CHAPTER 221 Intramedullary Spinal Cord Tumors in Children
Epidemiology
IMSCTs occur with a frequency equal to that of tumors in the rest of the central nervous system (CNS) when the risk is analyzed as a function of tissue volume, but the spinal cord represents just 4% of the CNS. Currently, the incidence of CNS tumors in children in North America is 4.5 per 100,000, or 3750 per year.1 Based on a survey of the cancer registries of 45 states for the year 2004, 5.1% of CNS tumors occur within the spinal cord or cauda equina.1 The obvious problem with this number is the fact that tumors of the cauda equina not involving the spinal cord are lumped with the pure spinal cord tumors. Series have separated tumors of the cauda from spinal cord tumors and have come up with a ratio of 4.6 spinal cord tumors to 1 cauda tumor. This would mean that 4.2% of tumors involving the CNS are IMSCTs. Consequently, we can expect to see roughly 170 to 180 IMSCTs in the United States every year. There does not seem to be a predilection for gender, and based on a limited data set, white individuals seem to be afflicted slightly more than blacks.
A dataset developed by Constantini and colleagues in which the outcomes of 164 children operated on for IMSCTs were analyzed and subsequently updated until the primary surgeon’s (Fred J. Epstein) retirement was reviewed for this chapter.2,3 A total of 241 children and adolescents were listed who underwent surgery for IMSCTs between 1978 and 2001 at an average age of 9.5 years. Thirty-one were infants aged 0 to 2 years, 130 were children aged 2 to 13 years, and 80 were adolescents aged 13 to 21. Discounting the second year of life, the age at diagnosis seemed fairly evenly distributed, with perhaps a tailing off in incidence in late adolescence—but this could have been confounded by the impact of the referral pattern early in the primary surgeon’s career. It is not clear why there was such a disparate number of infants who underwent surgery between the ages of 1 and 2 years. One hundred forty-one (58%) of the patients were male. Seventy-six (46%) of the children reviewed by Constantini had astrocytomas, 18 of which were malignant, and 58 (35%) children had a mixed glial-neuronal tumor, the majority of which were gangliogliomas as established by immunohistochemical staining.4 Nineteen (12%) were ependymomas. The remaining children had mixed gliomas5 or primitive neuroectodermal tumors.3 In a later review of this patient population that looked at 181 pediatric patients seen at the same practice between 1987 and 1998, 4 (2.2%) were found to have hemangioblastomas and 3 (1.7%) had cavernous malformations.6,7
Clinical Findings
The onset of symptoms in children harboring an IMSCT is usually realized in retrospect and predates the actual diagnosis by months if not a year or more. Constantini reported an average 11.6-month delay in diagnosis and a slightly shorter delay in children younger than 3 years (5.4 months).3,5 Frequently, an accident or other unrelated event leads the child to a physician. The ensuing examination shows an inconsistency between the initial complaint and the symptoms being exhibited, which leads to the radiographic study that makes the diagnosis. This all attests to the benign nature of the majority of these tumors and their insidious growth pattern. The history is substantially shorter in children with malignant tumors, and such a finding should warn the clinician of the aggressive nature of the tumor.
The most common complaint at initial encounter was motor regression, which occurred 65% of the time, followed by pain (48%), gait abnormality (37%), dysesthesia (32%), and progressive kyphoscoliosis (32%) in the largest reported series.3 Other series have reported the same symptoms and signs but with slightly differing incidences. Lunardi and coauthors also reported a 32% incidence of sphincter disturbance and a 48% incidence of sensory deficits.8 Muszynski and associates, in studying an updated version of the database developed for Constantini’s paper, found a 32% incidence of urinary retention and a 3% incidence of delay in milestones.3,9 On examination one can expect to find signs of myelopathy in most patients.8
Functional status at diagnosis varies. In reviewing 45 children with IMSCTs (29 with astrocytoma and 16 with ependymoma), Innocenzi and associates divided their patients into three groups based on their Karnosky Performance Status score.10 Group 1 had scores of 80 to 100; group 2, 60 to 80; and group 3, lower than 60. Twenty-two percent were in group 1, 56% in group 2, and 22% in group 3. Constantini and colleagues used a modified McCormick scale to assess the functional status of the patients.3,11 Fifteen of 164 (9%) were neurologically intact (grade 1), 76 (46%) were functionally independent with mild motor or sensory deficits (grade 2), 33 (20%) required an external assistive device to maintain functional independence and exhibited “moderate” deficits (grade 3), 22 (13%) had severe sensory or motor deficits, or both, rendering them functionally dependent (grade 4), and 18 (11%) had major plegia with only a flicker of movement below the level of paralysis (grade 5). Sixty of the 164 were seen at Dr. Epstein’s service with newly diagnosed disease but no previous treatment.2,3 Of these 60, 10 (17%) were grade 1, 32 (53%) were grade 2, 10 (17%) were grade 3, 6 (10%) were grade 4, and 2 (3%) were grade 5.
Symptoms and signs can vary according to the level of the spinal cord involved with tumor. Children with IMSCT at the cervicomedullary junction can have indications of brainstem involvement such as nausea and vomiting, a history of recurrent upper respiratory tract infections, and the development of a nasal quality in their speech. When the tumor primarily involves the cervical spinal cord, the child commonly complains of neck pain with or without radiation to one or both arms. This pain can be particularly intrusive at night.12 Loss of motor function seems to initially be limited to the upper extremities, with progression to involvement of the legs, bowel, and bladder occurring late.9 In younger children, the parents may report that their child has seemed to switch hand dominance, whereas parents of older children with IMSCTs note frequent tripping or falling. Sensory abnormalities trail the motor deficits and are more limited, typically being unilateral. When the sensory abnormalities are symmetrical, the diagnosis of ependymoma should be suspected.9
Hydrocephalus can be an unusual but real outcome in an individual with an IMSCT. Rifkinson-Mann and coauthors reported on 25 of 171 patients with IMSCTs operated on by Epstein in whom hydrocephalus was found to be present or develop later.13 This group of 171 patients included both children and adults. Eighty-eight children were operated on during this period. Two of the 25 patients reported by Rifkinson-Mann and colleagues were older than 21 years, with 1 having a malignant tumor and 1 a benign one. Consequently, there was a 26% incidence of hydrocephalus in these 88 children with IMSCT. Rifkinson-Mann and associates concluded that the incidence of hydrocephalus is much higher in children with malignant IMSCTs. They could not conclude why the hydrocephalus developed but did note that 12 of the patients (nearly half) had evidence of subarachnoid spread of their tumor, 12 (nearly half) had extension of the tumor or a cyst to the obex (inferring outlet obstruction of the fourth ventricle), and 1 had both. Interestingly, 6 of the children in this series were initially seen for treatment of intracranial hypertension and only later were found to have an IMSCT. Four of these tumors were malignant.
