CHAPTER 127 Intracranial Ependymomas in Adults
Intracranial ependymomas in adults are relatively rare brain tumors of neuroectodermal origin that account for 2% to 6% of all intracranial neoplasms and occur equally in males and females.1–6 These lesions are more common in children, in whom they account for 10% of brain tumors.7 Supratentorial ependymomas occur more frequently in adults, whereas infratentorial tumors are more common in the pediatric population (Table 127-1).7–15 Surgical resection is regarded as the standard treatment of these tumors, with a goal of gross total resection when safely feasible. The optimal therapeutic management of intracranial ependymomas in adults remains controversial because of the rarity of these lesions in this population and the limited number of studies pertaining to adults. The majority of studies on intracranial ependymomas have been conducted in the pediatric population. The low incidence of these tumors in adult patients has led to many published reports that are retrospective, included both intracranial and spinal ependymomas in some studies, combined data from children and adults in some series, and covered long periods of treatment, which confounds the interpretation of results because of changes in histologic grading, diagnosis, and therapy, all of which have contributed to the lack of ability to reach a consensus in optimally managing these lesions in adults. However, adjuvant postoperative radiotherapy (RT) is typically considered to play an important role in treatment, particularly in the care of patients with high-grade ependymomas, whereas its role in the management of low-grade tumors continues to remain controversial in adults.4,6,16,17 Furthermore, a role for adjuvant chemotherapy has not been adequately defined.
TABLE 127-1 Comparison of Intracranial Ependymomas in Adults and Children
| ADULT | PEDIATRIC | |
|---|---|---|
| Prevalence | 2%-6% of intracranial neoplasms | 10% of intracranial neoplasms |
| Location | Predominantly supratentorial | Predominantly infratentorial |
| Cerebrospinal fluid seeding | Less common | More common |
| Clinical studies | Paucity because of low incidence | Several because of higher incidence |
| 5-Year survival rate | 55%-90% | 40%-65% |
Although prospective studies are needed in adult patients harboring intracranial ependymomas, reports have found a trend for better survival in adults than in children.12,18–21
Pathology
Ependymal tumors are presumed to be derivatives of the neuroectodermal cell lineage that give rise to the ependymal cells lining the choroid plexus and white matter adjacent to the angulated ventricles, especially the regions adjacent to the trigone of the lateral ventricle and the foramen of Luschka, as well as the central canal of the spinal cord for tumors affecting this part of the neuraxis.1 These ependymal cells undergo neoplastic transformation leading to ependymomas. Ependymomas located in the brain parenchyma are thought to occur as a result of fetal ependymal cell rests remaining within the parenchyma during embryogenesis.22,23 It has been estimated that approximately 50% of supratentorial ependymomas originate in the brain parenchyma and the remaining tumors are derived from the lateral ventricles, with very few originating from the third ventricle.23 Although rare, supratentorial intracortical and intracranial ectopic ependymomas have been reported.24–34 Intracranial ectopic ependymomas occur where ependymal cells are typically absent and have been reported to involve the neurohypophysis, cranial nerve V, sella turcica, posterior fossa, falx, and cavernous sinus.27–34
The World Health Organization (WHO) has classified ependymal tumors into three grades according to cellular derivatives or degree of anaplasia: (1) myxopapillary ependymoma and subependymoma (WHO grade I), (2) ependymoma (WHO grade II), and (3) anaplastic ependymoma (WHO grade III).35–39 Gross inspection of ependymomas reveals them to be solid, well-delineated, dark, gray-red lesions. Myxopapillary ependymomas occur almost exclusively in the conus, cauda equina, and filum terminale regions, and subependymomas are benign, slow-growing tumors whose diagnosis portends a favorable prognosis. Histopathologic examination of ependymomas reveals that they are composed of uniform cuboidal and astrocyte-like fibrillary cells arranged into linear tubules in which the cells surround a central lumen and resemble ependymal epithelia. These histologic findings are known as rosettes. Immunostaining for glial fibrillary acidic protein (GFAP) is typically positive, and these GFAP-positive processes can be found aligned around blood vessels and are known as perivascular pseudorosettes.40 The rosettes and perivascular pseudorosettes are key histologic features of ependymomas.
Moreover, low-grade ependymomas, which are the prevalent histologic type in the adult population, are further stratified into four subtypes: (1) cellular, (2) papillary, (3) clear cell, and (4) tanycytic. Cellular ependymomas are hypercellular with rare or absent mitoses. Papillary ependymomas, which have a tubulovillous arrangement, have a very low incidence, and immunostaining is positive for GFAP and vimentin. The clear cell subtype is characterized by clear cytoplasm and can resemble oligodendrogliomas, central neurocytomas, hemangioblastomas, or metastatic clear cell carcinomas.35,41 Therefore, careful microscopic examination of the specimen is necessary. Tanycytic ependymomas may appear histologically similar to astrocytomas given that their perivascular pseudorosettes are less obvious and rosettes are usually absent. Microscopic examination demonstrates elongated paraventricular glial cells with cytoplasmic processes that extend to the ependymal surface.42
Anaplastic ependymomas are characterized by histologic features such as hypercellularity, frequent mitotic figures, pseudopalisading necrosis, vascular proliferation, and cellular and nuclear pleomorphism.43 Rosettes are usually absent or rare, perivascular pseudorosettes may be poorly demarcated, and there may be a reduction in immunostaining for GFAP when compared with conventional ependymomas. The diagnosis of poorly differentiated lesions may require electron microscopic analysis if determination of the pathology is difficult. These tumors are locally invasive and have a higher propensity to spread to other areas of the neuraxis via cerebrospinal fluid (CSF) pathways.
