[level-membership-for-neurosurgery-category]
Chapter 25 Intradural Extramedullary and Intramedullary Spinal Cord Tumors
• Surgery is the initial and primary treatment of choice in most spinal tumors.
• Intradural extramedullary spinal tumors, mostly benign peripheral nerve sheath tumors and meningiomas, carry an excellent prognosis. Surgery allows most of the time complete resection and neurological improvement or recovery.
• The main objective should be preserving the patient’s functional condition and making an accurate diagnosis. Postoperative results depend on the preoperative neurological status, the tumor grade, tumor location, and the surgical team’s experience.
• Many intramedullary spinal tumors, such as ependymomas, astrocytomas, and some of nonglial origin, have a favorable prognosis because they may be completely resected based on the favorable surgical plane between the tumor and spinal cord.
Intradural Extramedullary Tumors
Incidence and Types
Spinal intradural extramedullary tumors (SIEMTs) are primarily peripheral nerve sheath tumors (PNSTs) and meningiomas in similar numbers.1 In the largest reported study on SIEMTs, nerve sheath tumors and meningiomas accounted for almost 70% of all lesions.1 Other SIEMTs and lesions include arachnoid cysts, hamartomas, and ependymomas, with an incidence between 5.5-7.5% each.1 The differential diagnosis for SIEMTs and lesions is broad and includes angioblastoma, cavernoma, chordomas, dermoid, epidermoid, exophytic astrocytoma, germinoma, hemangiopericytoma, lipoma, lymphoma, malignant teratoma, medulloblastoma, melanocytoma, melanoma, metastasis, myxoma, neuroblastoma, and sarcoma.1,2
In about 50% of cases, SIEMTs are located at the thoracic level.1 Lesions with an extradural extension are seen at the cervical level in more than half of patients.1 Benign nerve sheath tumors are the most common SIEMT and are associated with extradural extension with an approximate incidence of 77%.1 Meningiomas, hamartomas, and sarcomas are other SIEMTs with extradural extension.1
Peripheral Nerve Sheath Tumors
Most schwannomas are purely intradural but 15% have an extradural component, being either solely extradural or both intra- and extradural. These tumors originate from the dorsal sensory nerve root.3 Typically, these lesions are well encapsulated and rarely include a cystic portion. In contrast, neurofibromas are not well encapsulated and rarely occur alone. Multiple plexiform neurofibromas are usually seen with neurofibromatosis type 1 (NF1), a genetic disorder associated with mutations of chromosome 17.4–6 Diffuse plexiform neurofibromas are detected clinically and radiologically, respectively, in 26.3% and 50% of the patients with NF1.6 They appear in early childhood and have a variable growth pattern. Lesions may grow rapidly until adolescence and then become quiescent.6 The presence of a tumor is not an indication for surgery. Resection is indicated in NF1 patients if the tumor is growing or is causing progressive neurological deficits. In 2% to 5% of the cases, malignant changes may be observed with rapid expansion of the tumor mass.6 Surgery can be challenging in malignant tumors when the tumor is attached and infiltrates surrounding soft tissue. Neurofibromatosis type 2 (NF2) is another genetic condition associated with chromosome 22 anomalies.6 The clinical picture of NF2 may include SIEMTs such as schwannomas, neurofibromas, and meningiomas in 30% of patients.6 Pathologically, nerve sheath tumors are subdivided according to the WHO (World Health Organization) classification into schwannomas (WHO grade I), neurofibromas (WHO grade I), and malignant peripheral nerve sheath tumors (WHO grade III/IV).
Meningiomas
Spinal meningiomas commonly occur in middle-aged patients with a predilection for women.7 Most of the sporadic meningiomas are solely intradural. Of those remaining, one third are intra- and extradural and two thirds are primarily extradural.8 These lesions have a strong tendency to develop at the thoracic level with a frequency reported in the literature between 55% and 83%.7,9–11
Clinical Picture
Most patients present with a long history of progressive symptoms and neurological deterioration because SIEMTs are slow-growing tumors. It is uncommon for a patient to abruptly deteriorate due to spontaneous tumor hemorrhage with spinal compression. SIEMTs appear with signs and symptoms indicative of upper and lower motor neuron involvement, related to the severity of the nerve root insult and spinal cord compression. In half of the cases, the first symptom reported by patients with SIEMTs is pain.1 The tenderness and pain are characteristically increased at night or in the supine position and are relieved after returning to an upright position. Pain is often described along a radicular dermatome but can also reach varying distribution. It is usually described as a dull, aching sensation, and less frequently as burning. Radicular involvement results in lower motor neuron dysfunction, with weakness, sensory disturbances, hyporeflexia, atrophy, and fasciculation at the affected level. Myelopathic symptoms are correlated with the severity, level, and site of spinal cord compression. Nerve sheath tumors typically compress the lateral aspect of the spinal cord and are therefore more likely to be associated with the development of a Brown-Sequard syndrome. Upper motor neuron deficits manifest by gait ataxia and spasticity of the affected extremities. Hyperreflexia as well as other pyramidal signs such as the Hoffman and Babinski signs may be detected upon clinical examination. Patients present with pain, gait ataxia, motor weakness, and sensory deficits in 70% to 77% of the cases, and dysesthesias and sphincter disturbances are found in about 40%.1
Preoperative Imaging
Magnetic resonance imaging (MRI) is currently the gold standard method for imaging. MRI provides accurate diagnoses and delineates the soft tissue components.12,13 Gadolinium administration highlights enhancing portions of the tumor. MR myelography is a special study that is particularly useful for showing small schwannomas.13
On MRI, schwannomas and meningiomas often appear as isointense to the spinal cord on T1-weighted images (WI). On T2-WI, schwannomas are most often hyperintense, as opposed to meningiomas that appear isointense.13 After gadolinium administration both lesions enhance intensely and homogeneously.13 A cystic component and hemorrhagic foci may be noted in schwannomas or neurofibromas, respectively, in 40% and 10% of the cases.13 Less frequently than with cranial lesions, there is often a characteristic dural tail associated with spinal meningiomas.3
The computed tomography (CT) scan enables detection of calcifications as well as of spinal canal and neural foramen enlargement in cases of extradural extension.13 CT angiography (CTA) and MRI angiography (MRA) provide relevant information for cervical dumbbell-shaped neurofibromas that develop in close proximity to the vertebral artery (VA).14 Conventional angiography has a few indications for some of these tumors. It is helpful for providing relevant information about the anterior spinal artery at the cervical level14 and the Adamkiewicz artery at the thoracolumbar junction. CT myelography is invasive and thus not as useful as MRI. It is no longer recommended as the primary imaging modality.13
Surgery
The surgical approach should be adjusted according to the presumed diagnosis and tumor location. The surgical strategy of a PNST is different from that with a meningioma. First, PNSTs most often develop on the anterolateral aspect of the spinal cord, mainly at the cervical level, inducing a posteromedial displacement of the neuraxis. Second, few of them are solely intradural/extramedullary lesions; numerous are dumbbell-shaped lesions extending into the vertebral foramen. Third, at the cervical spine level, their vascular supply from the vertebral artery (VA) requires special considerations.14
Peripheral Nerve Sheath Tumors
Several surgical techniques have been described for PNST resection. These lesions are accessed using a posterolateral approach, as described for meningiomas. The posterolateral approach through a laminectomy is adequate for the resection of the intradural component. In the case of cervical PNST extending through the vertebral foramen and in close proximity to the VA, the posterior approach has several drawbacks.14 First, a significant portion of the facet must be resected to reach the foraminal component of the tumor. This may potentially cause postoperative instability requiring fusion.15 Second, the VA providing the tumor blood supply is reached at the end of the surgical procedure, precluding any devascularization of the tumor supply before the final surgical step.14 Moreover, without proximal control of the VA, the risk of VA damage is higher.14 Finally, it often results in leaving tumor remnant that remains vascularized, and thus has a potential to grow in the future. For some anterior lesions, the anterolateral approach may be useful in select patients. It allows for complete devascularization of the tumor at the beginning of the procedure, cutting down on blood loss. Then, tumor resection can most often be completed in a single surgical step.14 The intradural component of the lesion may be accessed through the enlarged vertebral foramen that may be drilled out slightly. This tailored opening rarely destabilizes the spine and subsequent fusion is not uniformly required. The primary reason for a fusion is a history of previous laminectomy, which completely removes the facets. The anterolateral approach requires VA control, which requires experience. However, this approach is safe and associated with low morbidity rate in experienced hands;15,16 the incidence of permanent deficits drops to less than 3%.15,16 The main risk is a postoperative Horner syndrome.
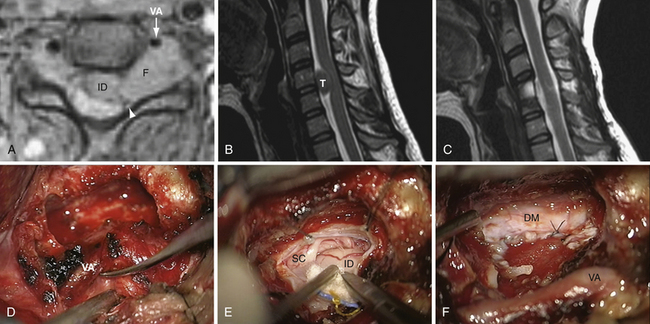
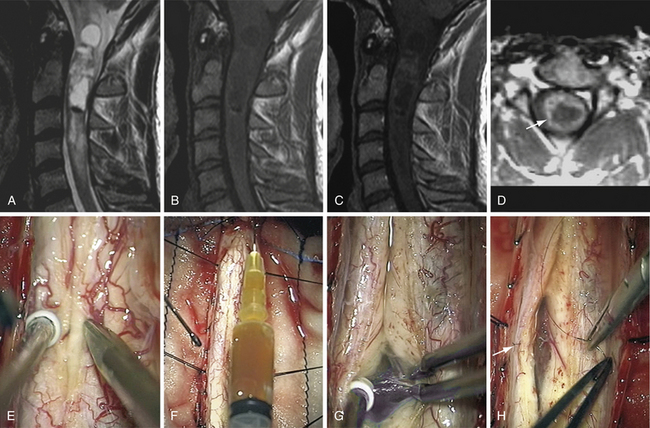
The Anterolateral Approach with Vertebral Artery Control for Cervical Dumbbell-Shaped Tumors (Fig. 25.1)
Preoperatively, the VA position has to be anticipated by an MRA or CTA: anatomical variations must be ruled out, as well as VA displacement induced by the tumor.17,18 The tumor often grows at the posterior VA aspect therefore pressing on the artery anteriorly or anteromedially. The patient is placed in the supine position, with head lying on a small cushion, slightly extended with a tape passing under the chin and turned to the contralateral side at 30 degrees.15,16 Fluoroscopy is used to check the correct level. Electrophysiological monitoring (consisting of motor and somatosensory evoked potentials [MEP, SSEP] and electromyography [EMG]) is maintained throughout the surgical procedure. The skin incision follows the medial border of the sternomastoid muscle. After division of the platysma, the dissection is pursued medially to the medial border of the sternomastoid muscle and laterally to the internal jugular vein. The carotid-jugular sheath is left undissected and is retracted medially. Dissection of the sympathetic chain is an important step in avoiding the main complication of this approach, which is Horner syndrome. The sympathetic chain must be differentiated from the longus colli muscle (LCM) and retracted laterally using stitches applied on the LCM aponeurosis that has been divided medially to the chain. At the C6-C7 level, the thoracic duct on the left side or the accessory thoracic duct on the right side must be preserved to avoid a leak.19,20 After its exposure, the LCM must be resected. In order to prevent VA injury, the LCM must be resected down to the transverse processes above and below the level of tumor. It is safe because, in the absence of anatomical variations, the VA is protected inside the bone of the transverse foramen.15–18 Afterward, the muscle is resected along the lateral border of the cervical vertebrae. From an anatomical point of view, it must be pointed out that, between C6 and the foramen magnum, a venous plexus enclosed inside a periosteal sheath surrounds the VA. The periosteal sheath is continuous with the periosteum of each transverse process. It means the periosteal sheath should be left intact inside the transverse foramen. It can be achieved only by performing a subperiosteal dissection of the anterior branch of the transverse process with a smooth spatula before resecting the bone with a Kerrison rongeur.15,16 In case of tearing of the periosteal sheath, troublesome venous bleeds may arise but can easily be controlled by direct bipolar cauterization of the sheath itself or packing with resorbable cellulose.15,16 At this point, the VA is controlled from above and below the tumor. The vertebral foramen, already enlarged by the tumor, may be made slightly larger by drilling. The tumor feeders will be cauterized during the separation between the anterior aspect of the nerve root and the VA posterior border, facilitating further tumor resection.15,16 In the case of a schwannoma, the tumoral nerve root fascicle may be separated from the normal nerve roots. The nerve root may be sacrificed if there is already a neurological deficit by nerve root involvement.14 Intraoperative stimulation may provide additional relevant information for this decision.14 Pursuing a lateromedial tumor resection will allow entrance into the intradural compartment. The dura mater opening through which the tumor has passed may be enlarged as much as necessary to provide the appropriate intradural exposure. The use of an ultrasonic aspirator (the Cavitron ultrasonic aspirator, CUSA) is helpful for debulking the lesion.14 The tumor should then be cautiously separated from the spinal cord using a microsurgical technique.
