[level-membership-for-neurosurgery-category]
Chapter 13 Intervertebral Disc Process of Degeneration
Physiology and Pathophysiology
In the United States alone, nearly 5.7 million people are diagnosed with intervertebral disc disorders each year.1,2 These disorders are responsible for widespread disability and account for tremendous costs associated with their treatment and loss of productivity. The intervertebral disc is a unique multifunctional structure. It provides a major stabilizing effect for the spine and allows for the dissipation of axial load, and yet is still flexible enough to confer significant mobility. Despite these essential roles in spinal mechanics, the disc also begins to show signs of biochemical and structural change almost immediately after birth, and signs of degeneration are present at an extremely young age.3–5 Unfortunately, these degenerative changes are often progressive, rendering the disc biochemically and biomechanically incapable of absorbing the forces necessary for normal physiologic spinal function and potentially leading to rapid degeneration of the spine.
Understanding the anatomy and function of the normal disc serves as the framework for comprehension of the complex processes of aging and the pathologic degeneration that afflict the disc. One of the most significant challenges to a complete understanding of disc disease is the lack of consistency with which radiographically detectable signs of disc degeneration translate to clinical symptomatology.6 As such, there is abundant evidence to suggest that aberrant disc morphology does not necessarily equate with back pain.6,7 This poses a significant challenge to researchers in this field because individual variation in disc degeneration makes it difficult to distinguish between age-related alterations and pathologic premature degenerative changes.5,8 In the future, the ability to make this distinction will lead to improved clarity regarding the complex process of disc degeneration.
Normal Intervertebral Disc
The intervertebral disc is composed of water, proteoglycan, collagen, and a small amount of noncollagenous proteins. There are three major regions to the intervertebral disc with differing structural compositions: the nucleus pulposus, anulus fibrosus, and cartilaginous end plates. Each has different functions. Of significant note, the intervertebral disc is one of the most hypocellular structures in the body9 (Fig. 13-1).
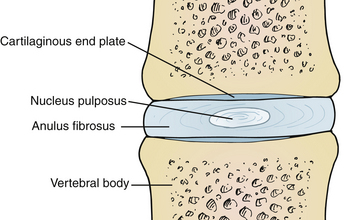
Anulus Fibrosus
The anulus fibrosus is the outer circumferential portion of the disc and is often subdivided into the inner and outer anulus. The outer anulus is a collection of highly organized, densely packed collagen fibrils, creating a lamellar structure of up to 25 lamellae in the mature disc.9,10 These collagen fibrils insert into the adjacent vertebral end plates and join them together to stabilize the spine. In addition to its predominantly collagenous structure, a small amount of elastin is present in the anulus, which helps it to restore and recoil after stretching in response to compression and tensile forces.9,11 The strong anulus provides the ability to absorb significant hoop stresses and maintain stiffness in the presence of the tensile forces induced by bending and twisting of the spine.12,13 The importance of this ability to resist hoop stress has been shown in large animal studies, which demonstrated that even a partial-thickness laceration into the anulus rapidly produced advanced disc degeneration.12,14 The outer anulus is populated with a low density of fibroblast-like cells.15 It is the only tissue in the mature disc that remains innervated and is capable of producing pain when exposed to noxious stimuli.16–19
The inner anulus appears to serve as a transition zone to the vastly different biochemical composition of the nucleus. It is less dense and lacks the organized lamellar structure of the outer anulus. It is populated with rounder, chondrocyte-like cells, and the concentration of proteoglycans and water content is increased.20
Nucleus Pulposus
The nucleus pulposus, the inner, less-organized region of the disc, occupies the middle and posterior thirds of the intervertebral disc.21 It consists of a rich proteoglycan matrix with a lesser amount of disorganized collagen than in the anulus.20,22 There is a significantly higher content of water in the nucleus. The large amount of proteoglycan and water present in the nucleus make it ideal for developing the “swelling pressures” necessary to resist the compressive forces of the spine. At birth, the nucleus is populated almost exclusively with notochordal cells. These cells rapidly begin to necrose shortly after birth and by the age of 4 to 10 years they are completely replaced by rounder, chondrocyte-like cells of mesenchymal origin9 (Table 13-1).
