[level-membership-for-neurosurgery-category]
CHAPTER 345 Intraoperative Cerebral Protection
Pathophysiology of Ischemic Injury
Cerebral Blood Flow
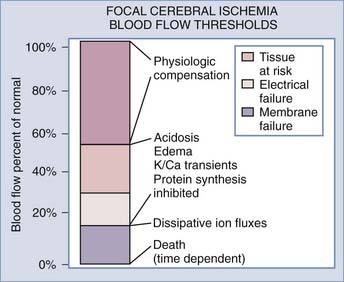
In 1948, Kety and Schmidt quantified normal cerebral blood flow (CBF) in “healthy normal men” to be 54 mL/100 g per minute.1 Sundt and others noted that a minimal CBF of 18 mL/100 g per minute is needed to maintain normal electroencephalographic (EEG) parameters during carotid endarterectomy (CEA).2,3 A further decrease in CBF causes neuronal electrical silence and decreased synaptic activity to preserve energy stores. Irreversible cellular damage occurs when CBF is below 10 mL/100 g per minute.4–8 In response to ischemia, the cerebral vessel autoregulatory mechanism induces vasodilation to increase collateral blood flow and thereby increase oxygen and glucose extraction for preservation of viable neurons (Fig. 345-1).
Ischemic Penumbra
Laboratory and clinical evidence demonstrates a spectrum of injured neurons when CBF is interrupted as a result of vessel occlusion. In the affected territory, there is a core of irreversibly damaged neurons surrounded by an area of electrically silent but viable neurons known as the ischemic penumbra.4,9–11 CBF in the penumbra has been determined to be between 10 and 20 mL/100 g per minute.7,12–16 These neurons may survive for hours, although the exact duration is unknown. Many factors may reduce the time for survivability of these marginal neurons. Hyperthermia of 1°C to 2°C and serum hyperglycemia have been shown to accelerate cell death.17,18 Restoration of CBF to the penumbra within the “critical hours” may salvage viable neurons and minimize the neurological deficits (Fig. 345-2). Without intervention, the core of the infarcted territory will subsume the penumbra as demonstrated by positron emission tomography (PET) and magnetic resonance imaging (MRI).19–21 The temporal and spatial evolution of the infarcted core is highly variable.19,21 Baron demonstrated by PET that the onset of the penumbra occurs as late as 16 hours after infarction.21 Susceptibility to ischemic injury also varies in different areas of the brain, with hippocampal CA1 and striatal neurons being more vulnerable than cortical neurons in ischemic models.22,23
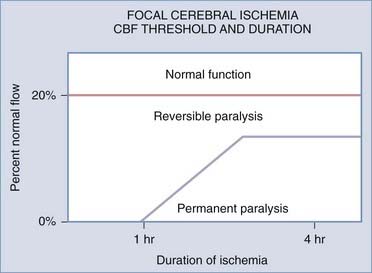
FIGURE 345-2 Relationship between time of ischemia and reversibility of neurological deficit. CBF, cerebral blood flow.
(Adapted from Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.)
The traditional concept of an infarcted core surrounding by a penumbra may be too simplistic. Many patients who improve with thrombolytic agents do not demonstrate a core surrounded by a larger penumbra on imaging. The core may be surrounded by heterogeneous regions of penumbra, with possibly separate areas of minimal flow or core within the penumbra as a result of variable degrees of collateral circulation.24
Energy Failure
The decrease in energy production from anaerobic metabolism (2 adenosine triphosphate [ATP] molecules) versus oxidative phosphorylation (32 ATP molecules) causes failure of the ionic electrochemical gradient established by the sodium-potassium adenosine triphosphatase (Na+,K+-ATPase) pump. Peri-infarct depolarization can be observed in the cortical penumbra with the use of direct current potential. These phenomena are associated with efflux of K+ and influx of Na+ and Ca2+.25–28 Biochemical analysis of PO2 and ATP in the penumbra confirms the depletion during depolarization,29,30 thus placing an additional burden on compromised tissue and increasing the size of the ischemic lesion by 30% to 100%.31–34 Ionic influx also results in oncosis,35 a condition characterized by swelling of cells and organelles, increased membrane permeability, and nonspecific DNA damage. Swelling of astrocytes causes large amounts of excitatory amino acids (EAAs) to be released through volume-activated anion channels.35 Although the death pathway of astrocytes is unclear, oncosis may contribute to the mechanism.
Altered Calcium Homeostasis and Excitotoxicity
Ischemia causes an increase in the intracellular calcium concentration from depolarization of voltage-gated channels, activation of ligand-gated channels, release of intracytoplasmic stores, and loss of ATP-dependent calcium extrusion pathways. In addition, there is an influx of calcium down an electrochemical gradient (Fig. 345-3).31 The calcium-mediated release of glutamate and EAAs from depolarized neurons plays a major role in ischemic cell death.36–38 In the presence of energy failure, depolarization of somatodendritic and presynaptic voltage-dependent calcium channels leads to excessive extracellular glutamate and EAAs (Fig. 345-4).39 When combined with failure of glutamate reuptake, toxic accumulation of extracellular EAAs causes excessive activation of the postsynaptic glutamate receptors N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoazolepropionic acid (AMPA). Overactivation of NMDA and AMPA receptors facilitates a massive influx of calcium and sodium ions, respectively.39 Animal models and microdialysis during neurosurgical procedures have shown glutamate and other EAAs to be markedly elevated over baseline levels.40–46 Accumulation of ions leads to cellular edema and peri-infarct depolarization.47,48 Such depolarization has been recorded for 6 to 8 hours after an initial ischemic insult.31 The depletion of energy stores needed for restoration of normal depolarization is thought to contribute to enlargement of an infarct. In addition, excessive intracellular calcium directly and indirectly initiates a series of detrimental cytoplasmic and nuclear cascades, including activation of Ca2+-dependent proteases and phospholipases, alterations in plasma membrane permeability, depolarization of mitochondria and release of mitochondrial proapoptotic proteins, production of reactive oxygen species (ROSs) and reactive nitrogen species (RNSs), and activation of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP).49–56
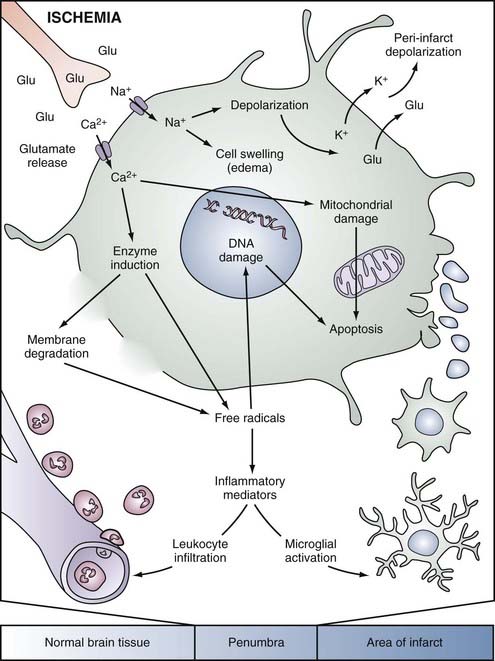
FIGURE 345-3 Simplified overview of pathophysiologic mechanisms in a focally ischemic brain. Energy failure leads to depolarization of neurons. Activation of specific glutamate receptors dramatically increases intracellular levels of Ca2+, Na+, and Cl−, whereas K+ is released into the extracellular space. Diffusion of glutamate (Glu) and K+ in the extracellular space can propagate a series of spreading waves of depolarization (peri-infarct depolarizations). Water shifts to the intracellular space via osmotic gradients, and cells swell (edema). The universal intracellular messenger Ca2+ overactivates numerous enzyme systems (e.g., proteases, lipases, endonucleases). Free radicals, which damage membranes (lipolysis), mitochondria, and DNA, are generated and in turn trigger caspase-mediated cell death (apoptosis). Free radicals also induce the formation of inflammatory mediators, which activate microglia and lead to the invasion of blood-borne inflammatory cells.31
(Used with permission from Barrow Neurological Institute.)
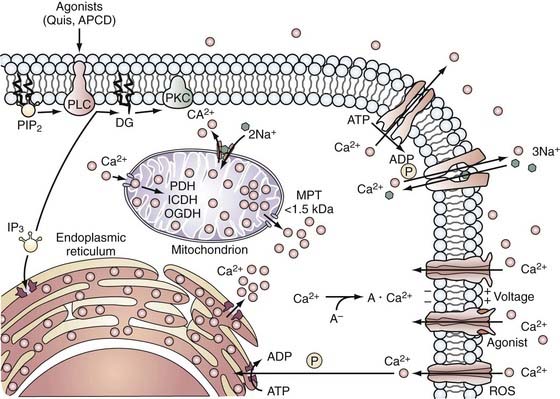
FIGURE 345-4 Calcium handling by cells. Calcium enters the cell through voltage- and agonist-operated channels and through nonspecific channels, such as those opened by reactive oxygen species (ROS), whereas Ca2+ is extruded from the cell by an adenosine triphosphate (ATP)-driven pump or by Ca2+-Na+ exchange. Cytosolic calcium (Ca2+) is sequestered by mitochondria and endoplasmic reticulum (ER) at the expense of respiratory energy or ATP, and it is bound to calcium-binding molecules. Ca2+ is released from the ER in response to agonist activation of receptors coupled to phospholipase C (PLC) and the formation of inositol 1,4,5-triphosphate (IP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). A rise in Ca2+ can cause translocation of protein kinase C (PKC) to membranes where it may be activated by diglycerides (DG) formed during hydrolysis of PIP2. Accumulation of Ca2+ in mitochondria can lead to the activation of a mitochondrial permeability transition (MPT) pore and result in the release of Ca2+ and other molecules with molecular weights of up to 1.5 kD from intramitochondrial compartments.39 ADP, adenosine diphosphate; APCD, 1-aminocyclopentane-1S,3R-dicarboxylic acid; ICDH, isocitrate dehydrogenase; OGDH, oxoglutarate dehydrogenase PDH, pyruvate dehydrogenase; Quis, quisqualate.
(Used with permission from Barrow Neurological Institute.)
Free Radicals
With its high oxygen consumption, the brain is especially susceptible to free radical damage. It also has high levels of iron and ascorbate,57–59 which can act as pro-oxidants under pathologic conditions, as well as low levels of antioxidant enzymes such as catalase, glutathione peroxidase, and superoxide dismutase.60 The cellular membranes are rich in polyunsaturated fatty acids, which are vulnerable to radical-induced peroxidation. The acidic environment from lactic acidosis during ischemia promotes dissociation of iron from protein and enhances the conversion of superoxide anion to more reactive radicals.58,61
ROSs and RNSs play significant roles in the pathogenesis and exacerbation of ischemic injury. It has been postulated that low levels of ROSs and RNSs trigger apoptosis and higher levels cause necrosis.62,63 During ischemia, nitric oxide (NO), a vasodilator, is generated by endothelial nitrous oxide synthase (NOS) to increase local CBF.64 Although this may be beneficial locally in the vasculature, a higher concentration of NO is produced by neuronal NOS and inducible NOS from inflammatory cells in the ischemic territory and may play a role in neuronal injury.64–68 NO and its derivative peroxynitrite, in addition to oxidizing effects on cellular structures, contribute to the oxidative stress by activating PARP, thus further depleting energy stores.69 PARP catalyzes the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD) to nuclear proteins. For every mole of ADP-ribose transferred, 1 mol of NAD is consumed. Four free energy equivalents of ATP are necessary to regenerate NAD.
Although ROSs are generated during ischemia, reperfusion after ischemia greatly increases ROS generation.57,70,71 Reoxygenation provides an excess supply of substrate for oxidation, thus producing more ROSs and RNSs. This further depletes endogenous antioxidants, including radical scavengers such as tocopherol (vitamin E) and carotene and enzymes such as superoxide dismutase and catalase. ROSs and RNSs are also produced in mitochondria during ischemia and reperfusion and are involved in the release of cytochrome c.58,72 Studies have shown that depletion of glutathione, a reductant, can cause a 100-fold increase in mitochondrial production of ROSs.60,73 Free radicals have been implicated in signaling pathways involving dephosphorylation and translocation of the proapoptotic Bad protein from the cytosol to the mitochondrial membrane.60 By affecting the redox state of neurons and glial cells, free radicals can alter their genetic expression, activate destructive proteins, and inhibit protective enzymes, which ultimately leads to cellular death and breakdown of the blood-brain barrier.
Free Fatty Acids
During ischemia, stimulation of glutamate receptors and an increase in intracellular calcium activate various subtypes of phospholipases, which causes a large amount of fatty acids, particularly arachidonic acid, to be released from the cellular membrane.74–76 With free radicals, arachidonic acid is further metabolized by cyclooxygenase/lipoxygenase to produce leukotriene C4, prostaglandin E2, and additional free radicals. These metabolites have been implicated in neuronal demise.77 Recent laboratory evidence suggests that arachidonic acid induces cytosolic and mitochondrial Na+ and Ca2+ overload via nonselective cation conductance, thereby leading to the formation of mitochondrial permeability transition pores, release of cytochrome c, and activation of caspase-3–dependent apoptosis.78
Inflammatory Response
Reperfusion facilitates the arrival of inflammatory cells at the ischemic zone, and postischemic inflammation contributes to ischemic injury in several ways.79 Inflammatory cells can aggravate ischemic injury by compromising blood flow in the microcirculation.80–82 These cells also stimulate platelet aggregation and the formation of microthrombi, which further propagates the ischemic injury. After adhesion to the endothelial walls, granulocytes migrate (diapedesis) into the parenchyma and can be seen in peri-ischemic areas within hours of arterial occlusion.83 Inflammatory cells are also responsible for the production of proteolytic enzymes and the generation of free radicals.84 Activation of metalloproteinases and proteases destroys surrounding tissues and the extracellular matrix, independent of the energy status of neurons.24,31 This inflammatory response is enhanced by proinflammatory chemokines such as interleukin-1β (IL-1β), IL-8, and tumor necrosis factor-α (TNF-α) and mediated by upregulation of adhesion molecules, including selectins, integrins (CD11/CD18), and the immunoglobulin supergene family.82,85–90
Cell Death
The two processes by which injured neurons die are necrosis and apoptosis, and these processes occur in both the infarcted core and the penumbra.53,91–95 When necrosis develops,91 cells swell in the first few hours with the formation of eosinophilic neurons. The cells then shrink and undergo pyknosis (condensation of nuclear chromatin). The number of necrotic neurons increases most markedly during the initial 6 to 12 hours. Phagocytosis of necrotic tissue takes place over a period of days. Pan-necrosis can occur in the infarcted core and give rise to a cystic cavity.25 In the penumbra, infarcted neurons are removed and reactive astrocytes proliferate, thereby creating an incomplete infarct.
Ischemic apoptosis may be triggered by oxidative stress and overactivation of glutamate receptors, although how ischemia triggers the apoptotic mechanism is unclear. The process begins with shrinkage of the nucleus and cytoplasm, followed by condensation of chromatin and nuclear fragmentation. The hallmark of apoptosis is a “laddering” pattern of DNA on gel electrophoresis caused by endonuclease cleavage of DNA into segments of nonrandom length.96,97 Eventually, the cells separate into small, pyknotic bodies that undergo phagocytosis with minimal associated inflammatory response.