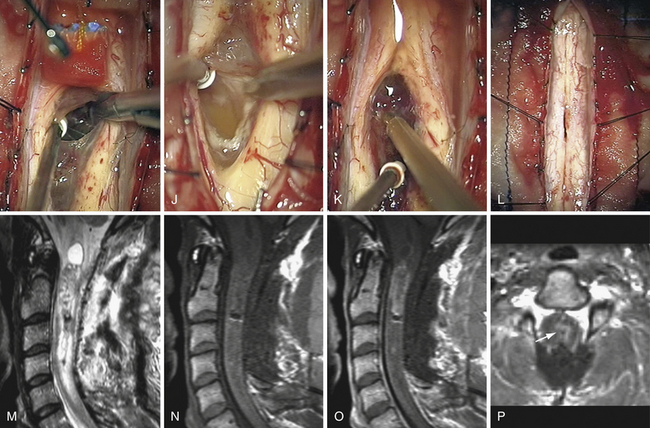
Meningiomas (Fig. 25.2)
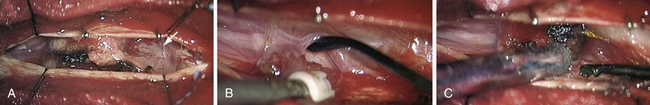
In the case of meningiomas, the tumor origin can be found anywhere along the surface of the dura. A general rule is that the neuraxis is displaced to the side opposite to the tumor origin. Therefore, posterior meningiomas put pressure on the spinal cord anteriorly and for this reason are better approached directly with a laminectomy. Posterolateral and lateral meningiomas displace the neuraxis anteromedially and medially, respectively. A posterolateral approach is advocated, by laminectomy possibly extended on the tumor side to the medial aspect of the facets. With anterolateral meningiomas, the posterior displacement of the neuraxis requires a more location-specific approach because surgical access is limited. In this situation, we still advocate a posterior approach, extended laterally on the side of the tumor, but the lateral surgical corridor must be enlarged by dividing dentate ligaments. With that maneuver, most lesions can be adequately resected. Rare reports have advocated the use of an anterior approach for anterior meningiomas.21 This anterior approach is considered safe and has several theoretical advantages: a large bony window of access, extradural cauterization of the anterior blood supply, direct visualization of the entire tumor ventralto the spinal cord, and tumor resection without spinal cord manipulation; but it is nevertheless associated with drawbacks, such as the need for fusion and a more challenging access route.21 It is nevertheless associated with drawbacks such as the need for fusion and a more challenging access route. The depth and narrowness of the working channel present a significant technical challenge. In addition, dural closure is also very difficult, resulting in a higher probability of CSF leakage.21
The Posterior Approach
The patient is placed in the prone position under continuous electrophysiological monitoring, especially for cervical lesions. At this level, we use a Mayfield head holder and are careful to position the head in a neutral position to avoid tumor-induced spinal cord compression. Fluoroscopy is used to accurately target the pathological levels. Usually, only two vertebrae must be exposed. The laminectomy may be extended laterally to facets on the side of the tumor. The dura mater is usually incised longitudinally over the tumor. The dural edges are retracted laterally with several 4-0 silk sutures. Lateral lesions may require section of the dentate ligaments to provide exposure. Sutures from dentate ligament to opposite dura keeps the surgical space open and offers good control of anterior dura as a result of gentle rotation of the spinal cord. Great care must be taken to avoid any traction or compression on the spinal cord itself. Cottonoids are placed at each end of the tumor to avoid intradural soiling. Most of the time, the arachnoid can be separated and the resection completed in the extra-arachnoidal plane. The tumor surface is cauterized and incised with a knife or microscissors. Samples are sent to the pathologist for immediate analysis. In lateral meningiomas, the tumor debulking is performed with either microinstruments or an ultrasonic aspirator, starting along the dural base if possible. Bleeding of feeding vessels is gently controlled with bipolar cauterization. In this way, the tumor is progressively devascularized and becomes mobile. The pressure at the tumor–spinal cord interface and at the dural insertion base is then released. Shrinkage of the tumor by electrocautery may also help to keep the plane open. Only vessels supplying the tumor should be sacrificed. Progressively, the dural base may be divided, resulting in tumor devascularization. Nevertheless, we advise keeping a small dural attachment until the tumor is completely released from the spinal cord. Otherwise, the lesion may become too mobile, thereby increasing difficulties and risks in completing the dissection of the spinal cord–tumor interface. At the completion of surgery, the dural base should be either cauterized or resected. The choice is guided by the location of the pathological dura and the possibilities of performing a watertight reconstruction. Resection and reconstruction with a patch graft is advised for posterior and posterolateral lesions. Contrarily, we advise only coagulating anterior or anterolateral dura mater portions. Cauterization instead of resection of the dura mater is not associated with a higher risk of recurrence.9,11,22 Finally, the intradural compartment is copiously washed with a normothermic saline solution. The dura mater is closed with a 5-0 nonabsorbable running watertight suture to prevent CSF leakage. The aponeurosis is also closed with a watertight running suture to avoid a CSF leak. Drains may be inserted at the level of the muscular layer.
Postoperative Outcome
Short-term outcome after the resection of SIEMTs is usually excellent, with the neurological condition generally improving significantly.2 Resolution of preoperative deficits has been noted in about 80% of the cases.7,8 The patient’s neurological condition remains stable in 14% of patients and few deteriorate.7 Several factors influence the outcome such as the preoperative neurological status, the duration of preoperative symptoms, and the type of surgical resection.7 The histological characteristics of the tumor may influence the long-term outcome: the psammomatous type is associated with a less favorable neurological outcome than the nonpsammomatous type. Psammomatous tumors consist essentially of acellular concretions, rendering blood vessels and spinal roots extremely adherent.8 Location of the tumor below C4 and posteriorly or laterally in the intradural compartment is, however, a favorable factor.8 Complete tumor resection is possible in 96% to 97% of the cases7,22 and is associated with a recurrence rate of 6% after 15 years of follow-up. Subtotal resection of a tumor resulted in recurrence in 17% of the cases.22 Serious complications may develop in case of perioperative vascular or medullar injuries but are unusual. Physical therapy hastens neurological recovery. Most patients can resume a normal or nearly normal neurological condition. A gratifying quick recovery may be observed within the first 6 weeks after surgery, but progressive recovery may be expected by 1 year after surgery. We see patients at 6 months, 1 year, and 2 years with performance of an MRI and then every 5 years in the absence of recurrence.
Radiation Therapy
When dealing with a malignant PNST, current consensus advocates postoperative radiation therapy (RT), started as soon as possible after surgery even if resection margins are tumor free.23,24 The role of radiotherapy in the management of spinal meningiomas is controversial. The slow indolent growth of these lesions and the risk associated with irradiating the vicinity of the spinal cord are confounding issues.7 Several indications for adjuvant radiation therapy have been proposed: early recurrence after total or subtotal resection, partial resection, and high-risk surgical treatment.7 If possible, we prefer repeat surgery over radiation for recurrence.
Intramedullary Spinal Cord Tumors
Surgery on spinal cord intramedullary tumors (SCIMTs) has been refined over the past few decades. The pioneering work of Elsberg25 and subsequently of many others such as Cooper,26 Epstein and Epstein,27 Guidetti and associates,28 McCormick and colleagues,29 Malis,30 Stein and McCormick,31,32 and many others33,34 have greatly contributed to advancing the microsurgical technique and success in removing spinal cord lesions. Several tools have added safety features to the microsurgical resection of these lesions. The advent of MRI has definitively improved diagnosis accuracy. The introduction of ultrasonic aspiration (CUSA) and intraoperative electrophysiological monitoring has modified the surgical approach and improved safety and outcome significantly.
Incidence and Types
With an incidence in adulthood of 2% to 4% of all central nervous system tumors, SCIMTs are considered rare.30,32,35–37 Three main groups of SCIMTs can be distinguished: tumors of glial origin, tumors of nonglial origin, and other mass lesions.
Tumors of Glial Origin
These tumors primarily consist of ependymomas and astrocytomas.13,38 Other glial tumors such as oligodendriogliomas are rare.39
Ependymomas
Spinal cord ependymomas are the most frequent SCIMTs in adult patients. They may reach considerable size prior to diagnosis. The majority of spinal cord ependymomas in adults have a good prognosis because usually they can be completely removed and are primarily grade II by the WHO classification. In the literature, the rate of complete removal varies from 69% to 97%.40–42 The vast majority of patients, 48% to 75%, are stabilized after the radical surgical resection, thereby highlighting the need for timely surgery in case of neurological deterioration.41–45 The overall rate of neurological improvement for this patient population varies between 10% and 40%.41–45 A rate of postoperative deterioration has been reported in the literature between 9% and 15%.41–45 Subtotal resection has been reportedly associated with a lack of clinical improvement,43 and incomplete resection is the most important factor correlating with tumor recurrence.41,46,47 The 5- and 10-year survival rates are respectively quoted between 83% to 96% and 80% to 91%.40,42,43,45,48,49
Astrocytomas
Spinal cord astrocytomas represent more than 80% of the SCIMTs in children,43 and in adults astrocytomas are the second most common type of tumor.43 Only 10% to 17% of astrocytomas in childhood are anaplastic lesions or glioblastomas, whereas in adults, the percentage is higher, 25% to 30%.28,34,43,50–53 In large studies, the proportion of pilocytic astrocytomas varies greatly from more than 30% to 60%.42,43 By their nature, grade II to IV tumors are infiltrating. Therefore, in contrast to ependymomas, radical resection of astrocytomas is not always possible. Surgery consists of removing as much tumor as possible. In some instances, one can only perform biopsy or decompression with duraplasty. In the literature, the rate of complete and subtotal removal varies between 11% to 31% and 21% to 62%, respectively.42,43,54 The pathological diagnosis of pilocytic astrocytoma and surgical experience are factors associated with higher rates of complete resection.42,43 Low spinal level (conus), malignancy, and adult onset are considered important factors associated with a higher tumor recurrence rate.43 The degree of resection, and spinal canal decompression via tumor debulking, have not been found to be directly related to progression-free survival or overall survival in many studies.26,32,43,50,52,55,56 In low-grade astrocytomas, the 5- and 10-year survival rates range from about 77% to 82%.43,56 These rates drop, respectively, to 27% and 14% for malignant astrocytomas.43,56
Gangliogliomas
Gangliogliomas are very uncommon lesions mainly reported as case reports and a few studies.57,58 These lesions are mainly encountered in children and young adults.57 In a large series of 56 patients, complete or subtotal resection has been achieved in 82%, and 18% of the patients had a 5-year survival rate of 88% and a progression-free survival rate of 67%.57
Factors Determining Outcome
The major determinant of long-term patient survival is based on the histological characteristics of the tumor. The 5-year progression-free survival (PFS) rate drops from 78% for low-grade gliomas to 30% for high-grade gliomas.50 A longitudinal database (Surveillance, Epidemiology, and End Results [SEER] database) encompassing 26% of the U.S. population was published in 2010 and included 1814 patients with spinal cord gliomas.59 In this study, age, histology, and grade were defined as significant predictors of outcome.59
Tumors of Nonglial Origin
Other lesions such as metastases, epidermoids, dermoid cysts, lipomas, intramedullary schwannomas, PNETs, and teratomas may also be encountered.13,43,55 In addition to these rare lesions, we have also encountered lymphomas and neuroglial cysts.
