[level-membership-for-pediatrics-category]
Fatty Liver Disease
Elizabeth M. Brunt
Dina G. Tiniakos
Nonalcoholic Fatty Liver Disease
Fatty Liver Disease in Various Patient Groups
Concurrent Alcoholic and Nonalcoholic Fatty Liver Disease
Fatty Liver Disease in Patients with Other Liver Disorders
Fatty Liver Disease in Children
Differentiating Alcohol-Induced from Nonalcoholic Fatty Liver Disease
Features that Occur More Frequently in Alcoholic Liver Disease
Features that Occur More Frequently in Nonalcoholic Fatty Liver Disease
Grading and Staging of Fatty Liver Disease
Other Forms of Fatty Liver Disease
Drug- and Toxin-Induced Fatty Liver Disease
Fatty Liver Disease after Transplantation
Miscellaneous Causes of Fatty Liver Disease
Introduction
Definitions
Steatosis, defined as the accumulation of triacylglycerides in hepatocytes, is a frequent finding in most liver biopsies.1 As much as 5% of the liver parenchyma may be composed of lipid. By convention, any quantity of lipid that occupies more than 5% of the liver mass is considered pathologic.
Macrovesicular steatosis is an accumulation of lipid droplets of various sizes in hepatocytes in which the cell nuclei are displaced peripherally. It is common to observe small and large droplets in the same cell and to find isolated droplets of lipid.
Microvesicular steatosis has a different appearance and clinical significance. The nuclei of affected hepatocytes are typically centrally located within abundant, foamy-appearing cytoplasm. Special stains, such as Oil Red O, may be necessary to confirm lipid accumulation, whereas in macrovesicular steatosis, the large and small lipid droplets are easily detectable by routine hematoxylin and eosin stain. Diseases associated with microvesicular steatosis are characterized by liver failure and hepatic encephalopathy rather than chronicity and potential fibrosis; the former features are the result of mitochondrial β-oxidation defects. The most common examples of purely microvesicular steatosis are acute fatty liver of pregnancy and Reye syndrome. This process has also been reported in the setting of drug toxicity. Drug-related entities are discussed in Chapter 48.
Alcohol-induced liver disease (ALD) refers to the spectrum of liver disorders directly related to excessive alcohol use. This chapter focuses primarily on the pathologic lesions of the ALD subset characterized by accumulation of fat in the liver parenchyma.
Nonalcoholic fatty liver disease (NAFLD) occurs without significant alcohol use. The clinicopathologic entity is characterized by a variety of hepatic parenchymal injury patterns with more than 5% macrovesicular steatosis.
The term nonalcoholic steatohepatitis (NASH) is in the same clinical setting as NAFLD, but NASH is associated with specific pathologic lesions in the liver. The lesions include various amounts of macrovesicular steatosis, hepatocellular injury (most commonly seen as ballooning), and inflammation in the hepatic lobules or portal tracts, or both. These lesions and others that may be seen in steatohepatitis are discussed in this chapter.
Nomenclature
The term ALD refers to a spectrum of liver diseases and includes large or mixed large and small droplet macrovesicular steatosis with or without lobular and portal inflammation; steatohepatitis with or without fibrosis; alcoholic hepatitis; alcoholic cirrhosis with or without steatosis, steatohepatitis, or alcoholic hepatitis; and alcoholic foamy degeneration. Hepatocellular carcinoma is a recognized consequence of ALD and cirrhosis.
NAFLD has a more limited pathologic spectrum than ALD. NAFLD may manifest as large droplet or mixed large and small droplet macrovesicular steatosis with or without lobular or portal inflammation; steatohepatitis with or without fibrosis; and cirrhosis with or without steatosis or steatohepatitis. All of the features of NAFLD may be seen in ALD, prompting use of the term nonalcoholic fatty liver disease.2,3 Hepatocellular carcinoma may arise as a consequence of NAFLD-induced cirrhosis and is therefore included in the spectrum of NAFLD lesions. Hepatocellular carcinoma is increasingly recognized in noncirrhotic NAFLD.4 However, the true incidence of this neoplastic complication in cirrhotic or noncirrhotic liver is less certain than with ALD.
Although NASH and NAFLD are commonly used terms, neither truly represents the underlying pathophysiologic disease process,5–7 and although there are histologic similarities with ALD, there are also dissimilarities between these disorders. The relationship between NAFLD and alcohol use may not be as straightforward as originally thought, because a few studies indicate that some patients may be at risk for liver disease from both causes simultaneously. Modest alcohol use also may protect against the effects of insulin resistance.8,9
For diagnostic pathologists, microscopically based diagnoses related to fatty liver disease are best rendered descriptively with terms such as steatosis or steatohepatitis, because in most circumstances, only careful clinical evaluation can determine the true underlying cause of the patient’s illness. Many different types of clinical situations may result in similar histologic findings, and in most cases, knowledge of the patient’s clinical information is essential to establish an accurate cause. This is similar to the situation for most cases of chronic hepatitis. The difference with fatty liver disease is that there are no serologic tests available to diagnose or exclude NAFLD with complete certainty.10
Alcohol-Induced Liver Disease
Clinical Features
The clinical features of ALD vary considerably. Table 49.1 highlights the primary clinical manifestations of the four main clinicopathologic subtypes of ALD: fatty liver, alcoholic hepatitis, alcoholic cirrhosis, and alcoholic foamy degeneration.
Table 49.1
Symptoms, Signs, and Laboratory Abnormalities of Alcohol-Induced Liver Disease
| Symptoms and Signs | Fatty Liver | Steatohepatitis and Alcoholic Hepatitis | Cirrhosis | Foamy Degeneration |
| Asymptomatic | +++ | + | Portal hypertension | − |
| Right upper quadrant discomfort | +++ | Pain, not mild | + to ++ | + |
| Hepatomegaly | ++ | ++ | + to ++ | + |
| Elevated AST, normal ALT | ++ | + | + (normal if abstinent) | ++ |
| AST/ALT ratio | AST > ALT | AST/ALT > 1-3 common | AST/ALT > 1 | AST > ALT |
| Marked AST elevation | <5 times normal | As much as 5 times normal | − | ++ |
| Marked alkaline phosphatase elevation | − | +/− | − | ++ |
| Bilirubin elevation | − | >10 mg/dL | − (unless advanced) | ++ |
| Jaundice | − | Cardinal sign | As above | ++ |
| Elevated white blood cell count | − | >10,000/mm3 | − (may be decreased) | − |
| Fever | − | +++ | + (with infection) | − |
| Ascites | − | +++ | + (advanced) | − |
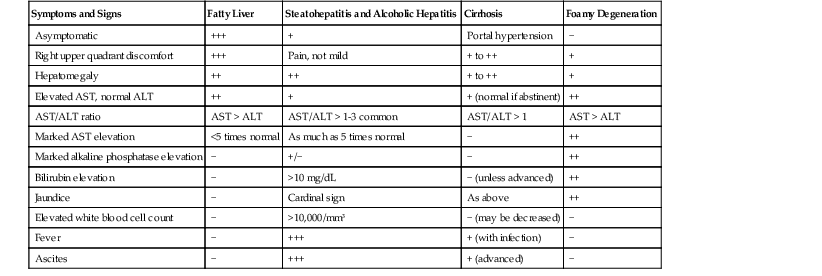
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; −, does not occur; +, occurs uncommonly; ++, common; +++, very common.
Patients with ALD and fatty liver may be asymptomatic or may have right upper quadrant discomfort at presentation (see Table 49.1). Patients with alcoholic hepatitis usually have abdominal pain and pancreatitis, whereas patients with cirrhosis and alcoholic foamy degeneration often experience only vague abdominal discomfort. In all forms of ALD, the liver is typically enlarged. Fever, an elevated white blood cell count, and jaundice occur in various degrees with the different forms of ALD and may be related to ALD (e.g., alcoholic hepatitis) or to a secondary infection (e.g., peritonitis caused by alcoholic cirrhosis).
At clinical presentation, patients may have elevated serum levels of aminotransferases (i.e., aspartate aminotransferase [AST] and alanine aminotransferase [ALT]), and a ratio of AST to ALT greater than 1 is useful in the evaluation of patients with ALD. AST levels are typically greater than ALT levels in all forms of ALD. Radiographic evaluation plays a role in screening for hepatocellular carcinoma in patients with cirrhosis and in the clinical care of critically ill patients with pancreatitis and suspected intestinal ileus.
Epidemiology and Prevalence
Worldwide, ALD has no social or ethnic barriers, but it is uncommon in very young and very elderly individuals. Both genders may be affected, although women are at higher risk. In the United States, 7.4% of the population meet the criteria for alcohol dependence or abuse. It is the leading cause of end-stage liver disease and liver-related mortality and follows only heart disease and cancer as the major health issue in the United States.11 The reported rates of per capita liters of alcohol consumed per year by individuals older than 15 years of age varies throughout the world: 13.9 in the Russian Federation; 12.9 in Germany, France, and the United Kingdom; 9.3 to 8.5 in North America, Japan, and Australia; 5.0 in China, Philippines, and Vietnam; 1.3 in Iran and Saudi Arabia; and 0.6 in Afghanistan and Pakistan.11 Alcohol use may combine synergistically with other forms of chronic liver disease to generate progressive disease, malignancy, and morbidity.12
Familial Associations
Family, twin, and ethnic studies have confirmed a genetic susceptibility to ALD.11 Factors under investigation include gender, genes involved in the metabolism of alcohol (particularly in East Asians), cytochrome P450 2E1 (CYP2E1) and other enzymes involved in the pathways of oxidative stress, various cytokines (e.g., tumor necrosis factor-α [TNF-α], interleukin 10 [IL10]), genes involved in the oxidation of hepatic fat, mechanisms of matrix deposition and degradation in fibrosis, and immune reactions to endotoxin and toxic adducts.13
Risk Factors
Liver disease does not develop in all individuals who abuse alcohol. The risk for ALD, even among heavy drinkers, is less than 10% to 15%. Known risk factors include the amount of alcohol consumed during a 10- to 20-year period (>60 g/day for men; 20 to 30 g/day for women), gender (female > male), central obesity, patterns of consumption (nonmealtime > mealtime; daily > weekend only; various types of beverages rather than one type), associated medications (acetaminophen), amount of coffee intake, and genes that regulate expression of proinflammatory cytokines and immune response mechanisms.13–18 A sequence variation in the patatin-like phospholipase 3 (PNPLA3) gene, which encodes adiponutrin, is associated with the development of cirrhosis in white alcoholics.19 The same gene variation was previously found to correlate with steatosis in different populations around the world and with features of progressive disease in NAFLD patients.20
Pathogenesis
Alcohol-induced liver damage is a multifactorial process that involves alterations of the gut, gut microbiota, and hepatocellular metabolic pathways and activation of the innate and adaptive immune systems. Resultant outcomes are activation of proinflammatory and profibrogenic molecular mechanisms in specific liver cells.
The primary mechanism of alcohol-induced liver injury is hepatocellular oxidative stress.21,22 Ethanol is metabolized to acetate in the liver mainly by two enzyme systems: the dehydrogenases (i.e., alcohol and aldehyde), which produce acetate and result in steatosis, and the microsomal ethanol-oxidizing system (MEOS). The direct hepatotoxic effects of alcohol are caused primarily by acetaldehyde. Oxidative stress and free radical production result in mitochondrial damage, depletion of the antioxidants such as reduced glutathione, toxicity by free radicals, and induction of lipid peroxidation. Acetaldehyde-formed toxic adducts bind to proteins and lead to additional hepatocellular injury, serve as neoantigens for initiation of the adaptive immune response, and promote collagen production by hepatic stellate cells. The net effect of acetaldehyde production in hepatocytes is the accumulation of intracellular proteins, lipids, water, and electrolytes with the loss of the hepatocyte structural keratins 8 (K8) and 18 (K18). Histologically, they manifest as cellular enlargement, ballooning, empty-appearing cells, steatosis, and loss of immunoreactivity for K8 and K18.23,24
The key enzyme of the MEOS is the ethanol-inducible CYP2E1, which is located primarily in the endoplasmic reticulum of acinar zone 3 hepatocytes. Activation of this enzyme system results in the production of reactive oxygen species, such as hydrogen peroxide (H2O2) and superoxide anions (O2−), which cause further lipid peroxidation of cell membranes. This enzyme system also metabolizes acetaminophen and produces the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI). Even small amounts of acetaminophen may augment depletion of the antioxidant capabilities of the liver in chronic alcoholism.
Other sources of ethanol-induced oxidative stress include endotoxin-activated Kupffer cells, functionally impaired mitochondria, and ferric iron accumulation. Oxidative stress promotes hepatocellular injury, apoptosis, and necrosis; proinflammatory cytokine production (e.g., TNF-α, IL1, IL6); and parenchymal inflammation with polymorphonuclear leukocytes (PMNs) and mononuclear cells and perisinusoidal fibrosis.25,26
Excessive alcohol intake leads to increased gut permeability and increased portal venous exposure to gut-derived endotoxins. This process results in Kupffer cell activation through the CD14/toll-like receptor 4 (TLR4) complex and activation of the innate immune response.22,26
Increased lactate production, another downstream effect of ethanol metabolism, manifests as hyperuricemia and impaired carbohydrate metabolism. Hypoglycemia may occur. Chronic alcohol use impairs protein production and secretion. Multiple aberrations of lipid metabolism result in increased triglyceride production and subsequent accumulation (i.e., steatosis).
The mechanisms of fibrosis are under active scientific investigation. Ultimately, there is an imbalance between collagen deposition and degradation by activated hepatic stellate cells and portal myofibroblasts. Unique to ALD is the additional involvement of hepatocytes and sinusoidal endothelial cells in fibrogenesis through the production of transforming growth factor-β (TGF-β) and fibronectin isoforms.27 Hypoxia induced by ethanol metabolism in zone 3 hepatocytes upregulates vascular endothelial growth factor.26,27
Pathology
Grossly, steatosis enlarges the liver and imparts a greasy appearance. Livers with advanced fibrosis or cirrhosis may be enlarged and firm and contain micronodules (≤3 mm in diameter) scattered throughout the parenchyma. In some cases, fibrotic livers may be small and firm. With time, the nodules may coalesce to form larger ones, at which point determination of the underlying cause may not be possible. Hepatocellular carcinomas typically stand out on background cirrhosis as raised, green-tinged or darker nodules that usually are larger than 8 mm in diameter.
The histologic pattern of tissue injury in patients with alcohol-induced fatty liver disease initially involves acinar zone 3 (roughly equivalent to perivenular) hepatocytes. In patients with cirrhosis, steatosis or steatohepatitis may or may not persist.
Steatosis
Steatosis, the earliest pathologic finding in ALD, is not consistently found in all forms of ALD.21,28 Alcoholic steatosis is typically macrovesicular and is characterized by large intracellular droplets of lipid in hepatocytes (single or multiple) that peripherally displace the cell nucleus. A minor component of lobular chronic inflammation or mild portal inflammation may be identified, but fibrosis is usually absent.
Steatohepatitis
Steatohepatitis is defined by the presence of both steatosis and hepatocyte injury. Injured hepatocytes may appear ballooned, apoptotic, or lytic. Confluent and bridging necrosis are rarely found in ALD.
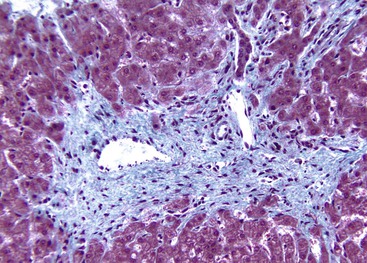
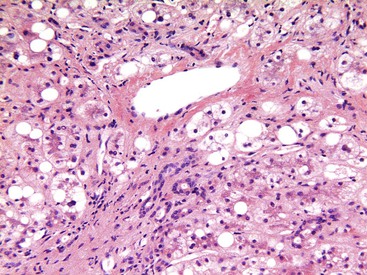
Hepatocellular ballooning is considered an essential feature of steatohepatitis by most hepatopathologists.7 Ballooned hepatocytes are enlarged cells with rarefied, reticulated cytoplasm (Fig. 49.1) that may or may not contain fat droplets and intracytoplasmic material referred to as Mallory-Denk bodies (MDBs) or Mallory hyaline. Ballooned hepatocytes are located predominantly in zone 3 and are commonly associated with some degree of perisinusoidal fibrosis (Fig. 49.2).29
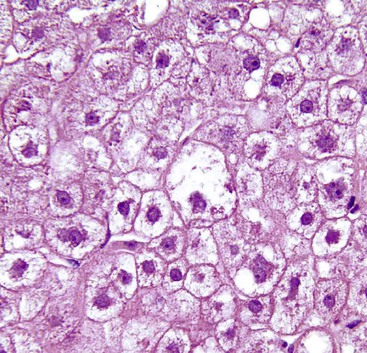
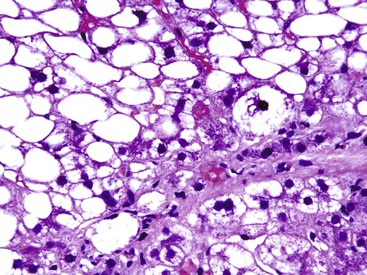
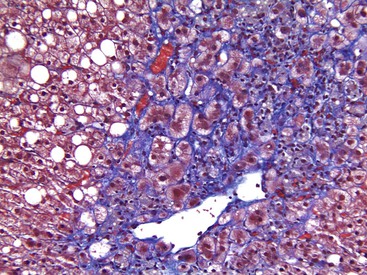
Apoptotic hepatocytes, also called acidophil bodies, are common in patients with alcoholic steatohepatitis. These round, eosinophilic fragments of cytoplasm located in the hepatic sinusoids may or may not retain nuclear pyknotic material. Lobular inflammation in patients with steatohepatitis is usually mild and consists of mixed acute and chronic or mainly chronic inflammation. Scattered lobular microgranulomas and lipogranulomas are often observed. Lobular lipogranulomas may consist of multiple or single fat droplets surrounded by mononuclear cells and eosinophils (Fig. 49.3). Large portal lipogranulomas are frequently observed in ALD.30 PMNs may be observed in small clusters adjacent to ballooned hepatocytes or shrunken, eosinophilic (apoptotic) hepatocytes that contain MDBs. When PMNs are found next to hepatocytes that contain MDBs, the lesion is often referred to as satellitosis (Fig. 49.4).
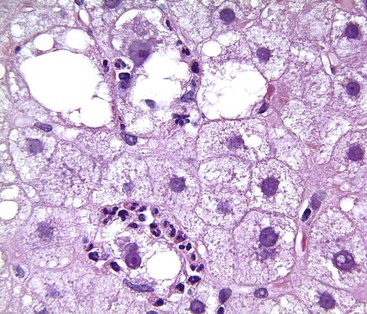
Although common, MDBs and satellitosis are not required for the diagnosis. MDBs are not specific to ALD; the structures are seen in NAFLD, chronic cholestatic liver disease, copper toxicity, and amiodarone toxicity. In steatohepatitis of any origin, MDBs are usually found in zone 3 hepatocytes (Fig. 49.5) and are more common in areas of perisinusoidal fibrosis, whereas in chronic cholestasis, copper toxicity, and amiodarone toxicity, MDBs are more common in periportal hepatocytes.1,6 MDBs may also occur in focal nodular hyperplasia, hepatocellular adenoma, and hepatocellular carcinoma.31 MDBs can often be visualized by trichrome stains (Fig. 49.6) and confirmed by the use of immunohistochemistry for K8, K18, ubiquitin, or dynactin 4 (DCTN4, formerly designated p62).32
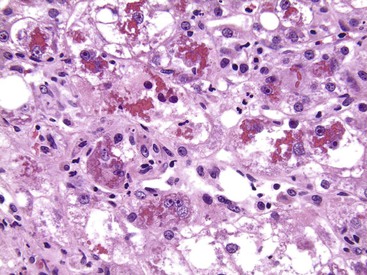
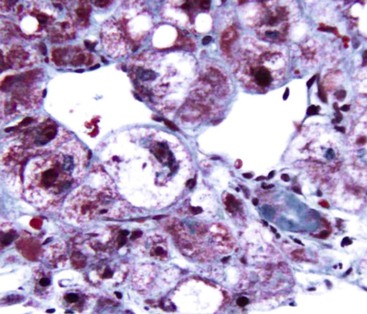
Some cases of ALD may show predominantly portal lymphocytic inflammation in the absence of serologic evidence of chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection. This feature may correlate with the degree of fibrosis33 and may reflect an associated autoimmune component to the underlying liver disease.34 Acute inflammation accompanied by a ductular reaction in portal tracts (i.e., pericholangitis) may represent concurrent pancreatitis, whereas increased numbers of portal macrophages may indicate recurrent partial biliary obstruction.30
Alcoholic Hepatitis
Patients with alcoholic hepatitis might not have steatosis. Histologic features of alcoholic hepatitis include hepatocyte ballooning, apoptotic bodies, MDBs with satellitosis, canalicular cholestasis, dense perisinusoidal fibrosis, and a perivenular lesion often referred to as sclerosing hyaline necrosis (i.e., MDBs, satellitosis, and obliterative fibrosis of the outflow vein). Portal hypertension may occur with sclerosing hyaline necrosis. Bridging fibrosis and a ductular reaction may be identified. The absence of steatosis does not rule out alcohol-induced hepatitis.