Diagnostic Evaluation
There is no question that magnetic resonance imaging (MRI) of the spine is the diagnostic procedure of choice when confronted with a patient with signs of myelopathy and an IMSCT is suspected. MRI provides excellent soft tissue imaging within the spinal column, and any intramedullary lesions, edema, and cysts can be visualized.14,15 It allows one to differentiate intramedullary lesions from extramedullary lesions, define the extent of the solid portion of the lesion, and with the aid of enhancement, differentiate tumoral cysts from nontumoral, reactive cysts. By using this information one can arrive at a reasonably certain diagnosis with regard to tissue type, which in turn will direct treatment strategies. Computed tomography and plain radiography are reserved for the evaluation of associated spinal deformities/instabilities, and myelography is generally reserved for situations when MRI cannot be performed.15
At minimum, a T1-weighted sequence, with and without gadolinium enhancement, and a T2-weighted sequence should be obtained in the axial and sagittal planes.14 Anything less will compromise one’s interpretation of the imaging. A gradient echo sequence and a fluid-attenuated inversion recovery (FLAIR) sequence can add additional information about hemosiderin deposits (gradient echo) or subtle intramedullary lesions (FLAIR). A diffusion-weighted image can be used when an ischemic infarct is suspected. One expects to see a significant mass when confronting an intramedullary tumor that will stand out on at least one of the sequences. When the size is subtle or not consistent with the degree of T2 signal hyperintensity (i.e., edema), one should consider an inflammatory process such as multiple sclerosis or transverse myelitis. Scanning of the brain and delayed, repeated imaging 3 to 6 months later will clarify the situation.14
Intramedullary astrocytomas are the most frequently encountered spinal cord tumors in children. On MRI they are seen to enlarge the cord’s diameter and on average extend over four spinal segments. Their epicenter is eccentric. On T1-weighted imaging, the majority (>80%) are hypointense with the rest being isointense (Fig. 221-1).16 Characteristically, the margins of the tumor blur into the surrounding cord parenchyma, and both tumoral cysts and reactive cysts can be present. The tumor enhances with gadolinium, as do the walls of tumoral cysts in all but the rarest of cases.17 On T2-weighted imaging they appear hyperintense, but unlike edema, this hyperintensity is inhomogeneous.14 Kulkarni and coworkers reviewed their experience with four children harboring malignant astrocytomas within the spinal cord.18 Apart from a higher incidence of subarachnoid dissemination, they found that the imaging characteristics of the tumor did not significantly differ from those expected with benign astrocytomas of the spinal cord.
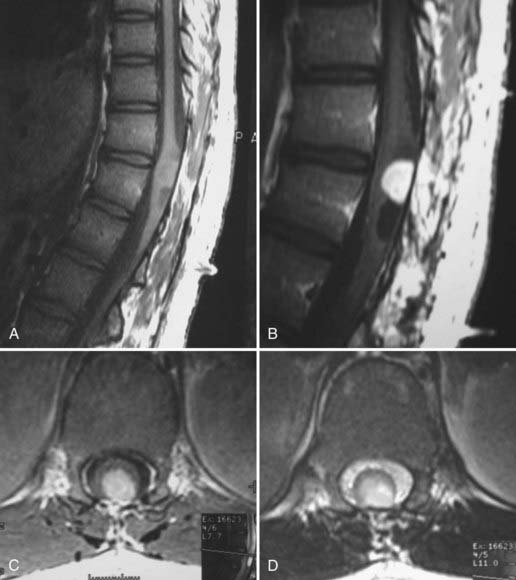
The second most common tumor of the spinal cord seen in children is ganglioglioma. Like astrocytomas, these tumors tend to arise eccentrically. When discovered, their average length was twice that of astrocytomas (eight versus four segments). Patel and colleagues postulated that many such large tumors that have previously been called astrocytomas (with the use of hematoxylin and eosin staining) might well be gangliogliomas. They based this on their finding that when they reanalyzed previously diagnosed spinal cord tumors with immunohistochemical stains for glial fibrillary acidic protein, synaptophysin, and vimentin, they found many to be gangliogliomas.16 In 67 patients with IMSCTs thus analyzed, 27 were reclassified as gangliogliomas, and all tumors that were holocord tumors proved to be gangliogliomas. On T1-weighted imaging the majority have mixed-intensity signals, with only a small percentage being hypointense or isointense, as opposed to astrocytomas.16 Patchy (65%) or focal (19%) enhancement is the rule after the administration of gadolinium, and importantly, 15% show no enhancement. On T2-weighted imaging, 60% have a homogeneous hyperintensity, whereas 40% exhibit a heterogeneous signal (Fig. 221-2). Tumor cysts are more common with gangliogliomas of the spinal cord than with astrocytomas or ependymomas and occur in 46% of patients.