Hemangioblastomas
Hemangioblastomas are highly vascular lesions that represent almost 2% to 15% of SCIMTs in some studies.42,43,60–63 These lesions consist of two main components: (1) large vacuolated stromal cells that have been identified as the neoplastic cell of origin and (2) a rich capillary network.64 Hemangioblastomas are often observed as encapsulated lesions abutting the pia, especially at the posterior or posterolateral aspect of the spinal cord close to the dorsal root entry zone. They may also be encountered anteriorly or in intramedullary sites.43,60,63 Radicular arteries or anterior branches feed these lesions.65 Despite being histologically benign, spinal hemangioblastomas may be associated with significant neurological morbidity, often secondary to associated cyst and syrinx (present in 80-90% of the cases) and edema.60 En bloc surgical resection is the treatment of choice, associated with excellent neurological outcome. Patients with incidental asymptomatic solitary lesions may be followed.60 Surgery is indicated as soon as symptoms develop or sequential MRI demonstrates tumor or cyst growth.43 For patients with von Hippel-Lindau (VHL) disease and multiple lesions, the timing of surgery remains open to debate.63 Some authors operate on asymptomatic patients if they exhibit radiological progression before significant neurological deficits occur.63 Follow-up of patients with VHL disease, and associated neoplasms such as pheochromocytoma and renal and pancreatic cancer, should be performed at regular intervals with spinal and brain MRI studies.43,66
Cavernomas
Intramedullary cavernomas accounted for 3% to 16% of intramedullary lesions in large studies.42,43,61,67 Cavernous malformations are well-circumscribed lesions consisting of closely packed, capillary-like vessels, without intervening brain or spinal tissue.67 The clinical presentation may vary. Some patients may present with gradual neurological deterioration explained by repeated hemorrhages. Others exhibit slow progressive deterioration induced by small hemorrhages and increasing gliosis, or with acute onset and significant neurological deterioration due to spontaneous hemorrhage.67–72 When an intraspinal cavernoma is discovered, a survey of the entire central nervous system is advised.67 Approximately, 40% of patients with a spinal cavernoma may also have a synchronous intracranial lesion.73 In our practice, removal of intramedullary cavernomas is indicated in two circumstances: (1) symptomatic lesions or (2) asymptomatic lesions, if the cavernoma is easily accessible, specifically posteriorly and/or abutting the pial surface. This surgical attitude is controversial for asymptomatic patients68,69 but it is supported by some in the literature.43,74 The annual rate of hemorrhage in symptomatic intramedullary cavernous malformations ranges from 1.4% to 4.5% but rises to 66% in the case of previous bleeding.72,75 Most lesions can be completely removed with concomitant clinical improvement. An improved postoperative outcome is obtained if symptoms have existed for less than 3 years.72
Associated Diseases
Most SCIMTs develop sporadically; only a minority is associated with genetic diseases. Hemangioblastomas are a component of von Hippel-Lindau disease. Ependymomas and astrocytomas are associated with neurofibromatosis type 1 and 2.43,60,63,76,77 Overall, SCIMTs are noted in these genetic disorders in approximately 20% of the patients.60,78 Intraspinal mass lesions such as hamartomas, dermoid cysts, and dermal sinus tracts are associated with spinal dysraphism.43,79–82 Demyelinating disease may present as a mass lesion with histological features identical to those seen in the brain. Sarcoidosis has also been described as a primary spinal cord lesion. Spinal cord involvement in sarcoidosis is rare, occurring in less than 1% of patients with sarcoidosis.83 Any isolated spinal cord intramedullary lesion that is diffusely enhanced and not associated with a mass effect should be investigated for inflammatory disease.
Clinical Expression
There is no typical presentation for spinal cord tumors (SCIMTs). An important characteristic in distinguishing them from demyelinating diseases should be addressed. Intermittent symptom regression and progression is never noted in SCIMTs.43 The clinical course is often insidious and progressive over years. In fact, in 80% of the patients, tumors grow slowly and most of them are benign lesions.13,43 Episodic deteriorations or abrupt onset may also be encountered especially as a result of hemorrhage. Spinal subarachnoid hemorrhage originates predominantly from arteriovenous malformations that mimic a tumor84 but also rarely from hemangioblastomas.85 Hydrocephalus induced by SCIMTs has been reported in the literature.86,87 We have also encountered a malignant intramedullary tumor with associated leptomeningeal dissemination.88 For this reason, a child presenting with hydrocephalus of unknown origin should have a spinal MRI to rule out an SCIMT.87
Adults more often complain of pain as a first symptom (35%), while children are more likely to present with motor weakness (42%).43 In descending order of frequency, patients may present with sensory disorders, gait ataxia, motor deficits, and sphincter disorders. In children under 3 years of age, in descending order, the clinical presentation may include progressive motor regression/weakness, torticollis, gait abnormalities, and kyphoscoliosis.89 The presentation is most often directly related to the level of the lesion. In large studies, the cervical cord is involved in 30% to 56% of the cases, the cervicothoracic cord in 17% to 25%, the thoracic cord in 17% to 49%, and the remaining ones occur in the conus area.42,61,90,91 However, it is possible for cervical intramedullary tumors to present without sensory or motor deficit in the upper extremities, and the patients with a conus tumor may not suffer from sphincter dysfunction. An aspecific presentation is inevitably associated with a diagnostic delay after the first symptom, with a mean time of 1 to 3 years depending on the tumor’s growth rate.13,43 The most useful clinical classification system is that reported by McCormick.29 In this clinical/functional classification system, grade I corresponds to a patient neurologically intact, with mild focal deficit not significantly affecting function of involved limb, with mild spasticity or reflex abnormality, or normal gait.29 In grade II, the patient presents with sensorimotor deficit affecting function of involved limbs, mild to moderate gait impairment, severe pain or dysesthetic syndrome impacting patient’s quality of life; he or she still functions and ambulates independently. In grade III, he or she suffers from more severe neurological deficit, requires cane/brace for ambulation or significant bilateral upper extremity impairment; he may or may not function independently.29 In grade IV, the patient has severe deficit and requires wheelchair or cane/brace with bilateral upper extremity impairment; he or she is usually not independent.29 This classification takes into account both sensory and motor functions to rate the patient’s clinical status at the time of the surgery and during follow-up.
Diagnosis
Preoperative Imaging
A complete MRI workup requires T2- and T1-WI with and without gadolinium sequences in at least two planes (sagittal and axial).13 The slice thickness should be of less than 3 mm and a large visual field should be used to encompass the entire spinal cord.13 Full spinal axis imaging allows ruling out transitional bone anomalies especially in thoracic tumors to accurately locate pathological levels under fluoroscopy at the beginning of the surgery. Several aspects of the tumor and its characteristics should be evaluated in order to determine the preliminary diagnosis and surgical plan. On axial images, it can be determined whether the lesion is centrally located or eccentric, with a possible exophytic component. The demarcation from the normal spinal cord tissue can also sometimes be determined. Enhancement after gadolinium administration directs the surgical trajectory and gives information on tumor vascularity as well as the spinal cord–tumor interface.
On MRI, intramedullary ependymomas appear as well-circumscribed lesions, typically located at the center of the spinal cord because they originate from ependymal cells lining the central canal.12,92,93 Their signal can be variable on T1-WI and hyperintense on T2-WI. Typically, ependymomas enhance homogeneously after gadolinium injection.13 Four patterns of contrast enhancement have been determined by Miyazawa and co-workers94: homogeneous pattern, heterogeneous pattern, heterogeneous with cyst wall enhancement, and enhanced nodule on the cyst wall. Associated satellite cysts are present in 60% of the cases.13,43 On T2 sequences, it is interesting to look for hypointense signal at both poles of the solid portion of the tumor, called the “cap-like” appearance. In our experience, although this finding is not pathognomonic, it is observed in only 30% of ependymomas and 12% of hemangioblastomas. It has been correlated histologically and intraoperatively with areas of chronic hemorrhage with hemosiderin deposits or high protein substances and sometimes is referred to as Froin’s syndrome.13
Astrocytomas often appear hypointense on T1-WI and hyperintense on T2-WI.13 FLAIR (fluid-attenuated inversion recovery) images and T2 sequences can be helpful in delineating the precise extension of an astrocytoma whose borders are not well demarcated due to its infiltrative nature. A cystic component is depicted in 27% to 42%13,43 of the tumors and is associated with hydromyelia in about half of the cases.13 Grade II astrocytomas generally do not enhance after gadolinium administration.13 Conversely, pilocytic astrocytomas and high-grade lesions that appear more heterogeneous with necrotic and cystic regions enhance intensely.13 Gradient echo T2-WI is a useful sequence for determining hemorrhagic zones within the lesion.13 Regardless of typical radiological appearances, the distinction between astrocytomas and ependymomas remains difficult and demands histological analysis. MRI can predict but not definitively ascertain the precise diagnosis. For this reason, the potential degree of resection cannot be predicted preoperatively.
On the contrary, MRI has a high degree of accuracy with nonglial tumors. The nodule of an hemangioblastoma appears classically iso- to hypointense on T1-WI and iso- to hyperintense on T2-WI.13 Homogeneous uptake is evident after gadolinium injection. Proton density imaging and T2-WI are appropriate for delineating enlarged feeding arteries, rich tumor vascular networks, and dilated draining veins.13 An associated cyst has been found in 88% of the cases in large studies.43 MRA with a contrast-enhanced bolus for three-dimensional T1-weighted sequences may currently identify some feeding arteries and large draining veins. Angiography is considered only when preoperative embolization is required to reduce the risk of bleeding during surgery.