Molecular Genetics
Molecular analysis has thus far demonstrated cytogenetic heterogeneity, although the data are limited. Aberrations involving chromosome 22 have been the most commonly implicated in ependymal tumorigenesis, including monosomy 22, various translocations, or the possible absence of tumor suppressor genes.44–47 The 22q region has been studied because it contains the neurofibromatosis type 2 (NF2) tumor suppressor gene. Patients with NF2 have an increased predilection for the development of ependymomas and meningiomas, and mutations in the NF2 gene have been detected in tumor specimens from these patients, in contrast to ependymomas observed in patients without NF2, in whom mutations in the NF2 gene have not been revealed.48–50 Studies have reported a loss of chromosome arm 22q in 50% to 60% of adult patients, and chromosome 22 has been implicated in the occurrence of familial intracranial ependymomas.46,51–60 In addition, abnormalities of chromosomes 1, 6, 7, 9, 10, 11, 12, 13, 16, 17, 19, and 20 have been associated with the development of ependymomas, although there are less data pertaining to them.61 Amplification of the gene MDM2 has been demonstrated in approximately 35% of ependymomas, but the role of its product, MDM2, has not been defined in the tumorigenesis of ependymomas.62 The development of new molecular techniques should aid in elucidation of the molecular mechanisms underlying the tumorigenesis of ependymomas.
Clinical Features
The clinical findings in patients with ependymomas are contingent on the size, location, and malignancy of the tumor. Ependymomas typically increase in size slowly and may reach a large dimension before detection.63 Anaplastic ependymomas may exhibit a more rapid onset of signs and symptoms. Although infratentorial lesions are more common in the pediatric population, they can occur in adults. Posterior fossa ependymomas may cause nausea, emesis, ataxia, hemiparesis, dizziness, nystagmus, and headaches.12,23 These symptoms are usually due to an increase in intracranial pressure as a consequence of obstructive hydrocephalus from filling of the fourth ventricle by the tumor or cerebellar compression, or both.
Supratentorial ependymomas are found more commonly in adults and tend to cause focal neurological deficits. Manifestation of the neurological deficits depends on the location of the tumor. These deficits may include motor weakness, aphasia, visual field deficits, behavioral changes, and memory impairment.8,64 Nausea, emesis, and headaches may occur as a result of an increase in intracranial pressure. Seizures may occur with extraventricular supratentorial tumors and have been reported to occur in approximately a third of patients.23 Although rare, supratentorial intracortical ependymomas have been reported in three adult patients, and all three were initially evaluated because of seizures.24,25 Parinaud’s syndrome may be observed in some patients with third ventricular ependymomas.1
Imaging
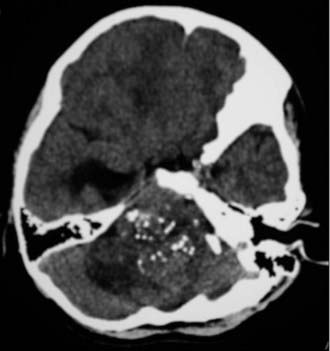
Ependymomas tend to be cystic, calcified, and well-circumscribed lesions. They may appear either isodense or hyperdense to brain parenchyma on computed tomography (CT). Administration of contrast material will typically demonstrate varying degrees of enhancement from mild to intense heterogeneous or homogeneous enhancement. CT imaging assists in the identification of any calcifications, which are present in 50% of supratentorial ependymomas and 46% of infratentorial ependymomas (Fig. 127-1).15 Imaging may be limited by artifact from the bony architecture of the posterior fossa.
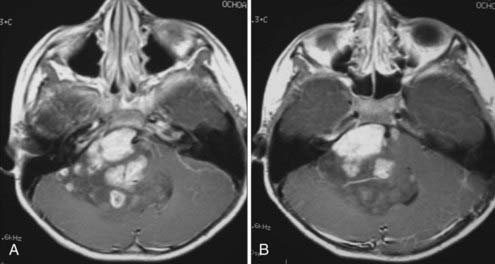
Magnetic resonance imaging (MRI) is the diagnostic modality of choice given its ability to provide greater anatomic detail. T1-weighted imaging demonstrates the tumor to be hypointense to isointense relative to white matter, whereas it is hyperintense to white matter on T2-weighted imaging. Similar to CT, administration of contrast material will typically demonstrate varying degrees of enhancement from mild to intense heterogeneous or homogeneous enhancement (Fig. 127-2). Calcium, hemosiderin, or necrosis may be demonstrated by hypointense foci on both T1- and T2-weighted images, and cystic changes are typically hyperintense on T2-weighted images (Fig. 127-3).65,66 Intratumoral hemorrhage has been observed in these lesions. Subsequently, the heterogeneity of signal characteristics on MRI could make the diagnosis of ependymoma difficult. Infratentorial ependymomas may extend from the fourth ventricle through the foramen of Luschka into the cerebellopontine cistern or downward through the foramen of Magendie into the cervical subarachnoid space.10,67,68 These characteristic findings lend support for the diagnosis of ependymoma.
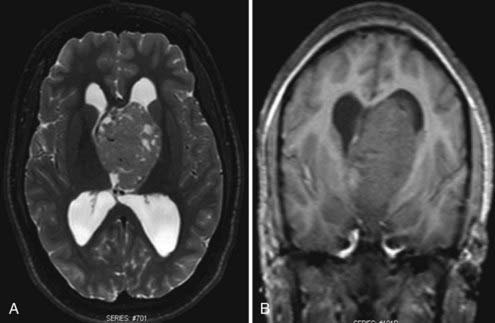
Staging
If an ependymoma is suspected on imaging of the brain or confirmed with biopsy or at surgery, MRI of the spine may be performed to rule out CSF seeding given the tumor’s predilection to disseminate throughout the CSF pathways. However, no conventional staging criteria exist. Metastatic seeding has been reported to occur more frequently with WHO grade III ependymomas, posterior fossa lesions, and an inability to achieve gross total resection, as well as in the pediatric population. CSF dissemination of ependymoma has been reported to occur less frequently in the adult population.69–74
Treatment
Surgery
Surgery is the mainstay of treatment of intracranial ependymomas, and reports have regarded the extent of tumor resection to weigh heavily in patient prognosis.12,14,15,67,75,76 An attempt at gross total resection, which may increase the chance for cure or longer disease-free survival, should always be made when technically feasible. Before the advent of microsurgical techniques, some authors reported high rates of morbidity and mortality related to the performance of surgery on ependymomas, especially for lesions located in the posterior fossa. Morbidity and mortality rates have been minimized with the introduction of microsurgical techniques, intraoperative neuronavigational modalities, intraoperative ultrasound, and intraoperative neurophysiologic monitoring, which contribute to the ability to achieve gross total or near-total resection of the ependymoma when technically possible. Complete resection of supratentorial ependymomas is more likely than for those located infratentorially because of the location of key structures, such as the brainstem, cranial nerve nuclei in the floor of the fourth ventricle, and vasculature, within a small, confined area.