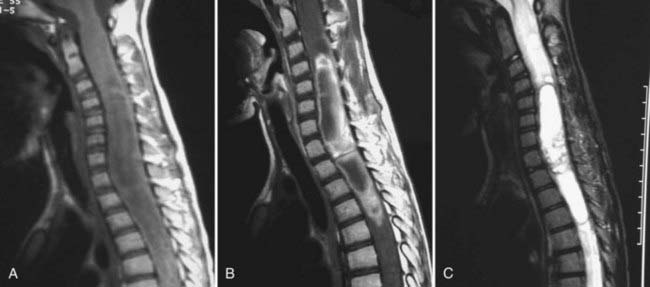
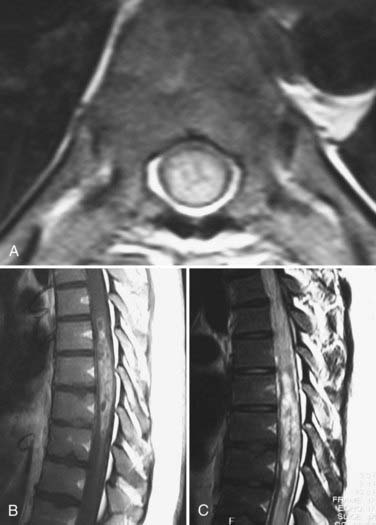
Unlike adults, in whom ependymoma is the most common intramedullary tumor, in children, ependymomas account for only 12% of intramedullary tumors.3 Their size at initial evaluation appears to be the same as that encountered with intramedullary astrocytomas (four vertebral levels).16 Unlike astrocytomas and gangliogliomas, ependymomas arise centrally and expand the cord symmetrically. The majority of nonmyxopapillary ependymomas are isointense or hypointense on T1-weighted imaging, as opposed to myxopapillary ependymomas, which are either isointense or hyperintense.19 T2-weighted imaging of both subtypes shows the tumor as a hyperintense signal (Fig. 221-3). Fourteen of 16 tumors in Kahan and colleagues’ series enhanced with gadolinium, with 5 showing a heterogeneous pattern, 6 a homogeneous pattern, and 3 only rim enhancement. One of their patients had minimal enhancement and one had none.19 Almost two thirds of their patients had cysts, with 8 being intratumoral, 6 rostral or caudal to the tumor, and 9 being a reactive dilation of the central canal. Twenty percent showed evidence of chronic hemorrhage. Choi and associates found that nearly 20% of their patients had some evidence of hemorrhage, with the majority showing hemosiderin susceptibility signals capping either the rostral or caudal pole.20 Fine and coauthors reported a similar finding,21 and Nemato and coworkers found evidence of hemorrhage in 60% of their patients when they first reported this phenomenon.22
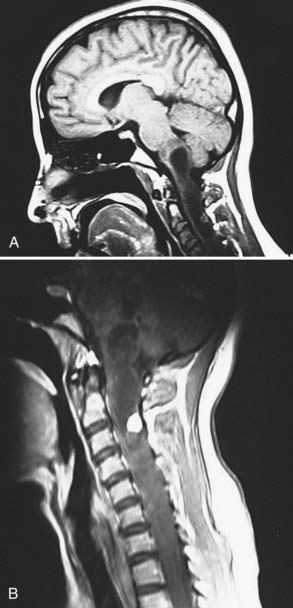
Hemangioblastomas are rare in children but do occur. Vougioukas and coauthors reported on 141 individuals with von Hippel-Lindau disease, 13 of whom were children and 5 had hemangioblastomas of the spinal cord.23 The report mentioned that the diagnosis of von Hippel-Lindau disease is more common in children with an intramedullary hemangioblastoma than in adults with the same tumor. The distribution of these tumors reflects the volume at risk. Chu and associates found that 50% of their patients’ tumors were within the thoracic cord and 30% in the cervical cord.24 Typically, these tumors extend to the pia, with most doing so dorsally.24 Most are 1 cm or less in diameter, but some can be much larger. T1-weighted imaging shows the tumors to be isointense or hypointense or have mixed (isointense and hypointense) signal intensity (Fig. 221-4). On T2-weigthed imaging they are usually hyperintense with a few being either isointense or of mixed signal intensity (hyperintense and isointense). Chu and colleagues found that 14 of 15 of their patients who had lesions that were 1 cm or less in diameter had lesions that were isointense on T1-weighted and hyperintense on T2-weighted images. Medium-size lesions (11 to 20 mm in diameter) were hypointense on T1 imaging and tumors larger than 20 mm in diameter were heterogeneous on T2 imaging.24 Syrinxes or cysts are common and occur in more than 50% of patients with hemangioblastomas of the spinal cord, and frequently the solid portion of the tumor appears as a nodule on the cyst wall.25 Hemangioblastomas in the spinal cord enhance vividly, with the enhancement being homogeneous in smaller tumors and heterogeneous in larger ones.7 Many appear as brightly enhancing nodules on the wall of a cyst. Not uncommonly, flow voids are seen in tumors larger than 1.5 cm. Some can also show extensive reactive edema.26 These tumors markedly enhance on angiography and show feeding arteries coming off the anterior and posterior spinal arteries and large draining veins. Because this information can be helpful in surgical planning for some tumors, angiography should be considered for a large tumor thought to possibly be a hemangioblastoma. Embolization is not indicated, however, because of the risk of interrupting the collateral circulation to the cord.7
Intramedullary cavernous malformations are easily diagnosed on MRI. They appear as small intra-axial lesions that are typically 1 cm or so in diameter.6 On T1-weighted images they appear hyperintense centrally with multiple areas of differing signal intensity consistent with multiple hemorrhages of differing ages.27,28 Surrounding this central hyperintense area is a region of hypointensity, which results in the characteristic target sign. On T2-weighted imaging the central portion is hypointense with small areas of hyperintensity. The surrounding hypointensity is due to hemosiderin deposits and is larger than that seen on the T1 images. The lesions may be associated with a venous angioma, which will appear as an adjacent flow void. Occasionally, these lesions may show evidence of fresh hemorrhage and clot, particularly if the patient has new neurological findings.