Cavernous malformations have a characteristic appearance on MRI. A cavernoma is composed of mixed signal intensities, surrounded by a hemosiderin hypointense ring. Completing the preoperative workup with a brain MRI is advocated because synchronous lesions are often noted and confirm the diagnosis.13
Magnetic Resonance Tractography
This method, which is based on diffusion tensor imaging techniques, is still in evolution. It holds potential for analyzing spinal cord fiber displacement or disruption from tumors or non-neoplastic lesions.95,96 Secondary alterations to white matter tracts can be reliably displayed and could therefore become of great value for planning treatment and follow-up.96
Preoperative Diagnostic Pitfalls
A wide differential diagnosis should be entertained in order to adopt the appropriate treatment strategy for lesions in the spinal cord. In some circumstances, associated conditions provide crucial information to focus the diagnosis and rule out an intramedullary tumor. Intramedullary bacterial, fungal, or parasistic abscesses should be in the differential diagnosis in the presence of systemic disease or immunocompromised patients. In contrast to SCIMTs, a rapid clinical deterioration is observed in infections.97 If symptoms develop abruptly, a medullary infarction is suspected.98 Multiple sclerosis can mimic an intramedullary tumor, especially active lesions that enhance on MRI.55 If there is any doubt, complete central nervous system screening for concomitant lesions should be performed by MRI. If other lesions are not discovered, but the clinical presentation is one of intermittent neurological deficits, demyelinating disease is not ruled out. Further testing is required.43 A cerebrospinal fluid examination to look for oligoclonal bands and serum markers may be helpful.55 If there is continuing doubt about a demyelinating diagnosis, and the neurological presentation is not progressive or atypical for neoplasm, a repeat MRI in a few weeks is a reasonable plan. Indeed, the signal characteristics of the lesion on MRI may change in the case of demyelinating or inflammatory disease. Neoplastic disease is unlikely to change in a short interval, unless there is a highly malignant tumor.43 Spinal cord edema and rim-like contrast enhancement may also be seen in the case of subacute necrotizing myelopathy. In this disorder, spinal cord atrophy develops over time.97
In a study of nine patients undergoing surgery for neoplastic-like lesions reported by Lee and associates,99 the histological examination of the surgical specimens showed demyelinating lesions, sarcoidosis, amyloid angiopathy, and a group of non-neoplastic inflammatory cells of unknown etiology. The most consistent finding that the author reported for differentiating non-neoplastic lesions from neoplastic ones was the absence of or minimal spinal cord expansion on preoperative MRI in non-neoplastic lesions.99
Biopsy
In some lesions, when the diagnosis cannot be defined based on clinical data, blood work or CSF samples, and MRI, a biopsy may be helpful for diagnosing granulomatous disease such as sarcoidosis, tuberculosis, brucellosis, and histoplasmosis, which can affect the spinal cord.99–103 These lesions often have systemic manifestations97 but even without them, the diagnosis cannot be made with an MRI alone, and thus, a biopsy is necessary.104 In these disorders, surgery should be limited to a biopsy; nodule resection may indeed be linked with neurological worsening.103 Then, a steroid-based treatment offers a good prognosis,105 although a relapse may occur when steroids are discontinued.106
Surgical Considerations
The most important factor in determining the long-term neurological and functional outcome following surgery is the patient’s preoperative neurological status.43,61,107 Therefore, we advise surgery in the presence of neurological deficits or decline. Serial monitoring for asymptomatic individuals is also critical. Patients with either no deficits or only mild deficits prior to surgery are unlikely to experience permanent deficits in a meticulously planned and executed surgery, reinforcing the importance of early diagnosis and treatment.50 Complete resection is attempted with ependymomas, hemangioblastomas, and cavernomas, as opposed to astrocytomas. Currently, the vast majority of well-demarcated tumors are potentially curable with a good quality of life. A carefully planned strategy should be adopted for the first surgical procedure. The patient’s prognosis is directly related to the results of the first surgery; a second surgery after an unsuccessful attempt usually results in neurological worsening due to damage caused by dissection of scar tissue or trying to discern unrecognizable surgical planes.
Intraoperative Monitoring
Intraoperative monitoring of the functional integrity of the spinal cord during intramedullary procedures has been recognized as a promising adjunct that may help in intraoperative decision making and in prediction of neurological outcome.108,109 Historically, somatosensory evoked potentials (SSEPs) were the first to be used, but motor evoked potentials (MEPs) have become more popular, useful, and technically accessible to surgeons. The continuation of the SSEPs should encourage pursuing an aggressive resection, but the loss of SSEPs is often of little value in anticipating postoperative motor deficits. In contrast, MEPs correlate more closely with the postoperative motor function. Loss of MEP waveform and a 50% reduction of the D wave seems to represent a critical points.61,109 In this situation, we wait for a few minutes to see if the waveform recovers, which is often the case, and then continue resection in another part of the tumor where there may be a good plane of cleavage. If the lesion is infiltrative or the MEP waveform does not improve or deteriorate, surgical judgment and experience becomes paramount in the decision on whether to curtail the operation or not.
Surgical Approach
Ependymomas and Astrocytomas (Figs. 25.3 and 25.4)
Intramedullary Dissection
At this point, it should be decided whether the approach will be midline between the posterior columns or directly on the lateral aspect of the spinal cord, such as the dorsal root entry zone. Most often, a standard midline dissection is preferred, but the strategy should be adapted to the tumor. Indeed, in gliomas, we almost always perform a midline approach with gentle separation of posterior columns in the posterior median sulcus,34,38,44 except on rare occasions when a lesion is located eccentrically and comes up to the pial surface of the cord, making entry much more direct.
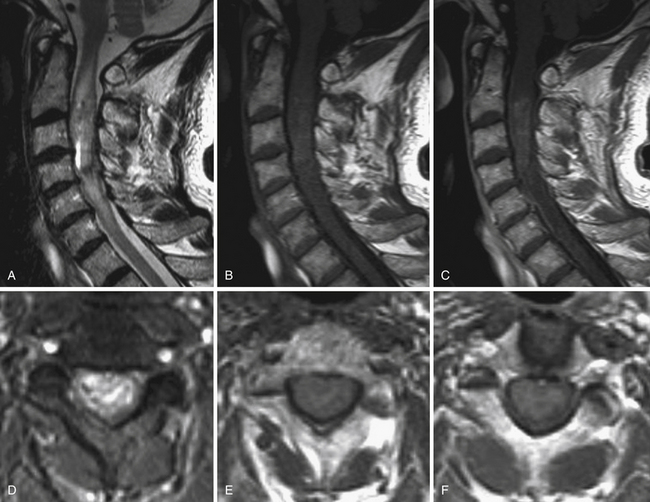
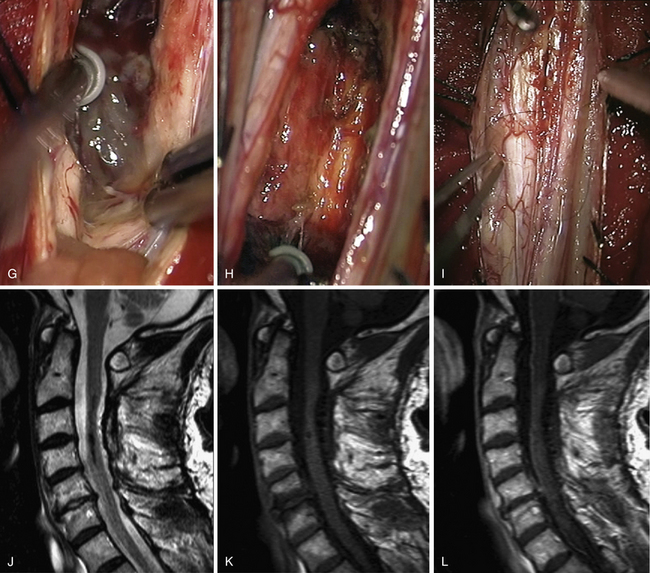
Identifying the midline can be challenging in some patients. It can be especially troublesome when working on astrocytomas that develop asymmetrically. If the problem cannot be solved, two pearls may be very helpful. First, the midline can be delineated after looking for it above or below the tumor where the spinal cord is normal. Second, the median sulcus can be recognized by the convergence of vessels toward the midline. Under high magnification with the microscope, the very tortuous posterior spinal vein running over the posterior median sulcus can be localized. Sometimes, vessels of varying size run longitudinally over the dorsal columns and have to be dissected and mobilized laterally to expose the sulcus. All of the narrowest arterial or venous structures in the sulcocommissural region should be dissected carefully and spared. Opening the midline implies careful retraction of the dorsal columns and progressive opening with microinstruments along the length of the solid portion of the tumor. These maneuvers open the spinal cord like the pages of a book. The process is pursued until the entire extent of the tumor is visualized as well as rostral and caudal cysts, if present.
Tumor Biopsy
As soon as the tumor is reached, a sufficient portion must be sampled with forceps and scissors, without applying cauterization. The pathologist immediately examines this biopsy. Then, we take some time before proceeding with surgery until we find out the results. Indeed, tumor removal should sometimes be discontinued early if there is an infiltrating or a malignant tumor, or a nontumoral process such as sarcoidosis.
Cleavage Plane Dissection
When the tumor has been debulked, it is essential to look for a tumor spinal cord cleavage plane. This plane exists in most ependymomas29,38,44,77,110 and in about 30% to 40% of astrocytomas.38,52,110,111 Finding the cleavage plane makes tumor removal more successful. The cleavage plane is observed as a color change between the tumor and the spinal cord. In our practice, the best dissection is preferably made with two microforceps. Very commonly, we dissect the spinal cord from the tumor, holding in the left hand an atraumatic sucker. We also regularly use cotton strips moistened with normothermic saline. If the tumor is encapsulated or not too friable, its margins can be grasped. Frequently, some parts of the tumor are more difficult to dissect than others. Instead of continuing, we recommend switching work areas. We recommend the same procedure when there is change or decrement in the SSEPs or MEPs waveforms. We will only come back to a tenacious area after further cytoreduction.
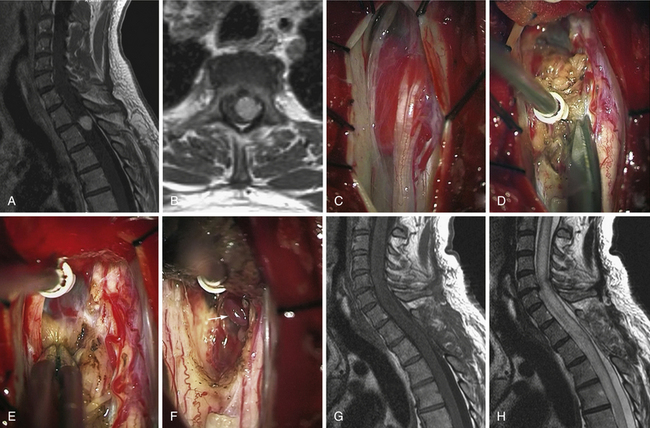
Hemangioblastomas (Fig. 25.5)
Approach
Hemangioblastomas are essentially located near the pial surface, lying over or inside the spinal cord. If the lesion is entirely inside the spinal cord, the lesion is accessed through the midline, as described previously. If the lesion is observed directly on the pia dorsally it is approached straight on.112 If present, we always take advantage of a huge and tense syrinx cyst associated with hemangioblastomas by gently puncturing the fluid with a 22-gauge needle; it collapses the spinal cord and gives ideal access to the solid tumor nodule through the transparent puncture site.113 In case of huge hemangioblastomas, the surgeon must be particularly cautious to avoid any cauterization of the draining vein at the beginning of the procedure. Interrupting venous outflow will induce hyperpressure inside the malformation and uncontrollable tumor bleeding. When the tumor is located ventrally, we use the same approach as described earlier for anterior/ventral meningiomas.
Resection
Biopsy or tumor debulking is to be avoided in hemangioblastoma to avoid massive bleeding, which could compromise the surgical procedure. Under microscopic magnification, tumor limits are defined. In fact, we look for a region on the surface with scarce vessels to be our starting dissection point. The cleavage plane remains distinct as long as bleeding does not interfere with the dissection. Slight tumor retraction obtained by gentle coagulation on its surface can be helpful for detaching the lesion from the cord. This maneuver is best completed with nonstick bipolar forceps or irrigating bipolars. After a step-by-step technique consisting of repeated cauterization and division of vascular connections, the lesion becomes devascularized prior to sacrifice of the draining vein. This vein should be interrupted only at the end of the procedure. We then finish the resection by carefully rolling out the tumor from its last spinal cord attachment. In this condition, we also advocate closing the arachnoid layer over the surgical field once the tumor is resected to prevent tethering the spinal cord against the dura.38
Cavernomas (Fig. 25.6)
Like hemangioblastomas, cavernomas are usually encountered close to the pial surface. If the lesion is located on the anterolateral aspect of the spinal cord, access becomes possible by dividing one or two dentate ligament attachments and gently rotating the spinal cord with 6-0 silk sutures applied to the ligaments. In this fashion, most anterolateral lesions are discovered through the transparent pia mater.38,114 We approach these lesions directly, through a limited opening of the pia, maintained if necessary with 8-0 silk sutures.
