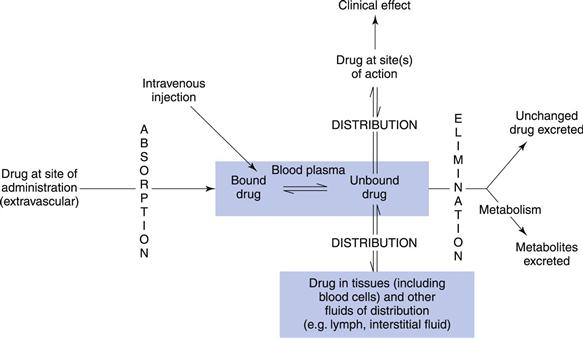
Apart from the intravenous route, where a drug is introduced directly into the blood stream, all other routes of administration, where the site of action is remote from the site of administration, involve the absorption of the drug into the blood. Once the drug reaches the blood it partitions between the plasma and the red blood cells, the erythrocytes. Drug in plasma partitions between the plasma proteins (mainly albumin) and the plasma water. It is the free or unbound drug in plasma water, and not the drug bound to the proteins, that passes out of the plasma through the capillary endothelium and to tissues and hence the site(s) of action.
A dynamic equilibrium exists between the concentration of the drug in the blood plasma and the drug at its site(s) of action. This is what is termed distribution, the degree of which will depend largely on the physicochemical properties of the drug, in particular its lipophilicity. As it is frequently difficult to access the drug at its site(s) of action, its concentration in the plasma is often taken as a surrogate for the concentration at its site(s) of action. Even though the unbound drug in the plasma would give a better estimate of the concentration of the drug at its site(s) of action, this requires much more complex and sensitive assays than a measurement of the total concentration of the drug (i.e. the sum of the bound and unbound drug) within the blood plasma. Thus it is this total drug concentration within the plasma that is usually measured for clinical purposes. Therefore, plasma protein binding is a critical parameter to consider when investigating the therapeutic effect of a drug molecule.
The concentration of the drug in blood plasma depends on numerous factors. These include the amount of an administered dose that is absorbed and reaches the systemic circulation; the extent of distribution of the drug between the systemic circulation and other tissues and fluids (which is usually a rapid and reversible process) and the rate of elimination of the drug from the body. The drug can either be eliminated unchanged, or be enzymatically cleaved or biochemically transformed, in which case it is said to have been metabolized. The study and characterization of the time course of drug absorption, distribution, metabolism and elimination (ADME) is termed pharmacokinetics. In contrast, pharmacodynamics is the study of the biochemical and physiological effects of the drug on the body. The majority of drugs either mimic normal physiological or biochemical processes or inhibit pathological processes. More simply; pharmacokinetics has also been defined as what the body does to the drug; whilst in contrast pharmacodynamics may be defined as what the drug does to the body. Pharmacokinetics can be used in the clinical setting to enhance the safe and effective therapeutic management of individual patients and increasingly pharmacodynamic markers are used to assess the success of therapy.
Figure 18.1 illustrates some of the factors that can influence the concentration of the drug in the blood plasma and also at its site(s) of action. Biopharmaceutics is concerned with the first stage – getting the drug from its route of administration into the blood stream or systemic circulation.
Concept of bioavailability
If a drug is given intravenously, it is administered directly into the blood and therefore we can be sure that all of the drug reaches the systemic circulation. The drug is therefore said to be 100% bioavailable. However, if a drug is given by another route there is no guarantee that the whole dose will reach the systemic circulation intact. The amount of an administered dose of the drug that does reach the systemic circulation in the unchanged form is known as the bioavailable dose. The percentage of an administered dose of a particular drug that reaches the systemic circulation intact is known as the bioavailability.
Bioavailability is defined in the FDA’s regulations as ‘the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action’. Absolute bioavailability compares the bioavailability of the unchanged drug in the systemic circulation following a non-intravenous dose, e.g. oral, rectal, transdermal, sub-lingual, intramuscular, subcutaneous, with the bioavailability of the same drug following intravenous administration. The bioavailability exhibited by a drug is thus very important in determining whether a therapeutically effective concentration will be achieved at the site(s) of action.
In defining bioavailability in these terms, it is assumed that the administered drug is the therapeutically active form. This definition would not be valid in the case of prodrugs, whose therapeutic action normally depends on their being converted into a therapeutically active form prior to, or on reaching the systemic circulation. It should also be noted that, in the context of bioavailability, the term ‘systemic circulation’ refers primarily to venous blood (excluding the hepatic portal vein, which carries blood from the gastrointestinal tract to the liver in the absorption phase) and the arterial blood, which carries the blood to the tissues.
Therefore, for a drug which is administered orally to be 100% bioavailable, the entire dose must move from the dosage form to the systemic circulation. The drug must therefore:
• be completely released from the dosage form
• be fully dissolved in the gastrointestinal fluids
• be stable in solution in the gastrointestinal fluids
• pass through the gastrointestinal barrier into the mesenteric circulation without being metabolized
• pass through the liver into the systemic circulation unchanged.