Pathology
As mentioned earlier, the bulk of IMSCTs in children and adolescents are of astrocytic or neuronal cell origin, as opposed to the ependymal origin seen in adults. Epstein,29 in one of the earliest published series of surgically managed spinal cord tumors in children, reported a 3:2 ratio of astrocytic to ependymal tumors. Hardison and colleagues in 1987 reported a series of 26 patients with IMSCTs in which 24 were of nonependymal glial origin and only 2 were of ependymal orgin.29a In 1994, O’Sullivan and coauthors reported on a Toronto experience consisting of 28 IMSCTs and found that that 16 were astrocytomas and 11 were ependymomas.30 Innocenzi and associates reported on the Rome experience in 1996 and found that 29 of 45 tumors were nonependymal gliomas and 16 were ependymomas.10 Miller, in reviewing the pathology of 117 of the 164 children reported by Constantini, found that 49 were of astrocytic, oligodendroglioma, or mixed origin and only 19 were of ependymal origin. Forty-one of the children had gangliogliomas or tumors containing other neuronal elements.3,31 This surprisingly high number of tumors containing neuronal elements was attributed to the authors’ having large specimens to analyze (the author postulated that neuronal elements could easily be missed when only a small biopsy sample was submitted) and to the introduction of immunohistochemical stains such as synaptophysin.31
The most common tumor encountered within the spinal cord is astrocytoma, with the majority being benign. Hardison and coauthors reported that of 23 astrocytomas, 17 were benign and 6 were malignant.29a O’Sullivan and coworkers found that of the 15 astrocytomas on which they operated, 12 were benign and 8 malignant.30 Thirty-six of the 49 specimens that Miller analyzed were benign, whereas 13 were malignant.31 Thus, of the 87 astrocytomas reported on in these three papers, 65 (75%) were benign. Historically, the bulk of these tumors have been thought to be pilocytic astrocytomas, but this was not found to be the case in the large series reviewed by Miller.31–33 It was Miller’s conclusion that there was a potential for misdiagnosis when only small biopsy samples were analyzed and that this accounted for the high number of pilocytic astrocytomas in series based on biopsy analysis. Astrocytomas are typically eccentric in position. Pilocytic astrocytomas contain cysts, which can coalesce to result in the typical cyst with a mural nodule as seen in their counterpart in the brain. The margins of astrocytomas are infiltrative, except for pilocytic astrocytomas, which have a sharp margin along the majority of its border and the normal cord parenchyma. Anaplastic astrocytomas and glioblastomas have broader zones of infiltration into relatively normal-functioning cord parenchyma. These facts are important to consider when planning resection, and frozen biopsy specimens can be quite helpful for grading purposes. Anaplastic astrocytomas typically account for a fourth of intramedullary spinal cord astrocytomas in children.31 Although spinal cord astrocytomas can occur in adults, they are far more common in children.9
Mixed glial-neuronal tumors have been found to be the second most common intraparenchymal spinal cord tumor in children and young adults. Their hallmark histologically is large neuronal cells with vesicles gathered about the nucleus that react with the immunoreactive synaptic vesicle membrane stain synaptophysin.34 By far and away the most common of these tumors is ganglioglioma, but Miller also found 10 children with tumors in the “neurocytoma” family.31 These neurocytoma-like tumors had small cells resembling oligodendroglioma cells except for the fact that they too exhibited synaptophysin immunoreactivity.31 Mixed glial-neuronal tumors, as with the astrocytomas, tend to arise in an eccentric fashion. Their margins are also infiltrative, and there is no “plane” between them and normal cord parenchyma. When encountered, they can be quite large and extend over multiple levels. None were found to be malignant in Miller’s series or in the larger series reported by Constantini and colleagues.3,31 They tend to occur in younger patients and are largely confined to the pediatric population, as opposed to astrocytomas.9
Oligodendrocytoma and mixed glioma (astrocytoma/oligodendroglioma) IMSCTs are encountered in small numbers in children.31 These tumors vary from having the typical appearance of an oligodendrocytoma to appearing as an astrocytic tumor with oligodendroglioma cells of varying density. Grossly and on radiographs they have an appearance similar to that of the other gliomas. The numbers currently described in the literature are too small to differentiate their biology from that of the other gliomas.
Although ependymomas are the most common intramedullary tumors found in adults, they are the minority in children and account for only 10% to 15%.9,31,35 When using World Health Organization grading, grades 1 (myxopapillary ependymoma) and 2 (ependymoma) are benign histologically, whereas grade 3 (anaplastic ependymoma) is not. There has been no established difference between the biology of these grades. The hallmark of intramedullary ependymomas is their central location and the fact that unlike gliomas, they are sharply demarcated from the surrounding parenchyma and tend to establish a plane between themselves and the surrounding spinal cord. This feature is of great surgical importance. The majority have the typical histologic feature of pseudorosettes. Myxopapillary ependymomas can also be found in children but are unusual; they accounted for only 4% in Miller’s series.31 Myxopapillary ependymomas arise in the conus and filum and have the potential to break through the pia and disseminate throughout the subarachnoid space, with nodular growth ensuing.
Hemangioblastomas are vascular tumors, and this is reflected in the findings on histologic examination. There are multiple, closely packed blood channels surrounding groups of cells containing eosinophilic cytoplasm that is richly vacuolated. The nuclei are hyperchromatic.26 Cavernous hemangiomas or cavernous malformations are purplish on gross inspection and have been described as mulberries because of their color and the irregular surface of the vascular sinusoids. Calcification or hematoma, or both, can be associated with them. On microscopic inspection, multiple, small vessels are found to be tightly opposed to one another without intervening tissue. The vessel walls are a single layer with thickness varying from that of capillaries to much thicker.36 The thick walls consist of only collagen fibers, with no evidence of elastic fibers or smooth muscle.