Alcoholic Foamy Degeneration
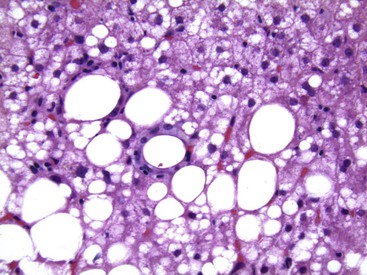
Alcoholic foamy degeneration is an unusual and often serious type of ALD associated with marked elevations of aminotransferases, γ-glutamyltransferase (GGT), alkaline phosphatase, and bilirubin.35 Histologically, it is characterized by diffuse, primarily microvesicular steatosis without inflammation or ballooning or by marked fibrosis with or without canalicular cholestasis (Fig. 49.7). MDBs are uncommon. Perivenular and perisinusoidal fibrosis may be evident. Clinically, the disorder mimics extrahepatic biliary obstruction, but the liver biopsy findings can be used to distinguish the two entities. Alcoholic foamy degeneration is reversible with abstinence from alcohol.
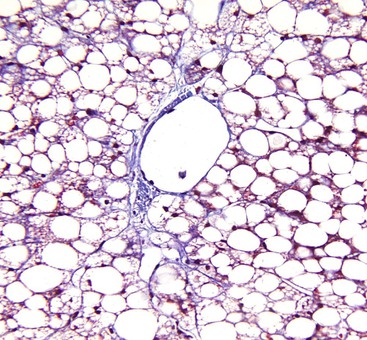
Fibrosis and Cirrhosis
The characteristic pattern of early fibrosis in patients with noncirrhotic alcoholic steatohepatitis or alcoholic hepatitis is pericellular or perisinusoidal. This pattern results from deposition of collagen in the space of Disse and is commonly referred to as chicken wire fibrosis (Fig. 49.8).
Fibrosis usually begins in zone 3 of the hepatic acini (Fig. 49.9). This type of fibrosis may be identified with or without steatohepatitis. However, when pericellular or perisinusoidal fibrosis occurs without steatohepatitis, it probably indicates that the patient had at least one prior episode of steatohepatitis. In ALD, perisinusoidal fibrosis may involve large portions of the lobules and become quite dense (Fig. 49.10). In these cases, the patient may also have clinical evidence of portal hypertension despite the absence of cirrhosis.
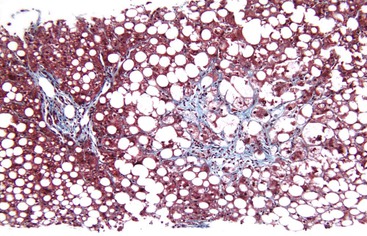
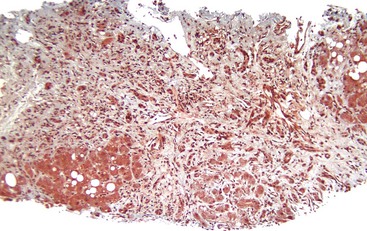
Perivenular fibrosis, a thickened sheath of collagen around the outflow veins in the absence of perisinusoidal fibrosis, has been identified in patients with steatosis that subsequently progressed to cirrhosis. This lesion is therefore considered a prognostic indicator of progression if alcohol exposure is continued.21
With disease progression, periportal fibrosis may develop (Fig. 49.11), followed in some cases by bridging fibrosis in a central-central, central-portal, or portal-portal pattern. In ALD, regions of parenchymal extinction (i.e., septa) may be quite broad (Fig. 49.12). After cirrhosis is established, areas of perisinusoidal fibrosis may not be evident. Periportal fibrosis and bridging fibrosis are often accompanied by a ductular (proliferative) reaction. In ALD, isolated portal fibrosis in the absence of portal inflammation is uncommon and may be a manifestation of recurrent pancreatitis or biliary obstruction.30 Itoh observed a pattern of portal fibrosis characteristic of ALD known as holly leaf.36 This pattern of fibrosis, which is commonly associated with hemochromatosis, is characterized by portal expansion and nonbridging, small, fibrous extensions or spikes into the surrounding parenchyma that resemble the outline of the holly leaf.
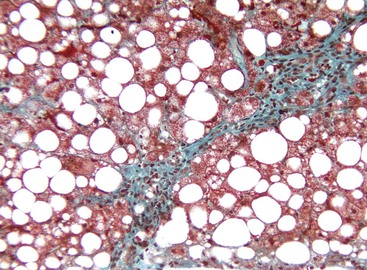
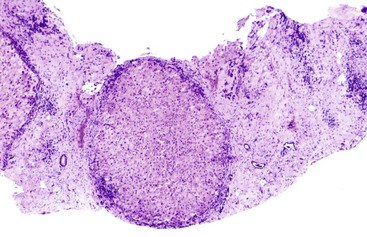
In patients with ALD with bridging fibrosis,37 periseptal ductular reaction may be prominent. The ductular reaction, characterized by proliferation of K7- and/or K19-positive ovoid cells forming ductular structures in an inflammatory, stromal matrix,38 less likely represents a cholestatic process in end-stage liver disease,26,30 but it is most likely the result of impaired regenerative activity of hepatocytes resulting from the anti-regenerative effects of alcohol.39
ALD-associated cirrhosis may be macronodular, micronodular, or mixed. Micronodular nodules are typically less than 3 mm in diameter. Larger nodules may develop when multiple nodules coalesce to form mixed micronodular and macronodular cirrhosis. Macronodular cirrhosis may be difficult to diagnose in a liver biopsy specimen because of the large size of the nodules; clues to abnormal architecture can be sought by use of a reticulin stain. Clusters of oncocytic hepatocytes suggest macronodular cirrhosis (see Chapter 50). Phlebosclerosis and occlusive disease of the terminal hepatic venules and sublobular veins may occur in patients with ALD-induced cirrhosis.40
In ALD-associated cirrhosis, periseptal hepatocellular copper granules and, uncommonly, α1-antitrypsin globules may be found. The role of heterozygosity for genes related to α1-antitrypsin in promoting damage in patients with ALD is a subject of ongoing debate.41 Increased iron levels in ALD is discussed later. Pseudotumoral nodules, which correspond to areas of active alcoholic hepatitis in cirrhotic livers, may be recognized on imaging studies and confused for hepatocellular carcinoma. These lesions may result from hypervascularity.42
Other Pathologic Features of Alcohol-Induced Liver Disease
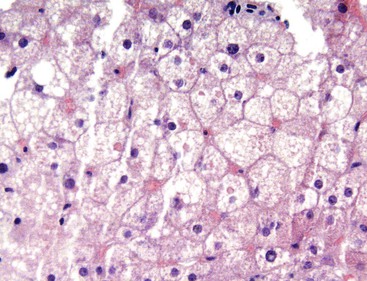
Megamitochondria.
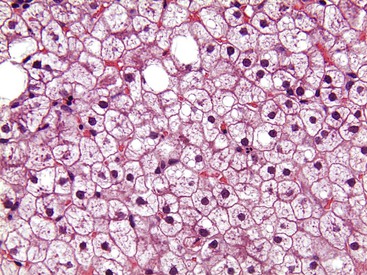
Megamitochondria (i.e., giant mitochondria) are identified by light microscopy as intracytoplasmic, round or cigar-shaped, eosinophilic structures in hepatocytes, and they are most readily seen in hepatocytes that contain microvesicular steatosis (Fig. 49.13). Megamitochondria may also occur in normal pregnancy, acute fatty liver of pregnancy, and Wilson disease.
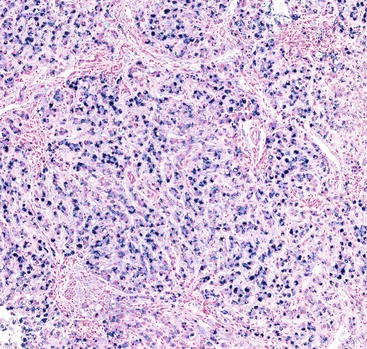
Iron Deposition.
Mildly increased iron deposition is common in most forms of ALD. However, in a minority of cases, it may be moderate or severe.23 Possible reasons for hemosiderosis in ALD include increased dysregulation of hepcidin production,43 intestinal iron absorption, iron in some types of alcoholic beverages, hemolysis related to spur cell anemia, and upregulation of the transferrin receptor.18 With a modified Perls stain, iron deposition is usually nonzonal.30,37 In ALD-related cirrhosis,37 iron deposition may vary in quantity between individual regenerative nodules (Fig. 49.14).
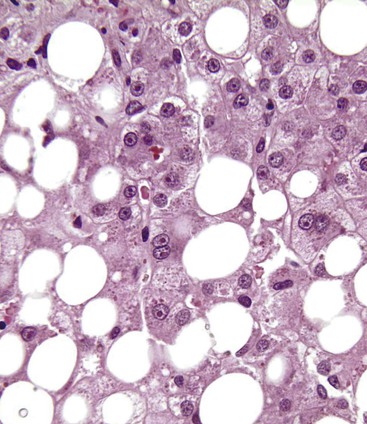
Acute and Chronic Cholestasis.
Any of the following pathologic processes related to ALD may result in intrahepatic cholestasis, which is characterized by intracanalicular bile plugs: severe fatty liver, alcoholic pancreatitis due to gallstones or alcohol, alcoholic foamy degeneration, alcoholic hepatitis, and decompensated alcoholic cirrhosis. Canalicular bile thrombi are observed in 15% to 32% of livers with ALD (Fig. 49.15). Severe fatty liver with portal tract features of cholangitis and cholestasis is a specific clinicopathologic cholestatic syndrome in patients with ALD. Bile stasis in ALD also may be a manifestation of superimposed viral hepatitis or drug-induced jaundice.21 In patients with established ALD-associated cirrhosis, a copper stain may show mild positivity, which represents chronic retention of bile caused by the cirrhotic remodeling.
In advanced cases of ALD, the cytoplasm of some hepatocytes may have a ground-glass appearance because of smooth endoplasmic reticulum proliferation, or it may be deeply eosinophilic, referred to as oxyphil or oncocytic change because of increased numbers of mitochondria. Both changes are considered adaptive. Historically, their presence was considered indicative of ongoing alcohol consumption, but one research group reported that they might also be observed after periods of prolonged abstinence.30
Vascular Lesions.
Sclerosing hyaline necrosis, which is a marker of severe alcoholic hepatitis, and obliterative vein lesions may be responsible for the development of noncirrhotic portal hypertension in patients with ALD. Sclerosing hyaline necrosis consists of a combination of acinar zone 3 lesions, including dense perivenular and perisinusoidal fibrosis (i.e., sclerosis), occlusion of terminal hepatic venules, MDBs, and liver cell necrosis and loss, which results in the formation of large perivenular scars. Lesions of the terminal hepatic venules and sublobular veins include perivenular fibrosis, phlebosclerosis, and, less frequently, lymphocytic phlebitis (Fig. 49.16).40
Treatment Effects.
Alcoholic steatosis may resolve completely within 4 weeks of alcohol abstinence.30 However, lesions associated with alcoholic hepatitis or steatohepatitis may persist for as long as 6 months after alcohol withdrawal, albeit with decreased severity.44 With abstinence, there is usually an increase in the degree of portal lymphocytic infiltration.44
Prognostic Lesions.
Perivenular fibrosis in steatosis in the absence of cirrhosis portends progression unless abstinence occurs. Acute cholestasis correlates with poor survival of hospitalized patients with ALD.21 These patients have a reduced 5-year survival rate (22%) compared with patients with ALD who have no or mild cholestasis (54%).23 The number of apoptotic bodies correlates positively with severe histologic and clinical disease activity.45,46 Alcoholic hepatitis, sclerosing hyaline necrosis, and severe steatosis represent poor-prognostic lesions in patients with ALD, particularly in those with cirrhosis.21,47 In noncirrhotic patients, mixed steatosis and the finding of megamitochondria in the initial liver biopsy of alcoholic patients without steatohepatitis correlate with an increased risk of subsequent fibrosis or cirrhosis on follow-up.48 In acute-on-chronic liver failure caused by ALD, marked ductular bilirubinostasis and many MDBs are significant predictors of in-hospital mortality.49
Lesions in the Allograft Liver.
Any of the previously described lesions may manifest as recurrent or de novo ALD in a liver allograft. Care is taken to evaluate other features of allografts, particularly acute and chronic rejection and recurrence of hepatitis C (see Chapter 46).
Natural History and Treatment
The financial and social burdens associated with ALD are significant.11,50 Alcoholic hepatitis is associated with a significant risk of hospitalization and loss of time from work, morbidity and short-term mortality, and progression to cirrhosis within 5 years for as many as 70% of individuals. Few affected individuals are able to reverse the disease process with complete abstinence.11 A 12-year historical study from a national registry of Danish subjects comparing biopsy-proven ALD with the general population showed that the cirrhosis risk was higher for affected women than men and twice as high for patients with steatohepatitis than those with pure steatosis.51
Chronic alcohol abuse is responsible for as many as 45% of cases of hepatocellular carcinoma in Western countries,52 a rate that is higher than that for chronic hepatitis C.18 Coexistent HCV infection has an additive effect on the risk of carcinoma,11 and other comorbid factors, such as chronic hepatitis B, hemochromatosis, obesity, and cigarette smoking, contribute to an increased risk of hepatocellular carcinoma and mortality in patients with ALD.53,54 Intrahepatic cholangiocarcinoma also has been linked to ALD.55
The mainstay of treatment for ALD is abstinence from alcohol. One of the pharmacologic methods of treatment of alcoholism, cyanamide, may produce isolated, nonzonal eosinophilic globules in hepatocytes. Treatment of hospitalized patients with alcoholic hepatitis includes steroids and the antioxidants pentoxifylline and S-adenosyl-l-methionine (SAMe). For abstinent patients with cirrhosis, liver transplantation is an accepted treatment option. More controversial is a practice of liver transplantation for patients with acute alcoholic hepatitis.56,57
Nonalcoholic Fatty Liver Disease
Clinical Features
Individuals with NAFLD may be asymptomatic or may show mild, nonspecific symptoms such as fatigue and right upper quadrant discomfort or pain (Table 49.2). Fever is not a feature of uncomplicated NAFLD; when present, it suggests a different or concomitant disorder.
Table 49.2
Symptoms, Signs, and Laboratory Abnormalities of Nonalcoholic Fatty Liver Disease
| Symptoms and Signs | Fatty Liver | Steatohepatitis | Cirrhosis |
| Asymptomatic | ++ | + | Portal hypertension |
| Right upper quadrant discomfort | ++ | ++ | + |
| Hepatomegaly | ++ | ++ | + |
| Elevated AST, normal ALT | − | − | − |
| AST/ALT ratio | AST/ALT < 1 | AST/ALT < 1 | AST/ALT ≥ 1 |
| Marked AST elevation | − | − | − |
| Marked alkaline phosphatase elevation | − | − | − |
| Bilirubin elevation | − | − | − |
| Jaundice | − | − | − |
| Elevated white blood cell count | − | − | − |
| Fever | − | − | − |
| Ascites | − | − | + (advanced) |
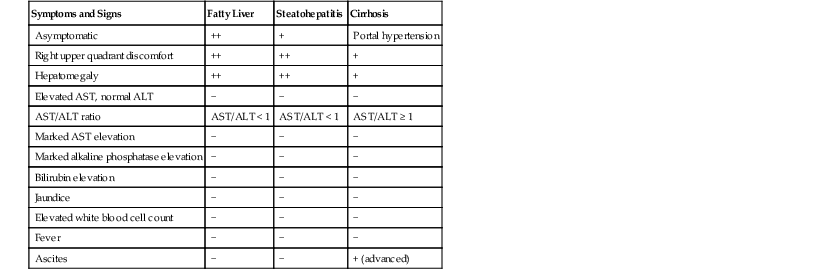
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; −, does not occur; +, occurs uncommonly; ++, common; +++, very common.
The most common clinical feature of NAFLD is central (truncal or abdominal) obesity, which is best assessed by measurement of waist circumference. For adult men, central obesity is a waist circumference greater than 102 cm, and for adult women, it is greater than 88 cm. Obesity is defined by body mass index (BMI), which is the weight in kilograms divided by the height in meters squared (kg/m2). Compared with waist circumference, it is less specific and does not assess the anatomic location of the metabolically unique subcutaneous and visceral fat depots or shape of the body habitus. For non-Asian adults, the current definitions used are overweight (BMI > 25 kg/m2) and obesity (BMI > 30 kg/m2). Because the Asian body habitus is deceptively thin and able to conceal visceral adiposity, lower BMI values for overweight and obesity have been recommended.58
Clinical evidence of hepatomegaly may be found in patients with an enlarged, fatty liver. Ascites and other features of portal hypertension may be identified in patients with NAFLD-associated cirrhosis. Obstructive sleep apnea and acanthosis nigricans are recognized associations of NAFLD.
Laboratory values commonly assessed for patients with NAFLD include ALT, AST, and GGT. Patients also undergo tests for hyperglycemia (glycated hemoglobin [HbA1c] is used to evaluate long-term glycemic control), tests for insulin resistance (based on calculations of fasting glucose and insulin levels by using quantitative insulin sensitivity check index [QUICKI] and homeostasis model assessment [HOMA-IR]), and tests of lipid parameters.21
The overall sensitivity of liver enzymes for detection of fatty liver disease is poor.21,59,60 For instance, patients with normal ALT levels may have steatosis or advanced steatohepatitis61 or silent cirrhosis.62,63 If elevated, the ALT value is greater than the AST value, and an ALT/AST ratio greater than 1 is often found in cases of precirrhotic NAFLD; the reverse situation occurs in ALD. Autoantibodies, particularly antinuclear antibody and rarely antimitochondrial antibody, have been reported in as many as 34% of individuals with NAFLD.64 In some, only further detailed clinical testing or liver biopsy can distinguish autoimmune hepatitis from concurrent autoimmune hepatitis with NAFLD or NAFLD alone.64 A possible association of autoantibodies with more severe disease has been suggested but not confirmed.65,66 In combination with the proven lack of reproducible gross evaluation of the liver by surgeons, authorities recommend a liver biopsy for all high-risk and bariatric patients,67–69 especially when other causes of steatosis have been excluded.59
Abnormal iron study results are a common feature in patients with NAFLD. For instance, elevated serum levels of ferritin (but not transferrin saturation) may be found in patients with NAFLD and may cause clinical concern about an iron overload disorder.70 The serum ferritin level in NAFLD patients has been reported as an independent predictor of the histologic severity of underlying liver disease.71 An elevated serum ferritin level may indicate hepatocyte necrosis and systemic inflammation, and it is a recognized feature of the metabolic syndrome (discussed later). However, iron overload itself is not a feature of type 2 diabetes mellitus. The associations of iron dysregulation, HFE (hemochromatosis) mutations, and fibrosis with the metabolic syndrome remain subjects of debate.70 A meta-analysis did not support an association between HFE genetic variants and NAFLD.72
The proinflammatory iron regulator hepcidin is expressed in the liver and adipose tissue of obese patients, and this finding correlates with low transferrin values in 68% and anemia in 24% of these individuals.73 Demonstration of hepcidin mRNA and protein in adipose tissue is a new finding and is the subject of iron and adipokine studies.73
Laboratory tests are indicative of the complexity of this condition. Markers of systemic inflammation, such as elevated levels of C-reactive protein, fibrinogen, and plasminogen activator inhibitor 1, are found in the setting of NASH and fibrosis.74 A depressed level of adiponectin, an endocrine product of visceral adipose tissue, is recognized as a pathologic feature of NAFLD or NASH.74 Some work has implicated additional factors as being proinflammatory (e.g., hemoglobin) in subsets of individuals with NAFLD but without the metabolic syndrome.75 In contrast with ALD, leukocytosis, cholestasis, and jaundice are not typical features of uncomplicated, noncirrhotic NASH.
By ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI), the characteristic finding in NAFLD is increased hepatic fat. The ultrasound finding is referred to as a bright liver. Newer types of imaging studies use methods to semiquantitate the lipid content of the liver.76 However, when steatosis involves less than 33% of the liver parenchyma, imaging tests may not detect it.77 Imaging is also not an adequate replacement for tissue analysis for determination of the degree of disease activity or the stage of fibrosis in patients with precirrhotic NASH.78
Epidemiology and Prevalence
The epidemiology and prevalence of NAFLD are understood in terms of its relationship with obesity and increased risks of stroke and heart disease, a group of associations referred to as metabolic syndrome. The common underlying feature of this syndrome is systemic insulin resistance. Studies have confirmed associations of NAFLD with hepatic insulin resistance, cardiovascular diseases,79 and the metabolic syndrome in adults80 and children.81 In one definition of metabolic syndrome, a patient must have at least two of the following disorders plus type 2 diabetes mellitus, insulin resistance, or glucose intolerance: obesity (BMI ≥ 30); arterial hypertension (≥140/90 mm Hg); dyslipidemia as low levels of high-density lipoprotein (<0.9 mmol/L) or hypertriglyceridemia (>1.7 mmol/L); and microalbuminuria (>20 µg/min).82 The sole finding of fatty liver may indicate progression of the metabolic syndrome83 and progression of liver disease.84
NAFLD is a worldwide disorder and has been documented in almost all cultures. This global phenomenon is closely related to acquisition of a Western diet and lifestyle, which includes less physical activity. Initially thought to be a disease predominantly of overweight women, the entire spectrum of NAFLD is now recognized as common among men of all ages, but severe NASH and advanced fibrosis remain more common among adult women.21,59 Several studies have confirmed an underrepresentation of this disease process in African Americans of all age groups and an overrepresentation in Hispanics.59
Once referred to as a disease of the affluent, NAFLD has become recognized as everyman’s disease.85 Future projections of the rates of obesity, type 2 diabetes mellitus, and their complications in the developing world for the year 2030 are staggering. They range from 18.6% in sub-Saharan Africa to 79.4% in India. The projected percent positive change for type 2 diabetes mellitus incidence is 32% in Europe, 72% in the United States, 150% in India, 162% in sub-Saharan Africa, and 164% in the Middle East.86
Having considered the worldwide and multicultural nature of NAFLD, some studies have evaluated certain ethnic proclivities for development of this disease. For instance, an image-based study of hepatic steatosis from a large, urban, multiethnic population in the United States76 found the following relative prevalence rates: Hispanic > white > African American. Results from a subsequent clinic-based study that used liver tests and liver biopsy results differed only slightly,87 showing the following ethnic prevalence rates: white > Hispanic> Asian. African Americans represented only 3% of patients in this cohort. Others have observed an apparent underrepresentation of African Americans in cohorts of patients with NAFLD.88 A small biopsy series from an inner-city hospital in the United States that primarily serves African Americans confirmed the relative scarcity of the disease in this patient population. Less than 2% of 320 liver biopsies showed clinicopathologic evidence of NAFLD, even though the BMI of the cohort was between 26.9 and 32.7.89
Unfortunately, most of the early prevalence studies were performed without a full understanding of the link between the metabolic syndrome and fatty liver disease. In later studies, surrogate markers such as elevated ALT levels and a bright liver on ultrasound were used to examine study populations. However, similar to pathologic studies, the results vary widely, which is partly related to the criteria used to define the positivity of test values. For instance, in studies that relied on elevated serum aminotransferase levels, recognized shortcomings included underrecognition of positive cases with normal test values, determination of criteria for normal values in an increasingly obese population,84 determination of reliable ALT values from frozen sera, and ascertainment of the levels of alcohol intake.90 Methods of assessment of body habitus (i.e., BMI, waist-to-hip ratio, midthigh measurements, and more sophisticated and expensive calculations) also vary.78 Commonly cited studies are listed in Table 49.3.
Table 49.3
Prevalence of Nonalcoholic Fatty Liver Disease among U.S. Adults Based on NHANES III Data*
| Study | Criteria | Prevalence of NAFLD |
| Clark et al, 2002274 | Any elevation of AST, ALT, GGT | 23% of all U.S. adults |
| 31% male | ||
| 16% female | ||
| 39% of obese men, 40% of obese women | ||
| Clark et al, 2003275 | ± patients with diabetes | AST > 37 U/L, ALT > 40 U/L (men) |
| AST, ALT > 31 U/L (women) | ||
| 7.9% of all U.S. adults; 69% unexplained (5.5% of adults) | ||
| Ruhl and Everhart, 2003276 | Elevated ALT only; excluded patients with diabetes | ALT > 43 U/L (men and women) |
| 2.8% elevated ALT, 65% explained by elevated body mass index |
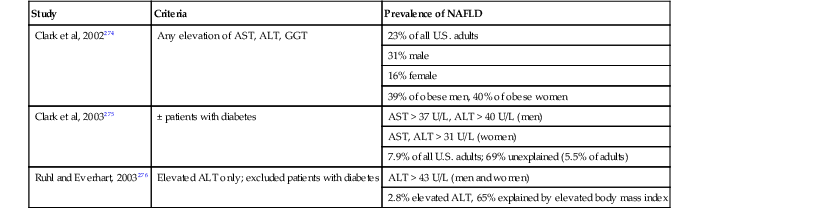
* Limitations of NHANES data: autoimmune hepatitis, primary biliary cirrhosis, Wilson’s disease, and α1-antitrypsin deficiency not excluded rigorously and absence of liver pathology for confirmation or exclusion.
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyltransferase; NAFLD, nonalcoholic fatty liver disease; NHANES III, Third National Health and Nutrition Examination Survey.
Validation of the findings of the Third National Health and Nutrition Examination Survey (NHANES III) has emerged from subsequent studies. One calculated hepatic lipid levels by proton magnetic spectroscopy in a multiethnic population in the southwestern United States and found that 31% of 2287 unselected individuals had a greater than 5.5% rate of hepatic steatosis. Of these, 79% had normal ALT values.76 A contemporaneous study from a large clinic population in California showed that of 742 individuals with newly diagnosed chronic liver disease, 21.4% had NAFLD.87 NAFLD was found to be the most common cause of incidental liver function test abnormalities (26.4%) in a U.K. prospective primary care cohort of 1118 adults by a combination of ultrasound and biomarker assays; ALD was a close second (25.3%). Of the NAFLD cases, 7.6% had evidence of previously undetected advanced fibrosis.91
A U.S. biopsy-based prevalence study of asymptomatic adults found that 46% of 156 adult, unselected patients who had steatosis on ultrasound had NAFLD diagnosed on biopsy. Of the biopsied NAFLD patients, 30% had diagnostic NASH, and 2.7% of those with NASH had advanced fibrosis (≥stage 2). This finding allowed speculation that more than 2 million of the U.S. adult population was at risk for advanced fibrosis because of NASH. These findings led the researchers to conclude that the prevalence of NAFLD was greater than that of hepatitis C and posed a serious concern for the future of health care.92
A study in a predominantly nonaffluent, nonobese population in India represented by 1911 inhabitants of a rural community showed the prevalence of NAFLD diagnosed by ultrasonography and CT to be 8.7%. NASH was seen in 31% of 36 biopsied persons with potentially significant disease, 4 of whom had cirrhosis.93,94
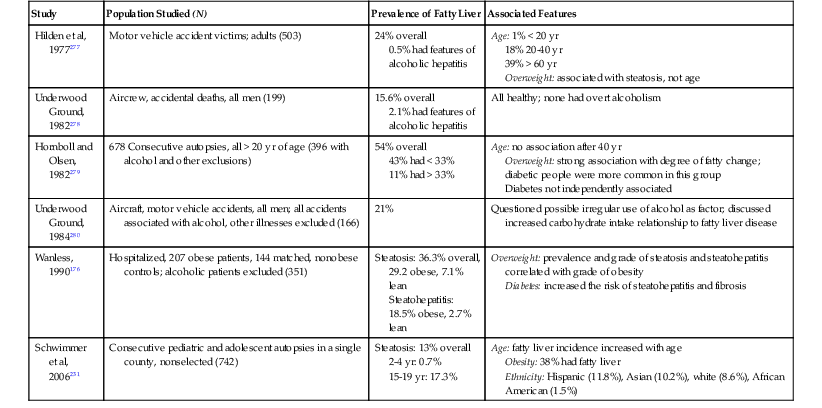
The estimated prevalence rate of NAFLD in the United States is greater than that of ALD (Table 49.4). It may affect as many as 5% of the adult population, with an even higher rate among patients with type 2 diabetes.59 Table 49.5 summarizes several published series focused on histologic documentation of fatty liver in adult autopsies. Correlations between body weight and diabetes were discussed in many of these studies long before the metabolic syndrome was identified as a specific entity. More than one investigator suggested that steatosis was a normal consequence of aging (see Pathogenesis).
Table 49.4
Prevalence Estimates of Fatty Liver Disease Compared with Other Chronic Liver Diseases
| Disorder | Prevalence |
| Nonalcoholic fatty liver disease | 25-75% of obese, diabetic adults 16% of 15- to 19-yr-old adolescents 38% of obese adolescents |
| Hepatitis C | 1.3-2.0% |
| Alcohol-induced liver disease | 1% |
| Hepatitis B | 0.3-0.4% |
| Hereditary hemochromatosis | 1/200-400 of northern European descent |
| Autoimmune hepatitis, primary biliary cirrhosis, primary sclerosing cholangitis | 9-17/100,000 |
| α1-Antitrypsin deficiency | 1/1500-7600 |
| Wilson disease | 1/30,000 |
Data from McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40(suppl 1):S17-S29 and Yu AS, Keeffe EB. Elevated AST or ALT to non-alcoholic fatty liver disease: accurate predictor of disease prevalence? Am J Gastroenterol. 2003;98:955-956.
Table 49.5
Prevalence of Fatty Liver Disease in Autopsy Studies
| Study | Population Studied (N) | Prevalence of Fatty Liver | Associated Features |
| Hilden et al, 1977277 | Motor vehicle accident victims; adults (503) | 24% overall 0.5% had features of alcoholic hepatitis |
Age: 1% < 20 yr 18% 20-40 yr 39% > 60 yr Overweight: associated with steatosis, not age |
| Underwood Ground, 1982278 | Aircrew, accidental deaths, all men (199) | 15.6% overall 2.1% had features of alcoholic hepatitis |
All healthy; none had overt alcoholism |
| Hornboll and Olsen, 1982279 | 678 Consecutive autopsies, all > 20 yr of age (396 with alcohol and other exclusions) | 54% overall 43% had < 33% 11% had > 33% |
Age: no association after 40 yr Overweight: strong association with degree of fatty change; diabetic people were more common in this group Diabetes not independently associated |
| Underwood Ground, 1984280 | Aircraft, motor vehicle accidents, all men; all accidents associated with alcohol, other illnesses excluded (166) | 21% | Questioned possible irregular use of alcohol as factor; discussed increased carbohydrate intake relationship to fatty liver disease |
| Wanless, 1990176 | Hospitalized, 207 obese patients, 144 matched, nonobese controls; alcoholic patients excluded (351) | Steatosis: 36.3% overall, 29.2 obese, 7.1% lean Steatohepatitis: 18.5% obese, 2.7% lean |
Overweight: prevalence and grade of steatosis and steatohepatitis correlated with grade of obesity Diabetes: increased the risk of steatohepatitis and fibrosis |
| Schwimmer et al, 2006231 | Consecutive pediatric and adolescent autopsies in a single county, nonselected (742) | Steatosis: 13% overall 2-4 yr: 0.7% 15-19 yr: 17.3% |
Age: fatty liver incidence increased with age Obesity: 38% had fatty liver Ethnicity: Hispanic (11.8%), Asian (10.2%), white (8.6%), African American (1.5%) |
In summary, the true prevalence rate of NAFLD remains unknown because there are no reliable or specific noninvasive tests available for an accurate diagnosis or exclusion of this process. A review of noninvasive markers in NAFLD, NASH, and fibrosis indicates the efforts in the field.21 Although liver biopsy evaluation is considered the gold standard of diagnostic tests, it cannot be considered an appropriate screening tool. Results from biopsy studies reinforce the observations that not all obese individuals have hepatic steatosis8,95,96 and that unexplained elevated ALT values cannot be assumed to indicate fatty liver disease. For example, Skelly and colleagues97 showed that as many as one third of 354 biopsies performed for unexplained elevated ALT levels were histologically normal or had abnormalities because of liver diseases other than NAFLD or NASH. Recommendations for the focused role of liver biopsy for patients with suspected NAFLD or NASH were discussed in the practice guidelines from the American Association for the Study of Liver Diseases59 and in a consensus conference of the European Association for the Study of the Liver.69
Familial Associations and Risk Factors
The genetic predispositions found in NAFLD are exemplified by differences in the incidence of other risk factors, such as obesity, diabetes, hypertension, and hyperlipidemia, among various ethnic populations. Studies have documented fatty liver disease and cirrhosis among kindreds.98 One study of 20 patients showed a trend toward familial clustering and maternal linkage for insulin resistance among patients with NAFLD.99
Type 2 diabetes and obesity are risk factors for progressive disease in clinical100 and histologic studies.8,101,102 Various possible environmental and genetic factors have been proposed for NAFLD,13,103 including the type of diet, exercise, smoking,104 the role of small bowel bacterial overgrowth, endotoxin and related immune or cytokine responses, polymorphisms for genes that control fatty acid oxidation (e.g., microsomal transfer protein), and fibrosis (i.e., angiotensinogen and TGF-β).
Broad, nonexclusive categories of genes under investigation as potential risk factors for NAFLD105 include those involved in the control of adiposity and insulin resistance (e.g., peroxisome proliferator–activated receptor-γ [PPAR-γ], central versus peripheral fat depots); determinants of hepatic steatosis (i.e., free fatty acid delivery and de novo lipogenesis, processing, and egress); hepatic fatty acid oxidation pathways (e.g., mitochondrial, peroxisomal, microsomal); hepatic oxidative stress mechanisms (e.g., production, defense); cytokine effects (e.g., TNF-α, IL6, adipokines); and determinants of progression of fibrosis. One genomic and proteomic study of 98 bariatric patients found downregulation of genes involved in defense against oxidative stress in NAFLD cases and upregulation of genes associated with fibrogenesis and apoptosis in steatohepatitis cases.106
Genome-wide association and candidate gene studies have identified a variant in PNPLA3, which encodes adiponutrin, an insulin-responsive phospholipase involved in lipolysis and lipogenesis that correlates with accumulation of hepatic triglyceride and some histologic features of severity of NASH with identical ethnic distributions in prevalence studies.107–112 This association is not limited to adults.112,113 The effects cannot be shown to be direct, and it is speculated the allelic variant results in sensitization of the liver to metabolic and environmental stressors.20
Pathogenesis
Certain similarities (i.e., steatosis, oxidative damage, and cytokine response) and dissimilarities between the pathogenesis of NAFLD and ALD are highlighted in Figure 49.17. The most commonly discussed driver of NAFLD is insulin resistance and the associated lipotoxicity (i.e., fat in nonfat depots) and complex pathways of systemic and local inflammation.114 The metabolic system of insulin involves signaling among skeletal muscle, adipose tissue, and liver.115–118
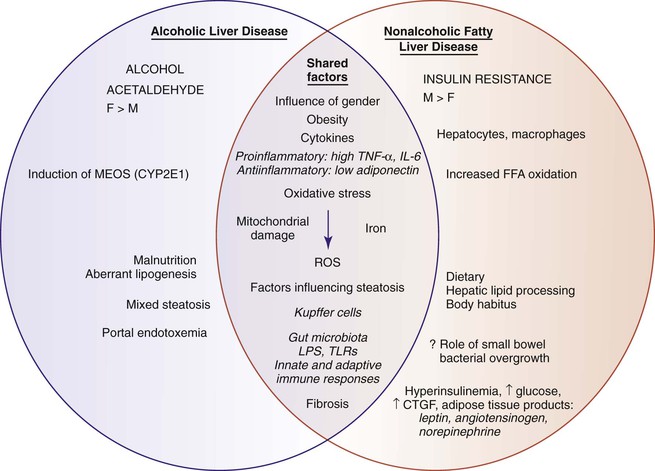
The manifestations of insulin resistance depend on the type of tissue involved. In visceral adipose tissue, there is unabated release of free fatty acids into portal venous blood. In peripheral muscle, insulin resistance manifests as an inability to use circulating glucose. In liver, continuation of glucose production (i.e., gluconeogenesis) combines with a lack of glycogen storage in the setting of hyperinsulinemia and hyperglycemia. Hepatic steatosis, which corresponds to an accumulation of triglycerides in hepatocytes, results from an imbalance between the delivery of free fatty acids and endogenous lipogenesis and fatty acid disposal through oxidation and packaging of esterified fatty acids and triglycerides for export by apolipoproteins as very-low-density lipoproteins. Each of these steps involves complex, genetically regulated pathways.
Current work shows that triglyceride accumulation is a mechanism of sequestration of potentially damaging free fatty acids and that nontriglyceride fatty acid metabolites likely drive subsequent necroinflammatory lesions recognized as NASH.119 Our understanding of pathogenesis of NAFLD and NASH also includes the roles of altered gut microbiota,120 intestinal permeability and lipopolysaccharide (LPS) exposure,121 differential macrophage activation in adipose tissue depots,122 innate immunity,123,124 and supporting activation by inflammasomes in obesity-related liver injury.125
Visceral (abdominal) adipose tissue is an energy storage depot and a source of highly active proteins, collectively referred to as adipokines or adipocytokines (Fig. 49.18). In NAFLD, adipokines such as TNF-α (elevated in NAFLD) and adiponectin (decreased in obesity and NAFLD), proinflammatory IL6, proinflammatory and profibrogenic derivatives of the renin-angiotensin system,126 and resistin may be involved in the pathogenesis of NAFLD. Studies have demonstrated production of selected chemokines by the epithelial components of the ductular reaction in advanced NAFLD.127 Leptin, another adipokine produced by peripheral adipose tissue, serves an important function in the control of satiety and appetite and is essential for fibrogenesis. Because of high levels of circulating leptin, NAFLD may represent a condition of central (brain) leptin resistance.
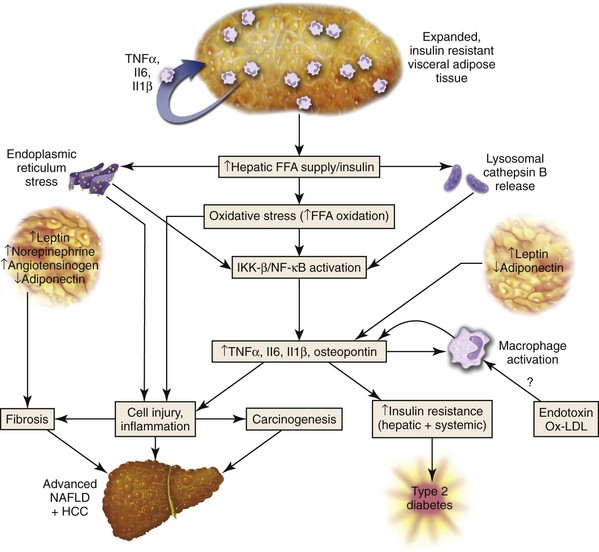
The amount of visceral adipose tissue increases with the age-related loss of estrogens and androgens in women and men. In men, this may be an early step in the alteration of insulin metabolism and progression to the metabolic syndrome.128 In several autopsy studies, increasing patient age correlated with development of hepatic steatosis.
Pathology
Grossly, noncirrhotic fatty livers are typically enlarged and yellow and have a greasy consistency. The cirrhotic liver may be small or enlarged. Fat may be observed in an irregular pattern in liver nodules as yellow areas compared with neighboring tan nodules. Cirrhotic livers in patients with NAFLD may not be as firm, may have the same stroma-to-parenchyma ratio, or may be as micronodular as in ALD.
Features of NAFLD in affected morbidly obese and superobese (BMI ≥ 40 kg/m2) individuals129 may not be the same as in non–morbidly obese patients. Most objective scientific studies are based on the latter, and the following discussion is based on that population unless stated otherwise.
The histologic injury pattern of noncirrhotic fatty liver disease in adults, regardless of cause, initially involves zone 3 hepatocytes (Fig. 49.19). In cirrhosis, steatosis or steatohepatitis may or may not persist. Table 49.6 outlines the key gross and microscopic features of NAFLD compared with ALD.