Evidence has shown that the Bcl-2 family of proteins plays a significant role in determining whether ischemic neurons resist or succumb to apoptosis.98,99 Proapoptotic Bcl-2 proteins, such as Bad and Bax, are translocated into the mitochondrial membrane, where they induce the formation of pores and release proapoptotic enzymes that lead to the activation of proteases and cell death.100,101 The antiapoptotic proteins Bcl-2 and Bcl-xL may prevent apoptosis by inhibiting the proapoptotic proteins from inducing mitochondrial pore formation (Fig. 345-5).102 Other proteins involved in the mitochondrial pore complex include pancortin-2 and the Wiskott-Aldrich syndrome protein verprolin homologous-1 (WAVE1).103,104 Further evidence in ischemic animal models has demonstrated that pancortin-2 sequesters Bcl-xL in association with WAVE1 to promote the translocation of Bax to mitochondria, which causes release of cytochrome c and apoptosis.103
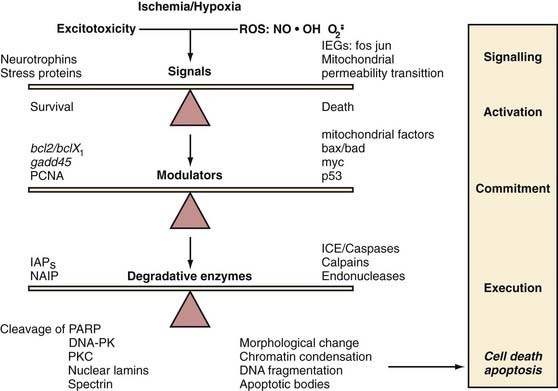
Other degradative enzymes activated during apoptosis include endonucleases, proteases, and caspases such as IL-1β–converting enzyme. Analysis of the DNA of apoptotic cells shows the laddering of approximately 200 base pairs of DNA fragments, which is specific for apoptosis rather than necrosis. These DNA fragments, indicative of neuronal death through apoptotic mechanisms, have been observed after focal ischemia by many investigators.93,105,106
Integration of Cellular Injury Mechanisms
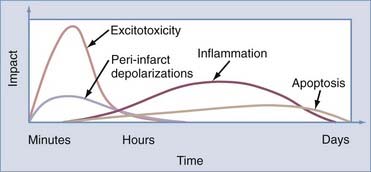
Ischemic injury causes dynamic and highly interrelated biochemical changes that have variable outcomes. In the absence of collateral circulation, neuronal tissue undergoes necrosis. When the collateral circulation is able to maintain cellular integrity, these neurons can survive for a finite period but are susceptible to injury leading to various end points, including necrosis, apoptosis, and reperfusion injury (Fig. 345-6).
Cerebroprotective Strategies for Focal Ischemia
Limiting the Duration of Ischemia
The use of temporary arterial occlusion for dissection of aneurysms and permanent clipping is now a common standard during aneurysm surgery, but it is crucial to establish a safe occlusion time with emphasis on the anatomic level of occlusion and the vascular territory occluded. The duration of focal ischemia that can be tolerated safely without clinically evident sequelae varies between individuals and vascular territories.107–111 The current consensus for temporary vessel occlusion is brief repetitive clipping periods, which provides increased safety and less risk for postoperative neurological deficit than a single episode of occlusion does, but definitive data have yet to confirm this consensus.
Laboratory studies of multiple ischemic events in focal models have demonstrated varying results.107,112–115 Sakaki and coworkers examined the response of three 20-minutes episodes of middle cerebral artery (MCA) occlusion in cats versus a single 1-hour period.112 They noted that intermittent occlusion produced less damage than single, longer occlusion did. This report did not specify the length of reperfusion between episodes of ischemia, so it is difficult to assess whether reperfusion injury could occur. Steinberg and colleagues used a rabbit model of multiple intracranial vessel occlusion and demonstrated a 59% decrease in the area of cortical ischemic neuronal damage but no difference in the extent of striatal ischemic damage with intermittent occlusion versus uninterrupted occlusion.115
Goldman and associates studied repetitive MCA occlusion in rats in a protocol designed to simulate intraoperative occlusion techniques.107 The total infarcted areas after 60, 90, and 120 minutes of uninterrupted occlusion were significantly greater than those after identical cumulative ischemic periods but with 5 minutes of reperfusion after each 10-minute ischemic period. In an investigation of normotensive rats undergoing MCA occlusion by Selman and colleagues, biochemical and pathologic evidence demonstrated that a single, prolonged episode of reversible ischemia was more deleterious than multiple occlusions of a similar total duration.113 Statistical differences were seen only when total single occlusion time reached 2 hours. To provide an infarct size that can be statistically evaluated in different treatments, the experimental models used to date have required total occlusion times longer than those generally needed in the clinical setting. The nature of these experimental paradigms and species differences must be kept in mind when attempting to generalize the results and apply them to clinical use.
Most reports on the use of temporary arterial occlusion in humans have been retrospective analyses of case series in which the use or nonuse of temporary occlusion was based on the experience and judgment of the surgeon.109,116–128 No randomized, prospective studies have been designed to compare the outcome of cerebrovascular procedures with temporary occlusion and those without local circulatory arrest. In 1961, Pool stated that bilateral anterior cerebral artery occlusion was safe for up to 20 minutes with the protective effects of hypothermia.129 Both Suzuki’s group127 and Ljunggren’s group119 estimated the maximum safe occlusion time of the MCA to be 20 minutes under normothermia. Other authors have recommended maintaining occlusion times at less than 15 minutes when possible,116,123,125,130 although some have reported occlusion lasting longer than 90 minutes without deficit.118
Samson and coworkers reviewed 121 patients from a group of 234 consecutive aneurysm patients.125 One hundred patients who did not experience intraoperative complications were analyzed. These patients underwent elective temporary occlusion under a standard neuroanesthetic regimen, including etomidate-induced burst suppression, normotension, and normothermia. Infarctions were noted in specific arterial territories as follows: basilar, 41%; middle cerebral, 26%; internal carotid, 7%; and anterior communicating, 16%. The authors identified the following factors as significant predictors of postoperative radiographic evidence of infarction: age (≥61 years), poor preoperative grade (Hunt and Hess grades III to IV), protracted duration of temporary occlusion, and the use of incomplete circulatory arrest.125 This series documents that temporary occlusion is not without risk; however, factors other than the duration of temporary occlusion, such as aneurysm configuration and the location of perforators, may be critical in determining this risk.131
In a nonconcurrent, prospective analysis, Ogilvy and colleagues studied the results of 132 consecutive aneurysm clipping procedures performed with temporary vascular occlusion in combination with mild hypothermia (33°C to 34°C), induced hypertension (systolic pressure of 150 mm Hg), and mannitol administration (100 g).126 Multivariate analysis demonstrated that intraoperative rupture and duration of clipping longer than 20 minutes were independently associated with stroke outcome. The average clip application time in patients with radiographic evidence of stroke was approximately 42 minutes as compared with 29 minutes in patients without radiographic evidence of stroke, whereas in patients with a clinically significant stroke, the average time was 50 minutes.126 The overall stroke rate in patients with an occlusion time of less than 20 minutes (1/67, 1.5%) was significantly less than that in patients with occlusion times longer than 20 minutes (12/65, 18.5%).126
Ferch and coauthors reported on 106 aneurysm patients retrospectively analyzed with regard to age, neurological status, aneurysm characteristics, intraoperative rupture, and duration and number of occlusive episodes.132 The overall symptomatic stroke rate attributed to temporary clip placement in this series was 17%. The incidence of stroke was 12% in patients with occlusion times of less than 10 minutes and 35% in patients with occlusion times longer than 10 minutes. In the group of patients who did not suffer stroke, the mean temporary occlusion time was 5.4 ± 5.2 minutes, whereas in those with stroke, it was 7.4 ± 8.1 minutes. In other series, the mean temporary occlusion time of the internal carotid artery without causing a stroke was 7.1 ± 3.8 minutes, and that for the MCA was 5.3 ± 3.9 minutes.132
Juvela and associates prospectively investigated temporary vessel occlusion times in 101 of 156 patients with ischemic lesions seen on computed tomography (CT) 3 months after subarachnoid hemorrhage.133 Thirty-five patients who underwent temporary vessel occlusion and had an ischemic lesion on follow-up CT had a total occlusion time of 8.5 ± 8.2 minutes versus 2.5 ± 2 minutes and a longer single occlusive episode of 6.1 ± 6.2 minutes versus 2.5 ± 2 minutes, with a trend toward significance in patients with a greater number of occlusive episodes lasting 1.78 ± 0.95 minute versus 1.14 ± 0.38 minute.133 Additional occlusion times of 25 to 42 minutes (mean, 33 ± 7 minutes) have been described in superficial temporal artery–MCA bypass patients without permanent neurological deficits.134
As alluded to in earlier studies, temporary clip placement and the duration of occlusion are not the only factors influencing postoperative ischemic injury. Other factors must also be considered, including patient age (>50 years), grade and time of the initial hemorrhage, poor clinical condition (Hunt and Hess grade III to IV), occlusion of perforators, intraoperative rupture, hypertension, the specific vascular territory, multiple aneurysms, and timing of surgery.132,133
Augmentation of Cerebral Blood Flow
Systemic arterial pressure can readily be manipulated in the setting of planned temporary ischemia. Symon and colleagues showed that MCA occlusion in primates results in loss of autoregulation in the ischemic region.135 Elevation of blood pressure should increase cerebral perfusion because of the passive nature of the vessels that have lost autoregulation in the ischemic territory. Other investigators have shown that the decrease in CBF with focal ischemia is less in animals with phenylephrine-induced hypertension than in controls.136–138 The beneficial effect of induced hypertension may, however, be limited to relatively brief episodes of ischemia. Smrcka and coworkers reported that hypertension reduced infarct size by 97% in rabbits subjected to 1 hour of arterial occlusion but achieved only a 45% reduction in animals with 2 hours of ischemia.139 In the clinical setting, use of hypertension is limited by the patient’s cardiac tolerance. Close monitoring of cardiac function with limitation of the elevation in blood pressure to approximately 10% above baseline is advisable.
Prevention of Iatrogenic Ischemia (Intraoperative Cerebral Blood Flow Monitoring)
Preoperative measurement of flow rates through cerebral vessels can be achieved with noninvasive studies such as quantitative magnetic resonance angiography (a noninvasive tool also used for bypass surveillance) and xenon-enhanced CT.140 These studies help the surgeon identify patients who have low cerebrovascular reserve and may be at higher risk for iatrogenic ischemia before entering the operating room.
The neurological examination provides a sensitive, qualitative assessment of CBF during cerebrovascular procedures but remains limited to CEA and by patient tolerance of an awake operation.141 Vessel patency after aneurysmal clip placement can be confirmed by a number of modalities, including direct microvascular Doppler (MVD) or transcranial Doppler (TCD) ultrasound, ultrasonic flow probe, intraoperative angiography, EEG studies, electrocorticography, multimodality evoked potential (MEP) and somatosensory evoked potential (SEP) monitoring, brain tissue oxygenation, and more recently, fluorescent angiography with fluorescein sodium or indocyanine green (ICG).
TCD can provide only a relative estimation of CBF based on normal ranges, and results vary according to vessel diameter and operator technique. TCD has been used extensively to monitor MCA CBF during CEA142–144 and postoperatively for the detection of vasospasm. MVD is a relatively fast and easy method of establishing vessel patency and aneurysm filling after clipping and can be used in patients undergoing multiple aneurysm clip applications. Use of the probe is limited by vessel depth and confounded by adjacent vascular tributaries. Unfortunately, these techniques do not determine whether flow is sufficient to prevent ischemia.
Direct intraoperative flow measurements can be made with the use of a microvascular ultrasonic flow probe. The device consists of an electronic flow detection unit and a flow-sensing perivascular probe. The probe uses the principle of ultrasonic transit time to assess intravascular flow without close vessel contact.145 The use of intraoperative angiography (IA) remains controversial; some reports recommend IA for complex aneurysms, such as giant aneurysms or those arising from specific locations, including the superior hypophysial, superior cerebellar, and posterior communicating arteries and the internal carotid artery bifurcation.146–149 The argument against IA includes increased operative time, poor image quality, exposure of personnel to radiation, and a high cost-benefit ratio. Lopez and coauthors reported on a prospective cohort of 191 patients with various cerebrovascular pathologies in whom 204 angiograms were performed.150 Intraoperative findings were positive in 23% of the patients (residual lesions in 12%, parent or vessel occlusion in 6%, vasospasm in 5%) and resulted in clip repositioning or additional clip placement in 8% of patients. Other studies have found that IA altered surgical treatment in 11% or 12% of cases and was associated with less than a 0.5% morbidity rate.151,152 Whether IA should be performed is ultimately left to the cerebrovascular surgeon’s discretion. Klopfenstein and colleagues prospectively assessed the cerebrovascular surgeon’s accuracy of predicting whether IA was necessary. In this series, 200 patients with 235 aneurysms underwent IA. The results led to changes in surgical treatment in 7% of cases. In 4.4% of these patients, the surgeon thought that IA was unnecessary.153
IA is safe and useful when treating patients with a wide variety of vascular pathologies and may be beneficial in patients for whom surgery has been prospectively deemed unnecessary by the surgeon. However, IA is limited in its evaluation of small perforating vessels, which cause 8% of iatrogenic clip infarcts.154 By no means should IA be a substitute for sound surgical technique and judgment.
EEG, electrocorticographic, SEP, and MEP studies are also used to evaluate CBF. EEG monitoring is commonplace for CEA, but its usefulness is limited with barbiturate use, hypothermia, and global ischemia and in the evaluation of subcortical structures. Intraoperative neurophysiologic SEP monitoring is sensitive in detecting cortical ischemia, but MEP monitoring provides more information on subcortical structures and the motor cortex and pathways.155,156 MEP has been found to be more sensitive than SEP monitoring in detecting insufficient blood flow in the MCA and anterior choroidal artery.157,158 MEP monitoring is limited by the fact that the patient is fixed in a head holder and any movement can be disruptive to the surgeon.
Angiography/fluorescence imaging with ICG, a near-infrared fluorescent tricarbocyanine dye used for several decades by cardiologists, is relatively new in neurosurgical application. An intravenous bolus of 0.2 to 0.5 mg/kg (maximum daily dose, 5 mg/kg) of ICG allows the surgeon to perform real-time evaluation of blood flow within vessels in the operative field. de Oliveira and associates performed a prospective study of 60 patients with 64 aneurysms; ICG angiography demonstrated vessel patency in 62 aneurysms.159 ICG angiography is safe and reliable for evaluation of perforating vessels in the surgical field but is limited in patients with deep-seated aneurysms, blood in the surgical field, complete occlusion, and clip obstruction. It is currently undergoing active evaluation in many neurovascular centers.