Supratentorial Ependymomas
Supratentorial intraventricular ependymomas can be approached through either a transcortical or interhemispheric transcallosal technique. The surgeon should have a sound knowledge base with regard to the different approaches to the ventricles. Preoperative MRI is used to assess a surgical corridor and formulate the most appropriate surgical approach based on tumor size, tumor origin, and visual angles within the ventricle. Tumor origin, in the case of ventricular tumors, refers to tumors that are primarily ventricular or parenchymal with major ventricular extension. The transcortical approaches predispose patients to cortical injury and seizures. Reports estimate the risk for postoperative seizures to range from 29% to 70% after a transcortical procedure, whereas the reported risk is 0% to 10% after a transcallosal approach.77–82 Disconnection syndrome may be observed after the interhemispheric posterior transcallosal approach.83–85
Infratentorial Ependymomas
MRI is performed within 48 hours postoperatively to assess the extent of surgical extirpation. In addition, the presence of hemorrhage, particularly in the ventricular system, is evaluated because this may exacerbate preexisting obstructive hydrocephalus. Emergency postoperative head CT should be performed for any new neurological deficits or alterations in mental status. Second-look surgery has been recommended to maximize the extent of resection in cases of unexpected residual tumor or when the decision for a staged operation has been made.86 The optimal timing of second-look surgery, however, is a subject of debate. Some authors prefer a delayed second-look surgery after adjuvant chemotherapy or RT because adjuvant therapy may reduce the tumor volume or alter its characteristics such that surgical outcomes are improved.86 Since it is unclear that adjuvant therapy is beneficial, in cases in which residual tumor is easily accessible with minimal morbidity, immediate second-look surgery might be an appropriate option to achieve radical resection of tumor.86
Neuroendoscopy
Neuroendoscopy has been well described for the treatment of hydrocephalus, sellar region tumors, and intracranial cysts, as well as for tumor biopsy procedures. Resection of intraventricular tumors by endoscopic techniques has typically been reserved for lesions with significant cystic components, and there are few reports demonstrating endoscopic resection of solid intraventricular tumors. This surgical modality is continually evolving, and authors have described successful resection of intraventricular ependymomas. Neuroendoscopy has been used to achieve complete resection of ependymomas of the posterior third ventricle and aqueduct of Sylvius.87,88 As more novel neuroendoscopic techniques are being developed, excision of solid intraventricular ependymomas will become more feasible.
Radiation Therapy
It has generally been agreed that RT plays an integral role in the management of patients with intracranial ependymomas, and the decision to administer this form of treatment is contingent on the extent of resection, tumor histology, and evidence of metastatic seeding. Very few retrospective studies have investigated this therapeutic modality solely in the adult population because of the low incidence in these patients. Other retrospective reports have focused on the pediatric population or contained a heterogeneous sample that included both children and adults. Therefore, data pertaining to irradiation in adults are lacking, and randomized prospective studies are needed. There appears to be a general consensus that postoperative RT is vital in the management of anaplastic ependymomas,18,72,73,89,90 but controversy remains with regard to the role of irradiation for WHO grade II ependymomas, especially when gross total resection has been achieved. Debate exists regarding whether one should manage a patient with grade II ependymoma conservatively after gross total resection with serial imaging to observe for any recurrence or provide immediate postoperative RT.
A small retrospective study of 34 adults with intracranial ependymoma demonstrated a positive outcome in which only 3 of 17 grade II patients were treated with RT. The 5-year overall survival rate was 87% in this subgroup.4 Another study of grade II posterior fossa ependymomas in which gross total resection was achieved found that adjuvant RT significantly improved tumor control but did not prolong survival.91 The recommended radiation dose ranges from 54 to 60 Gy in fractions, but this not based on any prospective randomized trials.
Furthermore, controversy surrounds the optimal field of RT for the treatment of ependymomas in terms of local field RT, whole-brain RT (WBRT), or craniospinal axis RT. The definition of “local field” is not well delineated, particularly for infratentorial ependymomas, where it can be defined as the tumor, tumor bed, or entire posterior fossa. A potential reason to use WBRT is for prevention of metastatic seeding via CSF pathways, but this potential risk is low in the adult population. Kovalic and colleagues demonstrated local RT to be more effective than WBRT with regard to tumor control and provided a 10-year survival rate advantage of 75% versus 28%.92 Other studies have likewise not found significant efficacy in the treatment of intracranial ependymomas.18,89,93 Reports have also demonstrated that craniospinal RT did not have a statistically significant effect on outcome and found that treatment failures were due to local tumor recurrence.2,18,74,89,93
Stereotactic radiosurgery (SRS) has been used for the management of recurrent and residual intracranial ependymomas. SRS provides a single high dose of radiation to the target in an attempt to achieve local control, but out-of-field and distant tumor progression are concerns. The standard therapeutic approach has been surgery followed by external beam RT (EBRT), so the results of small, limited studies have to weigh this factor into consideration. Jawahar and coauthors reported a 3-year local control rate of 62.3% in a combined series of adult and pediatric patients with recurrent or progressive anaplastic ependymomas treated by SRS.94 Another study reported a 3-year local control rate of 68% in 12 patients with 17 recurrent ependymomas, and distant failure developed in 2 patients.95 Moreover, the use of an SRS boost has been assessed in patients with ependymomas. In a retrospective analysis of 7 patients with low-grade ependymomas and 2 with anaplastic ependymomas, Mansur and colleagues demonstrated a 100% relapse-free survival rate in patients treated with an SRS boost after EBRT versus a 20% rate in patients treated to salvage local failure after EBRT.96 In a series of 8 patients with 13 ependymomas treated with SRS, Lo and coworkers treated 2 of these patients, who harbored fourth ventricular tumors, with an SRS boost after subtotal resection and EBRT.97 The patients were without tumor recurrence at 39.5 and 64.4 months, respectively.