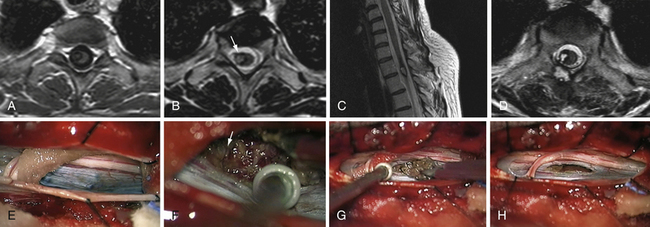
In hemangioblastomas and cavernomas, we do not close the spinal cord opening, but we do close the arachnoid layer.
Results
Functional Results
Prior to discharge, a basic functional evaluation is performed according to the four-tier system proposed by McCormick and colleagues29 to document and follow motor and sensory function over time. Immediately after surgery, it is not unusual for nearly all patients to worsen in comparison to their preoperative status. They should be aware that these deficits may be transitory and are not related to their long-term outcome. However, at 3 months, the neurological situation becomes more or less definitive.
 In preoperative grade I patients, 95% were unchanged and 5% worsened.
In preoperative grade I patients, 95% were unchanged and 5% worsened.
 In preoperative grade II patients, 7% improved, 86% were unchanged, and 7% worsened.
In preoperative grade II patients, 7% improved, 86% were unchanged, and 7% worsened.
 In preoperative grade III patients, 28% improved, 53% were unchanged, and 19% worsened.
In preoperative grade III patients, 28% improved, 53% were unchanged, and 19% worsened.
 In preoperative grade IV patients, none except one child improved. Most remained unchanged.
In preoperative grade IV patients, none except one child improved. Most remained unchanged.
Relevance of Radiotherapy
It is our opinion there is no indication for radiotherapy (RT) in benign SCIMTs even after incomplete removal, recurrence, or progression. Our personal opinion is based on a follow-up for over 5 years in 193 patients who had surgery either on a low-grade ependymoma (122 patients) or on a low-grade astrocytoma (71 patients), without adjunctive RT. We observed 5 recurrences (3 astrocytomas after respectively 5, 6, and 7 years and 2 ependymomas after 18 and 19 years) in those whose tumors were completely removed (110 ependymomas and 29 astrocytomas).
In fact, in our series, even low-grade astrocytomas that were partially removed have a very indolent evolution. Seventeen tumors remained stable in spite of partial removal; a few showed a slow MRI evolution without any clinical consequences. Furthermore, we have faced very difficult technical challenges when reoperating on several patients sent from other institutions who had previously received RT after biopsy or partial removal. All have worsened after our surgery in contrast to those who did not receive RT. When we are facing a recurrence or tumor progression in an astrocytoma, without any option for reoperation, we prefer to start with chemotherapy first (temozolamide). Unfortunately, in malignant gliomas the treatment is still only palliative such as with brain tumors. Contrary to low-grade spinal astrocytomas, in which we do not recommend postoperative adjunct RT, we recommend it, as do others,115 for malignant spinal gliomas. Regardless of which treatments are used—repeated surgery, radiation, or chemotherapy—the prognosis is poor and the disease is usually fatal in 9 months to 4 years.
Bruneau M., George B. Surgical technique for the resection of tumors in relation with the V1 and V2 segments of the vertebral artery. In: George B., Bruneau M., Spetzler R.F., editors. Pathology and Surgery Around the Vertebral Artery. Paris: Springer, 2010. Chap. 14
Fisher G., Brotchi J. Intramedullary Spinal Cord Tumors. New York: Thieme; 1996.
Klekamp J., Samii M. Extramedullary tumors. In: Klekamp J., Samii M., editors. Surgery of Spinal Tumors. Berlin: Springer; 2007:143-320.
Klekamp J., Samii M. Intramedullary tumors. In: Klekamp J., Samii M., editors. Surgery of Spinal Tumors. Berlin: Springer Verlag; 2007:120-131.
McCormick P.C., Torres R., Post K.D., et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72(4):523-532.
Please go to expertconsult.com to view a complete list of references.
1. Klekamp J., Samii M. Extramedullary tumors. In: Klekamp J., Samii M., editors. Surgery of Spinal Tumors. Berlin: Springer; 2007:143-320.
2. Brotchi J. Spinal intradural extramedullary tumors. In: Rengachary S.S., Ellenbogen R.G., editors. Principles of Neurosurgery. St Louis: Elsevier Mosby; 2005:681-688.
3. De Verdelhan O., Haegelen C., Carsin-Nicol B., et al. MR imaging features of spinal schwannomas and meningiomas. J Neuroradiol. 2005;32(1):42-49.
4. Viskochil D., Buchberg A.M., Xu G., et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62(1):187-192.
5. Wallace M.R., Marchuk D.A., Andersen L.B., et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181-186.
6. Ferner R.E. Neurofibromatosis 1 and neurofibromatosis 2: a twenty-first century perspective. Lancet Neurol. 2007;6(4):340-351.
7. Gezen F., Kahraman S., Canakci Z., et al. Review of 36 cases of spinal cord meningioma. Spine. 2000;25(6):727-731.
8. Schaller B. Spinal meningioma: relationship between histological subtypes and surgical outcome? J Neurooncol. 2005;75(2):157-161.
9. Levy W.J.Jr., Bay J., Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57(6):804-812.
10. Namer I.J., Pamir M.N., Benli K., et al. Spinal meningiomas. Neurochirurgia (Stuttgart). 1987;30(1):11-15.
11. Roux F.X., Nataf F., Pinaudeau M., et al. Intraspinal meningiomas: review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46(5):458-463. discussion 463-464
12. Parizel P.M., Baleriaux D., Rodesch G., et al. Gd-DTPA-enhanced MR imaging of spinal tumors. AJR Am J Roentgenol. 1989;152(5):1087-1096.
13. Balériaux D., Gultasli N. Intradural spinal tumors. In: Van Goethem J.W.M., van den Hauwe L., Parizel P.M., editors. Spinal Imaging. Berlin: Springer Verlag; 2007:417-460.
14. Bruneau M., George B. Surgical technique for the resection of tumors in relation with the V1 and V2 segments of the vertebral artery. In: George B., Bruneau M., Spetzler R.F., editors. Pathology and Surgery Around the Vertebral Artery. Paris: Springer, 2011. Chap. 14
15. Bruneau M., Cornelius J.F., George B. Anterolateral approach to the V2 segment of the vertebral artery. Neurosurgery. 2005;57(Suppl 4):262-267. discussion 262-267
16. Bruneau M., George B. The lateral approach to the V2 segment of the vertebral artery. In: George B., Bruneau M., Spetzler R.F., editors. Pathology and Surgery Around the Vertebral Artery. Paris: Springer, 2011. Chap. 13
17. Bruneau M., Cornelius J.F., Marneffe V., et al. Anatomical variations of the V2 segment of the vertebral artery. Neurosurgery. 2006;59(1 Suppl 1):ONS20-ONS24. discussion ONS20-24
18. Bruneau M., De Witte O., Regli L., George B. Anatomical variations. In: George B., Bruneau M., Spetzler R.F., editors. Pathology and Surgery Around the Vertebral Artery. Paris: Springer, 2011. Chap. 7
19. Bruneau M., Cornelius J.F., George B. Anterolateral approach to the V1 segment of the vertebral artery. Neurosurgery. 58(4 Suppl 2), 2006. ONS-215-219: discussion ONS-219
20. Bruneau M., George B. The lateral approach to the V1 segment of the vertebral artery. In: George B., Bruneau M., Spetzler R.F., editors. Pathology and Surgery Around the Vertebral Artery. Paris: Springer, 2011. Chap. 12
21. Payer M. The anterior approach to anterior cervical meningiomas: review illustrated by a case. Acta Neurochir (Wien). 2005;147(5):555-560. discussion 560
22. Solero C.L., Fornari M., Giombini S., et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25(2):153-160.
23. Kar M., Deo S.V., Shukla N.K., et al. Malignant peripheral nerve sheath tumors (MPNST)—clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol. 2006;4:55.
24. Casanova M., Ferrari A., Spreafico F., et al. Malignant peripheral nerve sheath tumors in children: a single-institution twenty-year experience. J Pediatr Hematol Oncol. 1999;21(6):509-513.
25. Elsberg C.A. Tumors of the Spinal Cord and the Symptoms of Irritation and Compression of the Spinal Cord and Nerve Roots: Pathology, Symptomatology, Diagnosis and Treatment. New York: Hoeber; 1925.
26. Cooper P.R. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25(6):855-859.
27. Epstein F., Epstein N. Surgical treatment of spinal cord astrocytomas of childhood. A series of 19 patients. J Neurosurg. 1982;57(5):685-689.
28. Guidetti B., Mercuri S., Vagnozzi R. Long-term results of the surgical treatment of 129 intramedullary spinal gliomas. J Neurosurg. 1981;54(3):323-330.
29. McCormick P.C., Torres R., Post K.D., et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72(4):523-532.
30. Malis L.I. Intramedullary spinal cord tumors. Clin Neurosurg. 1978;25:512-539.
31. Stein B.M. Surgery of intramedullary spinal cord tumors. Clin Neurosurg. 1979;26:529-542.
32. Stein B.M., McCormick P.C. Intramedullary neoplasms and vascular malformations. Clin Neurosurg. 1992;39:361-387.
33. Anson J.A., Spetzler R.F. Surgical resection of intramedullary spinal cord cavernous malformations. J Neurosurg. 1993;78(3):446-451.
34. Fisher G., Brotchi J. Intramedullary Spinal Cord Tumors. New York: Thieme; 1996.
35. Osborn A.G. Tumors, cysts and tumorlike lesions of the spine and spinal cord. In: Osborn A.G., editor. Diagnostic Neuroradiology. St. Louis: Mosby; 1994:876-918.
36. Sze G. Neoplastic disease of the spine and spinal cord. In: Atlas S.W., editor. Magnetic Resonance Imaging of the Brain and Spine. 2nd ed. Philadelphia: Lippincott-Raven; 1996:1339-1385.
37. Cooper P.R., Epstein F. Radical resection of intramedullary spinal cord tumors in adults. Recent experience in 29 patients. J Neurosurg. 1985;63(4):492-499.
38. Brotchi J. Intrinsic spinal cord tumor resection. Neurosurgery. 2002;50(5):1059-1063.
39. Guppy K.H., Akins P.T., Moes G.S., et al. Spinal cord oligodendroglioma with 1p and 19q deletions presenting with cerebral oligodendrogliomatosis. J Neurosurg Spine. 2009;10(6):557-563.
40. Ferrante L., Mastronardi L., Celli P., et al. Intramedullary spinal cord ependymomas—a study of 45 cases with long-term follow-up. Acta Neurochir (Wien). 1992;119(1-4):74-79.
41. Chang U.K., Choe W.J., Chung S.K., et al. Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol. 2002;57(2):133-139.
42. Raco A., Esposito V., Lenzi J., et al. Long-term follow-up of intramedullary spinal cord tumors: a series of 202 cases. Neurosurgery. 2005;56(5):972-981. discussion 972-981
43. Klekamp J., Samii M. Intramedullary tumors. In: Klekamp J., Samii M., editors. Surgery of Spinal Tumors. Berlin: Springer Verlag; 2007:120-131.
