[level-membership-for-neurosurgery-category]
Chapter 19 Intracranial Hypertension
• States of impaired consciousness are expressed as numerical scores on the Glasgow Coma Scale (GCS), which is used worldwide and has prognostic and management implications.
• The key clinical signs that help in clinical assessment and determination of prognosis are the presence or absence of certain brainstem reflexes such as pupillary response, corneal reflex, oculocephalic and oculovestibular reflexes, and gag reflex.
• Computed tomography (CT) scan of the head remains a key imaging modality in the rapid assessment of a patient with impaired consciousness. Mass lesions that are directly responsible for intracranial hypertension should be surgically removed.
• Intracranial pressure (ICP) is generally monitored when the GCS score is less than 8 with an abnormal CT scan, although there is no class I evidence that mandates use of ICP monitors worldwide. A fiberoptic monitor is a good tool for measuring and managing increased ICP in an unconscious patient. Fiberoptic ICP monitors have a low incidence of complication and infection in experienced hands. However, they do not give a therapeutic option for lowering ICP like a ventriculostomy catheter with external ventricular drainage does. Head of bed elevation, maintaining euthermia, sedation, analgesia, and mild hyperventilation can also have temporary therapeutic effects on elevated ICP.
• After conservative therapies have been exhausted, hypertonic saline may be as efficacious as mannitol and is associated with a lower overall incidence of comorbidity.
• Decompressive hemicraniectomy is a reasonable option in uncontrolled intracranial hypertension, especially if there is a mass lesion.
• Future studies are being undertaken to further elucidate the role of microdialysis and brain tissue oxygenation monitoring in the setting of intracranial hypertension.
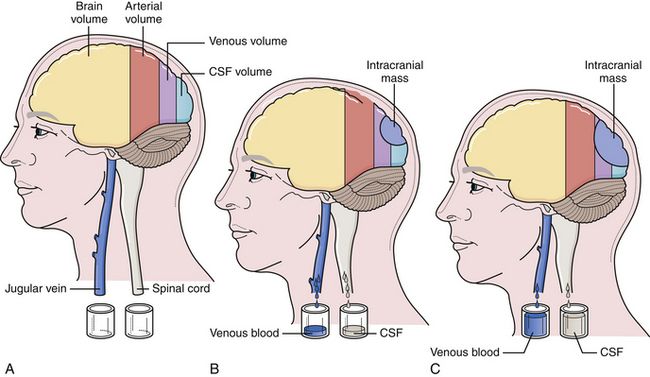
The cranium is a rigid structure. The major intracranial contents are the brain (to include the neuroglial elements and interstitial fluid), blood (arterial and venous), and cerebrospinal fluid (Table 19.1). When a new intracranial mass is introduced, a compensatory change in volume must occur through a reciprocal decrease in venous blood or cerebrospinal fluid (CSF) to keep the total intracranial volume constant. This is the Monro-Kellie doctrine (Fig. 19.1), which has been confirmed by many experimental and clinical observations. Only in children, with open fontanelles and whose sutures have not yet fused, can the cranium itself expand to physically accommodate extra volume.
TABLE 19.1 Intracranial Contents and Their Respective Volumes
| Component | Volume | Percentage of Total Volume |
|---|---|---|
| Brain (70%) and interstitial fluid (10%) | 1400 mL | 80% |
| Blood | 150 mL | 10% |
| Cerebrospinal fluid | 150 mL | 10% |
| TOTAL | 1700 mL | 100% |
Compliance (dV/dP) is the change in volume observed for a given change in pressure. This represents the accommodative potential of the intracranial space. In clinical practice, however, what is actually measured is elastance (dP/dV). Elastance is the inverse of compliance and is the change in pressure observed for a given change in volume. It represents the resistance to outward expansion of an intracranial mass. The elastance curve (not compliance) is what is plotted in Figure 19.2. Although technically a misnomer, the term compliance is actually entrenched in the literature to describe the aforementioned phenomenon instead of using the proper term elastance. Because of this we, too, will conform to the traditional nomenclature for the rest of this chapter.
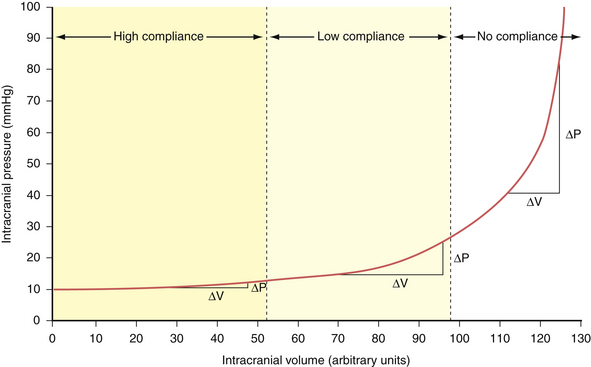
In accordance with the Monro-Kellie doctrine, several innate homeostatic mechanisms exist in an attempt to maintain intracranial pressure within the physiological range—generally considered less than 20 mm Hg in the acute inpatient setting. First, the venous system collapses easily and squeezes venous blood out through the jugular veins or through the emissary and scalp veins. Likewise, CSF can be displaced from the ventricular system through the foramina of Luschka and Magendie into the spinal subarachnoid space.1 When these compensatory mechanisms have been exhausted, small changes in volume produce precipitous increases in pressure. This effect can be demonstrated experimentally by inserting a Foley catheter into the epidural space of a rat and gradually inflating a balloon with increasing volumes. The curve produced by plotting intracranial pressure against volume is the so-called compliance curve in Figure 19.2. The innate homeostatic pressure-buffering mechanism offered by displacement of CSF and venous blood keeps this curve flat until a “critical volume” is reached. After this critical volume, small volumetric changes result in precipitous increases in pressure, and intracranial hypertension naturally ensues. Brain parenchyma and arterial blood do not participate, to any significant extent, in the innate intracranial pressure-buffering mechanisms.
Acute Intracranial Hypertention
Cerebral Blood Flow
Normal cerebral blood flow averages 55 to 60 mL/100 g brain tissue per minute. In the gray matter the blood flow is 75 mL/100 g brain tissue per minute, whereas in the white matter it is around 45 mL/100 g brain tissue per minute. This flow is sufficient to meet the metabolic needs of the brain. The brain usually begins to show signs of ischemia at 20 mL/100 g/minute and permanent damage usually results when the blood flow drops below 10 mL/100 g/minute in most models. The most significant factor that determines cerebral blood flow at any given time is the cerebral perfusion pressure (CPP). The CPP is the effective blood pressure gradient across the brain. CPP is the difference between the incoming mean arterial pressure (MAP) and the opposing intracranial pressure (ICP): CPP = MAP − ICP. The mean arterial pressure can be calculated in two ways: (1) the diastolic pressure plus one third of the pulse pressure (DP + 1⁄3PP) or (2) two thirds of the diastolic pressure plus one third of the systolic pressure (2⁄3DP + 1⁄3SP). The ICP has to be measured directly, which can be done with various devices and will be discussed later in this chapter. With increased ICP there is an obvious tendency for the cerebral perfusion pressure to decrease.