Chemotherapy
The efficacy of adjuvant chemotherapy in the treatment of intracranial ependymomas in adults has not been studied extensively, and consequently, there remains no definite role for it in the management of these patients. Few studies that focus solely on adults exist because of the low incidence of intracranial ependymomas in this patient population, in contrast to a number of studies conducted in the pediatric population.4,16,98–108 The few available reports describe small retrospective studies with heterogeneous chemotherapeutic regimens, include a limited number of patients, contain pediatric patients, and include data collected over long periods. Moreover, the data available from pediatric studies cannot be extrapolated to the adult population because adults harboring ependymomas tend to have a better prognosis than children do.18,20,109,110 Brandes and coworkers performed a retrospective study in adult patients with recurrent intracranial ependymomas and found a higher response rate with cisplatin-based chemotherapy, but the regimen did not prolong progression-free survival or overall survival.98
Prognosis
The outcome of adults with intracranial ependymomas has improved significantly during recent years, and the prognosis is more favorable than in children. A 5-year overall survival rate of 55% to 90% has been reported.2,4,18,20,90,111 Adult age, gross total resection, and supratentorial location have been identified as favorable prognostic indicators in some retrospective studies, whereas the effect of histopathology on outcome remains a controversial topic.
The extent of surgical resection has been shown to be the most significant prognostic factor in outcome for adults with intracranial ependymomas.12,72,92,112–118 The majority of studies report a trend toward higher survival rates 5 and 10 years after aggressive tumor extirpation. Vanuystel and associates noted a 5-year recurrence-free survival rate of 83% after gross total removal versus 47% for partially resected tumors.72 Another study observed a 10-year recurrence-free survival rate of 75% after complete resection versus 16.7% for partially resected ependymomas.119 Central nervous system dissemination may occur more often in patients with incomplete surgical resection than in those with complete resection, although metastatic seeding occurs at a lower rate in adults.70
Age of the patient has been shown to be an important independent factor in the prognosis. Adult patients appear to have a trend for a better prognosis than pediatric patients do, with 5-year survival rates of 55% to 90% versus 40% to 65%.9,12,20,21,101,108,120 It should be noted that final conclusions regarding retrospective studies are difficult because of variations in the definition of pediatric age, lower incidence in the adult population, and differences in the interpretations of histologic grading and location.72,89,90,92,93,121 It has been hypothesized that children have more immature neural tissue, which leads to more aggressive behavior of ependymomas.12,19 The higher incidence of anaplastic ependymomas and the predominance of the infratentorial location of ependymomas leading to more subtotal resections have been suggested as additional reasons for a poorer prognosis in children.
Retrospective studies have reported supratentorial ependymomas to have a better prognosis than those located in the infratentorial compartment.12,18,20,73,89,122 Infratentorial ependymomas may carry a worse prognosis as a result of the higher likelihood of subtotal resection because of possible invasion into the floor of the fourth ventricle, brainstem, or cranial nerves of the cerebellopontine angle through the foramen of Luschka.12,16,123
Controversy surrounds histopathology as a prognostic indicator. A possible explanation for the lack of histopathologic correlation to the outcome of intracranial ependymomas is the difficulty recognizing the anaplastic variant because the common criteria for anaplasia are not completely reliable for ependymomas. There is no consistency regarding histologic criteria for identifying anaplasia, determination of the anaplastic variant is not consistent for all pathologists, and there is a possibility that high-grade ependymomas may be overdiagnosed.3,5,11,12,73,111,120,124–127
Brandes AA, Cavallo G, Reni M, et al. A multicenter retrospective study of chemotherapy for recurrent intracranial ependymal tumors in adults by the Gruppo Italiano Cooperativo di Neuro-Oncologia. Cancer. 2005;104:143.
Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and Its Coverings, 3rd ed. New York: Churchill Livingstone; 1991.
Centeno RS, Lee AA, Winter J, et al. Supratentorial ependymomas. Neuroimaging and clinicopathological correlation. J Neurosurg. 1986;64:209.
Donahue B, Steinfeld A. Intracranial ependymoma in the adult patient: successful treatment with surgery and radiotherapy. J Neurooncol. 1998;37:131.
Guyotat J, Signorelli F, Desme S, et al. Intracranial ependymomas in adult patients: analyses of prognostic factors. J Neurooncol. 2002;60:255.
Healey EA, Barnes PD, Kupsky WJ, et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurgery. 1991;28:666.
Lamszus K, Lachenmayer L, Heinemann U, et al. Molecular genetic alterations on chromosomes 11 and 22 in ependymomas. Int J Cancer. 2001;91:803.
McLaughlin MP, Marcus RBJr, Buatti JM, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845.
Metellus P, Barrie M, Figarella-Branger D, et al. Multicentric French study on adult intracranial ependymomas: prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Brain. 2007;130:1338.
Paulino AC, Wen BC. The significance of radiotherapy treatment duration in intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2000;47:585.