Complex and giant aneurysms of the skull base and distal vessels present a unique challenge. These lesions may not lend themselves to open surgical clipping or endovascular coil embolization. Even in light of recent advances in endovascular therapy (i.e., stent-assisted coiling), parent vessel ligation and aneurysm trapping, in addition to bypass, may be the only alternative. Surgical success with selective bypass begins with adequate preoperative planning and implementation in appropriate patients. Flow-assisted surgery allows direct intraoperative flow measurement and may help ensure success of the bypass.160 This measurement can be quantified as a cerebral flow index (CFI): CFI = bypass flow/cut flow. With a CFI of 0.5 as a threshold value, the bypass patency rate was 92% in patients with CFIs greater than 0.5 versus 50% in patients with a CFI of less than 0.5.145,160,161
Reduction of Metabolic Activity
Hypothermia
Hypothermia has been shown to reduce neurological morbidity after cardiac arrest162,163 and neonatal hypoxic encephalopathy.164,165 Intraoperative mild hypothermia can be used during aneurysm surgery to reduce the ischemic injury induced by temporary vessel occlusion and brain retraction. Hypothermia ameliorates the primary and secondary mechanisms of ischemic injury, including metabolism, release of glutamate, generation of NO by NOS, and myeloperoxidase activity, in animal models of focal ischemia.166–170 Han and coworkers demonstrated that a decrease in NO produced a 40% reduction in infarct size, a 50% reduction in cells immunoreactive for inducible NOS and peroxynitrite—one of the most dangerous free radicals generated from NO—and the production of superoxide dismutase, thereby resulting in a reduction in the generation of oxygen free radicals.171
Both mild hypothermia (33°C to 34.5°C) and moderate hypothermia (27.5°C to 30°C) have been associated with decreased infarct size in experimental animals.172–176 Cooling animals beyond 33°C does not appear to provide greater benefit.177 Huh and coworkers demonstrated that both intra-ischemic and postischemic cooling to 32°C in rats with MCA occlusion reduced infarct size by 89% and 88%, respectively.178 Lo and Steinberg found an equivalent benefit of mild and moderate hypothermia as measured by the recovery of evoked potentials and magnetic resonance imaging parameters in rabbits after permanent focal ischemia.179 Karibe and coauthors reported that mild hypothermia was protective when delayed up to 30 minutes after the onset of ischemia but that hypothermia induced 60 minutes after the insult was not beneficial in the rat model.180 Karibe and colleagues also demonstrated an increase in ipsilateral frontal cortex CBF in 12 hypothermic patients versus 12 normothermic patients at day 4.181 Hypothermia ameliorated the focal ischemic injury caused by frontal brain retractor placement.
Conventional cooling has been compared with endovascular cooling. Steinberg and associates found that placement of an endovascular heat exchange catheter in the inferior vena cava via the femoral vein resulted in faster cooling (4.8°C/hr versus 0.9°C/hr with conventional cooling), and 99% of patients reached a target 33°C at the time of clipping as opposed to 20% cooled by conventional means.182
The safety of intraoperative moderate hypothermia has been evaluated. Kimme and coauthors reported on 326 patients who underwent 359 aneurysm-clipping operations.183 No significant difference in circulatory instability, coagulopathy, and infection was evident in comparison to previous studies. Pulmonary complications (ventilator dependency) were thought to be secondary to neurogenic pulmonary edema and could be ameliorated by rapid rewarming and early extubation.
Recently, the Intraoperative Hypothermia After Aneurysm Surgery Trial (IHAST2), an international double-blind trial in which 1001 grade I to III aneurysmal subarachnoid hemorrhage patients were subjected to mild hypothermia (33°C at clip placement) versus normothermia (36.5°C), was conducted. The study demonstrated no improvement with hypothermia, with 66% of hypothermia patients and 63% of normothermia patients reaching a Glasgow Outcome Scale score of 1.184 Hypothermia with circulatory arrest is most often used during aneurysm surgery for giant and complex posterior circulation aneurysms, including those not amenable to endovascular treatment; those with significant intra-aneurysmal thrombus, broad necks, or a projection endangering dissection and preservation of perforators; those adhering to vital structures; and fusiform aneurysms with a distal vessel not suitable for arterial bypass.
Anesthesia (Barbiturates, Etomidate, and Propofol)
Most drugs selected as anesthetics suppress neurotransmission. Such suppression reduces energy requirements, and a reduction in energy requirements should allow tissue to better preserve energy balance during transient interruption of substrate delivery. Because ATP synthesis recovers rapidly after restoration of substrate delivery, anesthetics would be expected to be protective if present during ischemia but not if given after restoration of substrate delivery. The beneficial effect of barbiturates in the treatment of experimental focal cerebral ischemia is well documented.118,185–189 The systemic cardiovascular effects and protracted depression of consciousness after barbiturate administration, as well as the potential need for prolonged postoperative mechanical ventilation, may, however, complicate the use of high-dose barbiturates in clinical situations.
Two groups of investigators have shown that rats anesthetized with barbiturates at doses producing mild EEG suppression have an equivalent reduction in infarct volume as those with barbiturate doses intended to maintain EEG burst suppression.190,191 The use of lower doses in the clinical setting may permit safer administration of barbiturates during cerebrovascular procedures.
Etomidate and propofol, short-acting anesthetics with EEG effects similar to those of barbiturates but without cardiovascular side effects, have been proposed for use in cerebrovascular procedures.128,192,193 Propofol has been shown to reduce intracranial pressure and cerebral swelling while lowering cerebral infusion pressure and CBF to a lesser extent than isoflurane or sevoflurane194 and causes fewer episodes of hypotension during elective intracranial surgery.195 In the past decade, propofol has been shown to be neuroprotective in vivo through its antioxidant protection against peroxynitrite by altering antiapoptotic gene expression and influencing the anti-inflammatory cytokine balance.196
Lavine and coauthors reported a retrospective analysis of 49 patients who underwent surgery for MCA aneurysms.197 Thirty-eight patients received intravenous pentobarbital, etomidate, or propofol, whereas the remaining 11 inhaled isoflurane. Postoperative radiographic evidence of infarction was present in 45.5% of patients receiving the inhaled agent as opposed to 15.8% of those who underwent intravenous anesthesia. Occlusion time was significantly longer in the pentobarbital group: 3.9 ± 2.2 minutes versus 13.6 ± 10.6 minutes in the no-stroke cohort. Although the retrospective, nonrandomized, historical control study design does not allow a definitive statement regarding the benefit of these agents or a comparison of them, it is the best evidence to date demonstrating a protective effect with their use during temporary intracranial vessel occlusion.
Serum Glucose Modulation
Glucose, the fundamental substrate of the central nervous system, is essential for appropriate neuron function, but hyperglycemia in the absence of oxygen results in anaerobic glycolysis and subsequent intracellular lactic acidosis after ischemia, which has been shown to increase ischemic injury by exacerbating damage to neurons and glia in the laboratory and clinical setting.198,199 Hyperglycemia has been shown to negatively influence outcomes in ischemic stroke patients.
Pasternak and colleagues examined post hoc data from 1000 IHAST patients with blood glucose levels higher than 129 mg/mL and found that they had worse neuropsychological outcomes at 3 months than did normoglycemic patients.200 In light of this finding, it is prudent to limit, monitor, and control the amount of glucose delivered in intravenous solutions during cerebrovascular procedures.
Cytoprotective Agents
Within the past few years, a variety of pharmacologic agents have been developed that interfere with biochemical mediators of the ischemic cascade. These compounds have been neuroprotective in experimental stroke models. Drug development has been directed toward pharmaceuticals that will protect patients with acute ischemic stroke. Unfortunately, agents that have survived phase III trials have failed to prove efficacious.201 Additionally, none have been systematically tested in the setting of iatrogenic focal ischemia. It seems intuitive, however, that if a drug proves beneficial when given several hours after the onset of ischemic stroke, the same drug might be protective if given at or just before the onset of focal ischemia during cerebrovascular surgery.
Calcium Channel Blockers
In animal studies of focal cerebral ischemia, a variety of calcium channel antagonists have been associated with reduced infarct volume, decreased peri-infarct edema, improved CBF, and improved neurological outcome.202–213 Nimodipine is a 1,4-dihydropyridine that is more lipophilic than nifedipine and dilates cerebral vessels at lower doses than those required for peripheral vasodilation.214 Nimodipine given within 12 hours of ischemic stroke has been associated with reduced mortality and greater neurological improvement in several studies215–218 but is not beneficial when given from 12 to 48 hours after the ictus.219–222 However, a trial of nimodipine administered within 6 hours of stoke demonstrated no benefit over placebo.223
Glutamate Antagonists
NMDA antagonists have shown beneficial effects in reducing the volume of infarction in a variety of experimental models.224–232 Those with potential clinical efficacy include dextrorphan, dextromethorphan, licostinel, and magnesium.228–235 Perhaps the most promising of these agents for intraoperative use is magnesium. Magnesium has reduced the extent of infarction in several experimental models and has a long history of safe use in humans.236–242 Marinov and coauthors reported that MgSO4 given intra-arterially to rats before temporary MCA occlusion resulted in a 19% to 28% reduction in the volume of infarction, depending on the duration of ischemia.239
Nitric Oxide Synthase Inhibitors
NOS inhibitors and free radical scavengers have provided significant neuroprotection in experimental animals subjected to focal ischemia.243–248 Tirilazad mesylate is a potent inhibitor of lipid peroxidation through suppression of inducible NOS.249 Most studies have shown a significant reduction in infarct volume in focal ischemia models236,250–255; however, human studies for the treatment of ischemic stroke have produced conflicting results.256,257 A multicenter, randomized, placebo-controlled study of tirilazad in patients with acute ischemic stroke was halted prematurely because of lack of benefit.258
Lubeluzole
Lubeluzole, a benzothiazole derivative, is a novel agent that may provide effective therapy for focal ischemia. The drug blocks the postischemic release of glutamate and taurine, blocks Ca2+ and Na+ channels, and inhibits the glutamate-activated NOS pathway.259–264 In focal ischemia models, lubeluzole reduced infarct volume by approximately 25%,265,266 and studies in humans have shown few side effects.267–269 Three randomized, placebo-controlled studies of patients with acute ischemic stroke have reported encouraging results.270–272 Unfortunately, the improvement was in a different outcome measure in each study. Additional clinical trials have failed to show efficacy in the treatment of acute stroke.264
Citicoline
Citicoline (cytidine 5-diphosphocholine) supplies choline and cytidine, both of which are substrates needed for the synthesis of phosphatidylcholine, a key membrane component. Administration of citicoline has been associated with reduced infarct volume in animal models of focal ischemia.273,274 In a multicenter, randomized, placebo-controlled, and blinded study of patients treated within 24 hours of ischemic stroke, citicoline was associated with improved neurologic, cognitive, and functional outcome.275 Most recently, Davalos and coworkers demonstrated in 1652 randomized patients that treatment with 2 g citicoline orally within 24 hours of moderate to severe stroke resulted in a 27.9% recovery rate at 3 months as opposed to 20.2% with placebo.201
Nicardipine
A dihydropyridine antagonist of voltage-sensitive Ca2+ channels, nicardipine has produced inconsistent favorable effects through its vasodilatory action and increase in CBF.276 After MCA occlusion in rats, Kittaka and colleagues demonstrated a direct neuroprotective effect of nicardipine consisting of a reduction in infarct size, improvement in neurological outcome, and a decrease in neuron-specific enolase plasma levels (marker of neuronal injury) without a change in laser Doppler flowmetry (CBF).277
Statins
3-Hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors prevent the first step in cholesterol synthesis. Ischemic injury has been shown to increase central nervous system glutamate release and subsequent excitotoxicity and neuronal cell death through release of calcium and excessive activation of the NMDA receptor. Recently, statins have been demonstrated to provide additional neuroprotection by preventing protected neurons from NMDA-induced neuronal death by 70% through protein rafts.278 In addition, statins have been shown to upregulate the cell survival Bcl-2 gene.279 Lovastatin has also been shown to protect cortical neurons against glutamate-mediated excitotoxicity. Lovastatin increased tumor necrosis factor receptor 2 (TNFR2) expression in cortical neurons. It was previously demonstrated that activation of TNFR2 signaling, which includes phosphorylation of protein kinase B/Akt and activation of nuclear factor κB, protects neurons against ischemic or excitotoxic insults.280
Other Cytoprotective Agents
Other classes of agents have shown promise in laboratory studies of focal ischemia but have undergone limited testing in the clinical setting. Such agents include antibodies designed to prevent leukocyte adhesion, growth factors, caspase inhibitors, DP-b99 (a zinc and calcium chelator), and NXY-059 (free radical scavenger). Antibodies to intercellular adhesion molecule-1 (ICAM-1), CD11/CD18 integrin, and P-selectin have reduced infarct volume in focal models of ischemia.281–283 An uncontrolled study of enlimomab (an anti–ICAM-1 antibody) in patients with ischemic stroke showed minimal side effects284; however, increased mortality and poorer functional outcome were reported in a placebo-controlled trial.285
In experimental models of ischemia, DP-b99 reduced infarct volume and neuron-specific enolase release and increased survival rates.286,287 Unfortunately, a randomized controlled trial by Diener and coworkers failed to demonstrate an improvement in the 90-day National Institutes of Health Stroke Scale (NIHSS) score in 150 stroke patients treated with a 4-day course of DP-b99 versus placebo.288 In addition, NXY-059, a nitrone compound with free radical trapping properties, reduced infarct volume in rats.289,290 In a clinical trial, 1722 patients received NXY-059 versus placebo within 6 hours of stroke (recombinant tissue plasminogen activator was given within 3 hours of ictus). Patients in the NXY-059 group showed an improvement in the modified Rankin score but not in the NIHSS score.291
Surgical Protocol
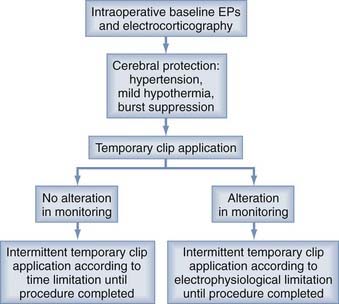
Before temporary artery occlusion, mild hypertension to approximately 10% above the patient’s baseline mean arterial pressure can be induced with phenylephrine, and an anesthetic regimen consisting of propofol and an inhaled agent is administered for general anesthesia. A loading dose of thiopental (10 mg/kg) can be given, followed by a maintenance infusion titrated to maintain EEG burst suppression (approximately 3 to 5 mg/kg per hour). If possible, occlusion is performed in an intermittent fashion on the basis of time limitations or electrophysiologic evidence of altered function (Fig. 345-7). After permanent clipping, the barbiturate administration is stopped and active rewarming is instituted as soon as blood flow is restored by removal of the temporary clip.
Amin-Hanjani S, Shin JH, Zhao M, et al. Evaluation of extracranial-intracranial bypass using quantitative magnetic resonance angiography. J Neurosurg. 2007;106:291-298.
Ashley WW, Amin-Hanjani S, Alaraj A, et al. Flow-assisted surgical cerebral revascularization. Neurosurg Focus. 2008;24(2):E20.
Bano D, Nicotera P. Ca2+ signals and neuronal death in brain ischemia. Stroke. 2007;38:674-676.
Cheng A, Arumugam TV, Liu D, et al. Pancortin-2 interacts with WAVE1 and Bcl-xL in a mitochondria-associated protein complex that mediates ischemic neuronal death. J Neurosci. 2007;27:1519-1528.
Chu X, Fu X, Zou L, et al. Oncosis, the possible cell death pathway in astrocytes after focal cerebral ischemia. Brain Res. 2007;1149:157-164.
de Oliveira JG, Beck J, Seifert V, et al. Assessment of flow in perforating arteries during intracranial aneurysm surgery using intraoperative near-infrared indocyanine green videoangiography. Neurosurgery. 2007;61:63-72.
Dolga AM, Nijholt IM, Ostroveanu A, et al. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J Alzheimers Dis. 2008;13:111-122.
Fang KM, Chang WL, Wang SM, et al. Arachidonic acid induces both Na+ and Ca2+ entry resulting in apoptosis. J Neurochem. 2008;104:1177-1189.
Hans P, Bonhomme V. Why we still use intravenous drugs as the basic regimen for neurosurgical anaesthesia. Curr Opin Anaesthesiol. 2006;19:498-503.
Hopwood SE, Parkin MC, Bezzina EL, et al. Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis. J Cereb Blood Flow Metab. 2005;25:391-401.
Johnson-Anuna LN, Eckert GP, Franke C, et al. Simvastatin protects neurons from cytotoxicity by up-regulating Bcl-2 mRNA and protein. J Neurochem. 2007;101:77-86.
Katz JM, et al. Is routine intraoperative angiography in the surgical treatment of cerebral aneurysms justified? A consecutive series of 147 aneurysms. Neurosurgery. 2006;58:719-727.