Three major factors regulate cerebral blood flow under physiological conditions: CPP, concentration of arterial CO2, and arterial PO2. The ability to maintain constant blood flow to the brain over a wide range of CPPs (50-150 mm Hg) is called cerebral autoregulation. When the CPP is low, the cerebral arterioles dilate to allow adequate flow at the decreased pressure. Conversely, an increase in CPP causes the arterioles to constrict and maintain the flow within physiological range (Fig. 19.3). Decreases in CO2 tension in the blood (i.e., hyperventilation) causes diffuse vasoconstriction.2 Vasoconstriction decreases both cerebral blood flow and cerebral blood volume in the brain. The risks and benefits of inducing hyperventilation are discussed under the treatment section at the end of this chapter. Lastly, severe hypoxia causes cerebrovascular dilatation. This effect only becomes apparent when the oxygen tension in the blood drops to 50 mm Hg and becomes maximal around 20 mm Hg.
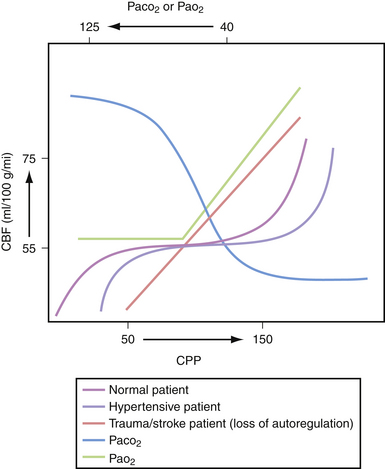
Under certain pathological conditions cerebral blood flow cannot always be autoregulated.3 When the CPP exceeds 150 mm Hg, such as in hypertensive crisis, the autoregulatory system fails. In this case there would be a passive increase in blood flow proportionate to the increase in systemic pressure, causing an exudation of fluid from the vascular system with resultant vasogenic edema.4 Additionally, certain toxins such as carbon dioxide can cause diffuse cerebrovascular dilatation and inhibit proper autoregulation.5 Lastly, during the first 4 to 5 days of head trauma, many patients can experience a disruption in cerebral autoregulation; this is often observed in the pediatric population and may result in hyperemia.6
Disruption of cerebral blood flow autoregulation in the setting of trauma could potentially lead to severe alterations in CPP when a patient’s brain is acutely injured.7 When disrupted, cerebral blood flow would be directly proportional to CPP under a much larger range than normal. This is likely one of the contributing reasons why even one episode of hypotension in the patient with acute head injury may lead to significantly worse outcomes.8 Thus, many institutions have adopted protocols to maintain a minimal CPP in the acute setting—usually around 50 to 60 mm Hg—and perhaps a maximum threshold of CPP if the intracranial hypertension can be linked directly to hyperemia.
Monitoring Intracranial Pressure
The most significant factor determining morbidity and mortality risk in patients with acute cranial neurosurgical disorders continues to be increased ICP. Continuous ICP monitoring is very useful for assessing intracranial dynamics in patients with suspected intracranial hypertension. There are no clinical indicators that can be used in the early stage of rising ICP to forestall a further rise. Classical clinical indicators described in the literature (cranial nerve abnormalities, posturing, etc.) occur in the end stage and they are not sensitive enough to show subtle changes in pressure. The two most common indications for ICP monitoring are closed head injury and spontaneous subarachnoid hemorrhage.
Indications for Monitoring Intracranial Pressure
The advent of continuous ICP monitoring has improved outcomes in traumatic brain injury9,10 by reducing the effects of secondary injury occurring after the initial brain insult. In the modern intensive care unit ICP monitoring in the adult patient with severe head injury is a recommended practice guideline based on available clinical evidence. Indications of ICP monitoring following head injury include a GCS score between 3 and 8 and an abnormal CT scan. Unfortunately, no class I evidence is available regarding the use of ICP monitors in pediatric head injury patients. Nevertheless, strong evidence supports the association between elevated ICP and poor outcomes,11 and aggressive treatment of elevated ICP is associated with the best clinical outcomes. ICP monitoring provides valuable data that can inform medical management.
Techniques of Monitoring Intracranial Pressure
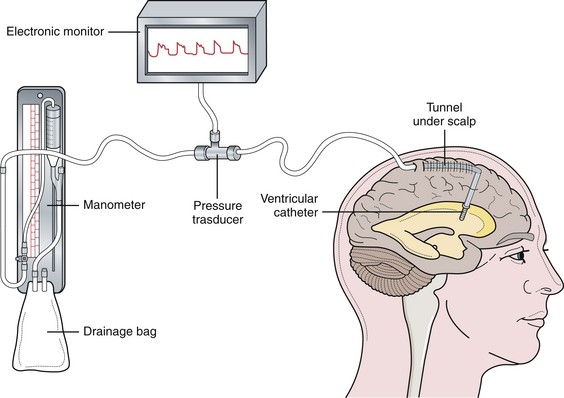
There are two commonly used pressure-monitoring systems in contemporary neurosurgical practice. An intraventricular catheter connected to a manometer and a drainage system is the standard against which all other systems are compared.12–14 The ventricular catheter ideally should be tunneled under the skin and brought out through a separate stab wound, well away from the ventricular entry site, to minimize the risk of infection. Infection is the most significant complication of intraventricular pressure monitoring. With the use of an electronic transducer, the waveform can also be monitored (Fig. 19.4). The major advantage of the method is that the ventricular catheter is used not only to measure the pressure, but also as a therapeutic modality allowing intermittent or continuous drainage of CSF when the pressure exceeds physiological limits.
A second method is the use of the fiberoptic transducer-tipped catheter system. The transducer-tipped catheter can be placed within the brain parenchyma or in the subdural space, depending on the surgeon’s choice and the clinical situation.15–17 The pressure monitor gives both digital readout and a waveform. The advantages of this system are that the zero point does not have to be reset with changes in head position because the pressure-sensing transducer is within the cranial cavity, and it is not susceptible to blockage by debris because it is not a fluid-coupled system. Also, insertion of a fiberoptic cable into the brain parenchyma is not affected by ventricular size or shift, which can make insertion of a ventricular catheter in the setting of intracranial hypertension quite trying. Lastly, the infection rates with fiberoptic probes are significantly lower than with ventriculostomy catheters, reported as less than 1% in one of the the largest reported series.18 The disadvantages of this system, however, are (1) higher cost and (2) baseline drift, which means there is evidence that with prolonged fiberoptic monitoring the indicated pressures may become less accurate in an unpredictable fashion.19,20
Intracranial Pressure Waveforms
The waveform of normal ICP typically shows three components: the percussion wave (P1), the tidal wave (P2), and finally the dicrotic notch (P3) (Fig. 19.5A). Under physiological conditions P1 is the tallest and is thought to represent the pressure at peak cardiac systole transmitted throughout the choroid plexus. P2 follows P1 and its resultant smaller peak is thought to represent the filling of the intracranial arteries with systolic blood and results in a rebound increase in ICP from this increase in intracranial volume. Last is the dicrotic wave.