| Component | Anulus Fibrosus | Nucleus Pulposus |
|---|---|---|
| Collagen (mainly types I and II; others present include types III, V, VI, IX, XI, XII, and XIV) | 70% of dry weight 80% type I collagen |
20% of dry weight 80% type II collagen |
| Proteoglycan | 25% of dry weight | 50%–70% of dry weight |
| Water | Lowest content | Highest content |
Cartilaginous End Plate
The cartilaginous end plate is a hyaline cartilage layer separating the disc from the adjacent bony vertebral end plate. It is usually less than 1 mm thick in the mature disc and lies immediately adjacent to the perforated bony end plate. It is loosely cemented to the underlying bony end plate by a thin layer of calcium that is absent near the perforated areas of the bony plate, which allows for nutrient diffusion from the vertebra to the disc.21
Matrix of the Intervertebral Disc
The matrix content of the intervertebral disc is critical to disc function. The uniquely hydrophilic nature of proteoglycans provides the swelling pressure required to increase the intradiscal water content necessary to resist compressive strains on the spine.9,23,24 The matrix composition changes with age, and the quantitative loss and qualitative deterioration of the proteoglycans is thought to represent the single largest biochemical contribution to disc degeneration.20,25
Proteoglycans are highly negatively charged molecules consisting of a core protein and chains of glycosaminoglycans radiating from the core. The chains are composed mainly of keratin and chondroitin sulfate. Multiple proteoglycans then are linked to hyaluronic acid to form large proteoglycan aggregates capable of producing even larger swelling pressures. The aggregates are held together by type II collagen and are cross-linked by type IX collagen.23,26
Aggrecan is the most abundant proteoglycan in the disc.20 It is responsible for 70% of the dry weight of the nucleus pulposus and 25% of the anulus fibrosus. Other proteoglycans present at lower levels include decorin, biglycan, fibromodulin, lumican, versican, and perlican.20,27
Nutrition of the Intervertebral Disc
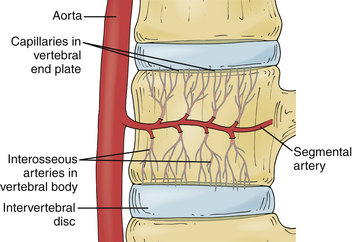
The intervertebral disc is the largest avascular tissue in the human body. Nutritional support for cell survival, matrix production, and disc homeostasis occurs through diffusion of nutrients through the vertebral end plates.28–30 In the newborn and young child, small blood vessels penetrate the cartilaginous end plates and enter the disc anulus and nucleus.21 These vessels rapidly become less efficient, yet remain partially functional until about 8 years of age. They then become completely nonfunctional over the next two decades of life.5,21,28,31,32
The vertebral body is supplied by an arterial network that drains into a subchondral venous plexus within the bone.28,33 Capillaries from this region penetrate into the subchondral plate and terminate in loops at the bony end plate–cartilaginous end plate junction.28,34,35 Through these capillaries, the nutrients necessary for disc metabolism are delivered and diffuse through the cartilaginous end plates to reach the center of the disc.28 The end plate acts as a semipermeable membrane, letting smaller neutral charges (water, amino acids, oxygen) through more easily than larger, positively charged molecules28,36–38 (Fig. 13-2).
Modulation and Regulation of Disc Matrix
Anabolic Influences
The intervertebral disc matrix is in a constant state of dynamic flux. Anabolic and catabolic influences on the disc matrix continuously interact with the disc to alter the physical composition of the matrix.23 Metalloproteinases (MMPs) modulate the matrix composition by breaking down aged proteoglycans under catabolic influences while newly synthesized matrix takes the place of the degraded proteoglycans.23,39 A very complex, multifactorial, poorly understood interaction of mechanical and biochemical factors defines whether a disc is capable of remodeling in response to loading conditions in order to maintain disc homeostasis.1 If the disc is unable to remodel and maintain a balance of anabolic and catabolic influences, then damage accumulates in a process that may eventually self-perpetuate into a state of continued degeneration.
The key anabolic modulating growth factors work by stimulating the chondrocyte-like cells to produce matrix while inhibiting the MMPs present in the disc.1 In addition to various growth factors, tissue inhibitors of MMPs (TIMPs) are present to suppress the activation of the MMPs23,40 (Table 13-2).