Table 49.6
Gross and Histologic Features of Alcoholic and Nonalcoholic Fatty Liver Disease in Patients with or without Cirrhosis
| Characteristics | Alcoholic Fatty Liver Disease | Nonalcoholic Fatty Liver Disease |
| Patients without Cirrhosis | ||
| Gross Features | Usually enlarged, soft, yellow, greasy liver | May be indistinguishable from noncirrhotic alcohol-induced liver disease |
| Histologic Features | ||
| Macrovesicular steatosis, zonal or diffuse | ± (Zone 3 or diffuse) | Required: zone 3 or diffuse in adults; diffuse or zone 1 in children |
| Mixed large and small droplet steatosis | ± | ± |
| Nonzonal patches of true microvesicular steatosis | ± | ± |
| Steatohepatitis | ± | ± |
| Hepatocellular ballooning | ± | ± |
| Acidophil bodies | + | + |
| Megamitochondria | ± | ± |
| Mallory-Denk bodies, zone 3 | ± | ± (in ballooned hepatocytes when present) |
| Thick, ropy | More likely | Less likely |
| Thin, wispy | Less likely | More likely |
| With satellitosis | More likely | Less likely |
| Portal chronic inflammation | ± | ± (may be prominent in resolution) |
| Portal acute inflammation | ± (accompanies ductular reaction, implies cholangiolitis, pancreatitis) | − |
| Lipogranulomas, portal or lobular | + | + |
| Glycogenated nuclei | ± | ± |
| Ductular reaction | Periportal, pericentral | ± |
| Iron: hepatocellular, zone 1 > zone 3 | ± (may be significant) | ± (usually mild) |
| Iron: RES cells, punctate, panacinar | ± | ± |
| Fibrosis: zone 3 perisinusoidal | ± | ± |
| Dense, diffuse | More likely | Less likely |
| Delicate | Less likely | More likely |
| Perivenular fibrosis | + | − |
| Periportal stellate fibrosis | + (with ductular reaction, acute inflammation) | − |
| Alcoholic foamy degeneration (pure microsteatosis) | + | − |
| Sclerosing hyaline necrosis, zone 3 | + | − |
| Veno-occlusive lesions | + | − |
| Canalicular cholestasis, cholestatic rosettes | + | − |
| Patients with Cirrhosis | ||
| Gross Features | Liver firm throughout; may be shrunken or quite large; may be cholestatic; grossly visible foci of parenchymal extinction | Not distinguishable from other hepatitic forms of cirrhosis |
| Histologic Features | ||
| May retain or lose active lesions of steatohepatitis | + | + |
| May be a cause of cryptogenic cirrhosis | + | + |
| Copper deposition, periseptal hepatocytes | ± | Uncommon |
| Hepatocellular iron, RES cell iron (patients without HFE mutation) | Common | ± |
| α1-Antitrypsin globules; increased MZ phenotype (?) | ± | − |
HFE, Hemochromatosis gene; MZ alleles, reduce α1-antitrypsin levels and impair liver function; RES, reticuloendothelial system; +, present; ±, present or absent; −, absent.
Steatosis
By definition, steatosis is identified in 100% of NAFLD cases. The minimum criterion for NAFLD is a finding of at least 5% macrosteatosis. In adults, steatosis commonly begins in zone 3 hepatocytes, which contrasts with the zone 1 steatosis in viral hepatitis C (Fig. 49.20) or the panacinar pattern seen in pediatric NAFLD.
Assessment of the degree of steatosis is often based on a semiquantitative scale by evaluating the percentage involvement of parenchyma, which is expressed as grades. This may also be accomplished by classifying the degree of involvement of the hepatic lobules using three grades (0% to 33%, 33% to 66%, and > 66%130) or by using a four-grade system to account for a 5% normal figure (<5%, 5% to 33%, 34% to 66%, and > 66%).131 Other quantitative methods proposed include digital image analysis,132 morphometry,133 and stereologic point counting134; these approaches are less practical for daily use.
Rarely, steatosis may occur without any inflammation; this condition is called isolated steatosis. More often, there are rare, scattered microgranulomas (i.e., Kupffer cell aggregates) or foci of chronic inflammation in the lobules, which may be called steatosis with inflammation.3 Hepatocellular injury, portal inflammation, and fibrosis raise considerations of additional processes.
Steatohepatitis
The minimal criteria for steatohepatitis are steatosis (≥5%), hepatocellular ballooning, and lobular inflammation (Fig. 49.21). Similar to other forms of chronic liver disease, fibrosis is not required to establish a diagnosis of steatohepatitis. In the initial phases, steatosis is centered in zone 3; with progression, steatosis may involve the entire hepatic lobule. Azonal steatosis may be seen in conjunction with advanced fibrosis.135 Nonzonal patches of microvesicular steatosis may occur in NAFLD (see Fig. 49.13),135 and this correlates with disease severity,136 as do the degree and zonality of steatosis.137
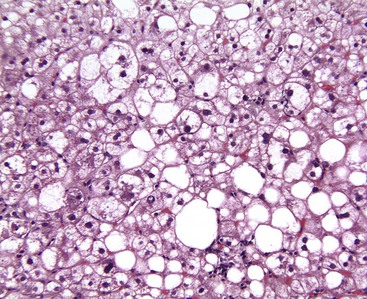
Several histologic types of hepatocellular injury may be seen with routine stains in specimens from patients with NAFLD. They include ballooned hepatocytes, apoptotic (acidophilic) bodies, or foci of spotty necrosis in which remnants of hepatocytes that have undergone lytic necrosis are surrounded by mononuclear cells and Kupffer cells. Areas of confluent and bridging necrosis are rare in NAFLD, which is rarely a cause of submassive or massive hepatic necrosis.138
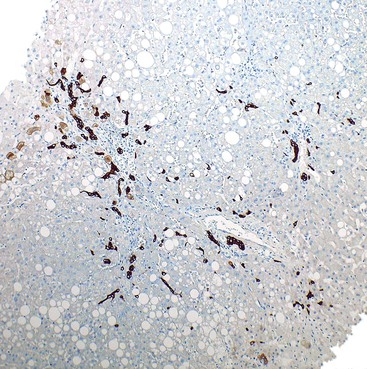
Hepatocyte ballooning is considered an essential, albeit challenging, histologic feature of steatohepatitis.7 Ballooned hepatocytes are enlarged hepatocytes with rarefied reticulated cytoplasm and with or without fat droplets and MDBs. Enlarged nuclei are common (see Fig. 49.1). The ballooned cells are located predominantly in zone 3. They are commonly associated with perisinusoidal fibrosis29,130 and secrete profibrogenic factors (see Fig. 49.2).139 Loss of K8 or K18 immunoreactivity with or without ubiquitin staining provides an objective histologic marker for ballooned hepatocytes in steatohepatitis (Fig. 49.22).23,24,29 PMNs may be associated with ballooned hepatocytes that contain MDBs. This satellitosis (see Fig. 49.4) is less common in NAFLD than in ALD. In rare cases, hepatocyte ballooning may occur in the absence of significant steatosis or inflammation.135 Specific immunophenotypes of activated lymphocytes correlate with disease activity.140
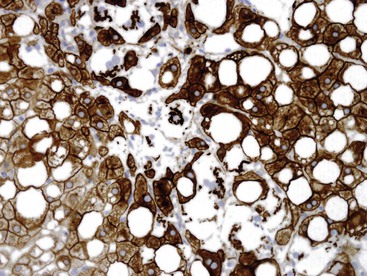
The number of apoptotic hepatocytes correlates with disease activity.141 Serum and tissue markers of apoptosis are being evaluated as potential noninvasive diagnostic tools for fibrosis and NASH.142 One study of NAFLD showed that positivity for the M30 antibody, which is directed against a caspase 3–cleaved K18 product, was identified in steatohepatitis but not in steatosis or in normal liver biopsies.143
The degree of lobular inflammation in NASH is commonly mild and can be mixed acute and chronic or predominantly chronic. Inflammation results from innate and adaptive immune responses.144 CD68-positive Kupffer cells are increased in zone 3.145 Infrequently, zone 3 chronic inflammation may obscure the outflow vein.135 Scattered lobular microgranulomas and lipogranulomas are commonly observed. The former consist of clusters of Kupffer cells that are positive for periodic acid–Schiff stain with diastase digestion (d-PAS) and CD68; the latter consist of fat droplets surrounded by mononuclear cells or eosinophilic PMNs (see Fig. 49.3). Large portal or perivenular lipogranulomas are common in NAFLD. Lipogranulomas are often associated with a small amount of fibrosis, which should not be included in the assessment of disease stage (discussed later). PMNs may be found in the lobules, and clusters near the terminal hepatic venule are common in surgically obtained biopsies, a condition referred to as surgical hepatitis.146
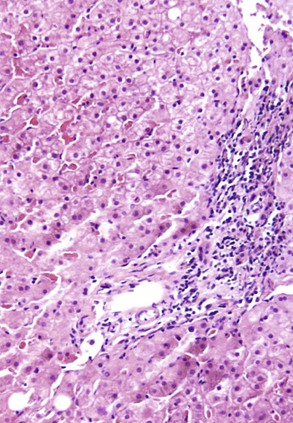
Mild to moderate portal chronic inflammation may be seen in the active and resolving phases of steatohepatitis (Fig. 49.23).130,147 However, when portal chronic inflammation is disproportionately greater than that in the lobules, the pathologist should suspect a superimposed chronic liver disease, such as chronic viral hepatitis.148,149 Increased portal inflammation in untreated patients with NAFLD correlates with increased histologic activity and clinical severity, and if more than mild, it may be considered a marker of advanced disease.150,151 Interpretation of more than mild portal chronic inflammation in NAFLD warrants seeking clinical information about possible prior treatment and concurrent disease. The finding of portal PMNs and cholangiolitis indicates alcohol-induced liver disease or biliary obstruction.21,37
Fibrosis and Cirrhosis
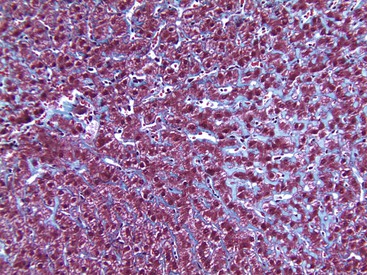
The characteristic pattern of fibrosis in adult patients with noncirrhotic steatohepatitis is zone 3 pericellular or perisinusoidal (i.e., chicken wire fibrosis), which results from deposition of collagen and extracellular matrix in the space of Disse (see Figs. 49.6 and 49.7). This type of fibrosis may be associated with steatohepatitis, and when found in isolation, it probably suggests healed prior episodes of steatohepatitis. Dense, extensive perisinusoidal fibrosis associated with portal hypertension, as occurs in patients with ALD, has not been described in patients with NAFLD. Diabetic hepatosclerosis, another change that correlates with severe extrahepatic organ damage in patients with type 1 diabetes, is not seen in NASH. It is characterized by patchy, dense perisinusoidal collagen and basement membrane deposition in the absence of steatosis (Fig. 49.24).152 With progression, fibrosis develops in the periportal region (see Fig. 49.9), followed in some cases by bridging fibrosis, which can be in a central-central, central-portal, or portal-portal pattern.
NAFLD-associated cirrhosis is most commonly macronodular or mixed. After cirrhosis is established, active features of steatohepatitis and perisinusoidal fibrosis may no longer be apparent. This fact has been well documented in studies of serial liver biopsies153 and supports the theory that NAFLD is a major cause of otherwise cryptogenic cirrhosis.154 Nevertheless, in some patients, active steatohepatitis may be observed in cases of established cirrhosis.155
Periportal and bridging fibrosis are accompanied by a ductular reaction,156,157 which consists of a proliferation of K7- and/or K19-positive hepatic progenitor cells, poorly formed ductular structures, increased stromal matrix, and usually mild acute inflammation (Figs. 49.25 and 49.26).158–160 Isolated portal fibrosis (i.e., portal or periportal fibrosis without evidence of coexisting zone 3 perisinusoidal fibrosis) has been described in a subset of morbidly obese adults who have undergone bariatric surgery,96,161 but whether this finding represents a lesion within the spectrum of NAFLD common to nonmorbidly obese patients or pathologic resolution of injury in morbidly obese individuals has not been explored.
Other Pathologic Features of Nonalcoholic Fatty Liver Disease
Mallory-Denk Bodies.
In NAFLD, MDBs are usually identified in ballooned hepatocytes, in which they often appear wispy, poorly formed, or delicate. Masson trichrome stain may be used to aid identification of MDBs (see Fig. 49.6). Immunohistochemical staining for K8 or K18 (see Fig. 49.22) ubiquitin may be positive in MDBs.32 Increased numbers of MDBs correlate with higher degrees of necroinflammatory activity in patients with NAFLD130 and confirm the diagnosis of steatohepatitis.60
Megamitochondria.
Megamitochondria (i.e., giant mitochondria) are identified by light microscopy as intracytoplasmic, round or cigar-shaped, eosinophilic structures in hepatocytes. They are often apparent in hepatocytes that also contain microvesicular steatosis (see Fig. 49.13). Ultrastructural studies of patients with NAFLD showed that paracrystalline inclusions, loss of cristae, and nonzonality of megamitochondria are characteristic features of NAFLD.162 Megamitochondria in NASH may represent a pathologic process or an adaptive change.163 Megamitochondria are not unique to ALD or NAFLD and can be seen in Wilson disease and pregnancy.
Iron Deposition.
A mild degree of iron deposition (1+) in periportal or periseptal hepatocytes and punctate reticuloendothelial iron may be identified in as many as 35% of NAFLD patients21 and may be related to the severity of disease.164,165 Iron accumulation is likely the result of dysmetabolic iron overload from altered regulation of iron transport associated with steatosis and insulin resistance.166
Glycogenated Nuclei.
Glycogenated nuclei are clear, vacuolated nuclei that are frequently found in NAFLD. They may occur as clusters of cells in zone 1 or scattered in the hepatocyte lobules.167 Glycogenated nuclei are also common in diabetes, Wilson disease, and normal liver biopsies from children. Glycogenated nuclei are of uncertain clinical significance,168 but they may indicate hepatocyte senescence.169
Ductular Reaction.
Ductular reaction is a result of most forms of acute or chronic liver injury.159 In NASH, the process correlates with the amount of portal-based fibrosis. The lesion has been associated with insulin resistance and with hepatocellular156 and ductular senescence.157,160 The ductular reaction is a metabolically active process157,160 that produces profibrogenic cytokines.127 An extensive ductular reaction accompanied by other features of chronic cholestasis, such as feathery degeneration of periportal hepatocytes, abundant deposition of periportal copper, or bile duct injury, should alert the pathologist to the possibility of a superimposed chronic biliary disease.
Hepatocellular Glycogen.
Hepatocellular glycogen can accumulate in the cytoplasm in NAFLD. Glycogen imparts a dull, grayish refractile appearance to the cytoplasm; this may occur in conjunction with steatosis or megamitochondria. In contrast to diffuse involvement by glycogenic hepatopathy, a process that occurs in poorly controlled type 1 diabetes (Fig. 49.27),170,171 the histologic change in NAFLD may affect only focal groups of hepatocytes.
Vascular Alterations.
A small intraacinar arteriolar branch in perivenular areas in zone 3 that was observed in NASH correlated with disease severity. When associated with dense perivenular inflammation, the lack of a visible bile duct helps to avoid misinterpretation of the lesion as an inflamed portal tract.172
Treatment Effects.
Resolution of steatosis, ballooning, lobular inflammation, steatohepatitis, fibrosis, and a decrease in the NAFLD activity score (NAS) have been reported as potential positive effects of treatment (e.g., diet and exercise, medical, surgical) in various clinical trials (see Fig. 49.23).173 Clinical trials have produced two additional observations—spontaneous resolution of the histologic features of NAFLD may occur, and even after clinically successful treatment, the degree of portal inflammation may increase.174 Laboratory values147 and histologic findings175 may provide evidence of recurrence several weeks after cessation of successful medical therapy, indicating the underlying systemic nature of the process.
Prognostic Lesions.
Table 49.7 summarizes the results of clinicopathologic studies that have correlated the histologic features of NAFLD with fibrosis.155,176–180 Using immunohistochemistry for activated hepatic stellate cells, others have demonstrated a correlation with advanced fibrosis in NAFLD biopsies.181–183 Practical application of this test awaits further validation. A meta-analysis of studies done with various methods identified lobular inflammation as a prognostic feature for fibrosis.179 Portal chronic inflammation is associated with advanced disease histologically and clinically.150,151 Other studies have highlighted microvesicular steatosis or the amount and location of steatosis.136,137 One immunohistochemical study highlighted the loss of K8 and K18 and positivity for ubiquitin in hepatocytes, which are markers of ballooning, as correlative with fibrosis.29
Table 49.7
Histologic Features of Nonalcoholic Fatty Liver Disease Reportedly Associated with Fibrosis or Cirrhosis
| Study | N | Steatosis | Lobular Inflammation | Ballooning/Mallory-Denk Bodies | Other |
| Wanless, 1990176 | 207 | Yes | NA | NA/NA | |
| Matteoni et al, 19993 | 98 | Yes | Yes | Yes/yes | |
| Angulo et al, 1999101 | 144 | No | No | Yes | |
| George et al, 1998177 | 51 | Yes | Yes | NA | Stainable iron; C282Y HFE mutation |
| Shimada et al, 2002155 | 81 | Yes | No | Yes | |
| Ratziu et al, 2000178 | 93/14* | Yes | Yes (with polymorphonuclear cells) | Yes | Four progressors: no evidence of portal fibrosis in first biopsy; necroinflammation in progressors only |
| Garcia-Monzon et al, 2000281 | 46 | Yes | Yes | NA | |
| Gramlich et al, 2004282 | 132 | Yes | Yes | Yes/yes | |
| Fassio et al, 2004283 | 106/22* | No | No | NA | Steatosis decreased or no change with fibrosis progression |
| Adams et al, 2005194 | 103/103* | No | No | No/no | Low initial fibrosis stage associated with higher rate of progression |
| Ekstedt et al, 2006195 | 129/68* | Yes (in first biopsy) | No | No/no | 100% negative predictive value for progression when less than stage 2 in first biopsy |
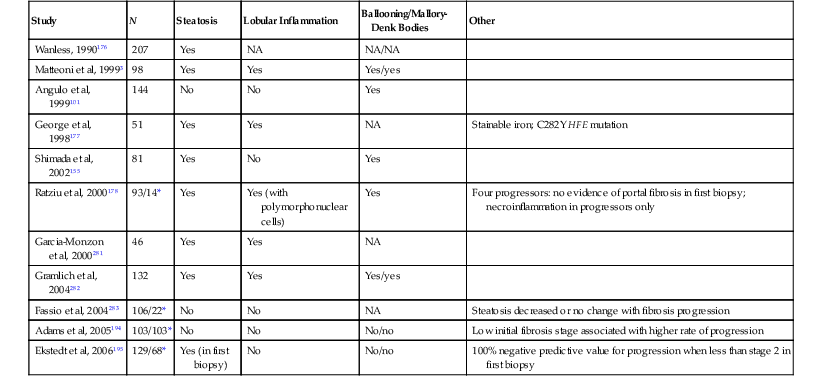
* Repeat biopsies.
HFE, Hemochromatosis gene; NA, not applicable.
There is strong interest in the development of serum biomarkers and noninvasive imaging tests to diagnose NASH and establish fibrosis in NAFLD.184 In patients with NAFLD, the metabolic syndrome was identified as a marker of histologic activity.185 Common features to many studies are older age, diabetes mellitus, degree of insulin resistance, and elevated aminotransferase levels in morbidly obese and nonmorbidly obese populations.8,178,179,186,187 The ratio of PMNs to lymphocytes correlates with histologic activity and fibrosis in liver biopsies.188
A proteomic approach has shown weight-dependent variations in diagnosed NAFLD and NASH.189 A meta-analysis of single nucleotide polymorphism (SNP) studies has shown that the I148M variant of the PNPLA3 gene is associated with increased histologic severity and fibrosis in NAFLD.20 This area of investigation will continue to grow because liver biopsy is not a practical screening approach.
Natural History and Treatment
Long-term studies of the natural history of NAFLD are challenging and few, but most authorities agree that steatosis alone is nonprogressive,190 whereas fibrosis progresses in 31% to 42% of individuals during a period of 3.2 to 13.7 years to cirrhosis and its complications of liver failure and hepatocellular carcinoma.191–193 In other patients, fibrosis remains stable or regresses.194,195 Some patients have had regression of NASH to NAFLD (i.e., steatosis) without specific treatment196 or after weight reduction.180
The literature is mixed in terms of the effect of NAFLD on overall survival. Some studies have shown no difference compared with the general population for individuals with cirrhosis from other forms of chronic liver disease.195,197 In contrast, studies from multiple countries have shown that patients with NAFLD have a worse overall survival rate.191,194,198 Compared with patients with advanced chronic hepatitis C, patients with NAFLD with advanced fibrosis or cirrhosis have lower rates of liver-related complications and hepatocellular carcinoma197 but similar overall mortality rates.199 Obesity,101 increasing patient age,101,178 and diabetes8,101,102,200 are risk factors for progression of steatosis to steatohepatitis or cirrhosis. Caffeine consumption is protective against the development of fibrosis.201,202 Cardiovascular disease may be the leading cause of death of patients with advanced NAFLD.195,203,204
Whether nonfibrotic, non-NASH NAFLD is necessarily a precursor that progresses or steatosis and NASH with fibrosis are separate entities is debated.119 Among patients with steatosis, only 2% to 20% have steatohepatitis or cirrhosis.92 Moreover, Cirrhosis (15% to 25%), liver failure, or hepatocellular carcinoma do not develop in all patients with steatohepatitis.205,206 Many succumb to cardiovascular complications.203
Problems related to biopsy sampling errors and the various definitions of steatosis, steatohepatitis, and NAFLD have created difficulties in natural history studies. NASH-related cirrhosis or liver failure is the third most common indication for liver transplantation in the United States (9.7% in 2009) and is on a trajectory to becoming the number one indication.207 NASH cirrhosis accounts for 30% to 40% of liver-related deaths,208 whereas steatosis without steatohepatitis or cirrhosis is considered a more benign process.192,194
The risk of hepatocellular carcinoma among patients with NASH is not always recognized in general practice,209 but it is a growing concern.4,205,206 Although it is lower than that for HCV-related cirrhosis in comparative series,197,210,211 screening only cirrhotics is recognized as inadequate, and several groups have reported hepatocellular carcinoma in noncirrhotic patients.4,206,212,213 Benign liver tumors such as adenoma and adenomatosis have been reported for patients with various degrees of NAFLD.214,215
Trials of NAFLD treatment have not been a great success, with the possible exception of weight loss surgery.216 Less drastic therapy and trials are aimed primarily at the presumed causes of disease and have included lifestyle intervention with weight loss and exercise, control of insulin sensitivity, and interventions broadly directed at inflammation, hepatocyte injury and fibrosis, and hyperlipidemia. Several reviews detail the trials and results.59,217,218
Some common forms of drug intervention include the insulin-sensitizing PPAR-γ agonist pioglitazone with or without antioxidants; antidiabetic drugs such as metformin and thiazolidinediones; the antioxidants betaine, ursodeoxycholic acid, vitamins E and C; antifibrotic angiotensin-converting enzyme inhibitors and receptor blockers; and the TNF-α antagonist pentoxifylline.