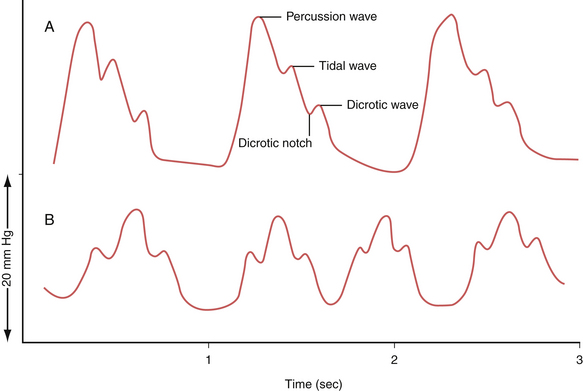
When the ICP rises and compliance decreases, one will observe P2 becoming taller than the P1. This is because a similar volume in intracranial arterial blood is now resulting in a higher change in ICP (Fig. 19.5B). Such alterations in the morphology can give the treating physician further clues regarding not only the compliance but also the autoregulatory capacity of the brain.
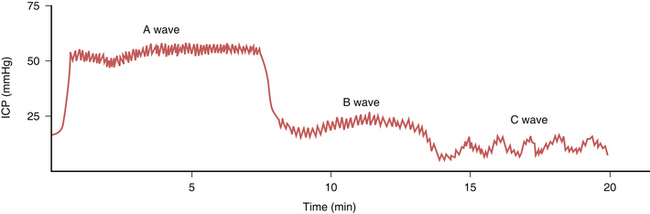
When the ICP waveforms are registered over a period of time certain trends may become apparent14 (Fig. 19.6). Plateau waves, or type A waves, are characterized by an abrupt elevation in ICP for 5 to 20 minutes followed by a rapid fall in the pressure to resting levels. The amplitude may reach as high as 50 to 100 mm Hg. Plateau waves may be clinically marked by a decreasing level of consciousness, restlessness, increased tone in the extremities, and tonic-clonic movements. Plateau waves may represent transient surges in ICP secondary to increased cerebral blood volume likely related to CO2 retention.
Types of Brain Herniation
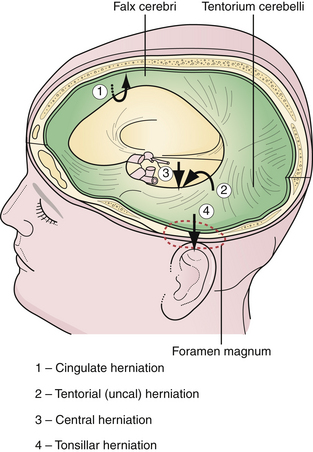
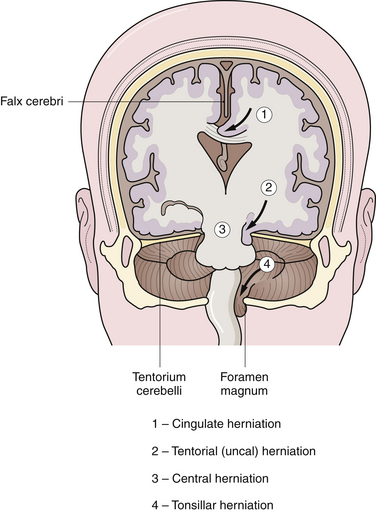
The brain is supported by dural folds that prevent undue movements of the brain within the cranial cavity (Fig. 19.7). There are two major dural folds—the falx cerebri and the tentorium cerebelli. The falx cerebri is a sickle-shaped dural fold in the midline that separates the two cerebral hemispheres. The tentorium cerebelli is a tent-shaped dural fold that separates the occipital lobes from the posterior fossa structures. The tentorial incisura, or hiatus, is the dural opening that surrounds the rostral brainstem. Care must be taken during ICP management so as to not to exacerbate the pressure differences between compartments and to inadvertently encourage herniation.
Cingulate Herniation
A focal mass lesion in the supratentorial compartment exerts progressive pressure locally on the ipsilateral hemisphere. The increase in ICP may not be uniform; it is greatest close to the mass, thus creating a pressure gradient. A supratentorial mass lesion may displace the cingulate gyrus, which is next to the free edge of the falx cerebri, and cause it to herniate under the falx to the opposite side. There is usually displacement of the ventricular system as well (Fig. 19.8). The anterior cerebral artery may be compromised by the tight, sharp edge of the falx cerebri. There are no clinical signs and symptoms specific to a cingulate herniation; however, an anterior cerebral artery stroke may result in a focal contralateral lower extremity plegia.
Uncal Herniation
Uncal herniation is the most dramatic and most common herniation syndrome observed clinically. Uncal herniation is often seen with lesions of the middle cranial fossa such as acute epidural hematoma, subdural hematoma, temporal lobe contusions, or temporal lobe neoplasms. An expansile mass of the middle fossa causes the uncus, the most inferomedial structure of the temporal lobe, to herniate between the rostral brainstem and the tentorial edge into the posterior fossa (see Fig. 19.8). If the vector of pressure is more posterior, then the parahippocampal gyrus can herniate as well. In postmortem studies, a deep groove may be noticed at the lateral margin of the uncus as a manifestation of the herniation. In some instances, the displacement of the brainstem by the uncus may cause compression of the contralateral cerebral peduncle against the opposite tentorial edge, producing a paradoxical indentation called Kernohan’s notch.
Central Transtentorial Herniation
In contrast to uncal herniation, which usually occurs from a mass lesion located near the tentorial hiatus, central transtentorial herniation occurs with mass lesions far removed from the tentorial hiatus, such as in frontal, parietal, or occipital areas. Bilateral mass lesions, such as subdural hematomas, can also cause central herniation. During central herniation, there is a downward displacement of the diencephalon and midbrain centrally through the tentorial incisura (see Fig. 19.8). The clinical syndrome in central herniation is not as easily recognizable as that of uncal herniation. The patients with central transtentorial herniation are obtunded, tend to have bilaterally small, reactive pupils, exhibit Cheyne-Stokes respiration, and may show loss of vertical gaze manifested as tonic downward gaze.
Tonsillar Herniation
Acute tonsillar herniation generally results from acute expansion of posterior fossa lesions. It may result from an ill-advised lumbar puncture in a patient with a mass lesion within the cranial cavity. The tonsils of the cerebellum herniate through the foramen magnum into the upper spinal canal, compressing the medulla (see Fig. 19.8). The manifestations of acute medullary compression are cardiorespiratory impairment, hypertension, high pulse pressure, Cheyne-Stokes respiration, neurogenic hyperventilation, and impaired consciousness. The patient may have a stiff neck or be in an opisthotonic position. Decorticate or decerebrate posturing may be present as well.