TABLE 13-2 Key Anabolic Modulators (Growth Factors) of the Intervertebral Disc
| Growth Factor | Role |
|---|---|
| Insulin-like growth factor-1 (IGF-1) | Stimulates proteoglycan synthesis Reduces cellular apoptosis |
| Epidermal growth factor (EGF) | Stimulates matrix synthesis Cellular proliferation |
| Basic fibroblast growth factor (bFGF) | Stimulates proteoglycan synthesis Cellular proliferation |
| Platelet-derived growth factor (PDGF) | Decreases cellular apoptosis |
| Transforming growth factor beta (TGF-β) | Greatly increased proteoglycan synthesis Cellular proliferation |
| Bone morphogenetic protein-2 (BMP-2) | Increased proteoglycan synthesis Cellular proliferation Increased mRNA for type II collagen, aggrecan, sox9, and osteocalcin Up-regulates gene expression of BMP-6, BMP-7, and TGF-β |
| Bone morphogenetic protein-7 (BMP-7; osteogenic protein-1 [OP-1]) | Increased proteoglycan synthesis Increased collagen synthesis Cellular proliferation |
| Link protein N-terminal peptide | In concentrations of 10–100 μg/mL stimulates matrix assembly |
From Masuda K, Oegema TJ, An H: Growth factors and treatment of intervertebral disc degeneration. Spine (Phila Pa 1976) 29:2757, 2004.
Catabolic Influences
Catabolic modulation in the disc occurs mainly through cytokine-induced and metalloproteinase degradation of matrix components.23 MMPs 1, 2, 3, 8, 9, and 13 are all present in the disc and serve to degrade collagen, aggrecan, versican, link protein, and the other proteoglycans.9,41–45 The aggrecanases ADAM-TS-4 and ADAM-TS-5, from the ADAM (a disintegrin and metalloproteinase) protein family, are also present in the disc and mainly function to degrade aggrecan and versican.9,46 These MMPs and aggrecanases allow for the large proteoglycans to be degraded into smaller components that may be more easily leached from the intervertebral disc by diffusion.20 In a pathologic catabolic state when the MMPs are working with little opposing matrix production, a significant net loss of proteoglycan and water content occurs, rendering the disc incapable of producing the swelling pressure necessary to resist compressive forces on the disc.20 Overproduction of MMPs can thus lead to increased forces on other spinal structures and contribute to disc and overall spinal degeneration.
In addition to the catabolic effect of MMPs, growing evidence suggests that inflammatory cytokines may have a significant catabolic influence on the disc.23,47,48 Animal studies have indicated that in the presence of injury to the intervertebral disc there are increased levels of interleukin (IL)-1, tumor necrosis factor alpha (TNF-α), and IL-8 near the injured regions of the disc.49 These cytokines have negative catabolic effects on the disc and also stimulate the disc cells to produce chemokines, which are responsible for macrophage attraction and stimulation, especially in the presence of disc herniation.48,50 Macrophages in the disc usually reflect an attempt to break up or remove herniated disc fragments, but their presence also leads to further inflammation and cytokine release, which may prove damaging to the disc.23,51 These inflammatory substances are also responsible for the activation of multiple cascades within the disc cell nuclei that lead to the production of metalloproteinases, which in turn contribute to matrix breakdown.48,52 In addition, these inflammatory cytokines are thought to contribute to the pain-producing effects of disc degeneration48,49,53,54 (Table 13-3).
TABLE 13-3 Key Catabolic Modulators of the Intervertebral Disc
| Catabolic Modulators | Actions |
|---|---|
| Interleukin-1 (IL-1) | Inhibits synthesis of matrix Promotes metalloproteinase production Stimulates nitric oxide synthetase to produce nitric oxide Enhances caseinase activity Triggers the translocation of transcription factors to cell nuclei that target genes that include collagenases and COX-2 |
| Tumor necrosis factor alpha (TNF-α) | Inhibits synthesis of matrix Promotes metalloproteinase production Stimulates nitric oxide synthetase to produce nitric oxide Triggers the translocation of transcription factors to cell nuclei that target genes that include collagenases and COX-2 |
| Nitric oxide | Produces free radicals that damage cell membranes and induce degenerative pathways Inhibits tissue inhibitors of metalloproteinases (TIMPs) |
| Interleukin-6 and interleukin-9 (IL-6, IL-9) fibronectin superoxide | Proinflammatory and pro-nociceptive effect stimulates proteoglycan degradation Degrades hyaluronic acid, causing aggregate breakdown |
COX-2, cyclooxygenase-2.