Klopfenstein JD, Spetzler RF, Kim LJ, et al. Comparison of routine and selective use of intraoperative angiography during aneurysm surgery: a prospective assessment. J Neurosurg. 2004;100:230-235.
Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49-59.
Lopez KA, Waziri AE, Granville R, et al. Clinical usefulness and safety of routine intraoperative angiography for patients and personnel. Neurosurgery. 2007;61:724-729.
Mongin AA. Disruption of ionic and cell volume homeostasis in cerebral ischemia: The perfect storm. Pathophysiology. 2007;14:183-193.
Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96-103.
Pasternak JJ, McGregor DG, Schroeder DR, et al. Hyperglycemia in patients undergoing cerebral aneurysm surgery: its association with long-term gross neurologic and neuropsychological function. Mayo Clin Proc. 2008;83:406-417.
Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281-1289.
Ponce J, de la Ossa NP, Hurtado O, et al. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39:1269-1275.
Saeed SA, Shad KF, Saleem T, et al. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp Brain Res. 2007;182:1-10.
Sattin JA, Zivin JA. Emerging therapies for acute ischemic stroke. Am J Ther. 2007;14:291-298.
Steinberg GK, Ogilvy CS, Shuer LM, et al. Comparison of endovascular and surface cooling during unruptured cerebral aneurysm repair. Neurosurgery. 2004;55:307-314.
Zhao H, Wang JQ, Shimohata T, et al. Conditions of protection by hypothermia and effects on apoptotic pathways in a rat model of permanent middle cerebral artery occlusion. J Neurosurg. 2007;107:636-641.
1 Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest. 1948;27:476-483.
2 Waltz AG, Wanek AR, Anderson RE. Comparison of analytic methods for calculation of cerebral blood flow after intracarotid injection of 133 Xe. J Nucl Med. 1972;13:66-72.
3 Sundt TMJr, Sharbrough FW, Anderson RE, et al. Cerebral blood flow measurements and electroencephalograms during carotid endarterectomy. J Neurosurg. 1974;41:310-320.
4 Astrup J, Symon L, Branston NM, et al. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke. 1977;8:51-57.
5 Astrup J, Blennow G, Nilsson B. Effects of reduced cerebral blood flow upon EEG pattern, cerebral extracellular potassium, and energy metabolism in the rat cortex during bicuculline-induced seizures. Brain Res. 1979;177:115-126.
6 Branston NM, Strong AJ, Symon L. Extracellular potassium activity, evoked potential and tissue blood flow. Relationships during progressive ischaemia in baboon cerebral cortex. J Neurol Sci. 1977;32:305-321.
7 Morawetz RB, Crowell RH, DeGirolami U, et al. Regional cerebral blood flow thresholds during cerebral ischemia. Fed Proc. 1979;38:2493-2494.
8 Meyer FB, Anderson RE, Sundt TMJr, et al. Intracellular brain pH, indicator tissue perfusion, electroencephalography, and histology in severe and moderate focal cortical ischemia in the rabbit. J Cereb Blood Flow Metab. 1986;6:71-78.
9 Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723-725.
10 Symon L, Branston NM, Strong AJ, et al. The concepts of thresholds of ischaemia in relation to brain structure and function. J Clin Pathol Suppl (R Coll Pathol). 1977;11:149-154.
11 Heiss WD. Flow thresholds of functional and morphological damage of brain tissue. Stroke. 1983;14:329-331.
12 Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.
13 Carter LP, Crowell RM, Sonntag VK, et al. Cortical blood flow during extracranial-intracranial bypass surgery. Stroke. 1984;15:836-839.
14 Gaines C, Carter LP, Crowell RM. Comparison of local cerebral blood flow determined by thermal and hydrogen clearance. Stroke. 1983;14:66-69.
15 Marcoux FW, Morawetz RB, Crowell RM, et al. Differential regional vulnerability in transient focal cerebral ischemia. Stroke. 1982;13:339-346.
16 Bandera E, Botteri M, Minelli C, et al. Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: a systematic review. Stroke. 2006;37:1334-1339.
17 Busto R, Dietrich WD, Globus MY, et al. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729-738.
18 Bruno A, Biller J, Adams HPJr, et al. Acute blood glucose level and outcome from ischemic stroke. Trial of ORG 10172 in Acute Stroke Treatment (TOAST) Investigators. Neurology. 1999;52:280-284.
19 Heiss WD, Huber M, Fink GR, et al. Progressive derangement of periinfarct viable tissue in ischemic stroke. J Cereb Blood Flow Metab. 1992;12:193-203.
20 Heiss WD, Graf R, Wienhard K, et al. Dynamic penumbra demonstrated by sequential multitracer PET after middle cerebral artery occlusion in cats. J Cereb Blood Flow Metab. 1994;14:892-902.
21 Baron JC. Mapping the ischaemic penumbra with PET: implications for acute stroke treatment. Cerebrovasc Dis. 1999;9:193-201.
22 Martone ME, Hu BR, Ellisman MH. Alterations of hippocampal postsynaptic densities following transient ischemia. Hippocampus. 2000;10:610-616.
23 Pisani A, Bonsi P, Calabresi P. Calcium signaling and neuronal vulnerability to ischemia in the striatum. Cell Calcium. 2004;36:277-284.
24 Lyden P. The ischemic penumbra and neuronal salvage. In: Lyden PD, editor. Current Clinical Neurology: Thrombolytic Therapy for Acute Stroke. Totowa, NJ: Humana Press; 2005:43-62.
25 Mongin AA. Disruption of ionic and cell volume homeostasis in cerebral ischemia: The perfect storm. Pathophysiology. 2007;14:183-193.
26 Hartley DM, Kurth MC, Bjerkness L, et al. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993-2000.
27 Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293-297.
28 Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369-379.
29 Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab. 1994;14:12-19.
30 Hopwood SE, Parkin MC, Bezzina EL, et al. Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis. J Cereb Blood Flow Metab. 2005;25:391-401.
31 Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-397.
32 Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399-415.
33 Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557-565.
34 Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431-1568.
35 Chu X, Fu X, Zou L, et al. Oncosis, the possible cell death pathway in astrocytes after focal cerebral ischemia. Brain Res. 2007;1149:157-164.
36 Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4:1884-1891.
37 Garthwaite G, Garthwaite J. Neurotoxicity of excitatory amino acid receptor agonists in rat cerebellar slices: dependence on calcium concentration. Neurosci Lett. 1986;66:193-198.
38 Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171-182.
39 Kristian T, Siesjö BK. Calcium in ischemic cell death. Stroke. 1998;29:705-718.
40 Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105-111.
41 Matsumoto K, Graf R, Rosner G, et al. Elevation of neuroactive substances in the cortex of cats during prolonged focal ischemia. J Cereb Blood Flow Metab. 1993;13:586-594.
42 Hillered L, Persson L, Ponten U, et al. Neurometabolic monitoring of the ischaemic human brain using microdialysis. Acta Neurochir (Wien). 1990;102:91-97.
43 Kanthan R, Shuaib A, Griebel R, et al. Intracerebral human microdialysis. In vivo study of an acute focal ischemic model of the human brain. Stroke. 1995;26:870-873.
44 Uchiyama-Tsuyuki Y, Araki H, Yae T, et al. Changes in the extracellular concentrations of amino acids in the rat striatum during transient focal cerebral ischemia. J Neurochem. 1994;62:1074-1078.
45 Hillered L, Hallstrom A, Segersvard S, et al. Dynamics of extracellular metabolites in the striatum after middle cerebral artery occlusion in the rat monitored by intracerebral microdialysis. J Cereb Blood Flow Metab. 1989;9:607-616.
46 Takagi K, Ginsberg MD, Globus MY, et al. Changes in amino acid neurotransmitters and cerebral blood flow in the ischemic penumbral region following middle cerebral artery occlusion in the rat: correlation with histopathology. J Cereb Blood Flow Metab. 1993;13:575-585.
47 Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536-537.
48 Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471-480.
49 O’Regan MH, Smith-Barbour M, Perkins LM, et al. A possible role for phospholipases in the release of neurotransmitter amino acids from ischemic rat cerebral cortex. Neurosci Lett. 1995;185:191-194.
50 Strijbos PJ, Leach MJ, Garthwaite J. Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci. 1996;16:5004-5013.
51 Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96-103.
52 Meli E, Pangallo M, Baronti R, et al. Poly(ADP-ribose) polymerase as a key player in excitotoxicity and post-ischemic brain damage. Toxicol Lett. 2003;139:153-162.
53 Nicotera P, Lipton SA. Excitotoxins in neuronal apoptosis and necrosis. J Cereb Blood Flow Metab. 1999;19:583-591.
54 Muralikrishna Adibhatla R, Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40:376-387.
55 Annunziato L, Amoroso S, Pannaccione A, et al. Apoptosis induced in neuronal cells by oxidative stress: role played by caspases and intracellular calcium ions. Toxicol Lett. 2003;139:125-133.
56 Bano D, Nicotera P. Ca2+ signals and neuronal death in brain ischemia. Stroke. 2007;38:674-676.
57 Figueroa S, Oset-Gasque MJ, Arce C, et al. Mitochondrial involvement in nitric oxide–induced cellular death in cortical neurons in culture. J Neurosci Res. 2006;83:441-449.
58 Hewett SJ, Uliasz TF, Vidwans AS, et al. Cyclooxygenase-2 contributes to N-methyl-D-aspartate–mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417-425.
59 Herrmann W. The importance of hyperhomocysteinemia as a risk factor for diseases: an overview. Clin Chem Lab Med. 2001;39:666-674.
60 Saeed SA, Shad KF, Saleem T, et al. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp Brain Res. 2007;182:1-10.
61 Costa D, Gomes A, Reis S, et al. Hydrogen peroxide scavenging activity by non-steroidal anti-inflammatory drugs. Life Sci. 2005;76:2841-2848.
62 Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2-14.
63 Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38:1433-1444.
64 Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997;28:1283-1288.
65 Chao CC, Hu S, Molitor TW, et al. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736-2741.
66 Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250-256.
67 Dawson VL, Brahmbhatt HP, Mong JA, et al. Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology. 1994;33:1425-1430.
68 Yun HY, Dawson VL, Dawson TM. Neurobiology of nitric oxide. Crit Rev Neurobiol. 1996;10:291-316.
69 Zhang J, Dawson VL, Dawson TM, et al. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687-689.
70 Fujimura M, Morita-Fujimura Y, Noshita N, et al. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817-2824.
71 Metodiewa D, Koska C. Reactive oxygen species and reactive nitrogen species: relevance to cyto(neuro)toxic events and neurologic disorders. An overview. Neurotox Res. 2000;1:197-233.
72 Manabe Y, Anrather J, Kawano T, et al. Prostanoids, not reactive oxygen species, mediate COX-2–dependent neurotoxicity. Ann Neurol. 2004;55:668-675.
73 Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med. 2001;30:433-446.
74 Siesjo BK, Ingvar M, Westerberg E. The influence of bicuculline-induced seizures on free fatty acid concentrations in cerebral cortex, hippocampus, and cerebellum. J Neurochem. 1982;39:796-802.
75 Dhillon HS, Dose JM, Scheff SW, et al. Time course of changes in lactate and free fatty acids after experimental brain injury and relationship to morphologic damage. Exp Neurol. 1997;146:240-249.
76 Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607-636.
77 Kaufmann WE, Andreasson KI, Isakson PC, et al. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601-624.
78 Fang KM, Chang WL, Wang SM, et al. Arachidonic acid induces both Na+ and Ca2+ entry resulting in apoptosis. J Neurochem. 2008;104:1177-1189.
79 Feuerstein GZ, Wang X, Barone FC. Inflammatory mediators and brain injury: The role of cytokines and chemokines in stroke and central nervous system disease. In: Ginsberg M, Bogousslavsky J, editors. Cerebrovascular Disease: Pathophysiology, Diagnosis and Management. Philadelphia: Blackwell Science; 1998:507-531.
80 Matsuo Y, Kihara T, Ikeda N, et al. Role of platelet-activating factor and thromboxane A2 in radical production during ischemia and reperfusion of the rat brain. Brain Res. 1995;709:296-302.
81 Nishigaya K, Yoshida Y, Sasuga M, et al. Effect of recirculation on exacerbation of ischemic vascular lesions in rat brain. Stroke. 1991;22:635-642.
82 Jean WC, Spellman SR, Nussbaum ES, et al. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382-1396.
83 Hallenbeck JM, Dutka AJ, Tanishima T, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17:246-253.
84 Menger MD, Lehr HA, Messmer K. Role of oxygen radicals in the microcirculatory manifestations of postischemic injury. Klin Wochenschr. 1991;69:1050-1055.
85 Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49-59.
86 Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233-1244.
87 Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932-940.
88 Yamasaki Y, Matsuura N, Shozuhara H, et al. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676-680.
89 Buttini M, Appel K, Sauter A, et al. Expression of tumor necrosis factor alpha after focal cerebral ischaemia in the rat. Neuroscience. 1996;71:1-16.
90 Sairanen TR, Lindsberg PJ, Brenner M, et al. Global forebrain ischemia results in differential cellular expression of interleukin-1beta (IL-1beta) and its receptor at mRNA and protein level. J Cereb Blood Flow Metab. 1997;17:1107-1120.
91 Brown AW, Brierley JB. The nature, distribution and earliest stages of anoxic-ischaemic nerve cell damage in the rat brain as defined by the optical microscope. Br J Exp Pathol. 1968;49:87-106.
92 Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491-498.
93 MacManus JP, Hill IE, Huang ZG, et al. DNA damage consistent with apoptosis in transient focal ischaemic neocortex. Neuroreport. 1994;5:493-496.
94 Tominaga T, Kure S, Narisawa K, et al. Endonuclease activation following focal ischemic injury in the rat brain. Brain Res. 1993;608:21-26.
95 Charriaut-Marlangue C, Margaill I, Represa A, et al. Apoptosis and necrosis after reversible focal ischemia: an in situ DNA fragmentation analysis. J Cereb Blood Flow Metab. 1996;16:186-194.
96 Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239-257.
97 Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251-306.
98 Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9:196-205.
99 Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281-1289.
100 Basanez G, Sharpe JC, Galanis J, et al. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem. 2002;277:49360-49365.
101 Zong WX, Lindsten T, Ross AJ, et al. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481-1486.
102 Ghribi O, Herman MM, Forbes MS, et al. GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis. 2001;8:764-773.
103 Cheng A, Arumugam TV, Liu D, et al. Pancortin-2 interacts with WAVE1 and Bcl-xL in a mitochondria-associated protein complex that mediates ischemic neuronal death. J Neurosci. 2007;27:1519-1528.
104 Danial NN, Gramm CF, Scorrano L, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952-956.
105 Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002-2008.
106 Li Y, Chopp M, Jiang N, et al. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389-397.
107 Goldman MS, Anderson RE, Meyer FB. Effects of intermittent reperfusion during temporal focal ischemia. J Neurosurg. 1992;77:911-916.
108 Symon L. Management of giant intracranial aneurysms. Clin Neurosurg. 1990;36:21-47.
109 Jabre A, Symon L. Temporary vascular occlusion during aneurysm surgery. Surg Neurol. 1987;27:47-63.
110 Buchthal A, Belopavlovic M, Mooij JJ. Evoked potential monitoring and temporary clipping in cerebral aneurysm surgery. Acta Neurochir (Wien). 1988;93:28-36.
111 Batjer H, Samson D. Intraoperative aneurysmal rupture: incidence, outcome, and suggestions for surgical management. Neurosurgery. 1986;18:701-707.