44. Brotchi J., Fischer G. Spinal cord ependymomas. Neurosurg Focus. 1998;4(5):e2.
45. Lonjon M., Goh K.Y., Epstein F.J. Intramedullary spinal cord ependymomas in children: treatment, results and follow-up. Pediatr Neurosurg. 1998;29(4):178-183.
46. Vijayakumar S., Estes M., Hardy R.W.Jr., et al. Ependymoma of the spinal cord and cauda equina: a review. Cleve Clin J Med. 1988;55(2):163-170.
47. Rawlings C.E.3rd, Giangaspero F., Burger P.C., et al. Ependymomas: a clinicopathologic study. Surg Neurol. 1988;29(4):271-281.
48. Shirato H., Kamada T., Hida K., et al. The role of radiotherapy in the management of spinal cord glioma. Int J Radiat Oncol Biol Phys. 1995;33(2):323-328.
49. Sgouros S., Malluci C.L., Jackowski A. Spinal ependymomas—the value of postoperative radiotherapy for residual disease control. Br J Neurosurg. 1996;10(6):559-566.
50. Constantini S., Miller D.C., Allen J.C., et al. Radical excision of intramedullary spinal cord tumors: surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg. 2000;93(suppl 2):183-193.
51. Epstein F.J., Farmer J.P. Pediatric spinal cord tumor surgery. Neurosurg Clin North Am. 1990;1(3):569-590.
52. Houten J.K., Cooper P.R. Spinal cord astrocytomas: presentation, management and outcome. J Neurooncol. 2000;47(3):219-224.
53. Innocenzi G., Raco A., Cantore G., et al. Intramedullary astrocytomas and ependymomas in the pediatric age group: a retrospective study. Childs Nerv Syst. 1996;12(12):776-780.
54. Kim M.S., Chung C.K., Choe G., et al. Intramedullary spinal cord astrocytoma in adults: postoperative outcome. J Neurooncol. 2001;52(1):85-94.
55. McCormick P.C., Stein B.M. Intramedullary tumors in adults. Neurosurg Clin North Am. 1990;1(3):609-630.
56. Jallo G.I., Danish S., Velasquez L., et al. Intramedullary low-grade astrocytomas: long-term outcome following radical surgery. J Neurooncol. 2001;53(1):61-66.
57. Jallo G.I., Freed D., Epstein F.J. Spinal cord gangliogliomas: a review of 56 patients. J Neurooncol. 2004;68(1):71-77.
58. Park C.K., Chung C.K., Choe G.Y., et al. Intramedullary spinal cord ganglioglioma: a report of five cases. Acta Neurochir (Wien). 2000;142(5):547-552.
59. Milano M.T., Johnson M.D., Sul J., et al. Primary spinal cord glioma: a surveillance, epidemiology, and end results database study. J Neurooncol. 2010;98(1):83-92.
60. Mandigo C.E., Ogden A.T., Angevine P.D., et al. Operative management of spinal hemangioblastoma. Neurosurgery. 2009;65(6):1166-1177.
61. Matsuyama Y., Sakai Y., Katayama Y., et al. Surgical results of intramedullary spinal cord tumor with spinal cord monitoring to guide extent of resection. J Neurosurg Spine. 2009;10(5):404-413.
62. Murota T., Symon L. Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery. 1989;25(5):699-707. discussion 708
63. Van Velthoven V., Reinacher P.C., Klisch J., et al. Treatment of intramedullary hemangioblastomas, with special attention to von Hippel-Lindau disease. Neurosurgery. 2003;53(6):1306-1313. discussion 1313-1314
64. Böhling T.P.K., Haltia M.J., Alitalo K., Neumann H.P.H. Von Hippel-Lindau disease and capillary haemangioblastoma. In: Kleihues P., Cavenee W.K., editors. Tumours of the Nervous System. Pathology and Genetics. Lyon: IARC Press; 2000:223-226.
65. Hurth M. [Intraspinal hemangioblastomas.]. Neurochirurgie. 1975;21(suppl 1):1-136.
66. Michels V.V. Investigative studies in von Hippel-Lindau disease. Neurofibromatosis. 1988;1(3):159-163.
67. Jallo G.I., Freed D., Zareck M., et al. Clinical presentation and optimal management for intramedullary cavernous malformations. Neurosurg Focus. 2006;21(1):e10.
68. Cantore G., Delfini R., Cervoni L., et al. Intramedullary cavernous angiomas of the spinal cord: report of six cases. Surg Neurol. 1995;43(5):448-451. discussion 451-452
69. Cristante L., Hermann H.D. Radical excision of intramedullary cavernous angiomas. Neurosurgery. 1998;43(3):424-430. discussion 430-431
70. Deutsch H., Jallo G.I., Faktorovich A., et al. Spinal intramedullary cavernoma: clinical presentation and surgical outcome. J Neurosurg. 2000;93(Suppl 1):65-70.
71. Ogilvy C.S., Louis D.N., Ojemann R.G. Intramedullary cavernous angiomas of the spinal cord: clinical presentation, pathological features, and surgical management. Neurosurgery. 1992;31(2):219-229. discussion 229-230
72. Zevgaridis D., Medele R.J., Hamburger C., et al. Cavernous haemangiomas of the spinal cord. A review of 117 cases. Acta Neurochir (Wien). 1999;141(3):237-245.
73. Cohen-Gadol A.A., Jacob J.T., Edwards D.A., et al. Coexistence of intracranial and spinal cavernous malformations: a study of prevalence and natural history. J Neurosurg. 2006;104(3):376-381.
74. Ojemann R.G., Crowell R.M., Ogilvy C.S. Management of cranial and spinal cavernous angiomas (honored guest lecture). Clin Neurosurg. 1993;40:98-123.
75. Sandalcioglu I.E., Wiedemayer H., Gasser T., et al. Intramedullary spinal cord cavernous malformations: clinical features and risk of hemorrhage. Neurosurg Rev. 2003;26(4):253-256.
76. Lee M., Rezai A.R., Freed D., et al. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38(1):32-37.
77. Schwartz T.H., McCormick P.C. Intramedullary ependymomas: clinical presentation, surgical treatment strategies and prognosis. J Neurooncol. 2000;47(3):211-218.
78. Parsa A.T., Fiore A.J., McCormick P.C., et al. Genetic basis of intramedullary spinal cord tumors and therapeutic implications. J Neurooncol. 2000;47(3):239-251.
79. Barkovich A.J., Edwards M., Cogen P.H. MR evaluation of spinal dermal sinus tracts in children. AJNR Am J Neuroradiol. 1991;12(1):123-129.
80. Gok A., Bayram M., Coskun Y., et al. Unusual malformations in occult spinal dysraphism. Turk J Pediatr. 1995;37(4):391-397.
81. Shen W.C., Chiou T.L., Lin T.Y. Dermal sinus with dermoid cyst in the upper cervical spine: case note. Neuroradiology. 2000;42(1):51-53.
82. Peter J.C., Sinclair-Smith C., de Villiers J.C. Midline dermal sinuses and cysts and their relationship to the central nervous system. Eur J Pediatr Surg. 1991;1(2):73-79.
83. Varron L., Broussolle C., Candessanche J.P., et al. Spinal cord sarcoidosis: report of seven cases. Eur J Neurol. 2009;16(3):289-296.
84. Lahanis S., Vlahos L., Gouliamos A., et al. Arteriovenous malformation of the spinal cord mimicking a tumour. Neuroradiology. 1993;35(8):598-599.
85. Sharma G.K., Kucia E.J., Spetzler R.F. Spontaneous intramedullary hemorrhage of spinal hemangioblastoma: case report. Neurosurgery. 2009;65(3):E627-E628. discussion E628
86. Rifkinson-Mann S., Wisoff J.H., Epstein F. The association of hydrocephalus with intramedullary spinal cord tumors: a series of 25 patients. Neurosurgery. 1990;27(5):749-754. discussion 754
87. Caviness J.A., Tucker M.H., Pia S.K., et al. Hydrocephalus as a possible early symptom in a child with a spinal cord tumor. Pediatr Neurol. 1998;18(2):169-171.
88. Galarza M., Peretta P., Gazzeri R., et al. Spinal cord gliomas and hydrocephalus: utility of neuroendoscopy. Minim Invasive Neurosurg. 2006;49(6):347-352.
89. Constantini S., Houten J., Miller D.C., et al. Intramedullary spinal cord tumors in children under the age of 3 years. J Neurosurg. 1996;85(6):1036-1043.
90. Fornari M., Pluchino F., Solero C.L., et al. Microsurgical treatment of intramedullary spinal cord tumours. Acta Neurochir Suppl (Wien). 1988;43:3-8.
91. Garces-Ambrossi G.L., McGirt M.J., Mehta V.A., et al. Factors associated with progression-free survival and long-term neurological outcome after resection of intramedullary spinal cord tumors: analysis of 101 consecutive cases. J Neurosurg Spine. 2009;11(5):591-599.
92. Fine M.J., Kricheff I.I., Freed D., et al. Spinal cord ependymomas: MR imaging features. Radiology. 1995;197(3):655-658.
93. Sun B., Wang C., Wang J., et al. MRI features of intramedullary spinal cord ependymomas. J Neuroimaging. 2003;13(4):346-351.
94. Miyazawa N., Hida K., Iwasaki Y., et al. MRI at 1.5 T of intramedullary ependymoma and classification of pattern of contrast enhancement. Neuroradiology. 2000;42(11):828-832.
95. Facon D., Ozanne A., Fillard P., et al. MR diffusion tensor imaging and fiber tracking in spinal cord compression. AJNR Am J Neuroradiol. 2005;26(6):1587-1594.
96. Vargas M.I., Delavelle J., Jlassi H., et al. Clinical applications of diffusion tensor tractography of the spinal cord. Neuroradiology. 2008;50(1):25-29.
97. Schwartz T.H., McCormick P. Non-neoplastic intramedullary pathology. Diagnostic dilemma: to Bx or not to Bx. J Neurooncol. 2000;47(3):283-292.
98. Andrews B.T., Kwei U., Greco C., et al. Infarct of the conus medullaris simulating a spinal cord tumor: case report. Surg Neurol. 1991;35(2):139-142.
99. Lee M., Epstein F.J., Rezai A.R., et al. Nonneoplastic intramedullary spinal cord lesions mimicking tumors. Neurosurgery. 1998;43(4):788-794. discussion 794-795
100. Levivier M., Brotchi J., Baleriaux D., et al. Sarcoidosis presenting as an isolated intramedullary tumor. Neurosurgery. 1991;29(2):271-276.
101. Rhoton E.L., Ballinger W.E.Jr., Quisling R., et al. Intramedullary spinal tuberculoma. Neurosurgery. 1988;22(4):733-736.
102. Voelker J.L., Muller J., Worth R.M. Intramedullary spinal Histoplasma granuloma. Case report. J Neurosurg. 1989;70(6):959-961.
103. Hitchon P.W., Haque A.U., Olson J.J., et al. Sarcoidosis presenting as an intramedullary spinal cord mass. Neurosurgery. 1984;15(1):86-90.
104. Levivier M., Baleriaux D., Matos C., et al. Sarcoid myelopathy. Neurology. 1991;41(9):1529-1530.
105. Sauter M.K., Panitch H.S., Kristt D.A. Myelopathic neurosarcoidosis: diagnostic value of enhanced MRI. Neurology. 1991;41(1):150-151.
106. Junger S.S., Stern B.J., Levine S.R., et al. Intramedullary spinal sarcoidosis: clinical and magnetic resonance imaging characteristics. Neurology. 1993;43(2):333-337.
107. Harrop J.S., Ganju A., Groff M., et al. Primary intramedullary tumors of the spinal cord. Spine. 2009;34(Suppl 22):S69-S77.
108. Kothbauer K.F. Intraoperative neurophysiologic monitoring for intramedullary spinal cord tumor surgery. Operative Techniques Neurosurg. 2003;6:2-8.