Treatment
Surgery
Initial management of IMSCTs is almost always surgical because of the obvious desire to confirm the radiologic diagnosis by histologic assessment of tissue obtained at surgery, as well as the therapeutic benefit derived from debulking of the tumor. More to the point, it is now known that 80% resection or greater imparts a 5-year event-free survival rate higher than 70%, an outcome that at least equals if not surpasses that of the other available therapies.3 Modern imaging gives a fairly reliable idea of the type of tumor present, thus allowing surgical planning and establishment of resection goals for intramedullary tumors. The morbidity associated with such surgery was historically the reason given for avoidance of aggressive surgery, but modern surgical equipment and technique can minimize this concern. There are even reports of operating on these tumors safely without the use of physiologic monitoring, lasers, or ultrasonic aspirators.3,4,37
The change in approach to IMSCTs and the resulting decrease in morbidity seen over the past several decades is largely the story of the coming of age of intraoperative physiologic monitoring or the real-time observation of nerve potentials conducted within the operative field during the surgery. Monitoring of somatosensory potentials was first introduced in the early 1970s.38 Motor evoked potentials were slower to evolve because of the difficulty of stimulating the motor cortex through the skull and generating action potentials within the motor pathways in an anesthetic environment that had been developed to not only keep patients asleep but also keep them from moving. Through dogged work during the 1980s, techniques evolved that resulted in the ability to reliably record evoked potentials descending in the spinal cord’s motor pathways. In 1998, criteria were reported that could be used to reliably predict permanent, postoperative motor deficits.39 Changes in upper motor potentials (D wave) were found to be 100% predictive of permanent postoperative loss of motor function. Sala described the results of a senior neurosurgeon with extensive experience in resecting IMSCTs and has made no stronger statement about the benefit of using physiologic monitoring.40 His center adopted the use of sensory and motor potentials after the return of Sala from an 18-month fellowship training in intraoperative physiologic monitoring. Sala introduced the same level of monitoring that evolved at our facility, and it was used on 50 consecutive patients undergoing surgery for IMSCTs. When compared with a historical group of the same surgeon’s patients who were matched in terms of preoperative functional levels, histology, tumor location, and extent of resection, there was a significantly better outcome (P < .0016) at the time of follow-up examination in those whose surgery included intraoperative monitoring. The recommended monitoring protocol is to record baseline motor (upper motoneuron or D wave and evoked muscle contraction or mMEPs) and sensory potentials before making an incision. Special attention is paid to the sensory evoked potentials during the initial myelotomy, and then observation of the motor potentials (D wave and mMEPs) is included during resection, with both types of potentials being checked every 2 minutes. The protocol can be adjusted at the direction of the surgeon to tailor the recordings to the particular pathways that are at risk during the various phases of the resection. Typically, a given motor potential can be updated in less than a second, and a complete circuit of the recordable potentials can be updated every few minutes. Therefore, when working immediately adjacent to the motor pathways, the protocol can be adjusted to provide nearly real-time information on how these pathways are tolerating the resection.
The approach to all these tumors is usually through a midline incision over the spine. When the lamina is intact, a laminotomy can be considered.41 The dura is then opened after placing epidural electrodes for monitoring. The amount of cord that is exposed is a function of the solid tumor within the cord and any associated cysts. Cysts do not need to be exposed unless they contain solid tumor that requires resection.
Deciding to operate on an intramedullary spinal cord cavernous angioma can be very problematic. Although the risk for hemorrhage with these lesions is small (0.5% to 1% per year), it does seem to be the most common reason for evaluation of children. When hemorrhage has occurred, the presence of a hematoma helps makes the decision to proceed with surgery because there is no better time for resection than when an associated clot approaches a pial surface.6,42 If surgery can be performed before resorption of the clot, a small myelotomy can gain access to the clot and removal of it will usually expose a portion of the cavernoma within the surgical bed. Removal then proceeds as one would with an intracranial cavernoma by cauterizing the surface of the cavernoma to shrink it away from the surrounding parenchyma. Care should be taken to confine cautery to the surface of the cavernoma to minimize collateral injury to the surrounding parenchyma.
Complications
Even though the expectation for successful outcomes in children and adolescents undergoing surgery for IMSCTs has changed dramatically over the past few decades, the potential for neurological injury and wound-healing complications still persists. The incidence of complications in patients undergoing surgery for spinal cord tumors in the United States between 1993 and 2002 was 17.5%.43 This information was obtained from the National Inpatient Sample maintained by the Agency for Healthcare Research and Quality. The proportion of patients younger than 18 years was 9.3%, and they experienced an 8.9% complication rate. For the group at large, urinary tract complications were the most common (3.7%), followed by wound hematoma (2.5%) and pulmonary complications (2.4%). In their review of 164 children and adolescents who underwent surgery for IMSCTs, Constantini and associates found no operative deaths and that 17.7% needed shunting for hydrocephalus that developed after surgery.44 Thirty-nine of the 164 patients (24%) deteriorated neurologically after their surgery as measured by the McCormick scale, but only 13 of these patients (7.9%) deteriorated by more than one grade.3 They found that the majority of patients experiencing deterioration in neurological function had significant neurological dysfunction before their surgery and that after 1987, no patient who was functioning normally (McCormick grade 1) deteriorated by more than one grade (i.e., they maintained their functional independence). Interestingly, 1987 was the year that monitoring of motor evoked potential was introduced to the surgical service treating these children. Urinary dysfunction occurred in 40.5%, and 26.6% were incontinent. More common but little discussed is the sensory deficits seen after surgery. In older children and adolescents, the dorsal columns are at risk for injury, and severe postoperative sensory deficits can and do occur. They can vary from mild, subdermatomal hypoesthesia to dense, functionally disabling proprioceptive deficits. When these deficits involve the arm, they can be particularly disabling functionally. Constantini and colleagues found that 61.4% of the patients reviewed experienced dysesthesia but only 6.9% were bothered by pain.44 Acupuncture can be quite useful in managing postoperative dysesthesias.
It is not uncommon to see scoliosis develop in children who have undergone surgery for IMSCTs.44–46 In a subsequent review of Constantini’s database review, 71.6% of the 164 patients reviewed had kyphoscoliosis and 37.1% required surgery for it.3,47 Kyphoscoliosis can also result from irradiation.48–50 The response to progressive scoliosis after surgery for IMSCTs in young patients is to first ensure that it is not due to tumor recurrence or progression and, if not, to treat it even if fusion is indicated.