Treatment
With almost no exception, all patients with acute elevated ICP can be managed initially by the same conservative steps. First, the importance of elevating the head of the bed to 30 to 45 degrees cannot be overemphasized. This posture promotes venous drainage to enhance the innate compensatory mechanisms of the CNS. The neck should be kept straight and not allowed to kink over to any side. Second, normothermia should be achieved because an elevated brain temperature can increase metabolism, which can not only elevate ICPs but also promote ischemia. Lastly, sedation and analgesia are invaluable tools in the management of acute ICP. Propofol is the most commonly used sedating agent not only because of its “quick on-quick off” effect but also because of its ability to lower cerebral metabolism. After these initial steps have been taken, further ICP therapy requires enhanced knowledge of physiology as described in the following section.
The Blood-Brain Barrier
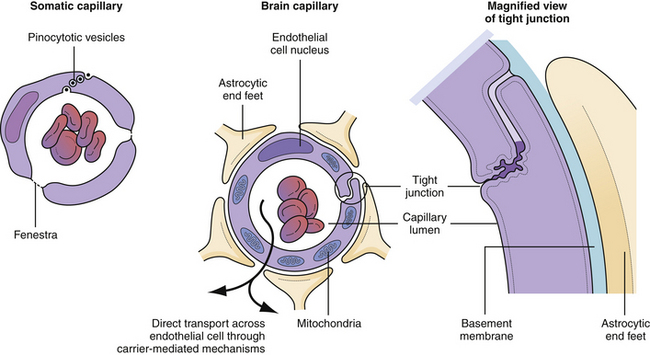
In order to understand the optimal way to treat intracranial hypertension, knowledge of the components of the blood-brain barrier is paramount. Not all substances that are carried in the blood can reach the neural tissue—a barrier composed of astrocytic foot process wrapping around a capillary endothelium composed of tight junctions4 (Fig. 19.9). These endothelial tight junctions are the barrier to the passive movement of many substances in order to protect the sensitive neural tissue from toxic materials. There are two mechanisms by which materials may be transported naturally across the endothelial cells. First, lipid-soluble substances can usually penetrate all capillary endothelial cell membranes in a passive manner.21 Second, amino acids and sugars are transported across the capillary endothelium by specific carrier-mediated mechanisms.22 The measurement of how easily something diffuses across the blood-brain barrier is called the reflection coefficient. A coefficient of 0 is complete passive diffusion; 1 indicates no passive diffusion.23,24
Decompressive Hemicraniotomy
The most direct way to normalize raised ICP to the physiological range is to eliminate its cause. If the increased pressure is the result of a mass effect, such as a blood clot, prompt evacuation of the offending lesion will restore ICP to normal more effectively than any other measure. The goal of a decompressive craniotomy is to perform a robust craniotomy that extends from the frontal lobe to the occipital lobe and includes the temporal lobe so that uncal herniation can be alleviated. After the bone is removed, the dura is often flapped open and the mass lesion evacuated. The brain can then be covered with a dural substitute to permit expansion. The bone flap can be placed back or kept off depending on intraoperative findings and personal surgical preferences. If the bone flap is kept off, the dural substitute is then covered with the scalp and a subgaleal drain is usually placed (Fig. 19.10). The bone flap may be either placed in a subcutaneous abdominal pocket, or more commonly at the present, storaged in a –80°C freezer in a sterile fashion. A cranioplasty or cranial bone replacement should be performed at a later date if the patient survives the acute injury.
The indications for performing this operation are most often (1) control of increased ICP that is recalcitrant to medical therapy and (2) progressively worsening neurological examination with evidence of massive hemispheric brain swelling, contusion, or intracranial blood or mass lesion. Despite the widespread use of decompressive hemicraniotomy worldwide,25–27 no level I data are available that show its superiority over aggressive medical treatment. In fact, a recently published randomized control trial evaluated patients with diffuse brain swelling (i.e., no mass effect) with refractory ICP.28 This study found that even though decompressive bifrontal craniectomy reduced ICP more than medical management, it did not translate in better outcomes and indeed appeared to actually worsen them in some circumstances. There are some critiques of this trial, including the definition of “refractory ICP,” but nonetheless, it reinforces that the use of decompressive hemicraniectomy in diffuse brain swelling is an area of controversy. In addition, surgery is obviously fraught with comorbidity, and thus, nonsurgical options is reasonable as a first-line therapy when there is no obvious mass lesion, contusion or intracranial blood. However, we still employ decompressive craniectomy in trauma patients who have failed medical therapy and have contusions and intracranial blood with elevated ICP. Indicated as a first-line therapy when there is no obvious mass lesion. These options are discussed in the following sections.
Mannitol
Mannitol is the osmotic agent universally used to treat cerebral edema.29–31 Other osmotic agents such as urea and glycerol are no longer used in contemporary neurosurgical practice because of side effects such as hemolysis. Mannitol decreases ICPs in two distinct mechanisms. First, when mannitol is initially delivered it creates an osmotic gradient from all body tissues into the intravascular space. This transiently increases intravascular volume and improves the rheology of cerebral blood. This transient increase in cerebral blood flow is thought to trigger the intrinsic autoregulatory mechanism of the intracranial vasculature (if it is intact) to vasoconstrict and decrease intracranial arterial blood volume. This decrease in blood volume translates into decreases in ICP, and is the mechanism that gives mannitol almost immediate efficacy in the setting of intracranial hypertension.
Hypertonic Saline
High-dose aliquots of hypertonic saline have recently emerged as a potential first-line therapy for elevated ICP. Thirty-milliliter boluses of 23.4% sodium chloride given through a central line over 15 minutes have been shown to be at least as efficacious as mannitol in multiple small prospective studies in lowering intracranial pressure.32,33 The mechanism by which 23.4% saline works is similar to that of mannitol, in that it creates an osmotic gradient to draw fluid from the interstitial space of the brain across the blood-brain barrier and into the intravascular space.
Loop Diuretics
Furosemide has been classically used as an adjunct to mannitol because it seems to have a synergistic effect. Furosemide alone, however, cannot be relied upon to reduce ICP. Because its primary action is on the kidney, it is not dependent on the intact blood-brain barrier for its effect. It is also possibly thought to reduce CSF production.
Subacute to Chronic Intracranial Hypertension
Causes of subacute to chronic intracranial hypertension are mass lesions (tumors, chronic subdural hematomas, etc.) that not only increase intracranial volume but also can induce interstitial edema. The most common symptom of subacute/chronic elevations is headache that is generalized in nature and is worse in the morning—presumably because of the increase in CO2 tension (from a physiological decrease in respiratory rate) and increased venous pressure from laying supine. The pain-sensitive structures within the cranial cavity are the dura and the blood vessels. Focal mass lesions that distort the dura and stretch the vessels tend to cause headache more frequently than diffuse generalized increases in ICP without focal mass effect, such as a pseudotumor cerebri or hydrocephalus. The headache, if present, may be associated with vomiting. The vomiting associated with increased ICP is usually not associated with nausea. Papilledema is another cardinal sign of increased ICP.34
Cerebral Edema
Cerebral edema is a prominent cause of subacute to chronic intracranial hypertension. It may be defined as a state of increased brain volume as a result of an increase in water content. There are three types of cerebral edema (Table 19.2).