Fatty Liver Disease in Various Patient Groups
Concurrent Alcoholic and Nonalcoholic Fatty Liver Disease
The coexistence of NAFLD and ALD has been recognized as a significant risk factor for progression.219 However, the overlapping contributory components cannot be histologically separated.
Alcohol use by individuals with NAFLD remains controversial. Modest alcohol intake protects against the effects of insulin resistance in morbidly obese individuals8 and is associated with a decreased prevalence of steatohepatitis in nonmorbidly obese patients with NAFLD.9 However, moderate alcohol consumption correlates with progression of fibrosis in NAFLD.220
Fatty Liver Disease in Patients with Other Liver Disorders
Clinical complications of acute viral hepatitis A, B, and C; chronic hepatitis C; and acetaminophen toxicity are seen in patients with alcoholism. These disorders require careful consideration by the pathologist when examining liver biopsy material with unusual findings.
The outcome and histologic features of ALD with concurrent acute viral hepatitis depend on the stage of ALD. Acetaminophen toxicity may not result in the characteristic features of zone 3 bridging necrosis in patients with ALD. The effects of chronic HCV infection are synergistic with those of fibrosis in patients with ALD.18 The histologic features of superimposed hepatitis caused by HCV are similar to those seen in nonalcoholic patients (see Chapter 46). Patients with ALD are also at risk for chronic pancreatitis and biliary obstruction, both of which may manifest histologically with cholangiolitis. The coexistence of ALD with hereditary hepatic iron overload and with α1-antitrypsin deficiency results in findings typical of those disorders in addition to those of ALD.
The coexistence of NAFLD with other chronic liver diseases is a growing area of interest because of the high prevalence of NAFLD and its possible influence on the progression of other liver diseases; the high prevalence of chronic HCV infection; the high incidence of steatosis among patients with HCV infection; and the challenges of diagnosing NAFLD in the setting of other chronic liver disorders.148,149,221 Several studies have shown a negative impact of steatosis and insulin resistance on the progression of disease in patients with HCV infection or hereditary hemochromatosis.222 In one series, 2.6% of more than 3000 nonallograft liver biopsies identified steatohepatitis (i.e., steatosis and zone 3 perisinusoidal fibrosis) with a non-ALD or non-NAFLD type of chronic liver disease, such as hepatitis C, hemochromatosis, α1-antitrypsin deficiency, or autoimmune hepatitis.148
Chronic hepatitis C is the most common chronic liver disease diagnosed with concurrent NASH (5% to 9% of chronic hepatitis C cases).148,223 Criteria for the diagnosis of NASH with chronic hepatitis C have not been established, but one recommendation relies on the unique characteristic of NASH zone 3 perisinusoidal fibrosis and hepatocyte ballooning with a zone 3 predilection in addition to steatosis and lobular inflammation.129,148
Rarely, clinical and histologic features of autoimmune hepatitis or primary biliary cirrhosis may be seen in patients with NAFLD.148 Diagnosis of these types of concurrent liver disorders requires more than elevated levels of serum autoantibodies (Fig. 49.28).66
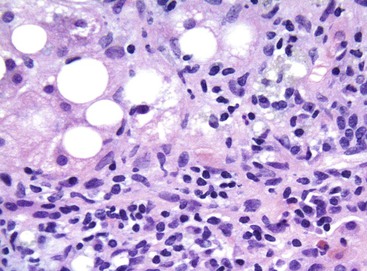
Fatty Liver Disease in Children
A variety of childhood genetic disorders are characterized by hyperinsulinemia, insulin resistance, and chronic liver injury. They include Bardet-Biedl, Alström, and Prader-Willi syndromes; lipodystrophy; and polycystic ovary syndrome.116 Histologic evaluation alone cannot define the cause of fatty liver disease in these cases. Other childhood disorders characterized by steatosis include HCV infection, rapid weight loss or starvation, and inborn errors of metabolism (e.g., urea cycle deficiency, fatty acid oxidation defects, organic acidemia, carnitine deficiency, cystic fibrosis, Wilson disease).
The growing epidemic of childhood obesity has led to increases in the prevalence of obesity-related conditions such as the metabolic syndrome.224,225 NAFLD, which strongly correlates with components of the metabolic syndrome such as visceral adiposity and insulin resistance,81,226 is considered the most common cause of chronic liver disease in children throughout the world.124 The prevalence of NAFLD among children increases with age, and it is higher among adolescent children.227 Changes in sex hormones, insulin sensitivity, and adipose tissue distribution and function228 during development and increased control over unhealthy food choices and increased sedentary lifestyle with age are considered influential factors.229 Equally disturbing for long-term liver disease is the convergence of binge alcohol drinking and adolescent obesity, both of which are increasing.230
A retrospective histologic study of autopsy livers from 742 unexplained deaths in individuals 2 to 19 years of age documented fatty liver disease in 13% overall, which corresponded to 17.3% of children in the 15- to 19-year age range and 38% of obese children.231 The prevalence of fatty liver disease in children varied according to ethnicity: Hispanic (11.8%), Asian (10.2%), white (8.6%), and African American (1.5%).231 This difference could be explained by the genetic background because a common variant of the PNPLA3 gene that is associated with NAFLD in obese children is more common in Hispanics than African Americans.113,232 NAFLD may result in cirrhosis in as many as 5% of affected children,21 and it is associated with shorter survival compared with age- and gender-matched control populations.233
The histologic features of pediatric NAFLD, especially in the younger children, are different from those of adults (Fig. 49.29).234,235 This fact has been documented repeatedly in studies from the United States,234 Europe,236 and Japan.237 For instance, compared with adults, liver biopsies from children with NAFLD show a predominance of portal-based chronic inflammation and fibrosis; more severe, predominantly panacinar (or zone 1) steatosis; lack of ballooning and MDBs; and lack of zone 3 perisinusoidal fibrosis.21 This pattern of injury was identified in 51% of patients in a study by Schwimmer and colleagues,234 who coined the term type 2 (pediatric-type) NAFLD. The histologic pattern of disease characterized by zone 3 accentuation of steatosis was called type 1 (adult-type) NAFLD and was found in only 17% of cases.234 The remainder of cases showed overlapping histologic features (Table 49.8).
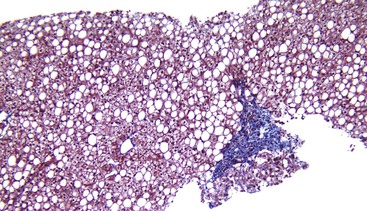
Table 49.8
Histologic Features of Adult and Pediatric Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis
| Histologic Feature | Adults | Children |
| Zone 3 predominance of histologic lesions | + | − |
| Portal predominance of histologic lesions | − | + |
| Steatosis | + | +++ |
| Lobular inflammation | + | ± |
| Portal inflammation | ± | + in most cases |
| Ballooning | ± | − |
| Mallory-Denk bodies | + | ± |
| Portal fibrosis | −* | + in most cases |
| Glycogenated nuclei | ± | ± |
| Zone 3 perisinusoidal fibrosis | ± | − |
* Isolated portal fibrosis occasionally is observed in morbidly obese adult patients with nonalcoholic fatty liver disease.
+, Present; +++, present and usually severe; ±, present or absent; −, absent.
In the study, the type 2 pattern of NAFLD was more common among obese boys, younger individuals, and those of nonwhite ethnicity.234 The switch of the histologic phenotype to an adult pattern in older children may reflect hormonal or endocrine changes related to puberty.227 Further studies from Italy236 and the United States238 confirmed that only a minority of pediatric NAFLD biopsies (2.4% and 6.2%, respectively) showed a zone 3 pattern of steatosis. However, in the same studies, 28.6% and 6.9%, respectively, had the portal-based (type 2) pattern of disease. Most patients (as many as 82% in the U.S. study) had overlapping features.236,238
The NASH Clinical Research Network (CRN) uses the diagnostic categories of definite steatohepatitis, borderline zone 1, borderline zone 3, and not steatohepatitis to cover diagnostic possibilities in NAFLD.135 The two borderline categories are broad enough to apply to pediatric biopsies. Hepatic progenitor cell activation and adipokine secretion by these same cells correlate with disease severity and portal-based fibrosis in pediatric NAFLD.127 Features of NAFLD in children and adults are summarized in Table 49.9.
Table 49.9
Clinical Features of Nonalcoholic Fatty Liver Disease in Adults and Children
| Feature | Adults | Children | Comments |
| Metabolic syndrome* | + | + | Sometimes called insulin resistance syndrome or syndrome X |
| Gender: M > F | Probably | + | Previously thought that F > M among adults |
| Central obesity | + | + | Central obesity is a greater risk because of various metabolic properties of visceral adipose tissue |
| Insulin resistance (calculated) or type 2 diabetes† | + | + | Linked to central obesity; in Asians, obesity measurements reflect different body habitus than in Westerners |
| Low adiponectin levels† | + | + | Nearly a universal association with NAFLD when assayed; adiponectin is lower in obese individuals with NAFLD compared with obese controls without NAFLD (adult and pediatric studies) |
| Ethnicity: Hispanic, Asian, white > African American | + | + | Several large series with different assays have confirmed these findings; poorly understood |
| Polycystic ovary syndrome† | + | + | Linked to insulin resistance |
| Lipodystrophy syndromes, acquired, congenital, and familial; generalized and partial†‡ | + | + | Some forms of lipodystrophy because of leptin deficiency, some to PPAR-γ mutations, and some acquired; all are characterized by different degrees and locations of subcutaneous fat loss, insulin resistance, acanthosis nigricans, polycystic ovary syndrome, and hepatic steatosis or steatohepatitis |
| Acanthosis nigricans† | Rare case reports | + (common) | Skin condition directly linked to insulin resistance |
| Sleep apnea† | + | Possible but not studied | Studies link sleep apnea to obesity, hypoxic events, and insulin resistance. |
| Pituitary or hypothalamic dysfunction† | + | + | Includes postsurgical or posttraumatic injury; adult-onset growth hormone deficiency |
| Petrochemical exposure | + | Well-documented case series from Brazil with biopsy evidence, rechallenge and long-term follow-up; insulin resistance in only 27% of cases | |
| Autoantibodies (ANA, ASMA, AMA)§ | ± | Not reported | ANA is most commonly documented (6-34%); AMA in 2% in only one series; ANA and ASMA in 0.4-15% |
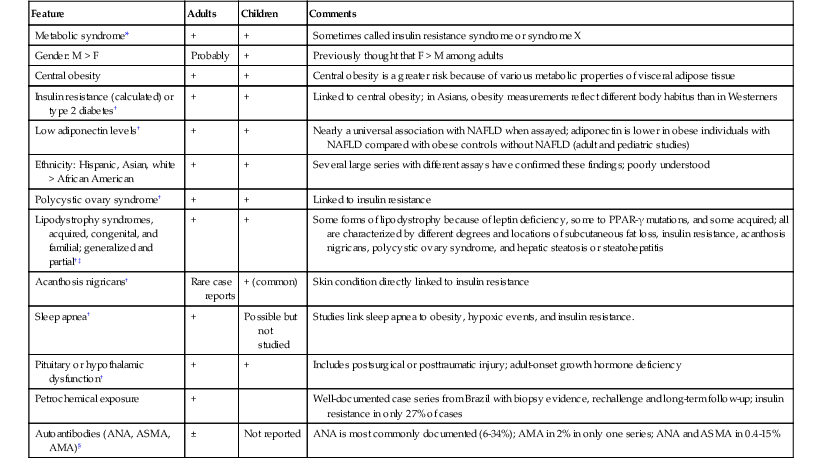
* Metabolic syndrome as defined by the Adult Treatment Panel III.
† Insulin resistance, type 2 diabetes mellitus, and obesity are associated features.
‡ May occur in HIV-positive subjects treated with protease inhibitors. For review of lipodystrophies, see Walker UA, Schott M, Schebaum WA, et al. Acquired and inherited lipodystrophies. N Engl J Med, 2004;351:103-104.
§ Topic reviewed in Brunt EM. Pathology of fatty liver disease. Mod Pathol. 2007;20(suppl 1):S40-S48.
AMA, Antimitochondrial antibody; ANA, antinuclear antibody; ASMA, anti–smooth muscle antibody; F, females; M, males; NAFLD, nonalcoholic fatty liver disease; PPAR-γ, peroxisome proliferator–activated receptor-γ; +, present; ±, present or absent; −, absent.
Differentiating Alcohol-Induced from Nonalcoholic Fatty Liver Disease
A clinical model was developed to help distinguish ALD from NAFLD based on a variety of parameters, such as mean corpuscular volume, AST/ALT ratio, BMI, and gender. An ALD/NAFLD index (ANI) greater than 0 favors ALD, whereas an ANI less than 0 favors NAFLD.239 The same group proposed the use of immunohistochemical markers. Decreased expression of the insulin receptor and increased expression of protein tyrosine phosphatase 1B (PTPN1), a protein that acts as a negative regulator of insulin resistance, was demonstrated in patients with NAFLD compared with those with ALD.240 Neither the clinical model nor the immunohistochemical marker combination have been validated.
Table 49.6 highlights some of the pathologic differences of ALD and NAFLD. Some data suggest that the degree of steatohepatitis is often more severe in ALD than in NAFLD. Numerous MDBs, increased lobular infiltration by PMNs with satellitosis, and acute cholestasis are features more suggestive of ALD than NAFLD.2,241,242 Reliable clinical information is ultimately essential in distinguishing ALD from NAFLD. Figure 49.30 shows their shared and distinct histologic features.
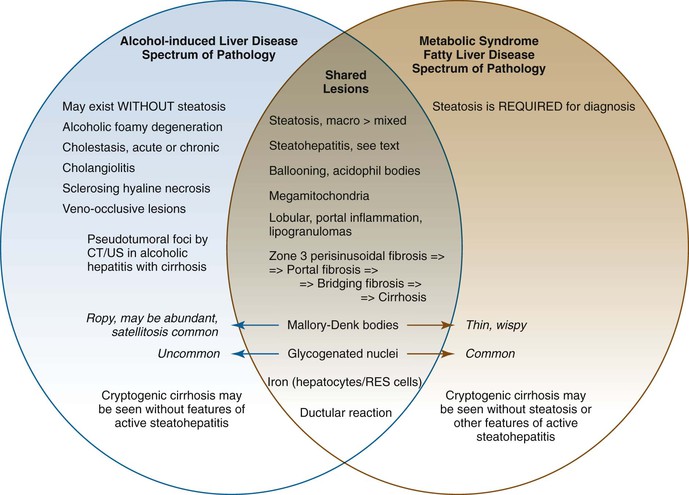
Features That Occur More Frequently in Alcoholic Liver Disease
Sclerosing hyaline necrosis, obliterative and inflammatory lesions of the hepatic outflow veins, alcoholic foamy degeneration, and acute cholestasis have been described in noncirrhotic ALD but not in patients with noncirrhotic NAFLD.129 In contrast to NAFLD, alcoholic hepatitis may occur in the absence of steatosis in ALD.21 Portal lipogranulomas are common in ALD.30 In cirrhosis caused by ALD, regions of parenchymal extinction (septa) are often quite broad, in contrast to cirrhosis caused by NAFLD (see Fig. 49.10).
Features That Occur More Frequently In Nonalcoholic Fatty Liver Disease
In comparative histologic studies, features that occur significantly more often in NAFLD than in ALD include marked steatosis, periportal hepatocellular glycogenated nuclei,36,243 and lipogranulomas.243 However, on an individual basis, the specificity of these histologic features is questionable. There may also be qualitative differences in the type of collagen deposition in NAFLD or ALD. For instance, type I collagen is more common in NAFLD-associated steatohepatitis, whereas type III collagen is more common in ALD.243
Grading and Staging of Fatty Liver Disease
A proposal for a novel histologic score with prognostic value for alcoholic hepatitis is based on the semiquantitative assessment of histologic parameters that are independently associated with short-term survival. They include lobular infiltration by PMNs, bilirubinostasis, megamitochondria, and stage of fibrosis. The score has been validated in two independent cohorts from Europe and the UnitedStates.244
Another study proposed a histologic grading system with predictive ability for alcoholic steatohepatitis based on semiquantification of hepatocyte ballooning and lobular inflammation, but it awaits validation.47 Although no formal semiquantitative pathologic scoring system has been accepted, adaptation of a system used in NAFLD may be applied to ALD.30,37
One popular semiquantitative grading and staging system for assessment of histologic lesions in NASH is based on evaluation of necroinflammatory activity and fibrosis, respectively130 (Tables 49.10 and 49.11). This grading system is based on a semiquantitative evaluation of the degree of steatosis, hepatocyte ballooning, and lobular and portal inflammation. The staging system takes into account the specific pattern of fibrosis unique to NAFLD and concomitant vascular architectural alterations.
Table 49.10
Brunt Grading System for Nonalcoholic Steatohepatitis
| Lesion Grade | Significant Histologic Variables | |||
| Steatosis | Ballooning | Inflammation | ||
| Mild (grade 1) | 1-2 | Minimal | Lobular: 1-2 | Portal: 0-1 |
| Moderate (grade 2) | 2-3 | Mild | Lobular: 1-2 | Portal: 1-2 |
| Severe (grade 3) | 2-3 | Marked | Lobular: 3 | Portal: 1-2 |
| Extent of Steatosis (1-3) | Extent of Lobular Inflammation (0-3) | Extent of Portal Inflammation (0-3) | |
| 1 ≤ 33% | 0 None | 0 None | |
| 2 33-66% | 1 <2 foci/20× field | 1 Mild | |
| 3 ≥ 66% | 2 2-4 foci/20× field | 2 Moderate | |
| 3 >4 foci/20× field | 3 Marked |

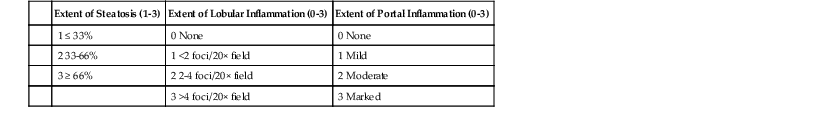
From Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467-2474.
Table 49.11
Brunt Staging System for Nonalcoholic Steatohepatitis
| Stage | Zone 3 Perisinusoidal Fibrosis | Periportal Fibrosis | Bridging Fibrosis | Cirrhosis |
| 1 | Focal or extensive | − | − | − |
| 2 | Focal or extensive | Focal or extensive | − | − |
| 3 | ± | ± | + | − |
| 4 | ± | ± | Extensive | + |
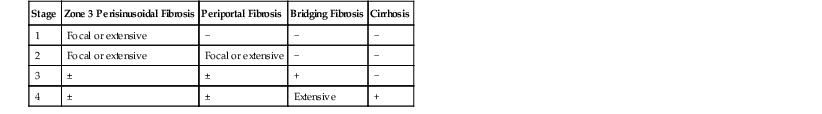
+, Present; ±, present or absent; −, absent.
From Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467-2474.
The NASH CRN, a multicenter consortium sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases, proposed and validated a feature-based scoring system for use in clinical trials (Table 49.12).131 The necroinflammatory grade is referred to as the NAFLD activity score (NAS). The NAS combines the unweighted scores of three individual histologic features: steatosis (0-3), ballooning (0-2), and lobular inflammation (0-2). The total score (0-8) encompasses the entire spectrum of NAFLD, from steatosis to steatohepatitis. Staging subdivides early fibrosis as follows: 1a for delicate zone 3 perisinusoidal fibrosis, 1b for dense zone 3 perisinusoidal fibrosis, and 1c for portal-only fibrosis, specifically for pediatric NAFLD and some cases of bariatric NAFLD. A concurrent validation study confirmed that a diagnosis of steatohepatitis correlated with a NAS of greater than 5 in most cases.