Rawlings CE3rd, Giangaspero F, Burger PC, et al. Ependymomas: a clinicopathologic study. Surg Neurol. 1988;29:271.
Reni M, Brandes AA. Current management and prognostic factors for adult ependymoma. Expert Rev Anticancer Ther. 2002;2:537.
Reni M, Brandes AA, Vavassori V, et al. A multicenter study of the prognosis and treatment of adult brain ependymal tumors. Cancer. 2004;100:1221.
Reni M, Gatta G, Mazza E, et al. Ependymoma. Crit Rev Oncol Hematol. 2007;63:81.
Rezai AR, Woo HH, Lee M, et al. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618.
Rogers L, Pueschel J, Spetzler R, et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosurg. 2005;102:629.
Roncaroli F, Consales A, Fioravanti A, et al. Supratentorial cortical ependymoma: report of three cases. Neurosurgery. 2005;57:E192.
Ruttledge MH, Sarrazin J, Rangaratnam S, et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6:180.
Schild SE, Nisi K, Scheithauer BW, et al. The results of radiotherapy for ependymomas: the Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42:953.
Schwartz TH, Kim S, Glick RS, et al. Supratentorial ependymomas in adult patients. Neurosurgery. 1999;44:721.
Shaw EG, Evans RG, Scheithauer BW, et al. Postoperative radiotherapy of intracranial ependymoma in pediatric and adult patients. Int J Radiat Oncol Biol Phys. 1987;13:1457.
Spagnoli D, Tomei G, Ceccarelli G, et al. Combined treatment of fourth ventricle ependymomas: report of 26 cases. Surg Neurol. 2000;54:19.
Stafford SL, Pollock BE, Foote RL, et al. Stereotactic radiosurgery for recurrent ependymoma. Cancer. 2000;88:870.
Wernicke C, Thiel G, Lozanova T, et al. Involvement of chromosome 22 in ependymomas. Cancer Genet Cytogenet. 1995;79:173.
1 Oppenheim JS, Strauss RC, Mormino J, et al. Ependymomas of the third ventricle. Neurosurgery. 1994;34:350.
2 Rawlings CE3rd, Giangaspero F, Burger PC, et al. Ependymomas: a clinicopathologic study. Surg Neurol. 1988;29:271.
3 Ernestus RI, Schroder R, Stutzer H, et al. The clinical and prognostic relevance of grading in intracranial ependymomas. Br J Neurosurg. 1997;11:421.
4 Guyotat J, Signorelli F, Desme S, et al. Intracranial ependymomas in adult patients: analyses of prognostic factors. J Neurooncol. 2002;60:255.
5 Korshunov A, Golanov A, Sycheva R, et al. The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer. 2004;100:1230.
6 Reni M, Brandes AA, Vavassori V, et al. A multicenter study of the prognosis and treatment of adult brain ependymal tumors. Cancer. 2004;100:1221.
7 Nazar GB, Hoffman HJ, Becker LE, et al. Infratentorial ependymomas in childhood: prognostic factors and treatment. J Neurosurg. 1990;72:408.
8 Applegate GL, Marymont MH. Intracranial ependymomas: a review. Cancer Invest. 1998;16:588.
9 Bouffet E, Perilongo G, Canete A, et al. Intracranial ependymomas in children: a critical review of prognostic factors and a plea for cooperation. Med Pediatr Oncol. 1998;30:319.
10 Duncan JA3rd. Brain tumors of childhood. Med Health R I. 1996;79:229.
11 Gerszten PC, Pollack IF, Martinez AJ, et al. Intracranial ependymomas of childhood. Lack of correlation of histopathology and clinical outcome. Pathol Res Pract. 1996;192:515.
12 Lyons MK, Kelly PJ. Posterior fossa ependymomas: report of 30 cases and review of the literature. Neurosurgery. 1991;28:659.
13 Palma L, Celli P, Cantore G. Supratentorial ependymomas of the first two decades of life. Long-term follow-up of 20 cases (including two subependymomas). Neurosurgery. 1993;32:169.
14 Robertson PL, Zeltzer PM, Boyett JM, et al. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg. 1998;88:695.
15 Sanford RA, Gajjar A. Ependymomas. Clin Neurosurg. 1997;44:559.
16 Reni M, Brandes AA. Current management and prognostic factors for adult ependymoma. Expert Rev Anticancer Ther. 2002;2:537.
17 Metellus P, Barrie M, Figarella-Branger D, et al. Multicentric French study on adult intracranial ependymomas: prognostic factors analysis and therapeutic considerations from a cohort of 152 patients. Brain. 2007;130:1338.
18 Stuben G, Stuschke M, Kroll M, et al. Postoperative radiotherapy of spinal and intracranial ependymomas: analysis of prognostic factors. Radiother Oncol. 1997;45:3.
19 Korshunov A, Golanov A, Timirgaz V. Immunohistochemical markers for intracranial ependymoma recurrence. An analysis of 88 cases. J Neurol Sci. 2000;177:72.
20 Garrett PG, Simpson WJ. Ependymomas: results of radiation treatment. Int J Radiat Oncol Biol Phys. 1983;9:1121.
21 Spagnoli D, Tomei G, Ceccarelli G, et al. Combined treatment of fourth ventricle ependymomas: report of 26 cases. Surg Neurol. 2000;54:19.
22 Centeno RS, Lee AA, Winter J, et al. Supratentorial ependymomas. Neuroimaging and clinicopathological correlation. J Neurosurg. 1986;64:209.
23 Dohrmann GJ. Ependymomas, 2nd ed. Wilikins RH, Rengachary SS, editors. Neurosurgery, Vol 1. New York: McGraw-Hill. 1996:1195.