Data from references 1, 9, 48; and Anderson DG, Tannoury C: Molecular pathogenic factors in symptomatic disc degeneration. Spine J 5(6 Suppl):260S, 2005.
Degeneration
It is very difficult to differentiate the normal degenerative changes of an aging disc from those of pathologic disc degeneration.5,8 It is also crucial to understand that even pathologic disc degeneration is not always accompanied by back pain, and therefore degeneration as discussed here does not necessarily translate to clinical symptomatology. The biochemical and cellular structure of the intervertebral disc begins changing almost immediately after birth, and the process progresses through life.3–5 It is useful to divide disc changes into the following two categories:
Gross and Histologic Changes
The hallmark histologic finding in intervertebral disc degeneration is the progressively diminishing vascular supply, which begins shortly after birth and leads to tissue breakdown as early as the second decade of life.5 Changes in the nucleus pulposus also begin immediately after birth. Within the first two decades of life the nucleus begins to undergo mucoid degeneration, increasing cleft formation, and granular changes consistent with granulation tissue formation at sites of disc injury. These clefts and granular changes eventually lead to frank tears that communicate with anular rim lesions.5,21
In the anulus, peripheral rim lesions appear late in the second decade of life and eventually progress to frank tears that communicate with the nuclear clefts. The end plate also shows degenerative changes that begin with the loss of the end plate vessels early in childhood. After the loss of these vessels, cartilage disorganization increases and cracks in the cartilaginous end plate begin to appear. Microfractures in the adjacent subchondral bone follow and new subchondral bone formation and eventual sclerosis of the plate occur. All of these changes eventually lead to a “burned-out” state,5 apparent in the seventh and later decades of life. At this time, all components of the disc begin to resemble scar tissue and there is no longer a distinction between the different regions of the disc5,21 (Table 13-4 and Figs. 13-3 to 13-6).
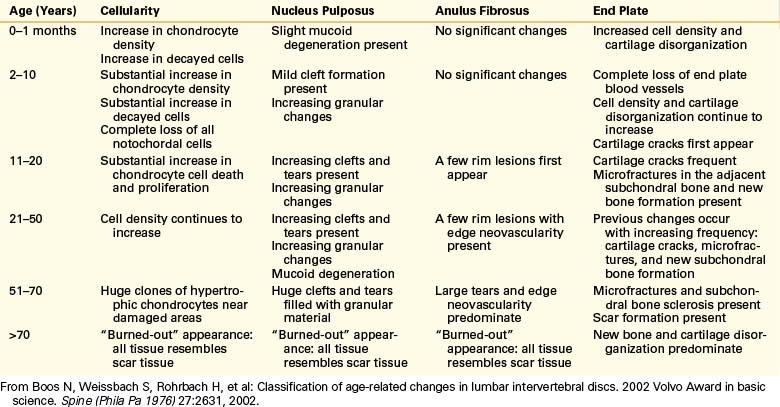

FIGURE 13-3 Sagittal section through a normal intervertebral disc.
(From Thalgott J, Albert T, Vaccaro A, et al: A new classification system for degenerative disc disease of the lumbar spine based on magnetic resonance imaging, provocative discography, plain radiographs and anatomic considerations. Spine J 4[6 Suppl]:167S, 2004.)

FIGURE 13-4 Sagittal section through a disc with mild degeneration.
(From Thalgott J, Albert T, Vaccaro A, et al: A new classification system for degenerative disc disease of the lumbar spine based on magnetic resonance imaging, provocative discography, plain radiographs and anatomic considerations. Spine J 4[6 Suppl]:167S, 2004.)


The cellular content of the disc also changes over time. The earliest changes occur shortly after birth and include replacement of the rapidly decaying notochordal cells with mesenchymal chondrocyte-like cells.9 Cell density in both the nucleus and the anulus also rapidly declines after birth. This occurs as the matrix grows along with the expanding volume of the intervertebral disc. The cell density in the nucleus pulposus shortly after birth is approximately 4 × 106 cells/cm3 and in the anulus fibrosus, 9 × 106 cells/cm3. This is significantly lower then the cellular density of even relatively acellular hyaline cartilage,9 which has about 1.4 × 107 cells/cm3. As the disc continues to age, cell death accelerates and large numbers of decaying cells can be found in clusters. By adulthood, more than 50% of the disc’s cells are necrotic55 (Fig. 13-7 and Table 13-5).