Anything which adversely affects either the release of the drug from the dosage form, its dissolution into the gastrointestinal fluids, its permeation through and stability in the gastrointestinal barrier or its stability in the hepatic portal circulation will influence the bioavailability exhibited by that drug from the dosage form in which it was administered.
Concept of biopharmaceutics
Many factors have been found to influence the rate and extent of absorption, and hence the time course of a drug in the plasma, and therefore at its site(s) of action. These include the foods eaten by the patient, the effect of the disease state on drug absorption, the age of the patient, the site(s) of absorption of the administered drug, the coadministration of other drugs, the physical and chemical properties of the administered drug, the type of dosage form, the composition and method of manufacture of the dosage form, the size of the dose and the frequency of administration.
Thus, a given drug may exhibit differences in its bioavailability if it is administered:
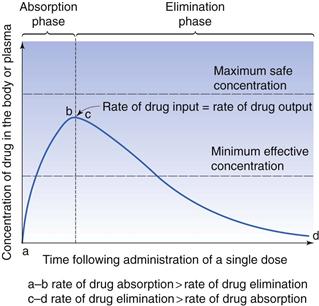
Variability in the bioavailability exhibited by a given drug from different formulations of the same type of dosage form, or from different types of dosage forms, or by different routes of administration, can cause the plasma concentration of the drug to be too high, and therefore cause side-effects, or too low, and therefore the drug will be ineffective. Figure 18.2 shows the plasma concentration-time curve following a single oral dose of a drug, indicating the parameters associated with a therapeutic effect. The therapeutic window is the drug concentrations which are above the minimum effective concentration and below the maximum safe concentration.
Poor biopharmaceutical properties may result in:
Summary
The following chapters (Chapters 19 and 20) deal in more detail with the physiological factors, dosage form factors and intrinsic properties of drugs that influence the rate and extent of absorption for oral drugs. Chapter 21 looks at means of assessing the biopharmaceutical properties of compounds.
A thorough understanding of the biopharmaceutical properties of a candidate drug is important both in the discovery setting, where potential drug candidates are being considered, and in the development setting, where it is important to anticipate formulation and manufacturing problems. The influence of variability and bioequivalence issues on clinical results must be studied to provide assurance to the regulatory authorities as to the robustness and quality of the drug substance and drug product.
Bibliography
1. Dressman J, Reppas C. Oral Drug Absorption: Prediction and Assessment Second Edition. USA: Informa Healthcare; 2008.
2. Rowland M, Tozer TN. Clinical Pharmacokinetcis and Pharmacodynamics: Concepts and Applications. Philadelphia: Lippincott Williams & Wilkins; 2011.
3. Van de Waterbeemed H, Testa B. Drug Bioavailability: Estimation of Solubility, Permeability and Absorption. Germany: Wiley-VDH; 2008.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Introduction to biopharmaceutics
Marianne Ashford
Chapter contents
Key points
• Pharmacodynamics is the study of the biochemical and physiological effects of the drug on the body.
What is biopharmaceutics?
Biopharmaceutics can be defined as the study of how the physicochemical properties of drugs, dosage forms and routes of administration affect the rate and extent of drug absorption.
The relationship between the drug, its dosage form and the route by which it is administered governs how much of the drug enters the systemic circulation and at what rate. For a drug to be effective, enough of it needs to reach its site(s) of action and stay there long enough to be able to exert its pharmacological effect. This is determined by the route of administration, the form in which the drug is administered and the rate at which it is delivered.
Background
Apart from the intravenous route, where a drug is introduced directly into the blood stream, all other routes of administration, where the site of action is remote from the site of administration, involve the absorption of the drug into the blood. Once the drug reaches the blood it partitions between the plasma and the red blood cells, the erythrocytes. Drug in plasma partitions between the plasma proteins (mainly albumin) and the plasma water. It is the free or unbound drug in plasma water, and not the drug bound to the proteins, that passes out of the plasma through the capillary endothelium and to tissues and hence the site(s) of action.
A dynamic equilibrium exists between the concentration of the drug in the blood plasma and the drug at its site(s) of action. This is what is termed distribution, the degree of which will depend largely on the physicochemical properties of the drug, in particular its lipophilicity. As it is frequently difficult to access the drug at its site(s) of action, its concentration in the plasma is often taken as a surrogate for the concentration at its site(s) of action. Even though the unbound drug in the plasma would give a better estimate of the concentration of the drug at its site(s) of action, this requires much more complex and sensitive assays than a measurement of the total concentration of the drug (i.e. the sum of the bound and unbound drug) within the blood plasma. Thus it is this total drug concentration within the plasma that is usually measured for clinical purposes. Therefore, plasma protein binding is a critical parameter to consider when investigating the therapeutic effect of a drug molecule.
The concentration of the drug in blood plasma depends on numerous factors. These include the amount of an administered dose that is absorbed and reaches the systemic circulation; the extent of distribution
[/not-level-membership-for-basic-science-category]