112 Sakaki T, Tsunoda S, Morimoto T, et al. Effects of repeated temporary clipping of the middle cerebral artery on pial arterial diameter, regional cerebral blood flow, and brain structure in cats. Neurosurgery. 1990;27:914-920.
113 Selman WR, Kiefer SP, Lust WD, et al. Metabolic evidence to support that single ischemic episodes are more devastating to the brain than are multiple insults of similar duration. J Cereb Blood Flow Metab. 1995;15(Suppl 1):S328.
114 Selman WR, Bhatti SU, Rosenstein CC, et al. Temporary vessel occlusion in spontaneously hypertensive and normotensive rats. Effect of single and multiple episodes on tissue metabolism and volume of infarction. J Neurosurg. 1994;80:1085-1090.
115 Steinberg GK, Panahian N, Sun GH, et al. Cerebral damage caused by interrupted, repeated arterial occlusion versus uninterrupted occlusion in a focal ischemic model. J Neurosurg. 1994;81:554-559.
116 Ogawa A, Sato H, Sakurai Y, et al. Limitation of temporary vascular occlusion during aneurysm surgery. Study by intraoperative monitoring of cortical blood flow. Surg Neurol. 1991;36:453-457.
117 Momma F, Wang AD, Symon L. Effects of temporary arterial occlusion on somatosensory evoked responses in aneurysm surgery. Surg Neurol. 1987;27:343-352.
118 McDermott MW, Durity FA, Borozny M, et al. Temporary vessel occlusion and barbiturate protection in cerebral aneurysm surgery. Neurosurgery. 1989;25:54-61.
119 Ljunggren B, Saveland H, Brandt L, et al. Temporary clipping during early operation for ruptured aneurysm: preliminary report. Neurosurgery. 1983;12:525-530.
120 Hosobuchi Y. Direct surgical treatment of giant intracranial aneurysms. J Neurosurg. 1979;51:743-756.
121 Heros RC, Nelson PB, Ojemann RG, et al. Large and giant paraclinoid aneurysms: surgical techniques, complications, and results. Neurosurgery. 1983;12:153-163.
122 Fox JL. Microsurgical treatment of ventral (paraclinoid) internal carotid artery aneurysms. Neurosurgery. 1988;22:32-39.
123 Charbel FT, Ausman JI, Diaz FG, et al. Temporary clipping in aneurysm surgery: technique and results. Surg Neurol. 1991;36:83-90.
124 Beck DW, Boarini DJ, Kassell NF. Surgical treatment of giant aneurysm of vertebral-basilar junction. Surg Neurol. 1979;12:283-285.
125 Samson D, Batjer HH, Bowman G, et al. A clinical study of the parameters and effects of temporary arterial occlusion in the management of intracranial aneurysms. Neurosurgery. 1994;34:22-28.
126 Ogilvy CS, Carter BS, Kaplan S, et al. Temporary vessel occlusion for aneurysm surgery: risk factors for stroke in patients protected by induced hypothermia and hypertension and intravenous mannitol administration. J Neurosurg. 1996;84:785-791.
127 Suzuki J, Kwak R, Okudaira Y. The safe time limit of temporary clamping of cerebral arteries in the direct surgical treatment of intracranial aneurysm under moderate hypothermia. Tohoku J Exp Med. 1979;127:1-7.
128 Batjer HH, Frankfurt AI, Purdy PD, et al. Use of etomidate, temporary arterial occlusion, and intraoperative angiography in surgical treatment of large and giant cerebral aneurysms. J Neurosurg. 1988;68:234-240.
129 Pool JL. Aneurysms of the anterior communicating artery. Bifrontal craniotomy and routine use of temporary clips. J Neurosurg. 1961;18:98-112.
130 Ohmoto T, Nagao S, Mino S, et al. Monitoring of cortical blood flow during temporary arterial occlusion in aneurysm surgery by the thermal diffusion method. Neurosurgery. 1991;28:49-54.
131 Solomon R. A clinical study of the parameters and effects of temporary arterial occlusion in the management of intracranial aneurysms. Comment on Samson D, Batjer HH, Bowman G, et al. Neurosurgery. 1994;34;:28-29.
132 Ferch R, Pasqualin A, Pinna G, et al. Temporary arterial occlusion in the repair of ruptured intracranial aneurysms: an analysis of risk factors for stroke. J Neurosurg. 2002;97:836-842.
133 Juvela S, Siironen J, Varis J, et al. Risk factors for ischemic lesions following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2005;102:194-201.
134 Horn P, Scharf J, Pena-Tapia P, et al. Risk of intraoperative ischemia due to temporary vessel occlusion during standard extracranial-intracranial arterial bypass surgery. J Neurosurg. 2008;108:464-469.
135 Symon L, Branston NM, Strong AJ. Autoregulation in acute focal ischemia. An experimental study. Stroke. 1976;7:547-554.
136 Cole DJ, Matsumura JS, Drummond JC, et al. Focal cerebral ischemia in rats: effects of induced hypertension, during reperfusion, on CBF. J Cereb Blood Flow Metab. 1992;12:64-69.
137 Chileuitt L, Leber K, McCalden T, et al. Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats. Surg Neurol. 1996;46:229-234.
138 Drummond JC, Oh YS, Cole DJ, et al. Phenylephrine-induced hypertension reduces ischemia following middle cerebral artery occlusion in rats. Stroke. 1989;20:1538-1544.
139 Smrcka M, Ogilvy CS, Crow RJ, et al. Induced hypertension improves regional blood flow and protects against infarction during focal ischemia: time course of changes in blood flow measured by laser Doppler imaging. Neurosurgery. 1998;42:617-624.
140 Zhao M, Amin-Hanjani S, Ruland S, et al. Regional cerebral blood flow using quantitative MR angiography. AJNR Am J Neuroradiol. 2007;28:1470-1473.
141 Friedman JA, Anderson RE, Meyer FB. Techniques of intraoperative cerebral blood flow measurement. Neurosurg Focus. 2000;9(5):e4.
142 Halsey JHJr. Risks and benefits of shunting in carotid endarterectomy. Stroke. 1992;23:1583-1587.
143 Lam AM, Newell DW. Intraoperative use of transcranial Doppler ultrasonography. Neurosurg Clin N Am. 1996;7:709-722.
144 Spencer MP. Transcranial Doppler monitoring and causes of stroke from carotid endarterectomy. Stroke. 1997;28:685-691.
145 Ashley WW, Amin-Hanjani S, Alaraj A, et al. Flow-assisted surgical cerebral revascularization. Neurosurg Focus. 2008;24(2):E20.
146 Katz JM. Is routine intraoperative angiography in the surgical treatment of cerebral aneurysms justified? A consecutive series of 147 aneurysms. Neurosurgery. 2006;58:719-727.
147 Origitano TC, Schwartz K, Anderson D, et al. Optimal clip application and intraoperative angiography for intensive care aneurysms. Surg Neurol. 1999;51:117-128.
148 Payner TD, Horner TG, Leipzig TJ, et al. Role of intraoperative angiography in the surgical treatment of cerebral aneurysms. J Neurosurg. 1998;88:441-448.
149 Rauzzino MJ, Quinn CM, Fisher WS3rd, et al. Angiography after aneurysm surgery: indications for “selective” angiography. Surg Neurol. 1998;49:32-40. discussion 40-41
150 Lopez KA, Waziri AE, Granville R, et al. Clinical usefulness and safety of routine intraoperative angiography for patients and personnel. Neurosurgery. 2007;61:724-729.
151 Chiang VL, Gailloud P, Murphy KJ, et al. Routine intraoperative angiography during aneurysm surgery. J Neurosurg. 2002;96:988-992.
152 Tang C, Cawley CM. Intraoperative angiography during aneurysm surgery: a prospective evaluation of efficacy. J Neurosurg. 2002;96:993-999.
153 Klopfenstein JD, Spetzler RF, Kim LJ, et al. Comparison of routine and selective use of intraoperative angiography during aneurysm surgery: a prospective assessment. J Neurosurg. 2004;100:230-235.
154 Friedman JA, Pichelmann MA, Piepgras DG, et al. Ischemic complications of surgery for anterior choroidal artery aneurysms. J Neurosurg. 2001;94:565-572.
155 Neuloh G, Schramm J. Monitoring of motor evoked potentials compared with somatosensory evoked potentials and microvascular Doppler ultrasonography in cerebral aneurysm surgery. J Neurosurg. 2004;100:389-399.
156 Pechstein U, Cedzich C, Nadstawek J, et al. Transcranial high-frequency repetitive electrical stimulation for recording myogenic motor evoked potentials with the patient under general anesthesia. Neurosurgery. 1996;39:335-343.
157 Horiuchi K, Suzuki K, Sasaki T, et al. Intraoperative monitoring of blood flow insufficiency during surgery of middle cerebral artery aneurysms. J Neurosurg. 2005;103:275-283.
158 Suzuki K, Kodama N, Sasaki T, et al. Intraoperative monitoring of blood flow insufficiency in the anterior choroidal artery during aneurysm surgery. J Neurosurg. 2003;98:507-514.
159 de Oliveira JG, Beck J, Seifert V, et al. Assessment of flow in perforating arteries during intracranial aneurysm surgery using intraoperative near-infrared indocyanine green videoangiography. Neurosurgery. 2007;61:63-72.
160 Amin-Hanjani S, Du X, Mlinarevich N, et al. The cut flow index: an intraoperative predictor of the success of extracranial-intracranial bypass for occlusive cerebrovascular disease. Neurosurgery. 2005;56:75-85.
161 Amin-Hanjani S, Shin JH, Zhao M, et al. Evaluation of extracranial-intracranial bypass using quantitative magnetic resonance angiography. J Neurosurg. 2007;106:291-298.
162 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
163 Hypothermia After Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
164 Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663-670.
165 Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574-1584.
166 Baker CJ, Fiore AJ, Frazzini VI, et al. Intraischemic hypothermia decreases the release of glutamate in the cores of permanent focal cerebral infarcts. Neurosurgery. 1995;36:994-1001.
167 Toyoda T, Suzuki S, Kassell NF, et al. Intraischemic hypothermia attenuates neutrophil infiltration in the rat neocortex after focal ischemia-reperfusion injury. Neurosurgery. 1996;39:1200-1205.
168 Kader A, Frazzini VI, Baker CJ, et al. Effect of mild hypothermia on nitric oxide synthesis during focal cerebral ischemia. Neurosurgery. 1994;35:272-277.
169 Kaibara T, Sutherland GR, Colbourne F, et al. Hypothermia: depression of tricarboxylic acid cycle flux and evidence for pentose phosphate shunt upregulation. J Neurosurg. 1999;90:339-347.
170 Winfree CJ, Baker CJ, Connolly ESJr, et al. Mild hypothermia reduces penumbral glutamate levels in the rat permanent focal cerebral ischemia model. Neurosurgery. 1996;38:1216-1222.
171 Han HS, Qiao Y, Karabiyikoglu M, et al. Influence of mild hypothermia on inducible nitric oxide synthase expression and reactive nitrogen production in experimental stroke and inflammation. J Neurosci. 2002;22:3921-3928.
172 Onesti ST, Baker CJ, Sun PP, et al. Transient hypothermia reduces focal ischemic brain injury in the rat. Neurosurgery. 1991;29:369-373.
173 Chen H, Chopp M, Zhang ZG, et al. The effect of hypothermia on transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1992;12:621-628.
174 Morikawa E, Ginsberg MD, Dietrich WD, et al. The significance of brain temperature in focal cerebral ischemia: histopathological consequences of middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1992;12:380-389.
175 Goto Y, Kassell NF, Hiramatsu K, et al. Effects of intraischemic hypothermia on cerebral damage in a model of reversible focal ischemia. Neurosurgery. 1993;32:980-984.
176 Zhao H, Wang JQ, Shimohata T, et al. Conditions of protection by hypothermia and effects on apoptotic pathways in a rat model of permanent middle cerebral artery occlusion. J Neurosurg. 2007;107:636-641.
177 Kader A, Brisman MH, Maraire N, et al. The effect of mild hypothermia on permanent focal ischemia in the rat. Neurosurgery. 1992;31:1056-1060.
178 Huh PW, Belayev L, Zhao W, et al. Comparative neuroprotective efficacy of prolonged moderate intraischemic and postischemic hypothermia in focal cerebral ischemia. Neurosurg Focus. 1999;7:E2.
179 Lo EH, Steinberg GK. Effects of hypothermia on evoked potentials, magnetic resonance imaging, and blood flow in focal ischemia in rabbits. Stroke. 1992;23:889-893.
180 Karibe H, Chen J, Zarow GJ, et al. Delayed induction of mild hypothermia to reduce infarct volume after temporary middle cerebral artery occlusion in rats. J Neurosurg. 1994;80:112-119.
181 Karibe H, Sato K, Shimizu H, et al. Intraoperative mild hypothermia ameliorates postoperative cerebral blood flow impairment in patients with aneurysmal subarachnoid hemorrhage. Neurosurgery. 2000;47:594-599.
182 Steinberg GK, Ogilvy CS, Shuer LM, et al. Comparison of endovascular and surface cooling during unruptured cerebral aneurysm repair. Neurosurgery. 2004;55:307-314.
183 Kimme P, Fridrikssen S, Engdahl O, et al. Moderate hypothermia for 359 operations to clip cerebral aneurysms. Br J Anaesth. 2004;93:343-347.
184 Todd MM, Hindman BJ, Clarke WR, et al. Mild intraoperative hypothermia during surgery for intracranial aneurysm. N Engl J Med. 2005;352:135-145.
185 Smith AL, Hoff JT, Nielsen SL, et al. Barbiturate protection in acute focal cerebral ischemia. Stroke. 1974;5:1-7.
186 Black KL, Weidler DJ, Jallad NS, et al. Delayed pentobarbital therapy of acute focal cerebral ischemia. Stroke. 1978;9:245-249.
187 Selman WR, Spetzler RF, Anton AH, et al. Management of prolonged therapeutic barbiturate coma. Surg Neurol. 1981;15:9-10.
188 Selman WR, Spetzler RF, Roessmann UR, et al. Barbiturate-induced coma therapy for focal cerebral ischemia. Effect after temporary and permanent MCA occlusion. J Neurosurg. 1981;55:220-226.
189 Selman WR, Spetzler RF, Roski RA, et al. Barbiturate coma in focal cerebral ischemia. Relationship of protection to timing of therapy. J Neurosurg. 1982;56:685-690.
190 Warner DS, Takaoka S, Wu B, et al. Electroencephalographic burst suppression is not required to elicit maximal neuroprotection from pentobarbital in a rat model of focal cerebral ischemia. Anesthesiology. 1996;84:1475-1484.
191 Schmid-Elsaesser R, Schroder M, Zausinger S, et al. EEG burst suppression is not necessary for maximum barbiturate protection in transient focal cerebral ischemia in the rat. J Neurol Sci. 1999;162:14-19.
192 Batjer HH. Cerebral protective effects of etomidate: experimental and clinical aspects. Cerebrovasc Brain Metab Rev. 1993;5:17-32.
193 Rosenwasser RH, Jimenez DF, Wending WW, et al. Routine use of etomidate and temporary vessel occlusion during aneurysm surgery. Neurol Res. 1991;13:224-228.
194 Petersen KD, Landsfeldt U, Cold GE, et al. Intracranial pressure and cerebral hemodynamic in patients with cerebral tumors: a randomized prospective study of patients subjected to craniotomy in propofol-fentanyl, isoflurane-fentanyl, or sevoflurane-fentanyl anesthesia. Anesthesiology. 2003;98:329-336.
195 Sneyd JR, Andrews CJ, Tsubokawa T. Comparison of propofol/remifentanil and sevoflurane/remifentanil for maintenance of anaesthesia for elective intracranial surgery. Br J Anaesth. 2005;94:778-783.