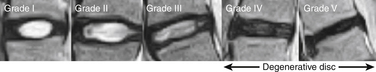
FIGURE 13-7 Midsagittal view on T2 density-weighted MRI of discs graded according to a modified Pfirrmann classification (see Table 13-5).
(From Hangai M, Kaneoka K, Kuno S, et al: Factors associated with lumbar intervertebral disc degeneration in the elderly. Spine J 8:732, 2008.)
TABLE 13-5 Modified Pfirrmann Classification as Evaluated on Midsagittal T2-Density-Weighted MRI
| Grade | Signal Intensity of Nucleus Pulposus | Distinction between Nucleus and Anulus |
| I | Hyperintense or isointense to CSF (bright white) and homogeneous | Clear |
| II | Hyperintense of isointense to CSF (white) and inhomogeneous | Clear |
| III | Intermediate to CSF (light gray) and inhomogeneous | |
| IV | Hypointense to CSF (dark gray) and inhomogeneous | |
| V | Low intense to CSF (black) and inhomogeneous |
CSF, cerebrospinal fluid.
Adapted from Hanagai M, Kaneoka K, Kuno S, et al: Factors associated with lumbar intervertebral disc degeneration in the elderly. Spine J 8:732, 2008.
Biochemical Changes
The most significant biochemical change in the degenerating disc is the net loss of proteoglycan.20,25 In addition to this quantitative loss of proteoglycan, the quality of the remaining proteoglycan is compromised.9,27,56 With the loss of large quantities of aggregated proteoglycan, the disc’s ability to create the swelling pressure necessary to absorb the forces imposed on it decreases. These changes also begin shortly after birth, when the rich proteoglycan matrix composition is altered. At birth, chondroitin sulfate is the predominant glycosaminoglycan in the disc, but as the blood supply is progressively lost and the disc volume increases, the disc contents become increasingly anaerobic. Because of the lack of oxygen, chondroitin sulfate is progressively replaced by keratan sulfate chains, which are created in the absence of oxygen.9,57–59
In addition to the changing glycosaminoglycan composition in the disc, the overall structure of the large proteoglycans changes. In the young disc, aggrecan is the predominant proteoglycan. Multiple aggrecan molecules bind to hyaluronan to form an even larger structure called an aggregate. In the fetal and newborn disc, aggregating proteoglycans predominate and are extremely effective in creating large swelling pressures.9,60,61 The amount of aggregating proteoglycan is rapidly diminished with age by proteolytic degradation of the large aggregates. By 6 months of age, only 50% of the anular and 30% of the nuclear proteoglycan content is in aggregated form. In the mature adult disc, only 10% of the proteoglycan is aggregated and these aggregates are smaller and have a higher keratan sulfate composition compared with their younger equivalents.27,62 These degenerative features of disc proteoglycans may make them less effective in creating the swelling pressure necessary to resist compressive forces9 (Fig. 13-8).
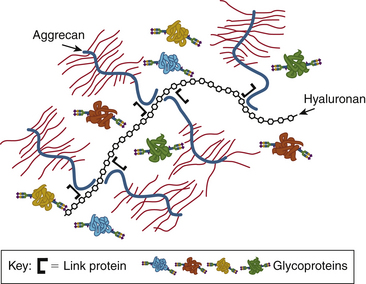
FIGURE 13-8 The large aggrecan molecule forms an aggregate with hyaluronan and link protein.
(From Esko JD, Kimata K, Lindahl U: Proteoglycans and sulfated glycosaminoglycans. In Varki A, Cummings RD, Esko JD, et al., editors: Essentials of glycobiology, ed 2, Cold Spring Harbor, NY, 2009, Cold Spring Harbor Laboratory Press. Online text version available at www.ncbi.nlm.nih.gov/books/NBK1900/.)