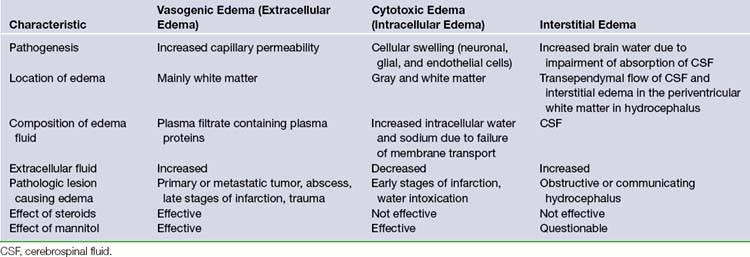
Vasogenic edema is the most common form of brain edema encountered in clinical practice (Fig. 19.11). It results from increased permeability of capillaries of the blood-brain barrier. The tight junctions between the endothelial cells become incompetent, allowing plasma filtrate to escape into the intercellular space35–37 (Fig. 19.12). The phenomenon of contrast enhancement in CT and magnetic resonance imaging (MRI) scans is, in part, because of the breakdown of the blood-brain barrier and resultant vasogenic edema. Vasogenic edema is most commonly seen with tumor, abscess, and chronic reaction to a blood clot. The edema is more marked in white matter than in gray matter.
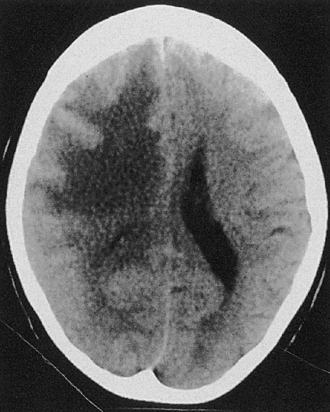
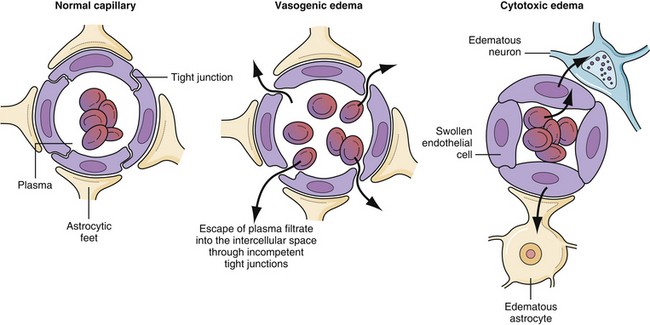
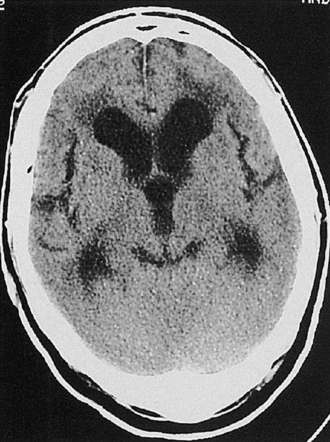
Cytotoxic edema most commonly results from hypoxia of the neural tissue. The hypoxia affects the adenosine triphosphate (ATP)-dependent sodium/potassium pump mechanism in the cell membrane, promoting an accumulation of intracellular sodium and the subsequent flow of water into the cell to maintain osmotic equilibrium. Thus, the edema is primarily intracellular and affects virtually all cells, including the endothelial cells, astrocytes, and neurons. Because of the swelling of these cells, the interstitial space is considerably narrowed (see Fig. 19.12). The two most common causes of cytotoxic edema are tissue hypoxia and water intoxication. The CT scan commonly shows only subtle initial changes or no changes at all, indicative of cytotoxic edema in the early phases of ischemic stroke.
Interstitial edema results from transudation of CSF in obstructive hydrocephalus. It is best observed on CT or MRI scan as periventricular low-density areas because of the retrograde transependymal flow of CSF into the interstitial space of the white matter (Fig. 19.13). Interstitial edema is most commonly observed in the frontal region. This finding generally indicates active hydrocephalus requiring surgical therapy.
Treatment
Craniotomy
As previously mentioned, the most direct way to normalize raised ICP to the physiological range is to eliminate its cause. If the increased pressure is the result of a mass effect, such as a tumor or chronic subdural hematoma, prompt evacuation of the offending lesion will restore ICP to normal more effectively than any other measure. Ventriculostomy may also be used to stabilize ICP.
Novel Adjunctive Therapies
Brain Tissue Oxygenation Monitoring
A polarographic cerebral oxygen monitor (Licox system) can be placed to the brain parenchyma itself to monitor brain tissue oxygenation levels (Fig. 19.14). Several retrospective studies correlated poor outcome with low cerebral oxygenation (jugular saturation) and low values for partial pressure of oxygen in brain tissue (PBTO2 (<10 mm Hg for >2 hours).38–4142 This finding opens up the possibility that therapeutric interventions to manipulate the PBTO2 value could potentially alter the traditionally poor outcome in traumatic brain injury, notably the 36% mortality rate.43,44 Indeed, several retrospective studies have suggested that active treatment of low PBTO2 (defined as <20 mm Hg) does improve outcomes as measured on the Glasgow Outcomes Scale.45,46 However, a few other papers suggest it does not and simply adds to the cost of care and stay in ICU.47 The precise role of PBTO2 measurement in management of ICP is evolving, and utility remains to be determined. However, its popularity in centers that manage a large population of head-injured patients has certainly expanded over the past decade.

Microdialysis
Brain microdialysis is being used in some centers as a research tool to investigate brain metabolism in patients with increased ICP, stroke, vasospasm, and other brain insults.48–52 The technique of cerebral microdialysis entails the implantation of specially designed catheters in the cerebral cortex to collect small-molecular-weight substances to help measure and identify neurotransmitters, peptides, and other substances. This research tool has clinical applications that are yet to be defined. The goal of this technique may be to identify the neurochemical milieu around the penumbra of trauma or stroke in order to devise better ways of protecting the undamaged cerebral cortex. Detailed discussion of this technique and its future uses is beyond the scope of this chapter.
Chemotherapy
Blood-brain barrier tight junctions can be transiently opened artificially by the intra-arterial injection of a bolus of an osmotic agent, such as mannitol, that dehydrates the endothelial cells or high-frequency ultrasound.53 During this brief interval, which lasts for a few hours, certain chemotherapeutic or other agents can be administered that would not otherwise be able to cross the blood-brain barrier to reach the glia.
8. Chesnut R.M., Marshall L.F., Klauber M.R., et al. The role of secondary brain injury determining outcome from severe head injury. J Trauma. 1993;34:216-222.
28. Cooper J.D., Rosenfeld J.V., Murray L., et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364(16):1493-1502.
33. Kerwin A., Schinco M.A., Tepas J.J.3rd, et al. The use of 23.4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: a pilot study. J Trauma. 2009;67:277-282.
14. Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Neurol Scand Suppl. 1960;149:1.