Outcome
Because nearly 80% of IMSCTs in children and adolescents will be benign, their treatment should both be benign and offer the greatest prospect for control over disease progression and functional decline. Five- and 10-year survival rates are 88% and 82%, respectively, for children operated on for benign IMSCTs.3 The event-free survival rate for these children when more than 80% of the tumor is resected is 70% at 10 years. This is in contradistinction to children with high-grade tumors, in whom 5- and 10-year survival rates were 18% and 12% and the 10-year event-free survival rate was less than 20%. Functionally, children with benign IMSCTs seemed to do reasonably well, with more than 60% functioning independently (McCormick grade 1 or 2) at a mean follow-up of 13 years. Twenty-three percent, however, were largely dependent on others for some or all daily needs (grade 4 or 5). Fifty-eight of 164 experienced recurrence of their tumor at a mean time of 37.9 months. Nearly half of these patients were treated with a repeat operation, and they experienced a similar type and frequency of morbidity with their second surgery as occurred with the initial operation.
Alternative and Adjuvant Therapies
Radiation Therapy
Traditionally, IMSCTs have been managed with radiation therapy possibly augmented by surgical biopsy and decompression.51 Reports that look at the experience in using radiation therapy to manage patients who have undergone conservative resection of their tumor show a 5-year survival rate of 75% to 85%.48,52–56 By the 1980s, concern was beginning to be raised about the deleterious effects of radiation on the young spine.57 As mentioned earlier, the 5- and 10-year event-free survival rates of children operated on for benign IMSCTs are 80% and 70%, respectively.3 One paper has reported on the long-term survival and event-free survival of children managed with surgery of varying extent followed by radiation therapy.30 Of the 22 children with histologically proven low-grade IMSCTs, 17 (77%) were reported as having a 10-year event free survival, and 18 (82%) were alive at 10 years. Four of the surviving patients experienced a second malignancy and 2 had late (6 and 14 years) occurrence of an anaplastic glioma in the irradiated bed as a result of either a second malignancy or malignant degeneration of their initial tumor. Sundaresan and coauthors reported on radiation-induced myelopathy in children who had undergone spinal cord irradiation.58 There are also reports of alteration in the growth of bones in children undergoing radiotherapy.59,60 Thus, it appears that the efficacy of aggressive resection (>80%) of IMSCTs essentially matches that of radiation therapy but that there is a risk of injury to the growing spine and the induction of second malignancies with radiation of the type used in the 1980s and earlier. What is unknown is whether newer forms of radiotherapy might bring these risks into a more acceptable range.
Chemotherapy
The utility of chemotherapy in the management of IMSCTs remains unclear, largely because of the small number of children whose treatment has been reported. With regard to the treatment of malignant spinal cord tumors with chemotherapy, these reports have uniformly concerned its use as an adjuvant to surgery and radiation therapy. The majority of these reports involve the treatment of a larger series of tumors containing a mixture of grades. There is one report of the use of so-called 8 in 1 chemotherapy (lomustine [CCNU], vincristine, hydroxyurea, procarbazine, cisplatin, cytosine arabinoside, and dacarbazine [DTIC]) for the treatment of high-grade gliomas of the spinal cord in 13 children.61 Ten of the 13 could be evaluated for treatment response: 5 were found to have had an objective response to the regimen, another 4 were reported to have experienced stabilization of their disease, and 1 experienced progression despite the treatment. Apart from this report, treatment of malignant spinal cord tumors with chemotherapy has largely been anecdotal.
The use of chemotherapy for low-grade IMSCTs has been limited over the past few decades, and the literature reflects this trend, consisting largely of single or small groups of cases. Resolution of symptoms with no radiographic or symptomatic progression after using vincristine and carboplatin in a 30-year-old with a cervical-thoracic astrocytoma has been reported by Bouffet and coworkers.62 In 1999, a multicenter trial was reported by the French Society of Pediatric Oncology in which carboplatin, procarbazine, etoposide, cisplatin, vincristine, and cyclophosphamide were used.63 In this series, eight children 3 months to 11 years of age were treated, with six having a benign IMSCT. Two of these children relapsed after completion of chemotherapy, one immediately on completion of chemotherapy and the other 22 months after completion. The overall efficacy of the chemotherapies used in the trial is difficult to interpret because of the other therapies used to manage these children. Another study of three children who experienced progressive growth of their benign IMSCTs after subtotal resection reported on the efficacy of treatment with carboplatin.64 One child had a complete remission and two had stabilization of their disease, but the follow-up observation period was short (17, 27, and 5 months). Recently, Mora and associates reported on their group’s experience in treating three infants 19 months or younger who exhibited growth of their tumors after either subtotal resection or biopsy followed by treatment with “carboplatin-based chemotherapy.”65 One infant was reported to be in remission 4 years after completion of therapy, one had a decreased tumor burden and was symptom free at 20 months, and the third had a decreased tumor burden and no symptoms at 12 months. Given the variable biology of IMSCTs that are low grade, the short follow-up of most patients treated in novel chemotherapy trials, and the small number of studies, it is impossible to compare the efficacy of conservative resection followed by chemotherapy with the efficacy of radical tumor resection in children with low-grade IMSCTs. These early results are intriguing, however, and should stimulate further research on this question.
Constantini S, Houten J, Miller DC, et al. Intramedullary spinal cord tumors in children under the age of 3 years. J Neurosurg. 1996;85:1036-1043.
Constantini S, Miller D, Allen J, et al. Radical excision of intramedullary spinal cord tumors: surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg Spine. 2000;93:183-193.
Kalangu K, Couto M. Radical resection of intramedullary spinal cord tumors without cavitron ultrasonic aspirator or CO2 laser: a “two stage” technique. Surg Neurol. 1996;46:310-316.