The collagen content of the disc is not immune to change, also exhibiting signs of degeneration. The disc collagen demonstrates evidence of proteolytic collagenase action, which accumulates and eventually contributes to the weakening mechanical properties of the disc.9,57,63 It is significant that the triple helix structure of disc collagen is more denatured and damaged than that of the collagen in articular cartilage, which may indicate less effective repair mechanisms in the disc.57 In addition, the collagen present in the disc appears to be nonenzymatically cross-linked by advanced glycation end products (AGEs).9,64–66
AGEs are protein modifiers formed through the Maillard reaction when an amino acid and a reducing sugar react.66 With age, AGEs accumulate in various tissues in the body such as the skin, myocardial cells, renal cells, chondrocytes, and intervertebral discs.66–70 AGEs originally were given substantial attention because of their increased presence in diabetes.71 After research elucidated the presence and negative effect of AGEs on articular cartilage, their presence and potential role in intervertebral disc degeneration were sought.66,72,73 in vitro and animal studies suggest that AGEs play a role in disc degeneration. Their increased concentration in diabetes may contribute to the increased relative risk for disc herniation seen in at least one study of patients with diabetes.74
AGEs likely influence disc degeneration through negative biologic and biomechanical contributions. Biomechanical studies have demonstrated that the accumulation of AGEs leads to alterations in the disc’s physical properties, the most significant of which is increased mechanical stiffness.75 This increased stiffness of the disc also likely leads to a decreased ability to produce the swelling pressure necessary for normal disc function.9,69 Biologic in vitro studies have also demonstrated that the accumulation of AGEs in intervertebral disc cells leads to a down-regulation in the production of aggrecan.66 In addition, a recent bovine study documented the increased expression of the receptor for AGEs (RAGE) in intervertebral discs in the presence of an inflammatory state with increased IL-1β. All of these studies noted a decrease in aggrecan production, which may at least partially explain the increased disc degeneration seen in the presence of chronic inflammation.48,76
Disc Material Properties and Disc Degeneration
The gross structural and biochemical changes associated with early degeneration lead to changes in the intervertebral disc’s mechanical properties, which make it less capable of performing its necessary functions.77 Cadaveric studies conducted on discs of various ages and degrees of degeneration have demonstrated an increased shear modulus in aged and degenerated discs, indicating a decreased ability of the disc to absorb energy.77–79 In essence, the discs become increasingly stiff and appear to undergo a transition from “fluid-like” behavior to “solid-like” behavior with increasing degeneration.80 Alterations in disc viscoelasticity have also been seen, including both increased creep and creep rate. In addition to these changes, which more drastically affect the nucleus, alterations in the anulus result in a decreased Poisson’s ratio as well as decreased radial permeability, which transforms the anisotropic hydraulic permeability of the normal disc to a more isotropic permeability in degenerated discs81 (Box 13-1).
BOX 13-1 Physical Properties of the Degenerated Disc
From Iatridis J, Maclean J, Roughley P, et al: Effects of mechanical loading on intervertebral disc metabolism in vivo. J Bone Joint Surg [Am] 88(Suppl 2):41, 2006.
With the compromised material properties demonstrated in degenerative models, abnormal motion segments are to be expected and have been demonstrated in cadaveric models.82 These studies suggest that early human disc degeneration and the changing physical properties of the disc may affect the overall mechanical function of the spine, leading to altered dynamics and increased intradiscal pressure as well as increased internal stress and strain patterns on the disc, which ultimately may contribute to further disc degeneration.82–84
Etiology of Disc Degeneration
We have discussed the basic components of the disc and the structural and biochemical changes that occur with disc degeneration; in the following sections, we examine the etiology of disc degeneration. As recently as the 1990s, the general belief was that age, sex, occupation, cigarette smoking, and vibration were the leading contributory factors to disc degeneration.85 Ironically, the contributions of height, weight, and genetics were less certain. Monozygotic twin studies performed over the past two decades have, however, provided tremendous insight into the etiology of disc degeneration and have strongly suggested that environmental and constitutional risk factors are only minor contributors to disc degeneration. Variations in disc degeneration can instead largely be explained by genetic factors.85–87
Even with the insight monozygotic twin studies have provided into the importance of the genetic contribution to disc degeneration, there is still a large amount of variation in the process that remains to be explained. This variation is probably the result of a complex, multifactorial set of gene-gene and gene-environment interactions that will not be amenable to linear regression modeling.85,88,89 The following sections discuss the proposed genetic and environmental influences on disc degeneration.