26. Polin R.S., Shaffey M.E., Bugaev C.A., et al. Decompressive bifrontal craniectomy in the treatment of severe refractory posttraumatic cerebral edema. Neurosurgery. 1997;41:84-100.
Please go to expertconsult.com to view the complete list of references.
1. Wanifuchi H., Schmzu T., Maruyama T. Age-related changes in the proportion of intracranial cerebrospinal fluid space measured using volumetric computerized tomography scanning. J Neurosurg. 2002;97:607-610.
2. Raichle M.E., Plum F. Hyperventilation and cerebral blood flow. Stroke. 1972;3:566-575.
3. Langfitt T.W., Weinstein J.D., Kassell N.F. Cerebral vasomotor paralysis produced by intracranial hypertension. Neurology. 1965;15:622-641.
4. Matsukado K., Inamura T., Nakaro S., et al. Enchanced tumor uptake of carboplatin and survival in glioma-bearing rats by intracarotid infusion of bradykinin analog RMP-7. Neurosurgery. 1996;39:125-147.
5. Ignelzi R.J. Cerebral edema: present perspectives. Neurosurgery. 1979;4:338-342.
6. Bouma G.J., Muizelaar J.P., Bandoh K., Marmarou A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992;77:15-19.
7. Bouma G.J., Muizelaar P., Choi S.C., et al. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991;75:685-693.
8. Chesnut R.M., Marshall L.F., Klauber M.R., et al. The role of secondary brain injury determining outcome from severe head injury. J Trauma. 1993;34:216-222.
9. Marmarou A., Anderson R.L., Ward J.D. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:s59-s66.
10. Bullock R., Chesnut R.M., et al. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2000;77:451-553.
11. Adelson P.D., Bratton S.L., Carney N.A., et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 5. Indications for intracranial pressure monitoring in pediatric patients with severe traumatic brain injury. Pediatr Crit Care Med. 2003;4(Suppl 3):S19-24.
12. Czosnyka M., Czosnyka Z.H., Richards H.K., Pickard J.D. Hydrodynamic properties of extraventricular drainage systems. Neurosurgery. 2003;52(3):619-623.
13. Kanter M.J., Narayan R.K. Intracranial pressure monitoring. Neurosurg Clin North Am. 1991;2:257-265.
14. Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Neurol Scand Suppl. 1960;149:1.
15. Raabe A., Stockel R., Hohrerin D., Schoche J. Reliability of intraventricular pressure measurement with fiber-optic or solid-state transducers: avoidance of methodological error. Neurosurgery. 1998;42:74-83.
16. Raabe A., Tatzauer R., Meyer O., et al. Reliability of epidural pressure measurement in clinical practice: behavior of three modern sensors during simultaneous ipsilateral intraventricular or intraparenchymal pressure measurement. Neurosurgery. 1998;43:306-315.
17. Gray W.P., Palmer J.D., Gill J., et al. A clinical study of parenchymal and subdural miniature strain-gauge transducers of monitoring intracranial pressure. Neurosurgery. 1996;39:927-934.
18. Gelabert-Gonzalez M., Ginesta-Galan V., Sernamito-García R., et al. The Camino intracranial pressure device in clinical practice. Assessment in a 1000 cases. Acta Neurochir (Wien). 2006;148(4):435-441.
19. Bavetta S., Norris J.S., Wyatt M., et al. Prospective study of zero drift in fiberoptic pressure monitors used in clinical practice. J Neurosurg. 1997;86:927-930.
20. Piper I., Barnes A., Smith D., Dunn L. The Camino intracranial pressure sensor: is it optimal technology? An internal audit with a review of current intracranial pressure monitoring technologies. Neurosurgery. 2001;49:1158-1169.
21. Kroll R.A., Neuwelt E.A. Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means. Neurosurgery. 1998;42:1083-1105.
22. Friden P.M. Receptor-mediated transport of therapeutics across the blood-brain barrier. Neurosurgery. 1994;35:294-301.
23. Neuwelt E.A., Abbott J.N., Drew L., et al. Neurosurgery. 1999;44:604-614.
24. Pollay M., Roberts P.A. Blood-brain barrier: a definition of normal and altered function. Neurosurgery. 1980;6:675-685.
25. Carter B.S., Ogilvy C.S., Candia G., et al. One year outcome after decompressive surgery for massive nondominant hemisphere infarction. Neurosurgery. 1997;40:1168-1179.
26. Polin R.S., Shaffey M.E., Bugaev C.A., et al. Decompressive bifrontal craniectomy in the treatment of severe refractory posttraumatic cerebral edema. Neurosurgery. 1997;41:84-100.
27. Munch E., Horn P., Schurer L., et al. Management of severe traumatic brain injury by decompressive craniectomy. Neurosurgery. 2000;47:315-326.
28. Cooper J.D., Rosenfeld J.V., Murray L., et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364(16):1493-1502.
29. Hartwell R.C., Sutton L.N. Mannitol, intracranial pressure and vasogenic edema. Neurosurgery. 1993;32:444-454.
30. Berger S., Schurer L., Hartl R., et al. Reduction of post-traumatic intracranial hypertension by hypertronic/hyperoncotic saline/dextran and hypertonic mannitol. Neurosurgery. 1995;7:98-109.
31. Al Qureshi, Wilson D.A., Traystman R.J. Treatment of elevated intracranial pressure in experimental intracerebral hemorrhage: comparison between mannitol and hypertonic saline. Neurosurgery. 1999;44(5):1055-1063. discussion 1063-1064
32. Rockswold J., Solid C.A., Paredes-Andrade E., et al. Hypertonic saline and its effect on the intracranial pressure, cerebral perfusion pressure and brain tissue oxygenation. Neurosurgery. 2009;65:1035-1042.
33. Kerwin A., Schinco M.A., Tepas J.J.3rd, et al. The use of 23.4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: a pilot study. J Trauma. 2009;67:277-282.
34. Fishman R.A. Brain edema. N Engl J Med. 1975;293:706-711.
35. Reulen H.J., Grahm R., Spatz M., Klatzo I. Role of pressure gradients and bulk flow in dynamics of vasogenic brain edema. J Neurosurg. 1977;46:24-35.
36. Ohata K., Marmarou A. Clearance of brain edema and macromolecules through the cortical extracellular space. J Neurosurg. 1992;77:387-396.
37. Reulen H.J., Tsuyumu M., Tack A., et al. Clearance of edema fluid into cerebrospinal fluid. J Neurosurg. 1978;48:754-764.
38. Stochetti N., Paparella A., Bridelli F., et al. A cerebral venous saturation studied with bilateral samples in the internal jugular vein. Neurosurgery. 1994;34:38-47.
39. Van Santbrink H., Maas A.I.R., Cees J.J., Avezaat C.J.J. Continuous monitoring of partial pressure of brain tissue oxygen in patients with severe head injury. Neurosurgery. 1996;38:21.
40. Zauner A., Bullock R., Di X., Young H.F. Brain oxygen CO2, pH, temperature monitoring: evaluation in feline brain. Neurosurgery. 1995;37:1168-1175.