Kothbauer K, Deletis V, Epstein F. Motor-evoked potential monitoring for intramedullary spinal cord tumor surgery: correlation of clinical and neurophysiological data in a series of 100 consectutive procedures. Neurosurg Focus. 1998;4(5):e1.
Miller D. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2006;47:189-194.
Nadkarni T, Rekate H. Pediatric intramedullary spinal cord tumors. Critical review of the literature. Childs Nerv Syst. 1999;15:17-28.
Sala F, Deletis V, Faccioli F, et al. Motor evoked potential monitoring improves outcome after surgery for intramedullary spinal cord tumors: a historical control study. Neurosurgery. 2006;58:1129-1143.
Smith J, Lury K, Castillo M. Imaging of spinal and spinal cord tumors. Semin Roentgenol. 2006;41:274-293.
1 CBTRUS. 2009-2010 CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in Eighteen States in 2002-2006. Hinsdale, IL: Central Brain Tumor registry of the United States www.cbtrus.org
2 Constantini S. Personal communication. New York. 2006.
3 Constantini S, Miller D, Allen J, et al. Radical excision of intramedulllary spinal cord tumors: surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg Spine. 2000;93:183-193.
4 Nadkarni T, Rekate H. Pediatric intramedullary spinal cord tumors. Critical review of the literature. Childs Nerv Syst. 1999;15:17-28.
5 Constantini S, Epstein F. Intraspinal tumors in infants and children, 4th ed. Youmans J, editor. Neurological Surgery, vol 4. Philadelphia: WB Saunders. 1996:3123-3133.
6 Deutsch H, Shrivistava R, Epstein F, et al. Pediatric intramedullary spinal cavernous malformations. Spine. 2001;26:E427-E431.
7 Roonprapunt C, Silvera V, Setton A, et al. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery. 2001;49:321-327.
8 Lunardi P, Licastro G, Missori P, et al. Management of intramedullary tumours in children. Acta Neurochir (Wien). 1993;120:59-65.
9 Muszynski C, Constantini S, Epstein F. Intraspinal intramedullary neoplasms. In: Albright A, Pollack I, Adelson P, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme, 1999.
10 Innocenzi G, Raco A, Cantore G, et al. Intramedullary astrocytomas and ependymomas in the pediatric age group: a retrospective study. Childs Nerv Syst. 1996;12:776-780.
11 McCormick P, Torres R, Post K, et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523-532.
12 Kothbauer K. Neurosurgical management of intramedullary spinal cord tumors in children. Pediatr Neurosurg. 2007;43:222-235.
13 Rifkinson-Mann S, Wisoff J, Epstein F. The association of hydrocephalus with intramedullary spinal cord tumors: a series of 25 patients. Neurosurgery. 1990;27:749-754.
14 Lowe G. Magnetic resonance imaging of intramedullary spinal cord tumors. J Neurooncol. 2000;47:195-210.
15 Smith J, Lury K, Castillo M. Imaging of spinal and spinal cord tumors. Semin Roentgenol. 2006;41:274-293.
16 Patel U, Pinto RS, Miller DC, et al. MR of spinal cord ganglioglioma. AJNR Am J Neuroradiol. 1998;19:879-887.
17 Lebovita Y, Pinto R, Lefton D, et al. Nonenhancing intramedullary spinal cord lesions. Paper presented at the 37th Annual Meeting of the American Society of Neuroradiologists, 1999.
18 Kulkarni A, Armstrong D, Drake J. MR characteristics of malignant spinal cord astrocytomas in children. Can J Neurol Sci. 1999;26:290-293.
19 Kahan H, Sklar E, Post J, et al. MR characteristics of histopathologic subtypes of spinal ependymoma. AJNR Am J Neuroradiol. 1996;17:143-150.
20 Choi J, Chang K, Yu I, et al. Intracranial and spinal ependymomas: review of MR images in 61 patients. Korean J Radiol. 2002;3:219-228.
21 Fine M, Kricheff I, Freed D, et al. Spinal cord ependymomas: MR imaging features. Radiology. 1995;197:655-658.
22 Nemato Y, Inoue Y, Tashiro T, et al. Intramedullary spinal cord tumors: significance of associated hemorrhage at MR imaging. Radiology. 1992;182:793-796.
23 Vougioukas V, Glasker S, Hubbe U, et al. Surgical treatment of hemangioblastomas of the central nervous system in pediatric patients. Childs Nerv Syst. 2005;22:1149-1153.
24 Chu B, Terae S, Hida K, et al. MR findings in spinal hemangioblastomas: correlation with symptoms and with angiographic and surgical findings. AJNR Am J Neuroradiol. 2001;22:206-217.
25 Miller D, McCutcheon I. Hemangioblastomas and other uncommon intramedullary tumors. J Neurooncol. 2000;47:253-270.
26 Wilkins R. A problem of cervical pain. Neurosurgery. 1995;36:158-165.
27 Ofntaine S, Melanson D, Cosgrove R, et al. Cavernous hemangiomas of the spinal cord: MR imaging. Radiology. 1988;166:839-841.
28 Gomori J, Grossman R, Goldberg H, et al. Occult cerebral vascular malformations: high-field MR imaging. Radiology. 1986;158:707-713.
29 Epstein F, Epstein N. Surgical treatment of spinal cord astrocytomas of childhood. A series of 19 patients. J Neurosurg. 1982;57:685-689.
29a Hardison H, Packer R, Rorke L, et al. Outcome of children with primary intramedullary spinal cord tumors. Childs Nerv Syst. 1987;3:89-92.
30 O’Sullivan C, Jenkin R, Doherty M, et al. Spinal cord tumors in children: long-term results of combined surgical and radiation treatment. J Neurosurg. 1994;81:507-512.
31 Miller D. Surgical pathology of intramedullary spinal cord neoplasms. J Neurooncol. 2000;47:189-194.
32 Minehan K, Shaw E, Scheithauer B, et al. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83:590-595.
33 Rossitch EJ, Zeidman S, Burger P, et al. Clinical and pathological analysis of spinal cord astrocytomas in children. Neurosurgery. 1990;27:193-196.