Genetic Influences
In 1995, Battié et al.86 published a study on the determinants of lumbar disc degeneration in which they looked at risk factors for disc degeneration in monozygotic male twins discordant for suspected environmental risk factors. Their findings provided strong support for the hypothesis that disc degeneration could largely be explained by a combination of genetic influences and still unidentified factors, which may represent complex, unpredictable genetic or other interactions. When looking at the upper lumbar region, they found that physical loading could explain only 7% of the variability in degeneration summary scores. Age could explain an additional 9%, but genetics could explain 61% of the variability. In the lower lumbar region, physical loading could explain 2%, age 7%, and genetics 34% of the variability.
With twin studies suggesting a strong genetic component to disc degeneration, further investigation to characterize the genetic influence on disc disease followed. At present, uncertainty remains as to whether a single gene has a predominant effect on disc degeneration or if degeneration is due to the smaller influence of many genes. Osteoarthritis, a similar degenerative condition of the musculoskeletal system, is considered to be an oligogenic, multifactorial genetic disease; disc degeneration is likely to be as well.85,90
To date, several gene loci have been identified as associated with disc degeneration or related pathology.91 These associations have been identified predominantly with small sample sizes and there has been significant variation in phenotypic definition. Despite small sample sizes and the challenges of phenotypic definition, there currently exists reasonable reproducible evidence among different ethnic populations to suggest the association of disc degeneration with the vitamin D receptor gene (VDR), the collagen IX genes (COL9A2, COL9A3), and the metalloproteinase-3 gene (MMP3).91 Despite their well-documented associations with disc degeneration, each of these genes has demonstrated only modest effects, suggesting that the most significant genetic susceptibilities have yet to be identified.91
The genetic contributions studied most frequently thus far relate to the biologic processes associated with matrix morphology, synthesis, and degradation, and the collagenous structures of the disc.85,91 Despite these probable genetic contributions to disc degeneration, other mechanisms and risk factors for disc degeneration likely exist. In recent studies by Videman et al.,92,93 some of the physical characteristics with genetic correlations were evaluated, including body height, weight, fat content, axial disc area, isokinetic lifting performance, and lifetime routine physical activities at work and leisure. Body weight, lifting strength, and axial disc area were more highly associated with disc degeneration than occupational and leisure physical activities, although all of them had minor influences on disc degeneration. In addition, higher body mass, greater lifting strength, and heavier work were all associated with increased disc height narrowing but less disc desiccation, confusing the matter. Smaller discs also appeared to have beneficial effects in these studies.
Videman et al.92,93 demonstrated the complexity of the genetic contribution to disc degeneration. As genetic research continues, gene-gene and gene-environment interactions will continue to be discovered, and a complete understanding of this complex process will be difficult to attain. For this reason, linear regression models will probably fail to capture the complexity of genetic influence on disc degeneration, and more advanced analytic techniques will be required.85,89 A summary of the major gene loci that have been associated with intervertebral disc degeneration is given in Table 13-6.
| Gene | Effect |
|---|---|
| Taq1 tt genotype of vitamin D receptor | Increased disc desiccation Increased anular tears |
| Taq1 TT genotype of vitamin D receptor | Increased disc bulging Increased osteophytes |
| Taq1 and Fok1 genotypes of vitamin D receptor | Increased bulging and disc height |
| Taq polymorphisms | Increased severity of osteophytosis Disc narrowing |
| 5A5A and 5A6A gentoypes of MMP3 gene | More degenerative findings than with the 6A6A genotype |
| COL9A2 and COL9A3 | Collagen IX genes associated with disc pathology and symptoms |
From Battié M, Videman T, Parent E: Lumbar disc degeneration: epidemiology and genetic influences. Spine (Phila Pa 1976) 29:2679, 2004.