Table 49.12
NASH Clinical Research Network Scoring System for Nonalcoholic Fatty Liver Disease
| Steatosis Grade | Lobular Inflammation | Hepatocellular Ballooning Fibrosis* |
| 0 <5% | 0 None | 0 None |
| 1 5-33% | 1 <2 foci/20× field | 1 Mild (few) |
| 2 34-66% | 2 2-4 foci/20× field | 2 Moderate-marked (many) |
| 3 >66% | 3 >4 foci/20× field | |
| NAFLD Activity Score (NAS) | ||
| 0-8 NAS = steatosis grade + inflammation grade + ballooning grade | ||
| Fibrosis Score* | ||
| 0 None | ||
| 1a Mild (delicate) zone 3 perisinusoidal fibrosis, requires Masson trichrome stain to identify | ||
| 1b Moderate (dense) zone 3 perisinusoidal fibrosis | ||
| 1c Portal fibrosis only | ||
| 2 Zone 3 perisinusoidal fibrosis with periportal fibrosis | ||
| 3 Bridging fibrosis | ||
| 4 Cirrhosis | ||
* Based on the use of Masson trichrome histochemical stain.
NAFLD, Nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
Data from Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313-1321.
NAS has been validated outside the CRN in a retrospective study of liver biopsies from adults with NAFLD.245 The use of the NAS for prediction of subsequent fibrosis has been questioned.246 The numbers generated by NAS were not meant to be a substitute for a histologic diagnosis made by a pathologist.131 Results from a NASH CRN study of 976 adult patients with NAFLD have shown that the diagnoses of definite NASH and semiquantitative NAS are distinct, with correlative but separate meanings, as are the classification systems for grading and staging chronic hepatitis.247 A recently developed scoring system (SAF score) allowing the use of an algorithm (FLIP algorithm)247a for the classification of liver injury in morbid obesity has been validated for use in NAFLD of diverse etiology.247b
Sampling error is an important issue in the histologic diagnosis and semiquantitative assessment of NAFLD.248,249 For example, studies that evaluated biopsies from the right lobe248 or from different lobes95 showed excellent concordance for the grade of steatosis, moderate concordance for assessment of the degree of fibrosis, and fair agreement for the degree of necroinflammatory features. In NASH, as in chronic viral hepatitis,250 the length of the tissue core affects the diagnostic yield, with definite NASH more commonly diagnosed and more extensive steatosis, lobular inflammation, and fibrosis observed in longer than shorter samples.251 Needle diameter also is important.249 Features of NAFLD, especially the degree of necroinflammation and fibrosis, may not be distributed uniformly throughout the liver parenchyma. Pathologists should encourage clinical colleagues to obtain adequate cores of liver tissue and preferably multiple cores. Caution is warranted in interpreting surgically obtained biopsies because the inflammatory foci of PMNs (i.e., surgical hepatitis) render semiquantitative assessment of inflammation challenging.96,146
Four histologic reproducibility studies that assessed interobserver and intraobserver variability in NAFLD in adults have shown good to excellent kappa scores for evaluation of the extent of steatosis, finding of perisinusoidal fibrosis, and degree of fibrosis.131,247b,252,253 In all four studies, the degree of lobular inflammation showed the lowest level of agreement, which is similar to the results of reproducibility studies of patients with chronic hepatitis.254 One study evaluated interobserver and intraobserver variability between a community general pathologist and an experienced hepatopathologist by using the NASH CRN scoring system. Although there was good intraobserver agreement, only fair to good agreement for steatosis, lobular inflammation, and a definite diagnosis of NASH and poor agreement for ballooning and fibrosis staging were found.255 As in chronic hepatitis C, the level of experience of pathologists may play a role in variability.256,247b
Other Forms of Fatty Liver Disease
Drug- and Toxin-Induced Fatty Liver Disease
Less than 2% of cases of steatohepatitis are estimated to be attributable to drug exposure.6 In some instances, presumed drug-related steatohepatitis may be a manifestation of an underlying preexisting disorder. Table 49.13 lists some of the manifestations of fatty liver in non-ALD, non-NAFLD settings. Wilson disease may result in some or all of the histopathologic manifestations of NAFLD or NASH in any age group.
Table 49.13
Fatty Liver Cases Reported in Non-ALD, Non-NAFLD Clinical Settings
| Clinical Setting and Medications | Mechanisms | Pathologic Findings | ||
| Steatosis* | Steatohepatitis | Fibrosis/Cirrhosis | ||
| Medications | ||||
| Nifedipine | Many patients also have features of MS | + | − | − |
| Diltiazem | + | − | − | |
| Coralgil | + | − | − | |
| Tamoxifen | + | + | +/+ | |
| Estrogens | + | − | − | |
| Corticosteroids | + | − | − | |
| Methotrexate | + | − | + (periportal)/+ | |
| Highly active antiretroviral therapy in HIV-positive patients | Lipodystrophy and IR are common; drugs may also cause mitochondrial damage | ++ | + | |
| Amiodarone | Interference with mitochondrial respiration | + (and phospholipidosis, cholestasis, zone 1) | ± (Mallory-Denk bodies) | + (periportal) |
| Toxins, industrial solvents | ± IR and MS | + (often zone 1) | ± | + |
| Childhood syndromes (Bardet-Biedl, Alström, Prader-Willi) | Each includes insulin resistance and hyperinsulinemia | + | + | +/+ |
| Allograft liver (de novo or recurrent disease) | Rejection therapy; inactivity; not well studied | + | + | +/+ |
| Starvation, protein-calorie malnutrition (Kwashiorkor) | Complex pathophysiologic process of lipids and carbohydrates | + (zone 1) | − | − |
| Other nutritional disorders (anorexia, bulimia, cachexia, massive weight loss; pancreatic insufficiency) | ± IR and MS | + | + | Possibly |
| Inflammatory bowel disease | Abnormal nutritional states | + | − | − |
| Celiac disease | Controversial | + | ± | Portal fibrosis, cirrhosis |
| Lipoprotein disorders | Disorders of lipoprotein metabolism | + | ||
| Liver diseases | ||||
| HCV | May cause IR or hepatic steatosis; may be caused by host and/or viral factors | + (as many as 70%; more common in genotype 3) | + (as a concurrent disease process) | +/+ (as a concurrent disease superimposed on HCV) |
| HBV | Not known to cause IR or MS | ± | − | − |
| HDV (Labrea hepatitis) | − | |||
| Wilson disease | ++ | |||
| Tyrosinemia | + | |||
| Galactosemia | + | |||
| Metabolic or endocrine disorders | Complex mechanisms not necessarily related to IR | + | − | − |
| Hypothyroidism | ||||
| Hereditary fructose intolerance | ||||
| Cystinuria | ||||
| Miscellaneous (ischemia, hepatic regeneration, aging) | Not well known | + (often zone 1) | − | − |
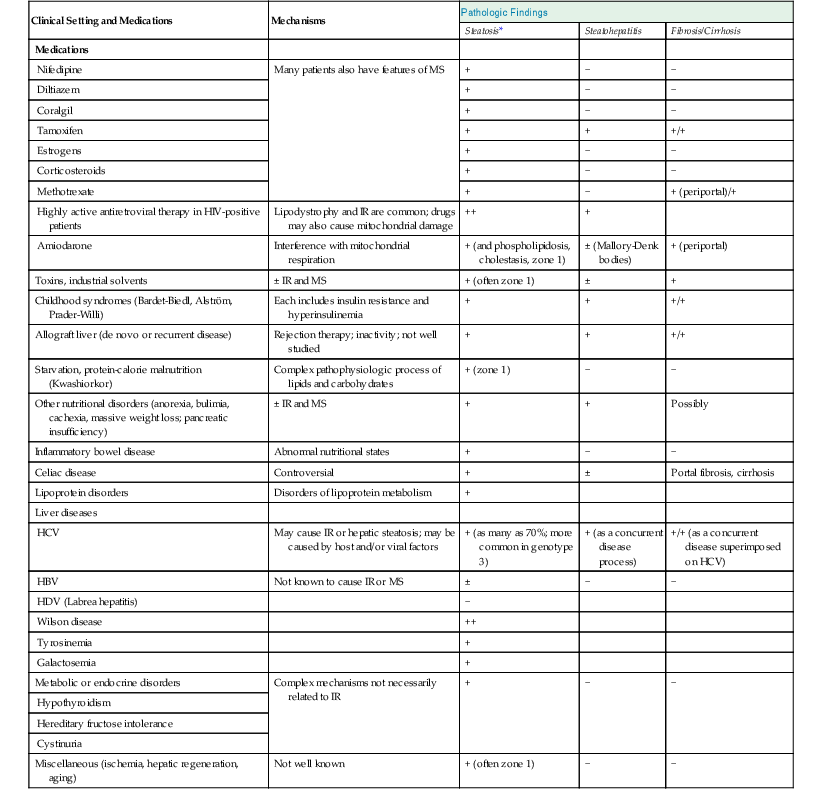
* Steatohepatitis usually includes steatosis and inflammation and may include ballooning and fibrosis.
ALD, Alcohol-induced liver disease; HBV, hepatitis B virus; HCV, hepatitis C virus, HDV, hepatitis D virus; HIV, human immunodeficiency virus; IR, insulin resistance; NAFLD, nonalcoholic fatty liver disease; MS, metabolic syndrome; −, does not occur; +, occurs uncommonly; ++, common; +++, very common; ±, may or may not occur.
Direct hepatotoxicity or mitochondrial injury resulting in steatohepatitis or phospholipidosis has been reported with the use of cardiac medications such as Coralgil, amiodarone, nifedipine, and diltiazem. Tamoxifen and related drugs for breast cancer, estrogens, and corticosteroids can aggravate obesity-related hyperinsulinemia or lipid abnormalities. Methotrexate-induced steatosis may be exacerbated by obesity. The drug’s role in causing steatohepatitis is no longer controversial because the pattern of fibrosis caused by methotrexate is typically periportal, not pericentral, and other common features of steatohepatitis, such as ballooning and MDBs, are not usually associated with this agent. Toxins other than alcohol that may result in steatosis or steatohepatitis include industrial solvents (e.g., paint thinners), petrochemicals, aflatoxins, arsenic compounds, amanitin, rapeseed cooking oil, and cocaine.21
Fatty Liver Disease after Transplantation
ALD and NAFLD may occur in the allograft liver as a recurrence of the original disease or in the form of de novo liver disease. For patients with ALD who continue to drink alcohol after transplantation, the full range of symptoms may occur.257
Determination of de novo or recurrent disease related to NAFLD is more complex. Steatosis may recur in 25% to 100% of patients who undergo transplantation for NASH-related cirrhosis or cryptogenic cirrhosis. NASH recurs in 4% to 37% of these cases and progresses to advanced fibrosis and cirrhosis in some.258–260 De novo steatohepatitis may occur in allograft livers, and in some affected patients, steatohepatitis may progress to advanced fibrosis and cirrhosis.261,262 Risk factors are similar to those for patients not undergoing transplantation. In concert with decreased physical activity levels, obesity, diabetes, hypertension, and hypercholesterolemia play a role in the development of fatty liver disease after transplantation.207,263
The risks of transplantation include the effects of systemic immunosuppression. For instance, bolus treatment with corticosteroids is a well-recognized risk factor for fatty liver disease in transplant recipients. Other antirejection agents, such as cyclosporine, are diabetogenic and promote hypertension and hypercholesterolemia.261 Recurrent HCV infection promotes insulin resistance and steatosis. The high frequency of steatosis (≤88%) and steatohepatitis among patients who undergo liver transplantation for cryptogenic cirrhosis favors NAFLD as a major cause of cryptogenic cirrhosis in the general population.259,264
Protein-Energy Malnutrition
Hepatic steatosis occurs only in the most severe form of protein-calorie starvation, such as kwashiorkor, a disease that primarily affects the visceral protein compartments of the body. Marasmus affects the somatic (skeletal muscle) protein compartments, but the liver is not affected. In kwashiorkor, steatosis develops initially in periportal (zone 1) hepatocytes and may progress to involve the entire acinus in severe cases. On reintroduction of appropriate dietary protein, zone 3 steatosis is the first to resolve. Necroinflammatory lesions, fibrosis, and cirrhosis do not occur in the absence of concomitant liver disease. Steatohepatitis is also highly unusual in this disorder.1
Some types of dietary disorders, such as anorexia, bulimia, cachexia, massive (rapid) weight loss, and uncontrolled inflammatory bowel disease, have been associated with various degrees of hepatic steatosis. However, steatohepatitis and fibrosis are uncommon in these conditions.1
Total Parenteral Nutrition
Steatosis, steatohepatitis, and progressive fibrosis are recognized side effects of total parenteral nutrition (TPN)265 that more commonly manifest in older children and adults than in infants or young children, in whom features of cholestatic liver disease more often develop.266 Steatosis that typically is most prominent in zone 1 hepatocytes develops in adults on TPN. Zone 3 cholestasis and ductopenia also may be identified.
Celiac Disease
The association of celiac disease with the development of fatty liver disease is controversial. Some patients with celiac disease may have elevated aminotransferase levels and hepatic steatosis, but they are typically underweight, not overweight. However, in a few reported cases of NAFLD with steatohepatitis patients had concurrent celiac disease in which the liver disease responded positively to a gluten-free diet.267 Increased intestinal permeability and small intestinal bacterial overgrowth may be the underlying factor for the development of NAFLD in these cases.268
Viral Hepatitides
Viral hepatitides, particularly HCV and hepatitis D virus (HDV) infections, are often associated with significant hepatic steatosis. Many papers discuss the variables associated with steatosis in hepatitis C, including the status of the patient (e.g., BMI, metabolic syndrome), HCV-induced insulin resistance, properties of certain HCV genotypes (particularly GT 3 and GT 1),223,269 and coinfection with human immunodeficiency virus (HIV).270 HCV has been strongly implicated in altered host lipid metabolism.271 Infection with HDV (i.e., Labrea hepatitis) is frequently associated with significant hepatic steatosis. HBV-related steatosis may be as common as in the general population and is related mainly to nonviral host factors.272
Miscellaneous Causes of Fatty Liver Disease
Macrovesicular steatosis may be found with inflammatory bowel disease,265 ischemia, heat stroke, liver regeneration, and aging. Tyrosinemia and galactosemia result in steatosis and fibrosis/cirrhosis in childhood. In all of these conditions, the steatotic changes are primarily located in zone 1 hepatocytes. Lipoprotein metabolism disorders such as abetalipoproteinemia, familial hypobetalipoproteinemia, Dorfman-Chanarin syndrome (i.e., abnormal lipid storage), and pseudoneonatal adrenoleukodystrophy are associated with the development of macrovesicular steatosis of the liver.21,273
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Fatty Liver Disease
Elizabeth M. Brunt
Dina G. Tiniakos
Nonalcoholic Fatty Liver Disease
Fatty Liver Disease in Various Patient Groups
Concurrent Alcoholic and Nonalcoholic Fatty Liver Disease
Fatty Liver Disease in Patients with Other Liver Disorders
Fatty Liver Disease in Children
Differentiating Alcohol-Induced from Nonalcoholic Fatty Liver Disease
Features that Occur More Frequently in Alcoholic Liver Disease
Features that Occur More Frequently in Nonalcoholic Fatty Liver Disease
Grading and Staging of Fatty Liver Disease
Other Forms of Fatty Liver Disease
Drug- and Toxin-Induced Fatty Liver Disease
Fatty Liver Disease after Transplantation
Miscellaneous Causes of Fatty Liver Disease
Introduction
Definitions
Steatosis, defined as the accumulation of triacylglycerides in hepatocytes, is a frequent finding in most liver biopsies.1 As much as 5% of the liver parenchyma may be composed of lipid. By convention, any quantity of lipid that occupies more than 5% of the liver mass is considered pathologic.
Macrovesicular steatosis is an accumulation of lipid droplets of various sizes in hepatocytes in which the cell nuclei are displaced peripherally. It is common to observe small and large droplets in the same cell and to find isolated droplets of lipid.
Microvesicular steatosis has a different appearance and clinical significance. The nuclei of affected hepatocytes are typically centrally located within abundant, foamy-appearing cytoplasm. Special stains, such as Oil Red O, may be necessary to confirm lipid accumulation, whereas in macrovesicular steatosis, the large and small lipid droplets are easily detectable by routine hematoxylin and eosin stain. Diseases associated with microvesicular steatosis are characterized by liver failure and hepatic encephalopathy rather than chronicity and potential fibrosis; the former features are the result of mitochondrial β-oxidation defects. The most common examples of purely microvesicular steatosis are acute fatty liver of pregnancy and Reye syndrome. This process has also been reported in the setting of drug toxicity. Drug-related entities are discussed in Chapter 48.
Alcohol-induced liver disease (ALD) refers to the spectrum of liver disorders directly related to excessive alcohol use. This chapter focuses primarily on the pathologic lesions of the ALD subset characterized by accumulation of fat in the liver parenchyma.
Nonalcoholic fatty liver disease (NAFLD) occurs without significant alcohol use. The clinicopathologic entity is characterized by a variety of hepatic parenchymal injury patterns with more than 5% macrovesicular steatosis.
The term nonalcoholic steatohepatitis (NASH) is in the same clinical setting as NAFLD, but NASH is associated with specific pathologic lesions in the liver. The lesions include various amounts of macrovesicular steatosis, hepatocellular injury (most commonly seen as ballooning), and inflammation in the hepatic lobules or portal tracts, or both. These lesions and others that may be seen in steatohepatitis are discussed in this chapter.
Nomenclature
The term ALD refers to a spectrum of liver diseases and includes large or mixed large and small droplet macrovesicular steatosis with or without lobular and portal inflammation; steatohepatitis with or without fibrosis; alcoholic hepatitis; alcoholic cirrhosis with or without steatosis, steatohepatitis, or alcoholic hepatitis; and alcoholic foamy degeneration. Hepatocellular carcinoma is a recognized consequence of ALD and cirrhosis.
NAFLD has a more limited pathologic spectrum than ALD. NAFLD may manifest as large droplet or mixed large and small droplet macrovesicular steatosis with or without lobular or portal inflammation; steatohepatitis with or without fibrosis; and cirrhosis with or without steatosis or steatohepatitis. All of the features of NAFLD may be seen in ALD, prompting use of the term nonalcoholic fatty liver disease.2,3 Hepatocellular carcinoma may arise as a consequence of NAFLD-induced cirrhosis and is therefore included in the spectrum of NAFLD lesions. Hepatocellular carcinoma is increasingly recognized in noncirrhotic NAFLD.4 However, the true incidence of this neoplastic complication in cirrhotic or noncirrhotic liver is less certain than with ALD.
Although NASH and NAFLD are commonly used terms, neither truly represents the underlying pathophysiologic disease process,5–7 and although there are histologic similarities with ALD, there are also dissimilarities between these disorders. The relationship between NAFLD and alcohol use may not be as straightforward as originally thought, because a few studies indicate that some patients may be at risk for liver disease from both causes simultaneously. Modest alcohol use also may protect against the effects of insulin resistance.8,9
For diagnostic pathologists, microscopically based diagnoses related to fatty liver disease are best rendered descriptively with terms such as steatosis or steatohepatitis, because in most circumstances, only careful clinical evaluation can determine the true underlying cause of the patient’s illness. Many different types of clinical situations may result in similar histologic findings, and in most cases, knowledge of the patient’s clinical information is essential to establish an accurate cause. This is similar to the situation for most cases of chronic hepatitis. The difference with fatty liver disease is that there are no serologic tests available to diagnose or exclude NAFLD with complete certainty.10
Alcohol-Induced Liver Disease
Clinical Features
The clinical features of ALD vary considerably. Table 49.1 highlights the primary clinical manifestations of the four main clinicopathologic subtypes of ALD: fatty liver, alcoholic hepatitis, alcoholic cirrhosis, and alcoholic foamy degeneration.
Table 49.1
Symptoms, Signs, and Laboratory Abnormalities of Alcohol-Induced Liver Disease
| Symptoms and Signs | Fatty Liver | Steatohepatitis and Alcoholic Hepatitis | Cirrhosis | Foamy Degeneration |
| Asymptomatic | +++ | + | Portal hypertension | − |
| Right upper quadrant discomfort | +++ | Pain, not mild | + to ++ | + |
| Hepatomegaly | ++ | ++ | + to ++ | + |
| Elevated AST, normal ALT | ++ | + | + (normal if abstinent) | ++ |
| AST/ALT ratio | AST > ALT | AST/ALT > 1-3 common | AST/ALT > 1 | AST > ALT |
| Marked AST elevation | <5 times normal | As much as 5 times normal | − | ++ |
| Marked alkaline phosphatase elevation | − | +/− | − | ++ |
| Bilirubin elevation | − | >10 mg/dL | − (unless advanced) | ++ |
| Jaundice | − | Cardinal sign | As above | ++ |
| Elevated white blood cell count | − | >10,000/mm3 | − (may be decreased) | − |
| Fever | − | +++ | + (with infection) | − |
| Ascites | − | +++ | + (advanced) | − |
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; −, does not occur; +, occurs uncommonly; ++, common; +++, very common.