24 Saito T, Oki S, Mikami T, et al. Supratentorial ectopic ependymoma: a case report. No Shinkei Geka. 1999;27:1139.
25 Roncaroli F, Consales A, Fioravanti A, et al. Supratentorial cortical ependymoma: report of three cases. Neurosurgery. 2005;57:E192.
26 Lehman NL, Jorden MA, Huhn SL, et al. Cortical ependymoma. A case report and review. Pediatr Neurosurg. 2003;39:50.
27 Cosgrove GR, Villemure JG, Robitaille Y, et al. Extraaxial ependymoma of the posterior fossa. Surg Neurol. 1985;24:433.
28 Donich D, Lee JH, Prayson R. Giant extra-axial cerebellopontine angle/cavernous sinus ependymoma: case report. Neurosurgery. 1999;44:195.
29 Fukui MB, Hogg JP, Martinez AJ. Extraaxial ependymoma of the posterior fossa. AJNR Am J Neuroradiol. 1997;18:1179.
30 Hanchey RE, Stears JC, Lehman RA, et al. Interhemispheric ependymoma mimicking falx meningioma. Case report. J Neurosurg. 1976;45:108.
31 Little NS, Morgan MK, Eckstein RP. Primary ependymoma of a cranial nerve. Case report. J Neurosurg. 1994;81:792.
32 Thomson S, Chakrabarty A, Marks P. Ependymoma of the neurohypophysis. Br J Neurosurg. 2001;15:277.
33 Winer JB, Lidov H, Scaravilli F. An ependymoma involving the pituitary fossa. J Neurol Neurosurg Psychiatry. 1989;52:1443.
34 Youkilis AS, Park P, McKeever PE, et al. Parasagittal ependymoma resembling falcine meningioma. AJNR Am J Neuroradiol. 2001;22:1105.
35 Rosenblum MK. Ependymal tumors: a review of their diagnostic surgical pathology. Pediatr Neurosurg. 1998;28:160.
36 Burger PC, Scheithauer BW, Vogel FS. Surgical Pathology of the Nervous System and Its Coverings, 3rd ed. New York: Churchill Livingstone; 1991.
37 Fuller GN, Burger PC. Gliomas: Pathology, 2nd ed. Wilkins RH, Rengachary SS, editors. Neurosurgery, Vol 1. New York: McGraw-Hill. 1996:735.
38 Kleihues P, Soylemezoglu F, Schauble B, et al. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211.
39 Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215.
40 Kimura T, Budka H, Soler-Federsppiel S. An immunocytochemical comparison of the glia-associated proteins glial fibrillary acidic protein (GFAP) and S-100 protein (S100P) in human brain tumors. Clin Neuropathol. 1986;5:21.
41 Min KW, Scheithauer BW. Clear cell ependymoma: a mimic of oligodendroglioma: clinicopathologic and ultrastructural considerations. Am J Surg Pathol. 1997;21:820.
42 Friede RL, Pollak A. The cytogenetic basis for classifying ependymomas. J Neuropathol Exp Neurol. 1978;37:103.
43 Kleihues P, Burger PC, Scheithauer BW. Histological typing of tumors of the central nervous system. In WHO Blue Book, 2nd ed. Berlin: Springer; 1993.
44 Lamszus K, Lachenmayer L, Heinemann U, et al. Molecular genetic alterations on chromosomes 11 and 22 in ependymomas. Int J Cancer. 2001;91:803.
45 Weremowicz S, Kupsky WJ, Morton CC, et al. Cytogenetic evidence for a chromosome 22 tumor suppressor gene in ependymoma. Cancer Genet Cytogenet. 1992;61:193.
46 Nijssen PC, Deprez RH, Tijssen CC, et al. Familial anaplastic ependymoma: evidence of loss of chromosome 22 in tumour cells. J Neurol Neurosurg Psychiatry. 1994;57:1245.
47 Ward S, Harding B, Wilkins P, et al. Gain of 1q and loss of 22 are the most common changes detected by comparative genomic hybridisation in paediatric ependymoma. Genes Chromosomes Cancer. 2001;32:59.
48 Rubio MP, Correa KM, Ramesh V, et al. Analysis of the neurofibromatosis 2 gene in human ependymomas and astrocytomas. Cancer Res. 1994;54:45.
49 Ruttledge MH, Sarrazin J, Rangaratnam S, et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6:180.
50 Slavc I, MacCollin MM, Dunn M, et al. Exon scanning for mutations of the NF2 gene in pediatric ependymomas, rhabdoid tumors and meningiomas. Int J Cancer. 1995;64:243.
51 Bijlsma EK, Voesten AM, Bijleveld EH, et al. Molecular analysis of genetic changes in ependymomas. Genes Chromosomes Cancer. 1995;13:272.
52 Kramer DL, Parmiter AH, Rorke LB, et al. Molecular cytogenetic studies of pediatric ependymomas. J Neurooncol. 1998;37:25.
53 Sawyer JR, Sammartino G, Husain M, et al. Chromosome aberrations in four ependymomas. Cancer Genet Cytogenet. 1994;74:132.
54 von Haken MS, White EC, Daneshvar-Shyesther L, et al. Molecular genetic analysis of chromosome arm 17p and chromosome arm 22q DNA sequences in sporadic pediatric ependymomas. Genes Chromosomes Cancer. 1996;17:37.
55 Wernicke C, Thiel G, Lozanova T, et al. Involvement of chromosome 22 in ependymomas. Cancer Genet Cytogenet. 1995;79:173.
56 Bromowicz J, Araszkiewicz H, Kinderman B. Familial occurrence of neoplasms of the central nervous system. Neurol Neurochir Pol. 1971;5:721.
57 Honan WP, Anderson M, Carey MP, et al. Familial subependymomas. Br J Neurosurg. 1987;1:317.
58 Sato T, Shimoda A, Takahashi T, et al. Congenital anaplastic ependymoma: a case report of familial glioma. Childs Brain. 1984;11:342.