109. Morota N., Deletis V., Constantini S., et al. The role of motor evoked potentials during surgery for intramedullary spinal cord tumors. Neurosurgery. 1997;41(6):1327-1336.
110. Brotchi J., Bruneau M., Lefranc F., et al. Surgery of intraspinal cord tumors. Clin Neurosurg. 2006;53:209-216.
111. Brotchi J. Intramedullary spinal cord astrocytomas: diagnosis and treatment. Crit Rev Neurosurg. 1997;7:83-88.
112. Roonprapunt C., Silvera V.M., Setton A., et al. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery. 2001;49(2):321-327. discussion 327–328
113. Lefranc F., Brotchi J. Surgical strategy in spinal cord hemangioblastomas. Operative Techniques Neurosurg. 2003;6:24-31.
114. Hsu F.P.K., Clatterbuck R.E., Kim L.J., Spetzler R.F. Intramedullary spinal cord cavernous malformations. Operative Techniques Neurosurg. 2003;6:32-40.
115. Benes V.3rd, Barsa P., Benes V.Jr., et al. Prognostic factors in intramedullary astrocytomas: a literature review. Eur Spine J. 2009;18(10):1397-1422.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 25 Intradural Extramedullary and Intramedullary Spinal Cord Tumors
• Surgery is the initial and primary treatment of choice in most spinal tumors.
• Intradural extramedullary spinal tumors, mostly benign peripheral nerve sheath tumors and meningiomas, carry an excellent prognosis. Surgery allows most of the time complete resection and neurological improvement or recovery.
• The main objective should be preserving the patient’s functional condition and making an accurate diagnosis. Postoperative results depend on the preoperative neurological status, the tumor grade, tumor location, and the surgical team’s experience.
• Many intramedullary spinal tumors, such as ependymomas, astrocytomas, and some of nonglial origin, have a favorable prognosis because they may be completely resected based on the favorable surgical plane between the tumor and spinal cord.
Intradural Extramedullary Tumors
Incidence and Types
Spinal intradural extramedullary tumors (SIEMTs) are primarily peripheral nerve sheath tumors (PNSTs) and meningiomas in similar numbers.1 In the largest reported study on SIEMTs, nerve sheath tumors and meningiomas accounted for almost 70% of all lesions.1 Other SIEMTs and lesions include arachnoid cysts, hamartomas, and ependymomas, with an incidence between 5.5-7.5% each.1 The differential diagnosis for SIEMTs and lesions is broad and includes angioblastoma, cavernoma, chordomas, dermoid, epidermoid, exophytic astrocytoma, germinoma, hemangiopericytoma, lipoma, lymphoma, malignant teratoma, medulloblastoma, melanocytoma, melanoma, metastasis, myxoma, neuroblastoma, and sarcoma.1,2
In about 50% of cases, SIEMTs are located at the thoracic level.1 Lesions with an extradural extension are seen at the cervical level in more than half of patients.1 Benign nerve sheath tumors are the most common SIEMT and are associated with extradural extension with an approximate incidence of 77%.1 Meningiomas, hamartomas, and sarcomas are other SIEMTs with extradural extension.1
Peripheral Nerve Sheath Tumors
Most schwannomas are purely intradural but 15% have an extradural component, being either solely extradural or both intra- and extradural. These tumors originate from the dorsal sensory nerve root.3 Typically, these lesions are well encapsulated and rarely include a cystic portion. In contrast, neurofibromas are not well encapsulated and rarely occur alone. Multiple plexiform neurofibromas are usually seen with neurofibromatosis type 1 (NF1), a genetic disorder associated with mutations of chromosome 17.4–6 Diffuse plexiform neurofibromas are detected clinically and radiologically, respectively, in 26.3% and 50% of the patients with NF1.6 They appear in early childhood and have a variable growth pattern. Lesions may grow rapidly until adolescence and then become quiescent.6 The presence of a tumor is not an indication for surgery. Resection is indicated in NF1 patients if the tumor is growing or is causing progressive neurological deficits. In 2% to 5% of the cases, malignant changes may be observed with rapid expansion of the tumor mass.6 Surgery can be challenging in malignant tumors when the tumor is attached and infiltrates surrounding soft tissue. Neurofibromatosis type 2 (NF2) is another genetic condition associated with chromosome 22 anomalies.6 The clinical picture of NF2 may include SIEMTs such as schwannomas, neurofibromas, and meningiomas in 30% of patients.6 Pathologically, nerve sheath tumors are subdivided according to the WHO (World Health Organization) classification into schwannomas (WHO grade I), neurofibromas (WHO grade I), and malignant peripheral nerve sheath tumors (WHO grade III/IV).
Meningiomas
Spinal meningiomas commonly occur in middle-aged patients with a predilection for women.7 Most of the sporadic meningiomas are solely intradural. Of those remaining, one third are intra- and extradural and two thirds are primarily extradural.8 These lesions have a strong tendency to develop at the thoracic level with a frequency reported in the literature between 55% and 83%.7,9–11
Clinical Picture
Most patients present with a long history of progressive symptoms and neurological deterioration because SIEMTs are slow-growing tumors. It is uncommon for a patient to abruptly deteriorate due to spontaneous tumor hemorrhage with spinal compression. SIEMTs appear with signs and symptoms indicative of upper and lower motor neuron involvement, related to the severity of the nerve root insult and spinal cord compression. In half of the cases, the first symptom reported by patients with SIEMTs is pain.1 The tenderness and pain are characteristically increased at night or in the supine position and are relieved after returning to an upright position. Pain is often described along a radicular dermatome but can also reach varying distribution. It is usually described as a dull, aching sensation, and less frequently as burning. Radicular involvement results in lower motor neuron dysfunction, with weakness, sensory disturbances, hyporeflexia, atrophy, and fasciculation at the affected level. Myelopathic symptoms are correlated with the severity, level, and site of spinal cord compression. Nerve sheath tumors typically compress the lateral aspect of the spinal cord and are therefore more likely to be associated with the development of a Brown-Sequard syndrome. Upper motor neuron deficits manifest by gait ataxia and spasticity of the affected extremities. Hyperreflexia as well as other pyramidal signs such as the Hoffman and Babinski signs may be detected upon clinical examination. Patients present with pain, gait ataxia, motor weakness, and sensory deficits in 70% to 77% of the cases, and dysesthesias and sphincter disturbances are found in about 40%.1
Preoperative Imaging
Magnetic resonance imaging (MRI) is currently the gold standard method for imaging. MRI provides accurate diagnoses and delineates the soft tissue components.12,13 Gadolinium administration highlights enhancing portions of the tumor. MR myelography is a special study that is particularly useful for showing small schwannomas.13
On MRI, schwannomas and meningiomas often appear as isointense to the spinal cord on T1-weighted images (WI). On T2-WI, schwannomas are most often hyperintense, as opposed to meningiomas that appear isointense.13 After gadolinium administration both lesions enhance intensely and homogeneously.13 A cystic component and hemorrhagic foci may be noted in schwannomas or neurofibromas, respectively, in 40% and 10% of the cases.13 Less frequently than with cranial lesions, there is often a characteristic dural tail associated with spinal meningiomas.3
The computed tomography (CT) scan enables detection of calcifications as well as of spinal canal and neural foramen enlargement in cases of extradural extension.13 CT angiography (CTA) and MRI angiography (MRA) provide relevant information for cervical dumbbell-shaped neurofibromas that develop in close proximity to the vertebral artery (VA).14 Conventional angiography has a few indications for some of these tumors. It is helpful for providing relevant information about the anterior spinal artery at the cervical level14 and the Adamkiewicz artery at the thoracolumbar junction. CT myelography is invasive and thus not as useful as MRI. It is no longer recommended as the primary imaging modality.13
Surgery
The surgical approach should be adjusted according to the presumed diagnosis and tumor location. The surgical strategy of a PNST is different from that with a meningioma. First, PNSTs most often develop on the anterolateral aspect of the spinal cord, mainly at the cervical level, inducing a posteromedial displacement of the neuraxis. Second, few of them are solely intradural/extramedullary lesions; numerous are dumbbell-shaped lesions extending into the vertebral foramen. Third, at the cervical spine level, their vascular supply from the vertebral artery (VA) requires special considerations.14
Peripheral Nerve Sheath Tumors
Several surgical techniques have been described for PNST resection. These lesions are accessed using a posterolateral approach, as described for meningiomas. The posterolateral approach through a laminectomy is adequate for the resection of the intradural component. In the case of cervical PNST extending through the vertebral foramen and in close proximity to the VA, the posterior approach has several drawbacks.14 First, a significant portion of the facet must be resected to reach the foraminal component of the tumor. This may potentially cause postoperative instability requiring fusion.15 Second, the VA providing the tumor blood supply is reached at the end of the surgical procedure, precluding any devascularization of the tumor supply before the final surgical step.14 Moreover, without proximal control of the VA, the risk of VA damage is higher.14 Finally, it often results in leaving tumor remnant that remains vascularized, and thus has a potential to grow in the future. For some anterior lesions, the anterolateral approach may be useful in select patients. It allows for complete devascularization of the tumor at the beginning of the procedure, cutting down on blood loss. Then, tumor resection can most often be completed in a single surgical step.14 The intradural component of the lesion may be accessed through the enlarged vertebral foramen that may be drilled out slightly. This tailored opening rarely destabilizes the spine and subsequent fusion is not uniformly required. The primary reason for a fusion is a history of previous laminectomy, which completely removes the facets. The anterolateral approach requires VA control, which requires experience. However, this approach is safe and associated with low morbidity rate in experienced hands;15,16 the incidence of permanent deficits drops to less than 3%.15,16 The main risk is a postoperative Horner syndrome.
The Anterolateral Approach with Vertebral Artery Control for Cervical Dumbbell-Shaped Tumors (Fig. 25.1)
Preoperatively, the VA position has to be anticipated by an MRA or CTA: anatomical variations must be ruled out, as well as VA displacement induced by the tumor.17,18 The tumor often grows at the posterior VA aspect therefore pressing on the artery anteriorly or anteromedially. The patient is placed in the supine position, with head lying on a small cushion, slightly extended with a tape passing under the chin and turned to the contralateral side at 30 degrees.15,16 Fluoroscopy is used to check the correct level. Electrophysiological monitoring (consisting of motor and somatosensory evoked potentials [MEP, SSEP] and electromyography [EMG]) is maintained throughout the surgical procedure. The skin incision follows the medial border of the sternomastoid muscle. After division of the platysma, the dissection is pursued medially to the medial border of the sternomastoid muscle and laterally to the internal jugular vein. The carotid-jugular sheath is left undissected and is retracted medially. Dissection of the sympathetic chain is an important step in avoiding the main complication of this approach, which is Horner syndrome. The sympathetic chain must be differentiated from the longus colli muscle (LCM) and retracted laterally using stitches applied on the LCM aponeurosis that has been divided medially to the chain. At the C6-C7 level, the thoracic duct on the left side or the accessory thoracic duct on the right side must be preserved to avoid a leak.19,20 After its exposure, the LCM must be resected. In order to prevent VA injury, the LCM must be resected down to the transverse processes above and below the level of tumor. It is safe because, in the absence of anatomical variations, the VA is protected inside the bone of the transverse foramen.15–18 Afterward, the muscle is resected along the lateral border of the cervical vertebrae. From an anatomical point of view, it must be pointed out that, between C6 and the foramen magnum, a venous plexus enclosed inside a periosteal sheath surrounds the VA. The periosteal sheath is continuous with the periosteum of each transverse process. It means the periosteal sheath should be left intact inside the transverse foramen. It can be achieved only by performing a subperiosteal dissection of the anterior branch of the transverse process with a smooth spatula before resecting the bone with a Kerrison rongeur.15,16 In case of tearing of the periosteal sheath, troublesome venous bleeds may arise but can easily be controlled by direct bipolar cauterization of the sheath itself or packing with resorbable cellulose.15,16 At this point, the VA is controlled from above and below the tumor. The vertebral foramen, already enlarged by the tumor, may be made slightly larger by drilling. The tumor feeders will be cauterized during the separation between the anterior aspect of the nerve root and the VA posterior border, facilitating further tumor resection.15,16 In the case of a schwannoma, the tumoral nerve root fascicle may be separated from the normal nerve roots. The nerve root may be sacrificed if there is already a neurological deficit by nerve root involvement.14 Intraoperative stimulation may provide additional relevant information for this decision.14 Pursuing a lateromedial tumor resection will allow entrance into the intradural compartment. The dura mater opening through which the tumor has passed may be enlarged as much as necessary to provide the appropriate intradural exposure. The use of an ultrasonic aspirator (the Cavitron ultrasonic aspirator, CUSA) is helpful for debulking the lesion.14 The tumor should then be cautiously separated from the spinal cord using a microsurgical technique.