41. Vandenbrink W.A., van Santbrink H., Steyerberg E.W., et al. Brain oxygen tension in severe head injury. Neurosurgery. 2000;46:868-882.
42. McCarthy M. Neurologic outcomes with cerebral oxygen monitoring in traumatic brain injury. Surgery. 2009;146(4):585-590.
43. Becker D.P., Miller J.D., et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg. 1977;47:491-502.
44. Marshall L.F., Eisenberg H.M., Jane J.A., et al. The outcome of severe closed head injury. J Neurosurg. 1991;75(Suppl):S28-S36.
45. Narotam P. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111:672-682.
46. Maloney-Wilensky E., Gracias V., Itkin A., et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med. 2009;37(6):2057-2063.
47. Martini R.P., Deem S., et al. Management guided by brain tissue oxygen monitoring and outcome following severe traumatic brain injury. J Neurosurg. 2009;111(4):644-649.
48. Unterberg A.W., Sakowitz O.W., Sarrafzadeh A.S., Benndorf G. Role of bedside microdialysis in the diagnosis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2001;94:740-749.
49. Hutchinson P.J., O’Connell M.T., Al-Rawj P.G., et al. Cerebral clinical microdialysis: a methodological study. J Neurosurg. 2000;93:37-43.
50. Reinstrup R., Stahls N., Mellergord P., et al. Intracerebral microdialysis in clinical practice: baseline values for chemical markers during wakefulness, anesthesia, and neurosurgery. Neurosurgery. 2000;47:701-713.
51. Zauner A., Doppenberg E.M.R., Woodward J.J., et al. Continuous monitoring of cerebral substrate delivery and clearance: initial experience in 24 patients with severe acute brain injuries. Neurosurgery. 1997;41:1082-1097.
52. Zauner A., Daughherty W.P., Bullock R., Warner D. Brain oxygenation and energy metabolism. Biological function and pathophysiology. Neurosurgery. 2002;51:289-299.
53. Mesiwala A., Farrell L., Wenzel H., et al. High intensity focused ultrasound selectively disrupts the blood-brain barrier in vivo. Ultrasound Med Biol. 2002;28:389-400.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 19 Intracranial Hypertension
• States of impaired consciousness are expressed as numerical scores on the Glasgow Coma Scale (GCS), which is used worldwide and has prognostic and management implications.
• The key clinical signs that help in clinical assessment and determination of prognosis are the presence or absence of certain brainstem reflexes such as pupillary response, corneal reflex, oculocephalic and oculovestibular reflexes, and gag reflex.
• Computed tomography (CT) scan of the head remains a key imaging modality in the rapid assessment of a patient with impaired consciousness. Mass lesions that are directly responsible for intracranial hypertension should be surgically removed.
• Intracranial pressure (ICP) is generally monitored when the GCS score is less than 8 with an abnormal CT scan, although there is no class I evidence that mandates use of ICP monitors worldwide. A fiberoptic monitor is a good tool for measuring and managing increased ICP in an unconscious patient. Fiberoptic ICP monitors have a low incidence of complication and infection in experienced hands. However, they do not give a therapeutic option for lowering ICP like a ventriculostomy catheter with external ventricular drainage does. Head of bed elevation, maintaining euthermia, sedation, analgesia, and mild hyperventilation can also have temporary therapeutic effects on elevated ICP.
• After conservative therapies have been exhausted, hypertonic saline may be as efficacious as mannitol and is associated with a lower overall incidence of comorbidity.
• Decompressive hemicraniectomy is a reasonable option in uncontrolled intracranial hypertension, especially if there is a mass lesion.
• Future studies are being undertaken to further elucidate the role of microdialysis and brain tissue oxygenation monitoring in the setting of intracranial hypertension.
The cranium is a rigid structure. The major intracranial contents are the brain (to include the neuroglial elements and interstitial fluid), blood (arterial and venous), and cerebrospinal fluid (Table 19.1). When a new intracranial mass is introduced, a compensatory change in volume must occur through a reciprocal decrease in venous blood or cerebrospinal fluid (CSF) to keep the total intracranial volume constant. This is the Monro-Kellie doctrine (Fig. 19.1), which has been confirmed by many experimental and clinical observations. Only in children, with open fontanelles and whose sutures have not yet fused, can the cranium itself expand to physically accommodate extra volume.
TABLE 19.1 Intracranial Contents and Their Respective Volumes
| Component | Volume | Percentage of Total Volume |
|---|---|---|
| Brain (70%) and interstitial fluid (10%) | 1400 mL | 80% |
| Blood | 150 mL | 10% |
| Cerebrospinal fluid | 150 mL | 10% |
| TOTAL | 1700 mL | 100% |
Compliance (dV/dP) is the change in volume observed for a given change in pressure. This represents the accommodative potential of the intracranial space. In clinical practice, however, what is actually measured is elastance (dP/dV). Elastance is the inverse of compliance and is the change in pressure observed for a given change in volume. It represents the resistance to outward expansion of an intracranial mass. The elastance curve (not compliance) is what is plotted in Figure 19.2. Although technically a misnomer, the term compliance is actually entrenched in the literature to describe the aforementioned phenomenon instead of using the proper term elastance. Because of this we, too, will conform to the traditional nomenclature for the rest of this chapter.
In accordance with the Monro-Kellie doctrine, several innate homeostatic mechanisms exist in an attempt to maintain intracranial pressure within the physiological range—generally considered less than 20 mm Hg in the acute inpatient setting. First, the venous system collapses easily and squeezes venous blood out through the jugular veins or through the emissary and scalp veins. Likewise, CSF can be displaced from the ventricular system through the foramina of Luschka and Magendie into the spinal subarachnoid space.1 When these compensatory mechanisms have been exhausted, small changes in volume produce precipitous increases in pressure. This effect can be demonstrated experimentally by inserting a Foley catheter into the epidural space of a rat and gradually inflating a balloon with increasing volumes. The curve produced by plotting intracranial pressure against volume is the so-called compliance curve in Figure 19.2. The innate homeostatic pressure-buffering mechanism offered by displacement of CSF and venous blood keeps this curve flat until a “critical volume” is reached. After this critical volume, small volumetric changes result in precipitous increases in pressure, and intracranial hypertension naturally ensues. Brain parenchyma and arterial blood do not participate, to any significant extent, in the innate intracranial pressure-buffering mechanisms.
Acute Intracranial Hypertention
Cerebral Blood Flow
Normal cerebral blood flow averages 55 to 60 mL/100 g brain tissue per minute. In the gray matter the blood flow is 75 mL/100 g brain tissue per minute, whereas in the white matter it is around 45 mL/100 g brain tissue per minute. This flow is sufficient to meet the metabolic needs of the brain. The brain usually begins to show signs of ischemia at 20 mL/100 g/minute and permanent damage usually results when the blood flow drops below 10 mL/100 g/minute in most models. The most significant factor that determines cerebral blood flow at any given time is the cerebral perfusion pressure (CPP). The CPP is the effective blood pressure gradient across the brain. CPP is the difference between the incoming mean arterial pressure (MAP) and the opposing intracranial pressure (ICP): CPP = MAP − ICP. The mean arterial pressure can be calculated in two ways: (1) the diastolic pressure plus one third of the pulse pressure (DP + 1⁄3PP) or (2) two thirds of the diastolic pressure plus one third of the systolic pressure (2⁄3DP + 1⁄3SP). The ICP has to be measured directly, which can be done with various devices and will be discussed later in this chapter. With increased ICP there is an obvious tendency for the cerebral perfusion pressure to decrease.