59 Sieb JP, Pulst SM, Buch A. Familial CNS tumors. J Neurol. 1992;239:343.
60 Yokota T, Tachizawa T, Fukino K, et al. A family with spinal anaplastic ependymoma: evidence of loss of chromosome 22q in tumor. J Hum Genet. 2003;48:598.
61 Reni M, Gatta G, Mazza E, et al. Ependymoma. Crit Rev Oncol Hematol. 2007;63:81.
62 Suzuki SO, Iwaki T. Amplification and overexpression of mdm2 gene in ependymomas. Mod Pathol. 2000;13:548.
63 Piepmeier JM. Tumors and approaches to the lateral ventricles. Introduction and overview. J Neurooncol. 1996;30:267.
64 Palma L. Ependymoma. J Neurosurg. 2006;105:503.
65 Spoto GP, Press GA, Hesselink JR, et al. Intracranial ependymoma and subependymoma: MR manifestations. AJR Am J Roentgenol. 1990;154:837.
66 Sun B, Wang CC, Wang J. MRI characteristics of midbrain tumours. Neuroradiology. 1999;41:158.
67 Epstein BS, Epstein JA, Carras R. Extension of posterior fossa tumors, particularly intraventricular fourth ventricle tumors, into the upper cervical spinal canal. Am J Roentgenol Radium Ther Nucl Med. 1970;110:31.
68 Lefton DR, Pinto RS, Martin SW. MRI features of intracranial and spinal ependymomas. Pediatr Neurosurg. 1998;28:97.
69 Salazar OM. A better understanding of CNS seeding and a brighter outlook for postoperatively irradiated patients with ependymomas. Int J Radiat Oncol Biol Phys. 1983;9:1231.
70 Rezai AR, Woo HH, Lee M, et al. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618.
71 Donahue B, Steinfeld A. Intracranial ependymoma in the adult patient: successful treatment with surgery and radiotherapy. J Neurooncol. 1998;37:131.
72 Vanuytsel LJ, Bessell EM, Ashley SE, et al. Intracranial ependymoma: long-term results of a policy of surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 1992;23:313.
73 McLaughlin MP, Marcus RBJr, Buatti JM, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845.
74 Read G. The treatment of ependymoma of the brain or spinal canal by radiotherapy: a report of 79 cases. Clin Radiol. 1984;35:163.
75 Duffner PK, Krischer JP, Sanford RA, et al. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr Neurosurg. 1998;28:215.
76 Foreman NK, Love S, Thorne R. Intracranial ependymomas: analysis of prognostic factors in a population-based series. Pediatr Neurosurg. 1996;24:119.
77 Asgari S, Engelhorn T, Brondics A, et al. Transcortical or transcallosal approach to ventricle-associated lesions: a clinical study on the prognostic role of surgical approach. Neurosurg Rev. 2003;26:192.
78 Bellotti C, Pappada G, Sani R, et al. The transcallosal approach for lesions affecting the lateral and third ventricles. Surgical considerations and results in a series of 42 cases. Acta Neurochir (Wien). 1991;111:103.
79 Kempe LG, Blaylock R. Lateral-trigonal intraventricular tumors. A new operative approach. Acta Neurochir (Wien). 1976;35:233.
80 Lapras C, Deruty R, Bret P. Tumors of the lateral ventricles. Adv Tech Stand Neurosurg. 1984;11:103.
81 Zuccaro G, Sosa F, Cuccia V, et al. Lateral ventricle tumors in children: a series of 54 cases. Childs Nerv Syst. 1999;15:774.
82 Shucart W. Anterior transcallosal and transcortical approaches. In: Apuzzo MLJ, editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1987:303.
83 Nakasu Y, Isozumi T, Nioka H, et al. Mechanism of mutism following the transcallosal approach to the ventricles. Acta Neurochir (Wien). 1991;110:146.
84 Geffen G, Walsh A, Simpson D, et al. Comparison of the effects of transcortical and transcallosal removal of intraventricular tumours. Brain. 1980;103:773.
85 Jeeves MA, Simpson DA, Geffen G. Functional consequences of the transcallosal removal of intraventricular tumours. J Neurol Neurosurg Psychiatry. 1979;42:134.
86 Foreman NK, Love S, Gill SS, et al. Second-look surgery for incompletely resected fourth ventricle ependymomas: technical case report. Neurosurgery. 1997;40:856.
87 Terasaki M, Uchikado H, Takeuchi Y, et al. Minimally invasive management of ependymoma of the aqueduct of Sylvius: therapeutic considerations and management. Minim Invasive Neurosurg. 2005;48:322.
88 Luther N, Souweidane MM. Neuroendoscopic resection of posterior third ventricular ependymoma. Case report. Neurosurg Focus. 2005;18(6A):E3.
89 Shaw EG, Evans RG, Scheithauer BW, et al. Postoperative radiotherapy of intracranial ependymoma in pediatric and adult patients. Int J Radiat Oncol Biol Phys. 1987;13:1457.
90 Schild SE, Nisi K, Scheithauer BW, et al. The results of radiotherapy for ependymomas: the Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42:953.
91 Rogers L, Pueschel J, Spetzler R, et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosurg. 2005;102:629.
92 Kovalic JJ, Flaris N, Grigsby PW, et al. Intracranial ependymoma long term outcome, patterns of failure. J Neurooncol. 1993;15:125.
93 Paulino AC, Wen BC. The significance of radiotherapy treatment duration in intracranial ependymoma. Int J Radiat Oncol Biol Phys. 2000;47:585.
94 Jawahar A, Kondziolka D, Flickinger JC, et al. Adjuvant stereotactic radiosurgery for anaplastic ependymoma. Stereotact Funct Neurosurg. 1999;73:23.
95 Stafford SL, Pollock BE, Foote RL, et al. Stereotactic radiosurgery for recurrent ependymoma. Cancer. 2000;88:870.
96 Mansur DB, Drzymala RE, Rich KM, et al. The efficacy of stereotactic radiosurgery in the management of intracranial ependymoma. J Neurooncol. 2004;66:187.