196 Hans P, Bonhomme V. Why we still use intravenous drugs as the basic regimen for neurosurgical anaesthesia. Curr Opin Anaesthesiol. 2006;19:498-503.
197 Lavine SD, Masri LS, Levy ML, et al. Temporary occlusion of the middle cerebral artery in intracranial aneurysm surgery: time limitation and advantage of brain protection. J Neurosurg. 1997;87:817-824.
198 Wass CT, Lanier WL. Glucose modulation of ischemic brain injury: review and clinical recommendations. Mayo Clin Proc. 1996;71:801-812.
199 Fukuda S, Warner DS. Cerebral protection. Br J Anaesth. 2007;99:10-17.
200 Pasternak JJ, McGregor DG, Schroeder DR, et al. Hyperglycemia in patients undergoing cerebral aneurysm surgery: its association with long-term gross neurologic and neuropsychological function. Mayo Clin Proc. 2008;83:406-417.
201 Davalos A, Castillo J, Alvarez-Sabin J, et al. Oral citicoline in acute ischemic stroke: an individual patient data pooling analysis of clinical trials. Stroke. 2002;33:2850-2857.
202 Benham CD, Brown TH, Cooper DG, et al. SB 201823-A, a neuronal Ca2+ antagonist is neuroprotective in two models of cerebral ischaemia. Neuropharmacology. 1993;32:1249-1257.
203 Harada K, Shiino A, Matsuda M, et al. Effects of a novel calcium antagonist, KB-2796, on neurologic outcome and size of experimental cerebral infarction in rats. Surg Neurol. 1989;32:16-20.
204 Kittaka M, Giannotta SL, Zelman V, et al. Attenuation of brain injury and reduction of neuron-specific enolase by nicardipine in systemic circulation following focal ischemia and reperfusion in a rat model. J Neurosurg. 1997;87:731-737.
205 Kondoh Y, Mizusawa S, Murakami M, et al. Fasudil (HA1077), an intracellular calcium antagonist, improves neurological deficits and tissue potassium loss in focal cerebral ischemia in gerbils. Neurol Res. 1997;19:211-215.
206 Ohtaki M, Tranmer B. Pretreatment of transient focal cerebral ischemia in rats with the calcium antagonist AT877. Stroke. 1994;25:1234-1239.
207 Perez-Pinzon MA, Yenari MA, Sun GH, et al. SNX-111, a novel, presynaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia in rabbits. J Neurol Sci. 1997;153:25-31.
208 Sakaki T, Tsujimoto S, Sasaoka Y, et al. The effect of a new calcium antagonist, TA3090 (clentiazem), on experimental transient focal cerebral ischemia in cats. Stroke. 1993;24:872-878.
209 Shiino A, Matsuda M, Susumu T, et al. Effects of the calcium antagonist nilvadipine on focal cerebral ischemia in spontaneously hypertensive rats. Surg Neurol. 1991;35:105-110.
210 Takizawa S, Matsushima K, Fujita H, et al. A selective N-type calcium channel antagonist reduces extracellular glutamate release and infarct volume in focal cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:611-618.
211 Germano IM, Bartkowski HM, Cassel ME, et al. The therapeutic value of nimodipine in experimental focal cerebral ischemia. Neurological outcome and histopathological findings. J Neurosurg. 1987;67:81-87.
212 Meyer FB, Anderson RE, Yaksh TL, et al. Effect of nimodipine on intracellular brain pH, cortical blood flow, and EEG in experimental focal cerebral ischemia. J Neurosurg. 1986;64:617-626.
213 Uematsu D, Greenberg JH, Hickey WF, et al. Nimodipine attenuates both increase in cytosolic free calcium and histologic damage following focal cerebral ischemia and reperfusion in cats. Stroke. 1989;20:1531-1537.
214 Scriabine A, van den Kerckhoff W. Pharmacology of nimodipine. A review. Ann N Y Acad Sci. 1988;522:698-706.
215 Gelmers HJ, Gorter K, de Weerdt CJ, et al. A controlled trial of nimodipine in acute ischemic stroke. N Engl J Med. 1988;318:203-207.
216 Paci A, Ottaviano P, Trenta A, et al. Nimodipine in acute ischemic stroke: a double-blind controlled study. Acta Neurol Scand. 1989;80:282-286.
217 Gelmers HJ, Hennerici M. Effect of nimodipine on acute ischemic stroke. Pooled results from five randomized trials. Stroke. 1990;21:IV81-84.
218 Mohr JP OJ, Harrison MJG, et al. Meta-analysis of oral nimodipine trials in acute ischemic stroke. Cerebrovasc Dis. 1994;4:197-203.
219 Randomised, double-blind, placebo-controlled trial of nimodipine in acute stroke. Trust Study Group. Lancet. 1990;336:1205-1209.
220 Clinical trial of nimodipine in acute ischemic stroke. The American Nimodipine Study Group. Stroke. 1992;23:3-8.
221 Kaste M, Fogelholm R, Erila T, et al. A randomized, double-blind, placebo-controlled trial of nimodipine in acute ischemic hemispheric stroke. Stroke. 1994;25:1348-1353.
222 Martinez-Vila E, Guillen F, Villanueva JA, et al. Placebo-controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction. Stroke. 1990;21:1023-1028.
223 Fisher M, Bogousslavsky J. Further evolution toward effective therapy for acute ischemic stroke. JAMA. 1998;279:1298-1303.
224 Minematsu K, Fisher M, Li L, et al. Effects of a novel NMDA antagonist on experimental stroke rapidly and quantitatively assessed by diffusion-weighted MRI. Neurology. 1993;43:397-403.
225 Park CK, Nehls DG, Graham DI, et al. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1988;24:543-551.
226 Bertorelli R, Adami M, Di Santo E, et al. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci Lett. 1998;246:41-44.
227 Maier CM, Sun GH, Kunis DM, et al. Neuroprotection by the N-methyl-D-aspartate receptor antagonist CGP 40116: in vivo and in vitro studies. J Neurochem. 1995;65:652-659.
228 Steinberg GK, Panahian N, Perez-Pinzon MA, et al. Narrow temporal therapeutic window for NMDA antagonist protection against focal cerebral ischaemia. Neurobiol Dis. 1995;2:109-118.
229 Britton P, Lu XC, Laskosky MS, et al. Dextromethorphan protects against cerebral injury following transient, but not permanent, focal ischemia in rats. Life Sci. 1997;60:1729-1740.
230 Steinberg GK, Kunis D, DeLaPaz R, et al. Neuroprotection following focal cerebral ischaemia with the NMDA antagonist dextromethorphan, has a favourable dose response profile. Neurol Res. 1993;15:174-180.
231 Takaoka S, Bart RD, Pearlstein R, et al. Neuroprotective effect of NMDA receptor glycine recognition site antagonism persists when brain temperature is controlled. J Cereb Blood Flow Metab. 1997;17:161-167.
232 Albers GW, Atkinson RP, Kelley RE, et al. Safety, tolerability, and pharmacokinetics of the N-methyl-D-aspartate antagonist dextrorphan in patients with acute stroke. Dextrorphan Study Group. Stroke. 1995;26:254-258.
233 Albers GW, Saenz RE, Moses JAJr. Tolerability of oral dextromethorphan in patients with a history of brain ischemia. Clin Neuropharmacol. 1992;15:509-514.
234 Albers GW, Saenz RE, Moses JAJr, et al. Safety and tolerance of oral dextromethorphan in patients at risk for brain ischemia. Stroke. 1991;22:1075-1077.
235 Albers GW, Clark WM, Atkinson RP, et al. Dose escalation study of the NMDA glycine-site antagonist licostinel in acute ischemic stroke. Stroke. 1999;30:508-513.
236 Schmid-Elsaesser R, Zausinger S, Hungerhuber E, et al. Neuroprotective effects of combination therapy with tirilazad and magnesium in rats subjected to reversible focal cerebral ischemia. Neurosurgery. 1999;44:163-171.
237 Izumi Y, Roussel S, Pinard E, et al. Reduction of infarct volume by magnesium after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 1991;11:1025-1030.
238 Tsuda T, Kogure K, Nishioka K, et al. Mg2+ administered up to twenty-four hours following reperfusion prevents ischemic damage of the Ca1 neurons in the rat hippocampus. Neuroscience. 1991;44:335-341.
239 Marinov MB, Harbaugh KS, Hoopes PJ, et al. Neuroprotective effects of preischemia intraarterial magnesium sulfate in reversible focal cerebral ischemia. J Neurosurg. 1996;85:117-124.
240 Muir KW, Lees KR. Dose optimization of intravenous magnesium sulfate after acute stroke. Stroke. 1998;29:918-923.
241 Muir KW. New experimental and clinical data on the efficacy of pharmacological magnesium infusions in cerebral infarcts. Magnes Res. 1998;11:43-56.
242 Muir KW, Lees KR. Clinical experience with excitatory amino acid antagonist drugs. Stroke. 1995;26:503-513.
243 Coert BA, Anderson RE, Meyer FB. A comparative study of the effects of two nitric oxide synthase inhibitors and two nitric oxide donors on temporary focal cerebral ischemia in the Wistar rat. J Neurosurg. 1999;90:332-338.
244 Imaizumi S, Woolworth V, Fishman RA, et al. Liposome-entrapped superoxide dismutase reduces cerebral infarction in cerebral ischemia in rats. Stroke. 1990;21:1312-1317.
245 Liu TH, Beckman JS, Freeman BA, et al. Polyethylene glycol–conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am J Physiol. 1989;256:H589-H593.
246 Martz D, Rayos G, Schielke GP, et al. Allopurinol and dimethylthiourea reduce brain infarction following middle cerebral artery occlusion in rats. Stroke. 1989;20:488-494.
247 Spinnewyn B, Cornet S, Auguet M, et al. Synergistic protective effects of antioxidant and nitric oxide synthase inhibitor in transient focal ischemia. J Cereb Blood Flow Metab. 1999;19:139-143.
248 Trifiletti RR. Neuroprotective effects of NG-nitro-L-arginine in focal stroke in the 7-day old rat. Eur J Pharmacol. 1992;218:197-198.
249 del Pilar Fernandez M, Meizoso MJ, Lodeiro MJ, et al. Effect of desmethyl tirilazad, dizocilpine maleate and nimodipine on brain nitric oxide synthase activity and cyclic guanosine monophosphate during cerebral ischemia in rats. Pharmacology. 1998;57:174-179.
250 Karki A, Westergren I, Widner H, et al. Tirilazad reduces brain edema after middle cerebral artery ligation in hypertensive rats. Acta Neurochir Suppl. 1994;60:310-313.
251 Wilson JT, Bednar MM, McAuliffe TL, et al. The effect of the 21-aminosteroid U74006F in a rabbit model of thromboembolic stroke. Neurosurgery. 1992;31:929-933.
252 Young W, Wojak JC, DeCrescito V. 21-Aminosteroid reduces ion shifts and edema in the rat middle cerebral artery occlusion model of regional ischemia. Stroke. 1988;19:1013-1019.
253 Schmid-Elsaesser R, Zausinger S, Hungerhuber E, et al. Monotherapy with dextromethorphan or tirilazad, but not a combination of both, improves outcome after transient focal cerebral ischemia in rats. Exp Brain Res. 1998;122:121-127.
254 Clark WM, Hotan T, Lauten JD, et al. Therapeutic efficacy of tirilazad in experimental multiple cerebral emboli: a randomized, controlled trial. Crit Care Med. 1994;22:1161-1166.
255 Hellstrom HO, Wanhainen A, Valtysson J, et al. Effect of tirilazad mesylate given after permanent middle cerebral artery occlusion in rat. Acta Neurochir (Wien). 1994;129:188-192.
256 Kassell NF, Haley ECJr, Apperson-Hansen C, et al. Randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in Europe, Australia, and New Zealand. J Neurosurg. 1996;84:221-228.
257 Haley ECJr, Kassell NF, Apperson-Hansen C, et al. A randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in North America. J Neurosurg. 1997;86:467-474.
258 A randomized trial of tirilazad mesylate in patients with acute stroke (RANTTAS). The RANTTAS Investigators. Stroke. 1996;27:1453-1458.
259 Ashton D, Willems R, Wynants J, et al. Altered Na+ channel function as an in vitro model of the ischemic penumbra: action of lubeluzole and other neuroprotective drugs. Brain Res. 1997;745:210-221.
260 Lesage AS, Peeters L, Leysen JE. Lubeluzole, a novel long-term neuroprotectant, inhibits the glutamate-activated nitric oxide synthase pathway. J Pharmacol Exp Ther. 1996;279:759-766.
261 Maiese K, TenBroeke M, Kue I. Neuroprotection of lubeluzole is mediated through the signal transduction pathways of nitric oxide. J Neurochem. 1997;68:710-714.
262 Marrannes R, De Prins E, Clincke G. Influence of lubeluzole on voltage-sensitive Ca++ channels in isolated rat neurons. J Pharmacol Exp Ther. 1998;286:201-214.
263 Scheller DK, De Ryck M, Kolb J, et al. Lubeluzole blocks increases in extracellular glutamate and taurine in the peri-infarct zone in rats. Eur J Pharmacol. 1997;338:243-251.
264 Diener HC, Cortens M, Ford G, et al. Lubeluzole in acute ischemic stroke treatment: A double-blind study with an 8-hour inclusion window comparing a 10-mg daily dose of lubeluzole with placebo. Stroke. 2000;31:2543-2551.
265 Culmsee C, Junker V, Wolz P, et al. Lubeluzole protects hippocampal neurons from excitotoxicity in vitro and reduces brain damage caused by ischemia. Eur J Pharmacol. 1998;342:193-201.
266 De Ryck M, Keersmaekers R, Duytschaever H, et al. Lubeluzole protects sensorimotor function and reduces infarct size in a photochemical stroke model in rats. J Pharmacol Exp Ther. 1996;279:748-758.
267 De Keyser J, Van de Velde V, Schellens RL, et al. Safety and pharmacokinetics of the neuroprotective drug lubeluzole in patients with ischemic stroke. Clin Ther. 1997;19:1340-1351.
268 Hacke W, Lees KR, Timmerhuis T, et al. Cardiovascular safety of lubeluzole (Prosynap(R)) in patients with ischemic stroke. Cerebrovasc Dis. 1998;8:247-254.
269 Hantson L, Tritsmans L, Crabbe R, et al. The safety and tolerability of single intravenous doses of lubeluzole (Prosynap) in healthy volunteers. Int J Clin Pharmacol Ther. 1997;35:491-495.
270 Diener HC, Hacke W, Hennerici M, et al. Lubeluzole in acute ischemic stroke. A double-blind, placebo-controlled phase II trial. Lubeluzole International Study Group. Stroke. 1996;27:76-81.
271 Diener HC. Multinational randomised controlled trial of lubeluzole in acute ischaemic stroke. European and Australian Lubeluzole Ischaemic Stroke Study Group. Cerebrovasc Dis. 1998;8:172-181.
272 Grotta J. Lubeluzole treatment of acute ischemic stroke. The US and Canadian Lubeluzole Ischemic Stroke Study Group. Stroke. 1997;28:2338-2346.
273 Schabitz WR, Weber J, Takano K, et al. The effects of prolonged treatment with citicoline in temporary experimental focal ischemia. J Neurol Sci. 1996;138:21-25.
274 D’Orlando KJ, Sandage BWJr. Citicoline (CDP-choline): mechanisms of action and effects in ischemic brain injury. Neurol Res. 1995;17:281-284.