Three major factors regulate cerebral blood flow under physiological conditions: CPP, concentration of arterial CO2, and arterial PO2. The ability to maintain constant blood flow to the brain over a wide range of CPPs (50-150 mm Hg) is called cerebral autoregulation. When the CPP is low, the cerebral arterioles dilate to allow adequate flow at the decreased pressure. Conversely, an increase in CPP causes the arterioles to constrict and maintain the flow within physiological range (Fig. 19.3). Decreases in CO2 tension in the blood (i.e., hyperventilation) causes diffuse vasoconstriction.2 Vasoconstriction decreases both cerebral blood flow and cerebral blood volume in the brain. The risks and benefits of inducing hyperventilation are discussed under the treatment section at the end of this chapter. Lastly, severe hypoxia causes cerebrovascular dilatation. This effect only becomes apparent when the oxygen tension in the blood drops to 50 mm Hg and becomes maximal around 20 mm Hg.
Under certain pathological conditions cerebral blood flow cannot always be autoregulated.3 When the CPP exceeds 150 mm Hg, such as in hypertensive crisis, the autoregulatory system fails. In this case there would be a passive increase in blood flow proportionate to the increase in systemic pressure, causing an exudation of fluid from the vascular system with resultant vasogenic edema.4 Additionally, certain toxins such as carbon dioxide can cause diffuse cerebrovascular dilatation and inhibit proper autoregulation.5 Lastly, during the first 4 to 5 days of head trauma, many patients can experience a disruption in cerebral autoregulation; this is often observed in the pediatric population and may result in hyperemia.6
Disruption of cerebral blood flow autoregulation in the setting of trauma could potentially lead to severe alterations in CPP when a patient’s brain is acutely injured.7 When disrupted, cerebral blood flow would be directly proportional to CPP under a much larger range than normal. This is likely one of the contributing reasons why even one episode of hypotension in the patient with acute head injury may lead to significantly worse outcomes.8 Thus, many institutions have adopted protocols to maintain a minimal CPP in the acute setting—usually around 50 to 60 mm Hg—and perhaps a maximum threshold of CPP if the intracranial hypertension can be linked directly to hyperemia.
Monitoring Intracranial Pressure
The most significant factor determining morbidity and mortality risk in patients with acute cranial neurosurgical disorders continues to be increased ICP. Continuous ICP monitoring is very useful for assessing intracranial dynamics in patients with suspected intracranial hypertension. There are no clinical indicators that can be used in the early stage of rising ICP to forestall a further rise. Classical clinical indicators described in the literature (cranial nerve abnormalities, posturing, etc.) occur in the end stage and they are not sensitive enough to show subtle changes in pressure. The two most common indications for ICP monitoring are closed head injury and spontaneous subarachnoid hemorrhage.
Indications for Monitoring Intracranial Pressure
The advent of continuous ICP monitoring has improved outcomes in traumatic brain injury9,10 by reducing the effects of secondary injury occurring after the initial brain insult. In the modern intensive care unit ICP monitoring in the adult patient with severe head injury is a recommended practice guideline based on available clinical evidence. Indications of ICP monitoring following head injury include a GCS score between 3 and 8 and an abnormal CT scan. Unfortunately, no class I evidence is available regarding the use of ICP monitors in pediatric head injury patients. Nevertheless, strong evidence supports the association between elevated ICP and poor outcomes,11 and aggressive treatment of elevated ICP is associated with the best clinical outcomes. ICP monitoring provides valuable data that can inform medical management.
Techniques of Monitoring Intracranial Pressure
There are two commonly used pressure-monitoring systems in contemporary neurosurgical practice. An intraventricular catheter connected to a manometer and a drainage system is the standard against which all other systems are compared.12–14 The ventricular catheter ideally should be tunneled under the skin and brought out through a separate stab wound, well away from the ventricular entry site, to minimize the risk of infection. Infection is the most significant complication of intraventricular pressure monitoring. With the use of an electronic transducer, the waveform can also be monitored (Fig. 19.4). The major advantage of the method is that the ventricular catheter is used not only to measure the pressure, but also as a therapeutic modality allowing intermittent or continuous drainage of CSF when the pressure exceeds physiological limits.
A second method is the use of the fiberoptic transducer-tipped catheter system. The transducer-tipped catheter can be placed within the brain parenchyma or in the subdural space, depending on the surgeon’s choice and the clinical situation.15–17 The pressure monitor gives both digital readout and a waveform. The advantages of this system are that the zero point does not have to be reset with changes in head position because the pressure-sensing transducer is within the cranial cavity, and it is not susceptible to blockage by debris because it is not a fluid-coupled system. Also, insertion of a fiberoptic cable into the brain parenchyma is not affected by ventricular size or shift, which can make insertion of a ventricular catheter in the setting of intracranial hypertension quite trying. Lastly, the infection rates with fiberoptic probes are significantly lower than with ventriculostomy catheters, reported as less than 1% in one of the the largest reported series.18 The disadvantages of this system, however, are (1) higher cost and (2) baseline drift, which means there is evidence that with prolonged fiberoptic monitoring the indicated pressures may become less accurate in an unpredictable fashion.19,20
Intracranial Pressure Waveforms
The waveform of normal ICP typically shows three components: the percussion wave (P1), the tidal wave (P2), and finally the dicrotic notch (P3) (Fig. 19.5A). Under physiological conditions P1 is the tallest and is thought to represent the pressure at peak cardiac systole transmitted throughout the choroid plexus. P2 follows P1 and its resultant smaller peak is thought to represent the filling of the intracranial arteries with systolic blood and results in a rebound increase in ICP from this increase in intracranial volume. Last is the dicrotic wave.
When the ICP rises and compliance decreases, one will observe P2 becoming taller than the P1. This is because a similar volume in intracranial arterial blood is now resulting in a higher change in ICP (Fig. 19.5B). Such alterations in the morphology can give the treating physician further clues regarding not only the compliance but also the autoregulatory capacity of the brain.
When the ICP waveforms are registered over a period of time certain trends may become apparent14 (Fig. 19.6). Plateau waves, or type A waves, are characterized by an abrupt elevation in ICP for 5 to 20 minutes followed by a rapid fall in the pressure to resting levels. The amplitude may reach as high as 50 to 100 mm Hg. Plateau waves may be clinically marked by a decreasing level of consciousness, restlessness, increased tone in the extremities, and tonic-clonic movements. Plateau waves may represent transient surges in ICP secondary to increased cerebral blood volume likely related to CO2 retention.
[/not-level-membership-for-neurosurgery-category]