Patients with ALD and fatty liver may be asymptomatic or may have right upper quadrant discomfort at presentation (see Table 49.1). Patients with alcoholic hepatitis usually have abdominal pain and pancreatitis, whereas patients with cirrhosis and alcoholic foamy degeneration often experience only vague abdominal discomfort. In all forms of ALD, the liver is typically enlarged. Fever, an elevated white blood cell count, and jaundice occur in various degrees with the different forms of ALD and may be related to ALD (e.g., alcoholic hepatitis) or to a secondary infection (e.g., peritonitis caused by alcoholic cirrhosis).
At clinical presentation, patients may have elevated serum levels of aminotransferases (i.e., aspartate aminotransferase [AST] and alanine aminotransferase [ALT]), and a ratio of AST to ALT greater than 1 is useful in the evaluation of patients with ALD. AST levels are typically greater than ALT levels in all forms of ALD. Radiographic evaluation plays a role in screening for hepatocellular carcinoma in patients with cirrhosis and in the clinical care of critically ill patients with pancreatitis and suspected intestinal ileus.
Epidemiology and Prevalence
Worldwide, ALD has no social or ethnic barriers, but it is uncommon in very young and very elderly individuals. Both genders may be affected, although women are at higher risk. In the United States, 7.4% of the population meet the criteria for alcohol dependence or abuse. It is the leading cause of end-stage liver disease and liver-related mortality and follows only heart disease and cancer as the major health issue in the United States.11 The reported rates of per capita liters of alcohol consumed per year by individuals older than 15 years of age varies throughout the world: 13.9 in the Russian Federation; 12.9 in Germany, France, and the United Kingdom; 9.3 to 8.5 in North America, Japan, and Australia; 5.0 in China, Philippines, and Vietnam; 1.3 in Iran and Saudi Arabia; and 0.6 in Afghanistan and Pakistan.11 Alcohol use may combine synergistically with other forms of chronic liver disease to generate progressive disease, malignancy, and morbidity.12
Familial Associations
Family, twin, and ethnic studies have confirmed a genetic susceptibility to ALD.11 Factors under investigation include gender, genes involved in the metabolism of alcohol (particularly in East Asians), cytochrome P450 2E1 (CYP2E1) and other enzymes involved in the pathways of oxidative stress, various cytokines (e.g., tumor necrosis factor-α [TNF-α], interleukin 10 [IL10]), genes involved in the oxidation of hepatic fat, mechanisms of matrix deposition and degradation in fibrosis, and immune reactions to endotoxin and toxic adducts.13
Risk Factors
Liver disease does not develop in all individuals who abuse alcohol. The risk for ALD, even among heavy drinkers, is less than 10% to 15%. Known risk factors include the amount of alcohol consumed during a 10- to 20-year period (>60 g/day for men; 20 to 30 g/day for women), gender (female > male), central obesity, patterns of consumption (nonmealtime > mealtime; daily > weekend only; various types of beverages rather than one type), associated medications (acetaminophen), amount of coffee intake, and genes that regulate expression of proinflammatory cytokines and immune response mechanisms.13–18 A sequence variation in the patatin-like phospholipase 3 (PNPLA3) gene, which encodes adiponutrin, is associated with the development of cirrhosis in white alcoholics.19 The same gene variation was previously found to correlate with steatosis in different populations around the world and with features of progressive disease in NAFLD patients.20
Pathogenesis
Alcohol-induced liver damage is a multifactorial process that involves alterations of the gut, gut microbiota, and hepatocellular metabolic pathways and activation of the innate and adaptive immune systems. Resultant outcomes are activation of proinflammatory and profibrogenic molecular mechanisms in specific liver cells.
The primary mechanism of alcohol-induced liver injury is hepatocellular oxidative stress.21,22 Ethanol is metabolized to acetate in the liver mainly by two enzyme systems: the dehydrogenases (i.e., alcohol and aldehyde), which produce acetate and result in steatosis, and the microsomal ethanol-oxidizing system (MEOS). The direct hepatotoxic effects of alcohol are caused primarily by acetaldehyde. Oxidative stress and free radical production result in mitochondrial damage, depletion of the antioxidants such as reduced glutathione, toxicity by free radicals, and induction of lipid peroxidation. Acetaldehyde-formed toxic adducts bind to proteins and lead to additional hepatocellular injury, serve as neoantigens for initiation of the adaptive immune response, and promote collagen production by hepatic stellate cells. The net effect of acetaldehyde production in hepatocytes is the accumulation of intracellular proteins, lipids, water, and electrolytes with the loss of the hepatocyte structural keratins 8 (K8) and 18 (K18). Histologically, they manifest as cellular enlargement, ballooning, empty-appearing cells, steatosis, and loss of immunoreactivity for K8 and K18.23,24
The key enzyme of the MEOS is the ethanol-inducible CYP2E1, which is located primarily in the endoplasmic reticulum of acinar zone 3 hepatocytes. Activation of this enzyme system results in the production of reactive oxygen species, such as hydrogen peroxide (H2O2) and superoxide anions (O2−), which cause further lipid peroxidation of cell membranes. This enzyme system also metabolizes acetaminophen and produces the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI). Even small amounts of acetaminophen may augment depletion of the antioxidant capabilities of the liver in chronic alcoholism.
Other sources of ethanol-induced oxidative stress include endotoxin-activated Kupffer cells, functionally impaired mitochondria, and ferric iron accumulation. Oxidative stress promotes hepatocellular injury, apoptosis, and necrosis; proinflammatory cytokine production (e.g., TNF-α, IL1, IL6); and parenchymal inflammation with polymorphonuclear leukocytes (PMNs) and mononuclear cells and perisinusoidal fibrosis.25,26
Excessive alcohol intake leads to increased gut permeability and increased portal venous exposure to gut-derived endotoxins. This process results in Kupffer cell activation through the CD14/toll-like receptor 4 (TLR4) complex and activation of the innate immune response.22,26
Increased lactate production, another downstream effect of ethanol metabolism, manifests as hyperuricemia and impaired carbohydrate metabolism. Hypoglycemia may occur. Chronic alcohol use impairs protein production and secretion. Multiple aberrations of lipid metabolism result in increased triglyceride production and subsequent accumulation (i.e., steatosis).
The mechanisms of fibrosis are under active scientific investigation. Ultimately, there is an imbalance between collagen deposition and degradation by activated hepatic stellate cells and portal myofibroblasts. Unique to ALD is the additional involvement of hepatocytes and sinusoidal endothelial cells in fibrogenesis through the production of transforming growth factor-β (TGF-β) and fibronectin isoforms.27 Hypoxia induced by ethanol metabolism in zone 3 hepatocytes upregulates vascular endothelial growth factor.26,27
Pathology
Grossly, steatosis enlarges the liver and imparts a greasy appearance. Livers with advanced fibrosis or cirrhosis may be enlarged and firm and contain micronodules (≤3 mm in diameter) scattered throughout the parenchyma. In some cases, fibrotic livers may be small and firm. With time, the nodules may coalesce to form larger ones, at which point determination of the underlying cause may not be possible. Hepatocellular carcinomas typically stand out on background cirrhosis as raised, green-tinged or darker nodules that usually are larger than 8 mm in diameter.
The histologic pattern of tissue injury in patients with alcohol-induced fatty liver disease initially involves acinar zone 3 (roughly equivalent to perivenular) hepatocytes. In patients with cirrhosis, steatosis or steatohepatitis may or may not persist.
Steatosis
Steatosis, the earliest pathologic finding in ALD, is not consistently found in all forms of ALD.21,28 Alcoholic steatosis is typically macrovesicular and is characterized by large intracellular droplets of lipid in hepatocytes (single or multiple) that peripherally displace the cell nucleus. A minor component of lobular chronic inflammation or mild portal inflammation may be identified, but fibrosis is usually absent.
Steatohepatitis
Steatohepatitis is defined by the presence of both steatosis and hepatocyte injury. Injured hepatocytes may appear ballooned, apoptotic, or lytic. Confluent and bridging necrosis are rarely found in ALD.
Hepatocellular ballooning is considered an essential feature of steatohepatitis by most hepatopathologists.7 Ballooned hepatocytes are enlarged cells with rarefied, reticulated cytoplasm (Fig. 49.1) that may or may not contain fat droplets and intracytoplasmic material referred to as Mallory-Denk bodies (MDBs) or Mallory hyaline. Ballooned hepatocytes are located predominantly in zone 3 and are commonly associated with some degree of perisinusoidal fibrosis (Fig. 49.2).29
Apoptotic hepatocytes, also called acidophil bodies, are common in patients with alcoholic steatohepatitis. These round, eosinophilic fragments of cytoplasm located in the hepatic sinusoids may or may not retain nuclear pyknotic material. Lobular inflammation in patients with steatohepatitis is usually mild and consists of mixed acute and chronic or mainly chronic inflammation. Scattered lobular microgranulomas and lipogranulomas are often observed. Lobular lipogranulomas may consist of multiple or single fat droplets surrounded by mononuclear cells and eosinophils (Fig. 49.3). Large portal lipogranulomas are frequently observed in ALD.30 PMNs may be observed in small clusters adjacent to ballooned hepatocytes or shrunken, eosinophilic (apoptotic) hepatocytes that contain MDBs. When PMNs are found next to hepatocytes that contain MDBs, the lesion is often referred to as satellitosis (Fig. 49.4).
Although common, MDBs and satellitosis are not required for the diagnosis. MDBs are not specific to ALD; the structures are seen in NAFLD, chronic cholestatic liver disease, copper toxicity, and amiodarone toxicity. In steatohepatitis of any origin, MDBs are usually found in zone 3 hepatocytes (Fig. 49.5) and are more common in areas of perisinusoidal fibrosis, whereas in chronic cholestasis, copper toxicity, and amiodarone toxicity, MDBs are more common in periportal hepatocytes.1,6 MDBs may also occur in focal nodular hyperplasia, hepatocellular adenoma, and hepatocellular carcinoma.31 MDBs can often be visualized by trichrome stains (Fig. 49.6) and confirmed by the use of immunohistochemistry for K8, K18, ubiquitin, or dynactin 4 (DCTN4, formerly designated p62).32
Some cases of ALD may show predominantly portal lymphocytic inflammation in the absence of serologic evidence of chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection. This feature may correlate with the degree of fibrosis33 and may reflect an associated autoimmune component to the underlying liver disease.34 Acute inflammation accompanied by a ductular reaction in portal tracts (i.e., pericholangitis) may represent concurrent pancreatitis, whereas increased numbers of portal macrophages may indicate recurrent partial biliary obstruction.30
Alcoholic Hepatitis
Patients with alcoholic hepatitis might not have steatosis. Histologic features of alcoholic hepatitis include hepatocyte ballooning, apoptotic bodies, MDBs with satellitosis, canalicular cholestasis, dense perisinusoidal fibrosis, and a perivenular lesion often referred to as sclerosing hyaline necrosis (i.e., MDBs, satellitosis, and obliterative fibrosis of the outflow vein). Portal hypertension may occur with sclerosing hyaline necrosis. Bridging fibrosis and a ductular reaction may be identified. The absence of steatosis does not rule out alcohol-induced hepatitis.
Alcoholic Foamy Degeneration
Alcoholic foamy degeneration is an unusual and often serious type of ALD associated with marked elevations of aminotransferases, γ-glutamyltransferase (GGT), alkaline phosphatase, and bilirubin.35 Histologically, it is characterized by diffuse, primarily microvesicular steatosis without inflammation or ballooning or by marked fibrosis with or without canalicular cholestasis (Fig. 49.7). MDBs are uncommon. Perivenular and perisinusoidal fibrosis may be evident. Clinically, the disorder mimics extrahepatic biliary obstruction, but the liver biopsy findings can be used to distinguish the two entities. Alcoholic foamy degeneration is reversible with abstinence from alcohol.
Fibrosis and Cirrhosis
The characteristic pattern of early fibrosis in patients with noncirrhotic alcoholic steatohepatitis or alcoholic hepatitis is pericellular or perisinusoidal. This pattern results from deposition of collagen in the space of Disse and is commonly referred to as chicken wire fibrosis (Fig. 49.8).
Fibrosis usually begins in zone 3 of the hepatic acini (Fig. 49.9). This type of fibrosis may be identified with or without steatohepatitis. However, when pericellular or perisinusoidal fibrosis occurs without steatohepatitis, it probably indicates that the patient had at least one prior episode of steatohepatitis. In ALD, perisinusoidal fibrosis may involve large portions of the lobules and become quite dense (Fig. 49.10). In these cases, the patient may also have clinical evidence of portal hypertension despite the absence of cirrhosis.
Perivenular fibrosis, a thickened sheath of collagen around the outflow veins in the absence of perisinusoidal fibrosis, has been identified in patients with steatosis that subsequently progressed to cirrhosis. This lesion is therefore considered a prognostic indicator of progression if alcohol exposure is continued.21
With disease progression, periportal fibrosis may develop (Fig. 49.11), followed in some cases by bridging fibrosis in a central-central, central-portal, or portal-portal pattern. In ALD, regions of parenchymal extinction (i.e., septa) may be quite broad (Fig. 49.12). After cirrhosis is established, areas of perisinusoidal fibrosis may not be evident. Periportal fibrosis and bridging fibrosis are often accompanied by a ductular (proliferative) reaction. In ALD, isolated portal fibrosis in the absence of portal inflammation is uncommon and may be a manifestation of recurrent pancreatitis or biliary obstruction.30 Itoh observed a pattern of portal fibrosis characteristic of ALD known as holly leaf.36 This pattern of fibrosis, which is commonly associated with hemochromatosis, is characterized by portal expansion and nonbridging, small, fibrous extensions or spikes into the surrounding parenchyma that resemble the outline of the holly leaf.
In patients with ALD with bridging fibrosis,37 periseptal ductular reaction may be prominent. The ductular reaction, characterized by proliferation of K7- and/or K19-positive ovoid cells forming ductular structures in an inflammatory, stromal matrix,38 less likely represents a cholestatic process in end-stage liver disease,26,30 but it is most likely the result of impaired regenerative activity of hepatocytes resulting from the anti-regenerative effects of alcohol.39
ALD-associated cirrhosis may be macronodular, micronodular, or mixed. Micronodular nodules are typically less than 3 mm in diameter. Larger nodules may develop when multiple nodules coalesce to form mixed micronodular and macronodular cirrhosis. Macronodular cirrhosis may be difficult to diagnose in a liver biopsy specimen because of the large size of the nodules; clues to abnormal architecture can be sought by use of a reticulin stain. Clusters of oncocytic hepatocytes suggest macronodular cirrhosis (see Chapter 50). Phlebosclerosis and occlusive disease of the terminal hepatic venules and sublobular veins may occur in patients with ALD-induced cirrhosis.40
In ALD-associated cirrhosis, periseptal hepatocellular copper granules and, uncommonly, α1-antitrypsin globules may be found. The role of heterozygosity for genes related to α1-antitrypsin in promoting damage in patients with ALD is a subject of ongoing debate.41 Increased iron levels in ALD is discussed later. Pseudotumoral nodules, which correspond to areas of active alcoholic hepatitis in cirrhotic livers, may be recognized on imaging studies and confused for hepatocellular carcinoma. These lesions may result from hypervascularity.42
Other Pathologic Features of Alcohol-Induced Liver Disease
Megamitochondria.
Megamitochondria (i.e., giant mitochondria) are identified by light microscopy as intracytoplasmic, round or cigar-shaped, eosinophilic structures in hepatocytes, and they are most readily seen in hepatocytes that contain microvesicular steatosis (Fig. 49.13). Megamitochondria may also occur in normal pregnancy, acute fatty liver of pregnancy, and Wilson disease.
Iron Deposition.
Mildly increased iron deposition is common in most forms of ALD. However, in a minority of cases, it may be moderate or severe.23 Possible reasons for hemosiderosis in ALD include increased dysregulation of hepcidin production,43 intestinal iron absorption, iron in some types of alcoholic beverages, hemolysis related to spur cell anemia, and upregulation of the transferrin receptor.18 With a modified Perls stain, iron deposition is usually nonzonal.30,37 In ALD-related cirrhosis,37 iron deposition may vary in quantity between individual regenerative nodules (Fig. 49.14).
Acute and Chronic Cholestasis.
Any of the following pathologic processes related to ALD may result in intrahepatic cholestasis, which is characterized by intracanalicular bile plugs: severe fatty liver, alcoholic pancreatitis due to gallstones or alcohol, alcoholic foamy degeneration, alcoholic hepatitis, and decompensated alcoholic cirrhosis. Canalicular bile thrombi are observed in 15% to 32% of livers with ALD (Fig. 49.15). Severe fatty liver with portal tract features of cholangitis and cholestasis is a specific clinicopathologic cholestatic syndrome in patients with ALD. Bile stasis in ALD also may be a manifestation of superimposed viral hepatitis or drug-induced jaundice.21 In patients with established ALD-associated cirrhosis, a copper stain may show mild positivity, which represents chronic retention of bile caused by the cirrhotic remodeling.
In advanced cases of ALD, the cytoplasm of some hepatocytes may have a ground-glass appearance because of smooth endoplasmic reticulum proliferation, or it may be deeply eosinophilic, referred to as oxyphil or oncocytic change because of increased numbers of mitochondria. Both changes are considered adaptive. Historically, their presence was considered indicative of ongoing alcohol consumption, but one research group reported that they might also be observed after periods of prolonged abstinence.30
Vascular Lesions.
Sclerosing hyaline necrosis, which is a marker of severe alcoholic hepatitis, and obliterative vein lesions may be responsible for the development of noncirrhotic portal hypertension in patients with ALD. Sclerosing hyaline necrosis consists of a combination of acinar zone 3 lesions, including dense perivenular and perisinusoidal fibrosis (i.e., sclerosis), occlusion of terminal hepatic venules, MDBs, and liver cell necrosis and loss, which results in the formation of large perivenular scars. Lesions of the terminal hepatic venules and sublobular veins include perivenular fibrosis, phlebosclerosis, and, less frequently, lymphocytic phlebitis (Fig. 49.16).40
Treatment Effects.
Alcoholic steatosis may resolve completely within 4 weeks of alcohol abstinence.30 However, lesions associated with alcoholic hepatitis or steatohepatitis may persist for as long as 6 months after alcohol withdrawal, albeit with decreased severity.44 With abstinence, there is usually an increase in the degree of portal lymphocytic infiltration.44
Prognostic Lesions.
Perivenular fibrosis in steatosis in the absence of cirrhosis portends progression unless abstinence occurs. Acute cholestasis correlates with poor survival of hospitalized patients with ALD.21 These patients have a reduced 5-year survival rate (22%) compared with patients with ALD who have no or mild cholestasis (54%).23 The number of apoptotic bodies correlates positively with severe histologic and clinical disease activity.45,46 Alcoholic hepatitis, sclerosing hyaline necrosis, and severe steatosis represent poor-prognostic lesions in patients with ALD, particularly in those with cirrhosis.21,47 In noncirrhotic patients, mixed steatosis and the finding of megamitochondria in the initial liver biopsy of alcoholic patients without steatohepatitis correlate with an increased risk of subsequent fibrosis or cirrhosis on follow-up.48 In acute-on-chronic liver failure caused by ALD, marked ductular bilirubinostasis and many MDBs are significant predictors of in-hospital mortality.49
Lesions in the Allograft Liver.
Any of the previously described lesions may manifest as recurrent or de novo ALD in a liver allograft. Care is taken to evaluate other features of allografts, particularly acute and chronic rejection and recurrence of hepatitis C (see Chapter 46).