97 Lo SS, Abdulrahman R, Desrosiers PM, et al. The role of Gamma Knife radiosurgery in the management of unresectable gross disease or gross residual disease after surgery in ependymoma. J Neurooncol. 2006;79:51.
98 Brandes AA, Cavallo G, Reni M, et al. A multicenter retrospective study of chemotherapy for recurrent intracranial ependymal tumors in adults by the Gruppo Italiano Cooperativo di Neuro-Oncologia. Cancer. 2005;104:143.
99 Needle MN, Goldwein JW, Grass J, et al. Adjuvant chemotherapy for the treatment of intracranial ependymoma of childhood. Cancer. 1997;80:341.
100 Needle MN, Molloy PT, Geyer JR, et al. Phase II study of daily oral etoposide in children with recurrent brain tumors and other solid tumors. Med Pediatr Oncol. 1997;29:28.
101 Souweidane MM, Bouffet E, Finlay J. The role of chemotherapy in newly diagnosed ependymoma of childhood. Pediatr Neurosurg. 1998;28:273.
102 Sutton LN, Goldwein J, Perilongo G, et al. Prognostic factors in childhood ependymomas. Pediatr Neurosurg. 1990;16:57.
103 Timmermann B, Kortmann RD, Kuhl J, et al. Combined postoperative irradiation and chemotherapy for anaplastic ependymomas in childhood: results of the German prospective trials HIT 88/89 and HIT 91. Int J Radiat Oncol Biol Phys. 2000;46:287.
104 Grill J, Kalifa C. High dose chemotherapy for childhood ependymoma. J Neurooncol. 1998;40:97.
105 Grill J, Kalifa C, Doz F, et al. A high-dose busulfan-thiotepa combination followed by autologous bone marrow transplantation in childhood recurrent ependymoma. A phase-II study. Pediatr Neurosurg. 1996;25:7.
106 Grill J, Le Deley MC, Gambarelli D, et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288.
107 Grill J, Pascal C, Chantal K. Childhood ependymoma: a systematic review of treatment options and strategies. Paediatr Drugs. 2003;5:533.
108 Evans AE, Anderson JR, Lefkowitz-Boudreaux IB, et al. Adjuvant chemotherapy of childhood posterior fossa ependymoma: cranio-spinal irradiation with or without adjuvant CCNU, vincristine, and prednisone: a Children’s Cancer Group study. Med Pediatr Oncol. 1996;27:8.
109 Gornet MK, Buckner JC, Marks RS, et al. Chemotherapy for advanced CNS ependymoma. J Neurooncol. 1999;45:61.
110 Davis C, Symon L. Posterior fossa ependymomas in adults. Acta Neurochir (Wien). 1986;82:115.
111 Ross GW, Rubinstein LJ. Lack of histopathological correlation of malignant ependymomas with postoperative survival. J Neurosurg. 1989;70:31.
112 Awaad YM, Allen JC, Miller DC, et al. Deferring adjuvant therapy for totally resected intracranial ependymoma. Pediatr Neurol. 1996;14:216.
113 Figarella-Branger D, Civatte M, Bouvier-Labit C, et al. Prognostic factors in intracranial ependymomas in children. J Neurosurg. 2000;93:605.
114 Oya N, Shibamoto Y, Nagata Y, et al. Postoperative radiotherapy for intracranial ependymoma: analysis of prognostic factors and patterns of failure. J Neurooncol. 2002;56:87.
115 Palma L, Celli P, Mariottini A, et al. The importance of surgery in supratentorial ependymomas. Long-term survival in a series of 23 cases. Childs Nerv Syst. 2000;16:170.
116 Paulino AC, Wen BC, Buatti JM, et al. Intracranial ependymomas: an analysis of prognostic factors and patterns of failure. Am J Clin Oncol. 2002;25:117.
117 Healey EA, Barnes PD, Kupsky WJ, et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurgery. 1991;28:666.
118 Ferrante L, Mastronardi L, Schettini G, et al. Fourth ventricle ependymomas. A study of 20 cases with survival analysis. Acta Neurochir (Wien). 1994;131:67.
119 Schwartz TH, Kim S, Glick RS, et al. Supratentorial ependymomas in adult patients. Neurosurgery. 1999;44:721.
120 Schiffer D, Giordana MT. Prognosis of ependymoma. Childs Nerv Syst. 1998;14:357.
121 Wallner KE, Wara WM, Sheline GE, et al. Intracranial ependymomas: results of treatment with partial or whole brain irradiation without spinal irradiation. Int J Radiat Oncol Biol Phys. 1986;12:1937.
122 Sala F, Talacchi A, Mazza C, et al. Prognostic factors in childhood intracranial ependymomas: the role of age and tumor location. Pediatr Neurosurg. 1998;28:135.
123 Pierre-Kahn A, Hirsch JF, Roux FX, et al. Intracranial ependymomas in childhood. Survival and functional results of 47 cases. Childs Brain. 1983;10:145.
124 Schiffer D, Chio A, Cravioto H, et al. Ependymoma: internal correlations among pathological signs: the anaplastic variant. Neurosurgery. 1991;29:206.
125 Schiffer D, Chio A, Giordana MT, et al. Histologic prognostic factors in ependymoma. Childs Nerv Syst. 1991;7:177.
126 Salazar OM, Castro-Vita H, VanHoutte P, et al. Improved survival in cases of intracranial ependymoma after radiation therapy. Late report and recommendations. J Neurosurg. 1983;59:652.
127 Kricheff II, Becker M, Schneck SA, et al. Intracranial ependymomas. A study of survival in 65 cases treated by surgery and irradiation. Am J Roentgenol Radium Ther Nucl Med. 1964;91:167.