275 Clark WM, Warach SJ, Pettigrew LC, et al. A randomized dose-response trial of citicoline in acute ischemic stroke patients. Citicoline Stroke Study Group. Neurology. 1997;49:671-678.
276 Tymianski M, Tutor CH. Normal and abnormal calcium homeostasis in neurons: A basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery. 1996;38:1176-1195.
277 Kittaka M, Giannotta SL, Zelman V, et al. Neuroprotective effects of nicardipine in a rat model of ischemia and reperfusion. Neurosurg Focus. 1997;2:E4.
278 Ponce J, de la Ossa NP, Hurtado O, et al. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39:1269-1275.
279 Johnson-Anuna LN, Eckert GP, Franke C, et al. Simvastatin protects neurons from cytotoxicity by up-regulating Bcl-2 mRNA and protein. J Neurochem. 2007;101:77-86.
280 Dolga AM, Nijholt IM, Ostroveanu A, et al. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J Alzheimers Dis. 2008;13:111-122.
281 Zhang RL, Chopp M, Li Y, et al. Anti–ICAM-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in the rat. Neurology. 1994;44:1747-1751.
282 Goussev AV, Zhang Z, Anderson DC, et al. P-selectin antibody reduces hemorrhage and infarct volume resulting from MCA occlusion in the rat. J Neurol Sci. 1998;161:16-22.
283 Chen H, Chopp M, Zhang RL, et al. Anti-CD11b monoclonal antibody reduces ischemic cell damage after transient focal cerebral ischemia in rat. Ann Neurol. 1994;35:458-463.
284 Schneider D, Berrouschot J, Brandt T, et al. Safety, pharmacokinetics and biological activity of enlimomab (anti–ICAM-1 antibody): an open-label, dose escalation study in patients hospitalized for acute stroke. Eur Neurol. 1998;40:78-83.
285 Investigators EAST. The enlimolab acute stroke trial: Final results [abstract]. Neurology. 1997;48:A270.
286 Angel I, Bar A, Horovitz T, et al. Metal ion chelation in neurodegenerative disorders. Drug Dev Res. 2002;56:300-309.
287 Krakovsky M, Polyak M, Angel I, et al. DP-B99: A novel membrane-targeted compound active against global and focal cerebral ischemia. In: Gjedde A, Hansen S, Knudsen G, et al, editors. Physiological Imaging of the Brain with PET. New York: Academic Press; 2001:347-352.
288 Diener HC, Schneider D, Lampl Y, et al. DP-b99, a membrane-activated metal ion chelator, as neuroprotective therapy in ischemic stroke. Stroke. 2008;39:1774-1778.
289 Marshall JW, Cummings RM, Bowes LJ, et al. Functional and histological evidence for the protective effect of NXY-059 in a primate model of stroke when given 4 hours after occlusion. Stroke. 2003;34:2228-2233.
290 Zhao Z, Cheng M, Maples KR, et al. NXY-059, a novel free radical trapping compound, reduces cortical infarction after permanent focal cerebral ischemia in the rat. Brain Res. 2001;909:46-50.
291 Sattin JA, Zivin JA. Emerging therapies for acute ischemic stroke. Am J Ther. 2007;14:291-298.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 345 Intraoperative Cerebral Protection
Pathophysiology of Ischemic Injury
Cerebral Blood Flow
In 1948, Kety and Schmidt quantified normal cerebral blood flow (CBF) in “healthy normal men” to be 54 mL/100 g per minute.1 Sundt and others noted that a minimal CBF of 18 mL/100 g per minute is needed to maintain normal electroencephalographic (EEG) parameters during carotid endarterectomy (CEA).2,3 A further decrease in CBF causes neuronal electrical silence and decreased synaptic activity to preserve energy stores. Irreversible cellular damage occurs when CBF is below 10 mL/100 g per minute.4–8 In response to ischemia, the cerebral vessel autoregulatory mechanism induces vasodilation to increase collateral blood flow and thereby increase oxygen and glucose extraction for preservation of viable neurons (Fig. 345-1).
Ischemic Penumbra
Laboratory and clinical evidence demonstrates a spectrum of injured neurons when CBF is interrupted as a result of vessel occlusion. In the affected territory, there is a core of irreversibly damaged neurons surrounded by an area of electrically silent but viable neurons known as the ischemic penumbra.4,9–11 CBF in the penumbra has been determined to be between 10 and 20 mL/100 g per minute.7,12–16 These neurons may survive for hours, although the exact duration is unknown. Many factors may reduce the time for survivability of these marginal neurons. Hyperthermia of 1°C to 2°C and serum hyperglycemia have been shown to accelerate cell death.17,18 Restoration of CBF to the penumbra within the “critical hours” may salvage viable neurons and minimize the neurological deficits (Fig. 345-2). Without intervention, the core of the infarcted territory will subsume the penumbra as demonstrated by positron emission tomography (PET) and magnetic resonance imaging (MRI).19–21 The temporal and spatial evolution of the infarcted core is highly variable.19,21 Baron demonstrated by PET that the onset of the penumbra occurs as late as 16 hours after infarction.21 Susceptibility to ischemic injury also varies in different areas of the brain, with hippocampal CA1 and striatal neurons being more vulnerable than cortical neurons in ischemic models.22,23
FIGURE 345-2 Relationship between time of ischemia and reversibility of neurological deficit. CBF, cerebral blood flow.
(Adapted from Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.)
The traditional concept of an infarcted core surrounding by a penumbra may be too simplistic. Many patients who improve with thrombolytic agents do not demonstrate a core surrounded by a larger penumbra on imaging. The core may be surrounded by heterogeneous regions of penumbra, with possibly separate areas of minimal flow or core within the penumbra as a result of variable degrees of collateral circulation.24
Energy Failure
The decrease in energy production from anaerobic metabolism (2 adenosine triphosphate [ATP] molecules) versus oxidative phosphorylation (32 ATP molecules) causes failure of the ionic electrochemical gradient established by the sodium-potassium adenosine triphosphatase (Na+,K+-ATPase) pump. Peri-infarct depolarization can be observed in the cortical penumbra with the use of direct current potential. These phenomena are associated with efflux of K+ and influx of Na+ and Ca2+.25–28 Biochemical analysis of PO2 and ATP in the penumbra confirms the depletion during depolarization,29,30 thus placing an additional burden on compromised tissue and increasing the size of the ischemic lesion by 30% to 100%.31–34 Ionic influx also results in oncosis,35 a condition characterized by swelling of cells and organelles, increased membrane permeability, and nonspecific DNA damage. Swelling of astrocytes causes large amounts of excitatory amino acids (EAAs) to be released through volume-activated anion channels.35 Although the death pathway of astrocytes is unclear, oncosis may contribute to the mechanism.
Altered Calcium Homeostasis and Excitotoxicity
Ischemia causes an increase in the intracellular calcium concentration from depolarization of voltage-gated channels, activation of ligand-gated channels, release of intracytoplasmic stores, and loss of ATP-dependent calcium extrusion pathways. In addition, there is an influx of calcium down an electrochemical gradient (Fig. 345-3).31 The calcium-mediated release of glutamate and EAAs from depolarized neurons plays a major role in ischemic cell death.36–38 In the presence of energy failure, depolarization of somatodendritic and presynaptic voltage-dependent calcium channels leads to excessive extracellular glutamate and EAAs (Fig. 345-4).39 When combined with failure of glutamate reuptake, toxic accumulation of extracellular EAAs causes excessive activation of the postsynaptic glutamate receptors N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoazolepropionic acid (AMPA). Overactivation of NMDA and AMPA receptors facilitates a massive influx of calcium and sodium ions, respectively.39 Animal models and microdialysis during neurosurgical procedures have shown glutamate and other EAAs to be markedly elevated over baseline levels.40–46 Accumulation of ions leads to cellular edema and peri-infarct depolarization.47,48 Such depolarization has been recorded for 6 to 8 hours after an initial ischemic insult.31 The depletion of energy stores needed for restoration of normal depolarization is thought to contribute to enlargement of an infarct. In addition, excessive intracellular calcium directly and indirectly initiates a series of detrimental cytoplasmic and nuclear cascades, including activation of Ca2+-dependent proteases and phospholipases, alterations in plasma membrane permeability, depolarization of mitochondria and release of mitochondrial proapoptotic proteins, production of reactive oxygen species (ROSs) and reactive nitrogen species (RNSs), and activation of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP).49–56
FIGURE 345-3 Simplified overview of pathophysiologic mechanisms in a focally ischemic brain. Energy failure leads to depolarization of neurons. Activation of specific glutamate receptors dramatically increases intracellular levels of Ca2+, Na+, and Cl−, whereas K+ is released into the extracellular space. Diffusion of glutamate (Glu) and K+ in the extracellular space can propagate a series of spreading waves of depolarization (peri-infarct depolarizations). Water shifts to the intracellular space via osmotic gradients, and cells swell (edema). The universal intracellular messenger Ca2+ overactivates numerous enzyme systems (e.g., proteases, lipases, endonucleases). Free radicals, which damage membranes (lipolysis), mitochondria, and DNA, are generated and in turn trigger caspase-mediated cell death (apoptosis). Free radicals also induce the formation of inflammatory mediators, which activate microglia and lead to the invasion of blood-borne inflammatory cells.31
(Used with permission from Barrow Neurological Institute.)
FIGURE 345-4 Calcium handling by cells. Calcium enters the cell through voltage- and agonist-operated channels and through nonspecific channels, such as those opened by reactive oxygen species (ROS), whereas Ca2+ is extruded from the cell by an adenosine triphosphate (ATP)-driven pump or by Ca2+-Na+ exchange. Cytosolic calcium (Ca2+) is sequestered by mitochondria and endoplasmic reticulum (ER) at the expense of respiratory energy or ATP, and it is bound to calcium-binding molecules. Ca2+ is released from the ER in response to agonist activation of receptors coupled to phospholipase C (PLC) and the formation of inositol 1,4,5-triphosphate (IP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). A rise in Ca2+ can cause translocation of protein kinase C (PKC) to membranes where it may be activated by diglycerides (DG) formed during hydrolysis of PIP2. Accumulation of Ca2+ in mitochondria can lead to the activation of a mitochondrial permeability transition (MPT) pore and result in the release of Ca2+ and other molecules with molecular weights of up to 1.5 kD from intramitochondrial compartments.39 ADP, adenosine diphosphate; APCD, 1-aminocyclopentane-1S,3R-dicarboxylic acid; ICDH, isocitrate dehydrogenase; OGDH, oxoglutarate dehydrogenase PDH, pyruvate dehydrogenase; Quis, quisqualate.
(Used with permission from Barrow Neurological Institute.)
Free Radicals
With its high oxygen consumption, the brain is especially susceptible to free radical damage. It also has high levels of iron and ascorbate,57–59 which can act as pro-oxidants under pathologic conditions, as well as low levels of antioxidant enzymes such as catalase, glutathione peroxidase, and superoxide dismutase.60 The cellular membranes are rich in polyunsaturated fatty acids, which are vulnerable to radical-induced peroxidation. The acidic environment from lactic acidosis during ischemia promotes dissociation of iron from protein and enhances the conversion of superoxide anion to more reactive radicals.58,61
ROSs and RNSs play significant roles in the pathogenesis and exacerbation of ischemic injury. It has been postulated that low levels of ROSs and RNSs trigger apoptosis and higher levels cause necrosis.62,63 During ischemia, nitric oxide (NO), a vasodilator, is generated by endothelial nitrous oxide synthase (NOS) to increase local CBF.64 Although this may be beneficial locally in the vasculature, a higher concentration of NO is produced by neuronal NOS and inducible NOS from inflammatory cells in the ischemic territory and may play a role in neuronal injury.64–68 NO and its derivative peroxynitrite, in addition to oxidizing effects on cellular structures, contribute to the oxidative stress by activating PARP, thus further depleting energy stores.69 PARP catalyzes the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD) to nuclear proteins. For every mole of ADP-ribose transferred, 1 mol of NAD is consumed. Four free energy equivalents of ATP are necessary to regenerate NAD.
Although ROSs are generated during ischemia, reperfusion after ischemia greatly increases ROS generation.57,70,71 Reoxygenation provides an excess supply of substrate for oxidation, thus producing more ROSs and RNSs. This further depletes endogenous antioxidants, including radical scavengers such as tocopherol (vitamin E) and carotene and enzymes such as superoxide dismutase and catalase. ROSs and RNSs are also produced in mitochondria during ischemia and reperfusion and are involved in the release of cytochrome c.58,72 Studies have shown that depletion of glutathione, a reductant, can cause a 100-fold increase in mitochondrial production of ROSs.60,73 Free radicals have been implicated in signaling pathways involving dephosphorylation and translocation of the proapoptotic Bad protein from the cytosol to the mitochondrial membrane.60 By affecting the redox state of neurons and glial cells, free radicals can alter their genetic expression, activate destructive proteins, and inhibit protective enzymes, which ultimately leads to cellular death and breakdown of the blood-brain barrier.
Free Fatty Acids
During ischemia, stimulation of glutamate receptors and an increase in intracellular calcium activate various subtypes of phospholipases, which causes a large amount of fatty acids, particularly arachidonic acid, to be released from the cellular membrane.74–76 With free radicals, arachidonic acid is further metabolized by cyclooxygenase/lipoxygenase to produce leukotriene C4, prostaglandin E2, and additional free radicals. These metabolites have been implicated in neuronal demise.77 Recent laboratory evidence suggests that arachidonic acid induces cytosolic and mitochondrial Na+ and Ca2+ overload via nonselective cation conductance, thereby leading to the formation of mitochondrial permeability transition pores, release of cytochrome c, and activation of caspase-3–dependent apoptosis.78
Inflammatory Response
Reperfusion facilitates the arrival of inflammatory cells at the ischemic zone, and postischemic inflammation contributes to ischemic injury in several ways.79 Inflammatory cells can aggravate ischemic injury by compromising blood flow in the microcirculation.80–82 These cells also stimulate platelet aggregation and the formation of microthrombi, which further propagates the ischemic injury. After adhesion to the endothelial walls, granulocytes migrate (diapedesis) into the parenchyma and can be seen in peri-ischemic areas within hours of arterial occlusion.83 Inflammatory cells are also responsible for the production of proteolytic enzymes and the generation of free radicals.84 Activation of metalloproteinases and proteases destroys surrounding tissues and the extracellular matrix, independent of the energy status of neurons.24,31 This inflammatory response is enhanced by proinflammatory chemokines such as interleukin-1β (IL-1β), IL-8, and tumor necrosis factor-α (TNF-α) and mediated by upregulation of adhesion molecules, including selectins, integrins (CD11/CD18), and the immunoglobulin supergene family.82,85–90
Cell Death
The two processes by which injured neurons die are necrosis and apoptosis, and these processes occur in both the infarcted core and the penumbra.53,91–95 When necrosis develops,91 cells swell in the first few hours with the formation of eosinophilic neurons. The cells then shrink and undergo pyknosis (condensation of nuclear chromatin). The number of necrotic neurons increases most markedly during the initial 6 to 12 hours. Phagocytosis of necrotic tissue takes place over a period of days. Pan-necrosis can occur in the infarcted core and give rise to a cystic cavity.25 In the penumbra, infarcted neurons are removed and reactive astrocytes proliferate, thereby creating an incomplete infarct.
Ischemic apoptosis may be triggered by oxidative stress and overactivation of glutamate receptors, although how ischemia triggers the apoptotic mechanism is unclear. The process begins with shrinkage of the nucleus and cytoplasm, followed by condensation of chromatin and nuclear fragmentation. The hallmark of apoptosis is a “laddering” pattern of DNA on gel electrophoresis caused by endonuclease cleavage of DNA into segments of nonrandom length.96,97 Eventually, the cells separate into small, pyknotic bodies that undergo phagocytosis with minimal associated inflammatory response.