Meningiomas (Fig. 25.2)
In the case of meningiomas, the tumor origin can be found anywhere along the surface of the dura. A general rule is that the neuraxis is displaced to the side opposite to the tumor origin. Therefore, posterior meningiomas put pressure on the spinal cord anteriorly and for this reason are better approached directly with a laminectomy. Posterolateral and lateral meningiomas displace the neuraxis anteromedially and medially, respectively. A posterolateral approach is advocated, by laminectomy possibly extended on the tumor side to the medial aspect of the facets. With anterolateral meningiomas, the posterior displacement of the neuraxis requires a more location-specific approach because surgical access is limited. In this situation, we still advocate a posterior approach, extended laterally on the side of the tumor, but the lateral surgical corridor must be enlarged by dividing dentate ligaments. With that maneuver, most lesions can be adequately resected. Rare reports have advocated the use of an anterior approach for anterior meningiomas.21 This anterior approach is considered safe and has several theoretical advantages: a large bony window of access, extradural cauterization of the anterior blood supply, direct visualization of the entire tumor ventralto the spinal cord, and tumor resection without spinal cord manipulation; but it is nevertheless associated with drawbacks, such as the need for fusion and a more challenging access route.21 It is nevertheless associated with drawbacks such as the need for fusion and a more challenging access route. The depth and narrowness of the working channel present a significant technical challenge. In addition, dural closure is also very difficult, resulting in a higher probability of CSF leakage.21
The Posterior Approach
The patient is placed in the prone position under continuous electrophysiological monitoring, especially for cervical lesions. At this level, we use a Mayfield head holder and are careful to position the head in a neutral position to avoid tumor-induced spinal cord compression. Fluoroscopy is used to accurately target the pathological levels. Usually, only two vertebrae must be exposed. The laminectomy may be extended laterally to facets on the side of the tumor. The dura mater is usually incised longitudinally over the tumor. The dural edges are retracted laterally with several 4-0 silk sutures. Lateral lesions may require section of the dentate ligaments to provide exposure. Sutures from dentate ligament to opposite dura keeps the surgical space open and offers good control of anterior dura as a result of gentle rotation of the spinal cord. Great care must be taken to avoid any traction or compression on the spinal cord itself. Cottonoids are placed at each end of the tumor to avoid intradural soiling. Most of the time, the arachnoid can be separated and the resection completed in the extra-arachnoidal plane. The tumor surface is cauterized and incised with a knife or microscissors. Samples are sent to the pathologist for immediate analysis. In lateral meningiomas, the tumor debulking is performed with either microinstruments or an ultrasonic aspirator, starting along the dural base if possible. Bleeding of feeding vessels is gently controlled with bipolar cauterization. In this way, the tumor is progressively devascularized and becomes mobile. The pressure at the tumor–spinal cord interface and at the dural insertion base is then released. Shrinkage of the tumor by electrocautery may also help to keep the plane open. Only vessels supplying the tumor should be sacrificed. Progressively, the dural base may be divided, resulting in tumor devascularization. Nevertheless, we advise keeping a small dural attachment until the tumor is completely released from the spinal cord. Otherwise, the lesion may become too mobile, thereby increasing difficulties and risks in completing the dissection of the spinal cord–tumor interface. At the completion of surgery, the dural base should be either cauterized or resected. The choice is guided by the location of the pathological dura and the possibilities of performing a watertight reconstruction. Resection and reconstruction with a patch graft is advised for posterior and posterolateral lesions. Contrarily, we advise only coagulating anterior or anterolateral dura mater portions. Cauterization instead of resection of the dura mater is not associated with a higher risk of recurrence.9,11,22 Finally, the intradural compartment is copiously washed with a normothermic saline solution. The dura mater is closed with a 5-0 nonabsorbable running watertight suture to prevent CSF leakage. The aponeurosis is also closed with a watertight running suture to avoid a CSF leak. Drains may be inserted at the level of the muscular layer.
Postoperative Outcome
Short-term outcome after the resection of SIEMTs is usually excellent, with the neurological condition generally improving significantly.2 Resolution of preoperative deficits has been noted in about 80% of the cases.7,8 The patient’s neurological condition remains stable in 14% of patients and few deteriorate.7 Several factors influence the outcome such as the preoperative neurological status, the duration of preoperative symptoms, and the type of surgical resection.7 The histological characteristics of the tumor may influence the long-term outcome: the psammomatous type is associated with a less favorable neurological outcome than the nonpsammomatous type. Psammomatous tumors consist essentially of acellular concretions, rendering blood vessels and spinal roots extremely adherent.8 Location of the tumor below C4 and posteriorly or laterally in the intradural compartment is, however, a favorable factor.8 Complete tumor resection is possible in 96% to 97% of the cases7,22 and is associated with a recurrence rate of 6% after 15 years of follow-up. Subtotal resection of a tumor resulted in recurrence in 17% of the cases.22 Serious complications may develop in case of perioperative vascular or medullar injuries but are unusual. Physical therapy hastens neurological recovery. Most patients can resume a normal or nearly normal neurological condition. A gratifying quick recovery may be observed within the first 6 weeks after surgery, but progressive recovery may be expected by 1 year after surgery. We see patients at 6 months, 1 year, and 2 years with performance of an MRI and then every 5 years in the absence of recurrence.
Radiation Therapy
When dealing with a malignant PNST, current consensus advocates postoperative radiation therapy (RT), started as soon as possible after surgery even if resection margins are tumor free.23,24 The role of radiotherapy in the management of spinal meningiomas is controversial. The slow indolent growth of these lesions and the risk associated with irradiating the vicinity of the spinal cord are confounding issues.7 Several indications for adjuvant radiation therapy have been proposed: early recurrence after total or subtotal resection, partial resection, and high-risk surgical treatment.7 If possible, we prefer repeat surgery over radiation for recurrence.
Intramedullary Spinal Cord Tumors
Surgery on spinal cord intramedullary tumors (SCIMTs) has been refined over the past few decades. The pioneering work of Elsberg25 and subsequently of many others such as Cooper,26 Epstein and Epstein,27 Guidetti and associates,28 McCormick and colleagues,29 Malis,30 Stein and McCormick,31,32 and many others33,34 have greatly contributed to advancing the microsurgical technique and success in removing spinal cord lesions. Several tools have added safety features to the microsurgical resection of these lesions. The advent of MRI has definitively improved diagnosis accuracy. The introduction of ultrasonic aspiration (CUSA) and intraoperative electrophysiological monitoring has modified the surgical approach and improved safety and outcome significantly.
Incidence and Types
With an incidence in adulthood of 2% to 4% of all central nervous system tumors, SCIMTs are considered rare.30,32,35–37 Three main groups of SCIMTs can be distinguished: tumors of glial origin, tumors of nonglial origin, and other mass lesions.
Tumors of Glial Origin
These tumors primarily consist of ependymomas and astrocytomas.13,38 Other glial tumors such as oligodendriogliomas are rare.39
Ependymomas
Spinal cord ependymomas are the most frequent SCIMTs in adult patients. They may reach considerable size prior to diagnosis. The majority of spinal cord ependymomas in adults have a good prognosis because usually they can be completely removed and are primarily grade II by the WHO classification. In the literature, the rate of complete removal varies from 69% to 97%.40–42 The vast majority of patients, 48% to 75%, are stabilized after the radical surgical resection, thereby highlighting the need for timely surgery in case of neurological deterioration.41–45 The overall rate of neurological improvement for this patient population varies between 10% and 40%.41–45 A rate of postoperative deterioration has been reported in the literature between 9% and 15%.41–45 Subtotal resection has been reportedly associated with a lack of clinical improvement,43 and incomplete resection is the most important factor correlating with tumor recurrence.41,46,47 The 5- and 10-year survival rates are respectively quoted between 83% to 96% and 80% to 91%.40,42,43,45,48,49
Astrocytomas
Spinal cord astrocytomas represent more than 80% of the SCIMTs in children,43 and in adults astrocytomas are the second most common type of tumor.43 Only 10% to 17% of astrocytomas in childhood are anaplastic lesions or glioblastomas, whereas in adults, the percentage is higher, 25% to 30%.28,34,43,50–53 In large studies, the proportion of pilocytic astrocytomas varies greatly from more than 30% to 60%.42,43 By their nature, grade II to IV tumors are infiltrating. Therefore, in contrast to ependymomas, radical resection of astrocytomas is not always possible. Surgery consists of removing as much tumor as possible. In some instances, one can only perform biopsy or decompression with duraplasty. In the literature, the rate of complete and subtotal removal varies between 11% to 31% and 21% to 62%, respectively.42,43,54 The pathological diagnosis of pilocytic astrocytoma and surgical experience are factors associated with higher rates of complete resection.42,43 Low spinal level (conus), malignancy, and adult onset are considered important factors associated with a higher tumor recurrence rate.43 The degree of resection, and spinal canal decompression via tumor debulking, have not been found to be directly related to progression-free survival or overall survival in many studies.26,32,43,50,52,55,56 In low-grade astrocytomas, the 5- and 10-year survival rates range from about 77% to 82%.43,56 These rates drop, respectively, to 27% and 14% for malignant astrocytomas.43,56
Gangliogliomas
Gangliogliomas are very uncommon lesions mainly reported as case reports and a few studies.57,58 These lesions are mainly encountered in children and young adults.57 In a large series of 56 patients, complete or subtotal resection has been achieved in 82%, and 18% of the patients had a 5-year survival rate of 88% and a progression-free survival rate of 67%.57
Factors Determining Outcome
The major determinant of long-term patient survival is based on the histological characteristics of the tumor. The 5-year progression-free survival (PFS) rate drops from 78% for low-grade gliomas to 30% for high-grade gliomas.50 A longitudinal database (Surveillance, Epidemiology, and End Results [SEER] database) encompassing 26% of the U.S. population was published in 2010 and included 1814 patients with spinal cord gliomas.59 In this study, age, histology, and grade were defined as significant predictors of outcome.59
Tumors of Nonglial Origin
Other lesions such as metastases, epidermoids, dermoid cysts, lipomas, intramedullary schwannomas, PNETs, and teratomas may also be encountered.13,43,55 In addition to these rare lesions, we have also encountered lymphomas and neuroglial cysts.
Hemangioblastomas
Hemangioblastomas are highly vascular lesions that represent almost 2% to 15% of SCIMTs in some studies.42,43,60–63 These lesions consist of two main components: (1) large vacuolated stromal cells that have been identified as the neoplastic cell of origin and (2) a rich capillary network.64 Hemangioblastomas are often observed as encapsulated lesions abutting the pia, especially at the posterior or posterolateral aspect of the spinal cord close to the dorsal root entry zone. They may also be encountered anteriorly or in intramedullary sites.43,60,
[/not-level-membership-for-neurosurgery-category]