Natural History and Treatment
The financial and social burdens associated with ALD are significant.11,50 Alcoholic hepatitis is associated with a significant risk of hospitalization and loss of time from work, morbidity and short-term mortality, and progression to cirrhosis within 5 years for as many as 70% of individuals. Few affected individuals are able to reverse the disease process with complete abstinence.11 A 12-year historical study from a national registry of Danish subjects comparing biopsy-proven ALD with the general population showed that the cirrhosis risk was higher for affected women than men and twice as high for patients with steatohepatitis than those with pure steatosis.51
Chronic alcohol abuse is responsible for as many as 45% of cases of hepatocellular carcinoma in Western countries,52 a rate that is higher than that for chronic hepatitis C.18 Coexistent HCV infection has an additive effect on the risk of carcinoma,11 and other comorbid factors, such as chronic hepatitis B, hemochromatosis, obesity, and cigarette smoking, contribute to an increased risk of hepatocellular carcinoma and mortality in patients with ALD.53,54 Intrahepatic cholangiocarcinoma also has been linked to ALD.55
The mainstay of treatment for ALD is abstinence from alcohol. One of the pharmacologic methods of treatment of alcoholism, cyanamide, may produce isolated, nonzonal eosinophilic globules in hepatocytes. Treatment of hospitalized patients with alcoholic hepatitis includes steroids and the antioxidants pentoxifylline and S-adenosyl-l-methionine (SAMe). For abstinent patients with cirrhosis, liver transplantation is an accepted treatment option. More controversial is a practice of liver transplantation for patients with acute alcoholic hepatitis.56,57
Nonalcoholic Fatty Liver Disease
Clinical Features
Individuals with NAFLD may be asymptomatic or may show mild, nonspecific symptoms such as fatigue and right upper quadrant discomfort or pain (Table 49.2). Fever is not a feature of uncomplicated NAFLD; when present, it suggests a different or concomitant disorder.
Table 49.2
Symptoms, Signs, and Laboratory Abnormalities of Nonalcoholic Fatty Liver Disease
| Symptoms and Signs | Fatty Liver | Steatohepatitis | Cirrhosis |
| Asymptomatic | ++ | + | Portal hypertension |
| Right upper quadrant discomfort | ++ | ++ | + |
| Hepatomegaly | ++ | ++ | + |
| Elevated AST, normal ALT | − | − | − |
| AST/ALT ratio | AST/ALT < 1 | AST/ALT < 1 | AST/ALT ≥ 1 |
| Marked AST elevation | − | − | − |
| Marked alkaline phosphatase elevation | − | − | − |
| Bilirubin elevation | − | − | − |
| Jaundice | − | − | − |
| Elevated white blood cell count | − | − | − |
| Fever | − | − | − |
| Ascites | − | − | + (advanced) |
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; −, does not occur; +, occurs uncommonly; ++, common; +++, very common.
The most common clinical feature of NAFLD is central (truncal or abdominal) obesity, which is best assessed by measurement of waist circumference. For adult men, central obesity is a waist circumference greater than 102 cm, and for adult women, it is greater than 88 cm. Obesity is defined by body mass index (BMI), which is the weight in kilograms divided by the height in meters squared (kg/m2). Compared with waist circumference, it is less specific and does not assess the anatomic location of the metabolically unique subcutaneous and visceral fat depots or shape of the body habitus. For non-Asian adults, the current definitions used are overweight (BMI > 25 kg/m2) and obesity (BMI > 30 kg/m2). Because the Asian body habitus is deceptively thin and able to conceal visceral adiposity, lower BMI values for overweight and obesity have been recommended.58
Clinical evidence of hepatomegaly may be found in patients with an enlarged, fatty liver. Ascites and other features of portal hypertension may be identified in patients with NAFLD-associated cirrhosis. Obstructive sleep apnea and acanthosis nigricans are recognized associations of NAFLD.
Laboratory values commonly assessed for patients with NAFLD include ALT, AST, and GGT. Patients also undergo tests for hyperglycemia (glycated hemoglobin [HbA1c] is used to evaluate long-term glycemic control), tests for insulin resistance (based on calculations of fasting glucose and insulin levels by using quantitative insulin sensitivity check index [QUICKI] and homeostasis model assessment [HOMA-IR]), and tests of lipid parameters.21
The overall sensitivity of liver enzymes for detection of fatty liver disease is poor.21,59,60 For instance, patients with normal ALT levels may have steatosis or advanced steatohepatitis61 or silent cirrhosis.62,63 If elevated, the ALT value is greater than the AST value, and an ALT/AST ratio greater than 1 is often found in cases of precirrhotic NAFLD; the reverse situation occurs in ALD. Autoantibodies, particularly antinuclear antibody and rarely antimitochondrial antibody, have been reported in as many as 34% of individuals with NAFLD.64 In some, only further detailed clinical testing or liver biopsy can distinguish autoimmune hepatitis from concurrent autoimmune hepatitis with NAFLD or NAFLD alone.64 A possible association of autoantibodies with more severe disease has been suggested but not confirmed.65,66 In combination with the proven lack of reproducible gross evaluation of the liver by surgeons, authorities recommend a liver biopsy for all high-risk and bariatric patients,67–69 especially when other causes of steatosis have been excluded.59
Abnormal iron study results are a common feature in patients with NAFLD. For instance, elevated serum levels of ferritin (but not transferrin saturation) may be found in patients with NAFLD and may cause clinical concern about an iron overload disorder.70 The serum ferritin level in NAFLD patients has been reported as an independent predictor of the histologic severity of underlying liver disease.71 An elevated serum ferritin level may indicate hepatocyte necrosis and systemic inflammation, and it is a recognized feature of the metabolic syndrome (discussed later). However, iron overload itself is not a feature of type 2 diabetes mellitus. The associations of iron dysregulation, HFE (hemochromatosis) mutations, and fibrosis with the metabolic syndrome remain subjects of debate.70 A meta-analysis did not support an association between HFE genetic variants and NAFLD.72
The proinflammatory iron regulator hepcidin is expressed in the liver and adipose tissue of obese patients, and this finding correlates with low transferrin values in 68% and anemia in 24% of these individuals.73 Demonstration of hepcidin mRNA and protein in adipose tissue is a new finding and is the subject of iron and adipokine studies.73
Laboratory tests are indicative of the complexity of this condition. Markers of systemic inflammation, such as elevated levels of C-reactive protein, fibrinogen, and plasminogen activator inhibitor 1, are found in the setting of NASH and fibrosis.74 A depressed level of adiponectin, an endocrine product of visceral adipose tissue, is recognized as a pathologic feature of NAFLD or NASH.74 Some work has implicated additional factors as being proinflammatory (e.g., hemoglobin) in subsets of individuals with NAFLD but without the metabolic syndrome.75 In contrast with ALD, leukocytosis, cholestasis, and jaundice are not typical features of uncomplicated, noncirrhotic NASH.
By ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI), the characteristic finding in NAFLD is increased hepatic fat. The ultrasound finding is referred to as a bright liver. Newer types of imaging studies use methods to semiquantitate the lipid content of the liver.76 However, when steatosis involves less than 33% of the liver parenchyma, imaging tests may not detect it.77 Imaging is also not an adequate replacement for tissue analysis for determination of the degree of disease activity or the stage of fibrosis in patients with precirrhotic NASH.78
Epidemiology and Prevalence
The epidemiology and prevalence of NAFLD are understood in terms of its relationship with obesity and increased risks of stroke and heart disease, a group of associations referred to as metabolic syndrome. The common underlying feature of this syndrome is systemic insulin resistance. Studies have confirmed associations of NAFLD with hepatic insulin resistance, cardiovascular diseases,79 and the metabolic syndrome in adults80 and children.81 In one definition of metabolic syndrome, a patient must have at least two of the following disorders plus type 2 diabetes mellitus, insulin resistance, or glucose intolerance: obesity (BMI ≥ 30); arterial hypertension (≥140/90 mm Hg); dyslipidemia as low levels of high-density lipoprotein (<0.9 mmol/L) or hypertriglyceridemia (>1.7 mmol/L); and microalbuminuria (>20 µg/min).82 The sole finding of fatty liver may indicate progression of the metabolic syndrome83 and progression of liver disease.84
NAFLD is a worldwide disorder and has been documented in almost all cultures. This global phenomenon is closely related to acquisition of a Western diet and lifestyle, which includes less physical activity. Initially thought to be a disease predominantly of overweight women, the entire spectrum of NAFLD is now recognized as common among men of all ages, but severe NASH and advanced fibrosis remain more common among adult women.21,59 Several studies have confirmed an underrepresentation of this disease process in African Americans of all age groups and an overrepresentation in Hispanics.59
Once referred to as a disease of the affluent, NAFLD has become recognized as everyman’s disease.85 Future projections of the rates of obesity, type 2 diabetes mellitus, and their complications in the developing world for the year 2030 are staggering. They range from 18.6% in sub-Saharan Africa to 79.4% in India. The projected percent positive change for type 2 diabetes mellitus incidence is 32% in Europe, 72% in the United States, 150% in India, 162% in sub-Saharan Africa, and 164% in the Middle East.86
Having considered the worldwide and multicultural nature of NAFLD, some studies have evaluated certain ethnic proclivities for development of this disease. For instance, an image-based study of hepatic steatosis from a large, urban, multiethnic population in the United States76 found the following relative prevalence rates: Hispanic > white > African American. Results from a subsequent clinic-based study that used liver tests and liver biopsy results differed only slightly,87 showing the following ethnic prevalence rates: white > Hispanic> Asian. African Americans represented only 3% of patients in this cohort. Others have observed an apparent underrepresentation of African Americans in cohorts of patients with NAFLD.88 A small biopsy series from an inner-city hospital in the United States that primarily serves African Americans confirmed the relative scarcity of the disease in this patient population. Less than 2% of 320 liver biopsies showed clinicopathologic evidence of NAFLD, even though the BMI of the cohort was between 26.9 and 32.7.89
Unfortunately, most of the early prevalence studies were performed without a full understanding of the link between the metabolic syndrome and fatty liver disease. In later studies, surrogate markers such as elevated ALT levels and a bright liver on ultrasound were used to examine study populations. However, similar to pathologic studies, the results vary widely, which is partly related to the criteria used to define the positivity of test values. For instance, in studies that relied on elevated serum aminotransferase levels, recognized shortcomings included underrecognition of positive cases with normal test values, determination of criteria for normal values in an increasingly obese population,84 determination of reliable ALT values from frozen sera, and ascertainment of the levels of alcohol intake.90 Methods of assessment of body habitus (i.e., BMI, waist-to-hip ratio, midthigh measurements, and more sophisticated and expensive calculations) also vary.78 Commonly cited studies are listed in Table 49.3.
Table 49.3
Prevalence of Nonalcoholic Fatty Liver Disease among U.S. Adults Based on NHANES III Data*
| Study | Criteria | Prevalence of NAFLD |
| Clark et al, 2002274 | Any elevation of AST, ALT, GGT | 23% of all U.S. adults |
| 31% male | ||
| 16% female | ||
| 39% of obese men, 40% of obese women | ||
| Clark et al, 2003275 | ± patients with diabetes | AST > 37 U/L, ALT > 40 U/L (men) |
| AST, ALT > 31 U/L (women) | ||
| 7.9% of all U.S. adults; 69% unexplained (5.5% of adults) | ||
| Ruhl and Everhart, 2003276 | Elevated ALT only; excluded patients with diabetes | ALT > 43 U/L (men and women) |
| 2.8% elevated ALT, 65% explained by elevated body mass index |
* Limitations of NHANES data: autoimmune hepatitis, primary biliary cirrhosis, Wilson’s disease, and α1-antitrypsin deficiency not excluded rigorously and absence of liver pathology for confirmation or exclusion.
ALT, Alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyltransferase; NAFLD, nonalcoholic fatty liver disease; NHANES III, Third National Health and Nutrition Examination Survey.
Validation of the findings of the Third National Health and Nutrition Examination Survey (NHANES III) has emerged from subsequent studies. One calculated hepatic lipid levels by proton magnetic spectroscopy in a multiethnic population in the southwestern United States and found that 31% of 2287 unselected individuals had a greater than 5.5% rate of hepatic steatosis. Of these, 79% had normal ALT values.76 A contemporaneous study from a large clinic population in California showed that of 742 individuals with newly diagnosed chronic liver disease, 21.4% had NAFLD.87 NAFLD was found to be the most common cause of incidental liver function test abnormalities (26.4%) in a U.K. prospective primary care cohort of 1118 adults by a combination of ultrasound and biomarker assays; ALD was a close second (25.3%). Of the NAFLD cases, 7.6% had evidence of previously undetected advanced fibrosis.91
A U.S. biopsy-based prevalence study of asymptomatic adults found that 46% of 156 adult, unselected patients who had steatosis on ultrasound had NAFLD diagnosed on biopsy. Of the biopsied NAFLD patients, 30% had diagnostic NASH, and 2.7% of those with NASH had advanced fibrosis (≥stage 2). This finding allowed speculation that more than 2 million of the U.S. adult population was at risk for advanced fibrosis because of NASH. These findings led the researchers to conclude that the prevalence of NAFLD was greater than that of hepatitis C and posed a serious concern for the future of health care.92
A study in a predominantly nonaffluent, nonobese population in India represented by 1911 inhabitants of a rural community showed the prevalence of NAFLD diagnosed by ultrasonography and CT to be 8.7%. NASH was seen in 31% of 36 biopsied persons with potentially significant disease, 4 of whom had cirrhosis.93,94
The estimated prevalence rate of NAFLD in the United States is greater than that of ALD (Table 49.4). It may affect as many as 5% of the adult population, with an even higher rate among patients with type 2 diabetes.59 Table 49.5 summarizes several published series focused on histologic documentation of fatty liver in adult autopsies. Correlations between body weight and diabetes were discussed in many of these studies long before the metabolic syndrome was identified as a specific entity. More than one investigator suggested that steatosis was a normal consequence of aging (see Pathogenesis).
Table 49.4
Prevalence Estimates of Fatty Liver Disease Compared with Other Chronic Liver Diseases
| Disorder | Prevalence |
| Nonalcoholic fatty liver disease | 25-75% of obese, diabetic adults 16% of 15- to 19-yr-old adolescents 38% of obese adolescents |
| Hepatitis C | 1.3-2.0% |
| Alcohol-induced liver disease | 1% |
| Hepatitis B | 0.3-0.4% |
| Hereditary hemochromatosis | 1/200-400 of northern European descent |
| Autoimmune hepatitis, primary biliary cirrhosis, primary sclerosing cholangitis | 9-17/100,000 |
| α1-Antitrypsin deficiency | 1/1500-7600 |
| Wilson disease | 1/30,000 |
Data from McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40(suppl 1):S17-S29 and Yu AS, Keeffe EB. Elevated AST or ALT to non-alcoholic fatty liver disease: accurate predictor of disease prevalence? Am J Gastroenterol. 2003;98:955-956.
Table 49.5
Prevalence of Fatty Liver Disease in Autopsy Studies
| Study | Population Studied (N) | Prevalence of Fatty Liver | Associated Features |
| Hilden et al, 1977277 | Motor vehicle accident victims; adults (503) | 24% overall 0.5% had features of alcoholic hepatitis |
Age: 1% < 20 yr 18% 20-40 yr 39% > 60 yr Overweight: associated with steatosis, not age |
| Underwood Ground, 1982278 | Aircrew, accidental deaths, all men (199) | 15.6% overall 2.1% had features of alcoholic hepatitis |
All healthy; none had overt alcoholism |
| Hornboll and Olsen, 1982279 | 678 Consecutive autopsies, all > 20 yr of age (396 with alcohol and other exclusions) | 54% overall 43% had < 33% 11% had > 33% |
Age: no association after 40 yr Overweight: strong association with degree of fatty change; diabetic people were more common in this group Diabetes not independently associated |
| Underwood Ground, 1984280 | Aircraft, motor vehicle accidents, all men; all accidents associated with alcohol, other illnesses excluded (166) | 21% | Questioned possible irregular use of alcohol as factor; discussed increased carbohydrate intake relationship to fatty liver disease |
| Wanless, 1990176 | Hospitalized, 207 obese patients, 144 matched, nonobese controls; alcoholic patients excluded (351) | Steatosis: 36.3% overall, 29.2 obese, 7.1% lean Steatohepatitis: 18.5% obese, 2.7% lean |
Overweight: prevalence and grade of steatosis and steatohepatitis correlated with grade of obesity Diabetes: increased the risk of steatohepatitis and fibrosis |
| Schwimmer et al, 2006231 | Consecutive pediatric and adolescent autopsies in a single county, nonselected (742) | Steatosis: 13% overall 2-4 yr: 0.7% 15-19 yr: 17.3% |
Age: fatty liver incidence increased with age Obesity: 38% had fatty liver Ethnicity: Hispanic (11.8%), Asian (10.2%), white (8.6%), African American (1.5%) |
In summary, the true prevalence rate of NAFLD remains unknown because there are no reliable or specific noninvasive tests available for an accurate diagnosis or exclusion of this process. A review of noninvasive markers in NAFLD, NASH, and fibrosis indicates the efforts in the field.21 Although liver biopsy evaluation is considered the gold standard of diagnostic tests, it cannot be considered an appropriate screening tool. Results from biopsy studies reinforce the observations that not all obese individuals have hepatic steatosis8,95,96 and that unexplained elevated ALT values cannot be assumed to indicate fatty liver disease. For example, Skelly and colleagues97 showed that as many as one third of 354 biopsies performed for unexplained elevated ALT levels were histologically normal or had abnormalities because of liver diseases other than NAFLD or NASH. Recommendations for the focused role of liver biopsy for patients with suspected NAFLD or NASH were discussed in the practice guidelines from the American Association for the Study of Liver Diseases59 and in a consensus conference of the European Association for the Study of the Liver.69
Familial Associations and Risk Factors
The genetic predispositions found in NAFLD are exemplified by differences in the incidence of other risk factors, such as obesity, diabetes, hypertension, and hyperlipidemia, among various ethnic populations. Studies have documented fatty liver disease and cirrhosis among kindreds.98 One study of 20 patients showed a trend toward familial clustering and maternal linkage for insulin resistance among patients with NAFLD.99
Type 2 diabetes and obesity are risk factors for progressive disease in clinical100 and histologic studies.8,101,102 Various possible environmental and genetic factors have been proposed for NAFLD,13,103 including the type of diet, exercise, smoking,104 the role of small bowel bacterial overgrowth, endotoxin and related immune or cytokine responses, polymorphisms for genes that control fatty acid oxidation (e.g., microsomal transfer protein), and fibrosis (i.e., angiotensinogen and TGF-β).
Broad, nonexclusive categories of genes under investigation as potential risk factors for NAFLD105 include those involved in the control of adiposity and insulin resistance (e.g., peroxisome proliferator–activated receptor-γ [PPAR-γ], central versus peripheral fat depots); determinants of hepatic steatosis (i.e., free fatty acid delivery and de novo lipogenesis, processing, and egress); hepatic fatty acid oxidation pathways (e.g., mitochondrial, peroxisomal, microsomal); hepatic oxidative stress mechanisms (e.g., production, defense); cytokine effects (e.g., TNF-α, IL6, adipokines); and determinants of progression of fibrosis. One genomic and proteomic study of 98 bariatric patients found downregulation of genes involved in defense against oxidative stress in NAFLD cases and upregulation of genes associated with fibrogenesis and apoptosis in steatohepatitis cases.106
Genome-wide association and candidate gene studies have identified a variant in PNPLA3, which encodes adiponutrin, an insulin-responsive phospholipase involved in lipolysis and lipogenesis that correlates with accumulation of hepatic triglyceride and some histologic features of severity of NASH with identical ethnic distributions in prevalence studies.107–112 This association is not limited to adults.112,113 The effects cannot be shown to be direct, and it is speculated the allelic variant results in sensitization of the liver to metabolic and environmental stressors.20
Pathogenesis
Certain similarities (i.e., steatosis, oxidative damage, and cytokine response) and dissimilarities between the pathogenesis of NAFLD and ALD are highlighted in Figure 49.17. The most commonly discussed driver of NAFLD is insulin resistance and the associated lipotoxicity (i.e., fat in nonfat depots) and complex pathways of systemic and local inflammation.114 The metabolic system of insulin involves signaling among skeletal muscle, adipose tissue, and liver.115–118
The manifestations of insulin resistance depend on the type of tissue involved. In visceral adipose tissue, there is unabated release of free fatty acids into portal venous blood. In peripheral muscle, insulin resistance manifests as an inability to use circulating glucose. In liver, continuation of glucose production (i.e., gluconeogenesis) combines with a lack of glycogen storage in the setting of hyperinsulinemia and hyperglycemia. Hepatic steatosis, which corresponds to an accumulation of triglycerides in hepatocytes, results from an imbalance between the delivery of free fatty acids and endogenous lipogenesis and fatty acid disposal through oxidation and packaging of esterified fatty acids and triglycerides for export by apolipoproteins as very-low-density lipoproteins. Each of these steps involves complex, genetically regulated pathways.
Current work shows that triglyceride accumulation is a mechanism of sequestration of potentially damaging free fatty acids and that nontriglyceride fatty acid metabolites likely drive subsequent necroinflammatory lesions recognized as NASH.119 Our understanding of pathogenesis of NAFLD and NASH also includes the roles of altered gut microbiota,120 intestinal permeability and lipopolysaccharide (LPS) exposure,121 differential macrophage activation in adipose tissue depots,122 innate immunity,123,124 and supporting activation by inflammasomes in obesity-related liver injury.125
Visceral (abdominal) adipose tissue is an energy storage depot and a source of highly active proteins, collectively referred to as adipokines or adipocytokines (Fig. 49.18
[/not-level-membership-for-pediatrics-category]