34 Lang FF, Epstein FJ, Ransohoff J, et al. Central nervous system gangliogliomas. Part 2: Clinical outcome. J Neurosurg. 1993;79:867-873.
35 DeSousa A, Kalsbeck J, Mealey J, et al. Intraspinal tumors in children. A review of 81 cases. J Neurosurg. 1979;51:437-445.
36 Ogilvy C, Louis D, Ojemann R. Intramedullary cavernous angiomas of the spinal cord: clinical presentation, pathologic features, and surgical management. Neurosurgery. 1992;31:219-230.
37 Kalangu K, Couto M. Radical resection of intramedullary spinal cord tumors without cavitron ultrasonic aspirator or CO2 laser: A “two stage” technique. Surg Neurol. 1996;46:310-316.
38 Tamaki T, Kiubota S. History of the development of intraoperative spinal cord monitoring. Eur Spine J. 2007;16(suppl 2):S140-S146.
39 Kothbauer K, Deletis V, Epstein F. Motor-evoked potential monitoring for intramedullary spinal cord tumor surgery: correlation of clinical and neurophysiological data in a series of 100 consecutive procedures. Neurosurg Focus. 1998;4(5):e1.
40 Sala F, Deletis V, Faccioli F, et al. Motor evoked potential monitoring improves outcome after surgery for intramedullary spinal cord tumors: a historical control study. Neurosurgery. 2006;58:1129-1143.
41 Abbott R, Feldstein N, Wisoff J, et al. Osteoplastic laminotomy in children. Pediatr Neurosurg. 1992;18:153-156.
42 Hsu P, Hsu B, Rigamonti D, et al. Epidemiology of cavernous malformations. In: Awad I, Barrow D, editors. Cavernous Malformations. Rolling Meadows, IL: American Association of Neurological Surgeons, 1993.
43 Patil C, Patil T, Lad S, et al. Complications and outcomes after spinal cord tumor resection in the United States from 1993 to 2002. Spinal Cord. 2007;46:1-5.
44 Constantini S, Houten J, Miller DC, et al. Intramedullary spinal cord tumors in children under the age of 3 years. J Neurosurg. 1996;85:1036-1043.
45 Allen S, Kahn E. A case of scoliosis produced by spinal cord tumor. J Nerv Ment Dis. 1933;77:53-55.
46 Kiwak K, Deray M, Shields W. Torticollis in three children with syringomyelia and spinal cord tumor. Neurology. 1983;33:946-948.
47 Yao KC, McGirt J, Chaichana KL, et al. Risk factors for progressive spinal deformity following resection of intramedullary spinal cord tumors in children: an analysis of 161 consecutive cases. J Neurosurg. 2007;107(6 suppl):463-468.
48 Abdel-Wahab M, Corn B, Wolfson A, et al. Prognostic factors and survival in patients with spinal glioma after radiation therapy. Am J Clin Oncol. 1999;22:344-351.
49 Katzman H, Waug T, Berdon W. Skeletal changes following irradiation of childhood tumors. J Bone Joint Surg Am. 1969;51:825-842.
50 Riseborough E, Grabias S, Burton R, et al. Skeletal alterations following irradiation for Wilms’ tumor. J Bone Joint Surg Am. 1976;58:183-193.
51 Wood E, Berne A, Taveras J. The value of radiation therapy in the management of intrinsic tumors of the spinal cord. Radiology. 1954;63:11-24.
52 Huddart R, Traish D, Ashley S, et al. Management of spinal astrocytoma with conservative surgery and radiotherapy. Br J Neurosurg. 1993;7:473-481.
53 Jyothirmayi R, Madhavan J, Nair M, et al. Conservative surgery and radiotherapy in the treatment of spinal cord astrocytoma. J Neurooncol. 1997;33:205-211.
54 Mansur D, Hekmatpanah J, Wollman R, et al. Low grade gliomas treated with adjuvant radiation therapy in the modern imaging era. Am J Clin Oncol. 2000;23:222-226.
55 Rodrigues G, Waldron J, Wong C, et al. A retrospective analysis of 52 cases of spinal cord gliomas managed with radiation therapy. Int J Radiat Oncol Biol Phys. 2000;48:837-842.
56 Sandler H, Papadopoulos S, Thorton AJ, et al. Spinal cord astrocytomas: results of therapy. Neurosurgery. 1992;30:490-493.
57 Stein B. Intramedullary spinal cord tumors. Clin Neurosurg. 1983;30:717-741.
58 Sundaresan N, Gutierrez F, Larsen M. Radiation myelopathy in children. Ann Neurol. 1978;4:47-50.
59 Mitchell M, Logan P. Radiation-induced changes in bone. Radiographics. 1998;18:1125-1136.
60 Sklar C. Growth and neuroendocrine dysfunction following therapy for childhood cancer. Pediatr Clin North Am. 1997;44:489-503.
61 Allen J, Aviner S, Yates A, et al. Treatment of high-grade spinal cord astrocytoma of childhood with “8 in 1” chemotherapy and radiotherapy: a pilot study of CCG-945. J Neurosurg. 1998;88:215-220.
62 Bouffet E, Amat D, Devaux Y, et al. Chemotherapy for spinal cord astrocytoma. Med Pediatr Oncol. 1997;29:560-562.
63 Dolreau V, Grill J, Zerah M, et al. Chemotherapy for unresectable and recurrent intramedullary glial tumours in children. Br J Cancer. 1999;81:835-840.
64 Hassal T, Mitchell A, Ashley D. Carboplatin chemotherapy for progressive intramedullary spinal cord low-grade gliomas in children: three case studies and a review of the literature. J Neurooncol. 2001;3:251-257.
65 Mora J, Cruz O, Gala S, et al. Successful treatment of childhood intramedullary spinal cord tumors with irinotecan and cisplatin. Neuro Oncol. 2006;9:39-46.