Evidence has shown that the Bcl-2 family of proteins plays a significant role in determining whether ischemic neurons resist or succumb to apoptosis.98,99 Proapoptotic Bcl-2 proteins, such as Bad and Bax, are translocated into the mitochondrial membrane, where they induce the formation of pores and release proapoptotic enzymes that lead to the activation of proteases and cell death.100,101 The antiapoptotic proteins Bcl-2 and Bcl-xL may prevent apoptosis by inhibiting the proapoptotic proteins from inducing mitochondrial pore formation (Fig. 345-5).102 Other proteins involved in the mitochondrial pore complex include pancortin-2 and the Wiskott-Aldrich syndrome protein verprolin homologous-1 (WAVE1).103,104 Further evidence in ischemic animal models has demonstrated that pancortin-2 sequesters Bcl-xL in association with WAVE1 to promote the translocation of Bax to mitochondria, which causes release of cytochrome c and apoptosis.103
Other degradative enzymes activated during apoptosis include endonucleases, proteases, and caspases such as IL-1β–converting enzyme. Analysis of the DNA of apoptotic cells shows the laddering of approximately 200 base pairs of DNA fragments, which is specific for apoptosis rather than necrosis. These DNA fragments, indicative of neuronal death through apoptotic mechanisms, have been observed after focal ischemia by many investigators.93,105,106
Integration of Cellular Injury Mechanisms
Ischemic injury causes dynamic and highly interrelated biochemical changes that have variable outcomes. In the absence of collateral circulation, neuronal tissue undergoes necrosis. When the collateral circulation is able to maintain cellular integrity, these neurons can survive for a finite period but are susceptible to injury leading to various end points, including necrosis, apoptosis, and reperfusion injury (Fig. 345-6).
Cerebroprotective Strategies for Focal Ischemia
Limiting the Duration of Ischemia
The use of temporary arterial occlusion for dissection of aneurysms and permanent clipping is now a common standard during aneurysm surgery, but it is crucial to establish a safe occlusion time with emphasis on the anatomic level of occlusion and the vascular territory occluded. The duration of focal ischemia that can be tolerated safely without clinically evident sequelae varies between individuals and vascular territories.107–111 The current consensus for temporary vessel occlusion is brief repetitive clipping periods, which provides increased safety and less risk for postoperative neurological deficit than a single episode of occlusion does, but definitive data have yet to confirm this consensus.
Laboratory studies of multiple ischemic events in focal models have demonstrated varying results.107,112–115 Sakaki and coworkers examined the response of three 20-minutes episodes of middle cerebral artery (MCA) occlusion in cats versus a single 1-hour period.112 They noted that intermittent occlusion produced less damage than single, longer occlusion did. This report did not specify the length of reperfusion between episodes of ischemia, so it is difficult to assess whether reperfusion injury could occur. Steinberg and colleagues used a rabbit model of multiple intracranial vessel occlusion and demonstrated a 59% decrease in the area of cortical ischemic neuronal damage but no difference in the extent of striatal ischemic damage with intermittent occlusion versus uninterrupted occlusion.115
Goldman and associates studied repetitive MCA occlusion in rats in a protocol designed to simulate intraoperative occlusion techniques.107 The total infarcted areas after 60, 90, and 120 minutes of uninterrupted occlusion were significantly greater than those after identical cumulative ischemic periods but with 5 minutes of reperfusion after each 10-minute ischemic period. In an investigation of normotensive rats undergoing MCA occlusion by Selman and colleagues, biochemical and pathologic evidence demonstrated that a single, prolonged episode of reversible ischemia was more deleterious than multiple occlusions of a similar total duration.113 Statistical differences were seen only when total single occlusion time reached 2 hours. To provide an infarct size that can be statistically evaluated in different treatments, the experimental models used to date have required total occlusion times longer than those generally needed in the clinical setting. The nature of these experimental paradigms and species differences must be kept in mind when attempting to generalize the results and apply them to clinical use.
Most reports on the use of temporary arterial occlusion in humans have been retrospective analyses of case series in which the use or nonuse of temporary occlusion was based on the experience and judgment of the surgeon.109,116–128 No randomized, prospective studies have been designed to compare the outcome of cerebrovascular procedures with temporary occlusion and those without local circulatory arrest. In 1961, Pool stated that bilateral anterior cerebral artery occlusion was safe for up to 20 minutes with the protective effects of hypothermia.129 Both Suzuki’s group127 and Ljunggren’s group119 estimated the maximum safe occlusion time of the MCA to be 20 minutes under normothermia. Other authors have recommended maintaining occlusion times at less than 15 minutes when possible,116,123,125,130 although some have reported occlusion lasting longer than 90 minutes without deficit.118
Samson and coworkers reviewed 121 patients from a group of 234 consecutive aneurysm patients.125 One hundred patients who did not experience intraoperative complications were analyzed. These patients underwent elective temporary occlusion under a standard neuroanesthetic regimen, including etomidate-induced burst suppression, normotension, and normothermia. Infarctions were noted in specific arterial territories as follows: basilar, 41%; middle cerebral, 26%; internal carotid, 7%; and anterior communicating, 16%. The authors identified the following factors as significant predictors of postoperative radiographic evidence of infarction: age (≥61 years), poor preoperative grade (Hunt and Hess grades III to IV), protracted duration of temporary occlusion, and the use of incomplete circulatory arrest.125 This series documents that temporary occlusion is not without risk; however, factors other than the duration of temporary occlusion, such as aneurysm configuration and the location of perforators, may be critical in determining this risk.131
In a nonconcurrent, prospective analysis, Ogilvy and colleagues studied the results of 132 consecutive aneurysm clipping procedures performed with temporary vascular occlusion in combination with mild hypothermia (33°C to 34°C), induced hypertension (systolic pressure of 150 mm Hg), and mannitol administration (100 g).126 Multivariate analysis demonstrated that intraoperative rupture and duration of clipping longer than 20 minutes were independently associated with stroke outcome. The average clip application time in patients with radiographic evidence of stroke was approximately 42 minutes as compared with 29 minutes in patients without radiographic evidence of stroke, whereas in patients with a clinically significant stroke, the average time was 50 minutes.126 The overall stroke rate in patients with an occlusion time of less than 20 minutes (1/67, 1.5%) was significantly less than that in patients with occlusion times longer than 20 minutes (12/65, 18.5%).126
Ferch and coauthors reported on 106 aneurysm patients retrospectively analyzed with regard to age, neurological status, aneurysm characteristics, intraoperative rupture, and duration and number of occlusive episodes.132 The overall symptomatic stroke rate attributed to temporary clip placement in this series was 17%. The incidence of stroke was 12% in patients with occlusion times of less than 10 minutes and 35% in patients with occlusion times longer than 10 minutes. In the group of patients who did not suffer stroke, the mean temporary occlusion time was 5.4 ± 5.2 minutes, whereas in those with stroke, it was 7.4 ± 8.1 minutes. In other series, the mean temporary occlusion time of the internal carotid artery without causing a stroke was 7.1 ± 3.8 minutes, and that for the MCA was 5.3 ± 3.9 minutes.132
Juvela and associates prospectively investigated temporary vessel occlusion times in 101 of 156 patients with ischemic lesions seen on computed tomography (CT) 3 months after subarachnoid hemorrhage.133 Thirty-five patients who underwent temporary vessel occlusion and had an ischemic lesion on follow-up CT had a total occlusion time of 8.5 ± 8.2 minutes versus 2.5 ± 2 minutes and a longer single occlusive episode of 6.1 ± 6.2 minutes versus 2.5 ± 2 minutes, with a trend toward significance in patients with a greater number of occlusive episodes lasting 1.78 ± 0.95 minute versus 1.14 ± 0.38 minute.133 Additional occlusion times of 25 to 42 minutes (mean, 33 ± 7 minutes) have been described in superficial temporal artery–MCA bypass patients without permanent neurological deficits.134
As alluded to in earlier studies, temporary clip placement and the duration of occlusion are not the only factors influencing postoperative ischemic injury. Other factors must also be considered, including patient age (>50 years), grade and time of the initial hemorrhage, poor clinical condition (Hunt and Hess grade III to IV), occlusion of perforators, intraoperative rupture, hypertension, the specific vascular territory, multiple aneurysms, and timing of surgery.132,133
Augmentation of Cerebral Blood Flow
Systemic arterial pressure can readily be manipulated in the setting of planned temporary ischemia. Symon and colleagues showed that MCA occlusion in primates results in loss of autoregulation in the ischemic region.135 Elevation of blood pressure should increase cerebral perfusion because of the passive nature of the vessels that have lost autoregulation in the ischemic territory. Other investigators have shown that the decrease in CBF with focal ischemia is less in animals with phenylephrine-induced hypertension than in controls.136–138 The beneficial effect of induced hypertension may, however, be limited to relatively brief episodes of ischemia. Smrcka and coworkers reported that hypertension reduced infarct size by 97% in rabbits subjected to 1 hour of arterial occlusion but achieved only a 45% reduction in animals with 2 hours of ischemia.139 In the clinical setting, use of hypertension is limited by the patient’s cardiac tolerance. Close monitoring of cardiac function with limitation of the elevation in blood pressure to approximately 10% above baseline is advisable.
Prevention of Iatrogenic Ischemia (Intraoperative Cerebral Blood Flow Monitoring)
Preoperative measurement of flow rates through cerebral vessels can be achieved with noninvasive studies such as quantitative magnetic resonance angiography (a noninvasive tool also used for bypass surveillance) and xenon-enhanced CT.140 These studies help the surgeon identify patients who have low cerebrovascular reserve and may be at higher risk for iatrogenic ischemia before entering the operating room.
The neurological examination provides a sensitive, qualitative assessment of CBF during cerebrovascular procedures but remains limited to CEA and by patient tolerance of an awake operation.141 Vessel patency after aneurysmal clip placement can be confirmed by a number of modalities, including direct microvascular Doppler (MVD) or transcranial Doppler (TCD) ultrasound, ultrasonic flow probe, intraoperative angiography, EEG studies, electrocorticography, multimodality evoked potential (MEP) and somatosensory evoked potential (SEP) monitoring, brain tissue oxygenation, and more recently, fluorescent angiography with fluorescein sodium or indocyanine green (ICG).
TCD can provide only a relative estimation of CBF based on normal ranges, and results vary according to vessel diameter and operator technique. TCD has been used extensively to monitor MCA CBF during CEA142–144 and postoperatively for the detection of vasospasm. MVD is a relatively fast and easy method of establishing vessel patency and aneurysm filling after clipping and can be used in patients undergoing multiple aneurysm clip applications. Use of the probe is limited by vessel depth and confounded by adjacent vascular tributaries. Unfortunately, these techniques do not determine whether flow is sufficient to prevent ischemia.
Direct intraoperative flow measurements can be made with the use of a microvascular ultrasonic flow probe. The device consists of an electronic flow detection unit and a flow-sensing perivascular probe. The probe uses the principle of ultrasonic transit time to assess intravascular flow without close vessel contact.145 The use of intraoperative angiography (IA) remains controversial; some reports recommend IA for complex aneurysms, such as giant aneurysms or those arising from specific locations, including the superior hypophysial, superior cerebellar, and posterior communicating arteries and the internal carotid artery bifurcation.146–149 The argument against IA includes increased operative time, poor image quality, exposure of personnel to radiation, and a high cost-benefit ratio. Lopez and coauthors reported on a prospective cohort of 191 patients with various cerebrovascular pathologies in whom 204 angiograms were performed.150 Intraoperative findings were positive in 23% of the patients (residual lesions in 12%, parent or vessel occlusion in 6%, vasospasm in 5%) and resulted in clip repositioning or additional clip placement in 8% of patients. Other studies have found that IA altered surgical treatment in 11% or 12% of cases and was associated with less than a 0.5% morbidity rate.151,152 Whether IA should be performed is ultimately left to the cerebrovascular surgeon’s discretion. Klopfenstein and colleagues prospectively assessed the cerebrovascular surgeon’s accuracy of predicting whether IA was necessary. In this series, 200 patients with 235 aneurysms underwent IA. The results led to changes in surgical treatment in 7% of cases. In 4.4% of these patients, the surgeon thought that IA was unnecessary.153
IA is safe and useful when treating patients with a wide variety of vascular pathologies and may be beneficial in patients for whom surgery has been prospectively deemed unnecessary by the surgeon. However, IA is limited in its evaluation of small perforating vessels, which cause 8% of iatrogenic clip infarcts.154 By no means should IA be a substitute for sound surgical technique and judgment.
EEG, electrocorticographic, SEP, and MEP studies are also used to evaluate CBF. EEG monitoring is commonplace for CEA, but its usefulness is limited with barbiturate use, hypothermia, and global ischemia and in the evaluation of subcortical structures. Intraoperative neurophysiologic SEP monitoring is sensitive in detecting cortical ischemia, but MEP monitoring provides more information on subcortical structures and the motor cortex and pathways.155,156 MEP has been found to be more sensitive than SEP monitoring in detecting insufficient blood flow in the MCA and anterior choroidal artery.157,158 MEP monitoring is limited by the fact that the patient is fixed in a head holder and any movement can be disruptive to the surgeon.
Angiography/fluorescence imaging with ICG, a near-infrared fluorescent tricarbocyanine dye used for several decades by cardiologists, is relatively new in neurosurgical application. An intravenous bolus of 0.2 to 0.5 mg/kg (maximum daily dose, 5 mg/kg) of ICG allows the surgeon to perform real-time evaluation of blood flow within vessels in the operative field. de Oliveira and associates performed a prospective study of 60 patients with 64 aneurysms; ICG angiography demonstrated vessel patency in 62 aneurysms.159 ICG angiography is safe and reliable for evaluation of perforating vessels in the surgical field but is limited in patients with deep-seated aneurysms, blood in the surgical field, complete occlusion, and clip obstruction. It is currently undergoing active evaluation in many neurovascular centers.
Complex and giant aneurysms of the skull base and distal vessels present a unique challenge. These lesions may not lend themselves to open surgical clipping or endovascular coil embolization. Even in light of recent advances in endovascular therapy (i.e., stent-assisted coiling), parent vessel ligation and aneurysm trapping, in addition to bypass, may be the only alternative. Surgical success with selective bypass begins with adequate preoperative planning and implementation in appropriate patients. Flow-assisted surgery allows direct intraoperative flow measurement and may help ensure success of the bypass.160 This measurement can be quantified as a cerebral flow index (CFI): CFI = bypass flow/cut flow. With a CFI of 0.5 as a threshold value, the bypass patency rate was 92% in patients with CFIs greater than 0.5 versus 50% in patients with a CFI of less than 0.5.145,160,161
Reduction of Metabolic Activity
Hypothermia
Hypothermia has been shown to reduce neurological morbidity after cardiac arrest162,163 and neonatal hypoxic encephalopathy.164,165 Intraoperative mild hypothermia can be used during aneurysm surgery to reduce the ischemic injury induced by temporary vessel occlusion and brain retraction. Hypothermia ameliorates the primary and secondary mechanisms of ischemic injury, including metabolism, release of glutamate, generation of NO by NOS, and myeloperoxidase activity, in animal models of focal ischemia.166–170 Han and coworkers demonstrated that a decrease in NO produced a 40% reduction in infarct size, a 50% reduction in cells immunoreactive for inducible NOS and peroxynitrite—one of the most dangerous free radicals generated from NO—and the production of superoxide dismutase, thereby resulting in a reduction in the generation of oxygen free radicals.171
[/not-level-membership-for-neurosurgery-category]