Nutritional Influences
Significant evidence exists to suggest a nutritional contribution to disc degeneration.5,30,94–96 In the young disc, as previously discussed, a vascular supply penetrates the vertebral end plate and is in close contact with the disc’s cartilaginous end plate. By maturity, only small blood vessels that reach the outer 1 to 2 mm of the disc periphery are present. This decreased vascular supply is likely a large contributor to disc degeneration.5,21
The inevitable decreased oxygen content that follows the loss of these blood vessels may explain the loss of the notochordal cells early in life, which require higher oxygen concentrations for survival.9 Decreasing oxygenation may also explain the increased substitution of keratan sulfate for chondroitin sulfate on aggrecan molecules, as well as leading to the increasingly glycolytic metabolism of the disc cells. In addition to the loss of the vascular supply, sclerosis of the subchondral plate negatively affects its porosity, leading to poor diffusion to the relatively avascular disc.28,30,94,97–99
Because of the low oxygen concentration within the disc, disc cell metabolism is anaerobic and thus consumes glucose and produces lactic acid. In low glucose states, the disc cells will eventually die. In addition, when cell pH drops below 6.4, cellular viability is compromised.28 In low pH states, cellular homeostasis is altered and a catabolic state is favored.28 Cell matrix production is inhibited while production of MMPs continues.100 These conditions, coupled with the decreased capability to diffuse lactic acid and other waste products, are thought to contribute to disc degeneration. Decreased diffusion also leads to difficulty with removal of degraded proteoglycans from the disc, both stiffening the disc structure and negatively affecting its ability to produce the swelling pressures necessary to function properly.9
Further support is garnered for the nutritional contribution to disc degeneration in specific pathologic states such as atherosclerotic disease, avascular necrosis due to decompression sickness, Gaucher disease, and nicotine exposure, which are all detrimental to the local blood supply to the vertebral body. The capillaries of the vertebral end plate in these disease states are narrowed and eventually blocked. This inhibits the transport of oxygen and other nutrients to the disc as well as limits the escape of lactic acid and proteolytic degradation products from the disc.28,101,102
Mechanical Influences
The mechanical influence on the metabolic state of the disc has long been debated. Occupational medicine literature has favored the catabolic effects of occupational loading on the intervertebral disc, whereas the sports and exercise physiology literature has often viewed mechanical loading of the disc as beneficial and anabolic.86,103–109 Monozygotic twin studies have also revealed that variations in mechanical loading of the spine play a relatively small role in explaining variations in disc degeneration.86 Over the past decade, the concept of the “threshold of stimuli” has developed, which may partially explain the possible anabolic and catabolic effects seen with disc loading at various magnitudes and frequencies.77,110,111
With this concept in mind, multiple small rodent studies have been conducted that have shown anabolic effects when tail discs have been loaded within a certain threshold, including increased messenger RNA expression, increased up-regulation of disc proteins, increased matrix production, and down-regulation of proteolytic enzymes. Outside the threshold of loading ranges (i.e., too little or too great a stimulus), negative effects are seen.78,110,111
The intervertebral disc experiences substantial forces and frequent loading on a daily basis, including fluid and osmotic pressures, interstitial fluid flow, and compressive, tensile, and shear stresses. These almost constant forces place a significant mechanical demand on the disc and have been thought to contribute to the tissue failure—anular and nuclear tears, as well as end plate damage—associated with disc aging and degeneration.109
Significant injury to the intervertebral disc has been associated with catastrophic disc overload.78,112–114 In cadaver models, when the motion-restricting effect of the facet joints is removed by facetectomy, torsional stress on the anulus results in posterolateral anular tears that radiate centrally. Changes such as disc narrowing, anular fissuring, and nuclear migration have all been seen in this setting. Although disc overload has been shown to lead to significant damage to the disc, it remains uncertain whether the daily wear and tear associated with normal physiologic loads leads to increased degneneration.23,115,116
In an effort to evaluate the daily stresses experienced by the intervertebral disc and to assess the cumulative injury model, a recent monozygotic twin study was conducted by Videman et al. on twins with an 8-kg or greater weight discordance.92 They discovered that higher body mass was not harmful to the intervertebral disc. In fact, the higher body mass actually resulted in a slight delay in lumbar disc desiccation. These findings significantly challenge the common belief that increased weight and the resulting increased loading of the intervertebral disc lead to increased disc degeneration. The findings of this study support the notion that “physical activity strengthens both the vertebrae and the discs,”117 and are also supportive of the threshold of stimuli model.93
Whole-body vibration has been incriminated in the pathogenesis of disc degeneration in the past. Both animal and in vitro studies have suggested a negative impact of vibration on the disc.23 Epidemiologic studies have also provided evidence to suggest that vibration plays a negative role in disc health.23,118–120 Data also suggest that helicopter pilots, truck drivers, and tractor operators have a high rate of back pain.23,121–124 Despite these findings, a monozygotic twin spine study has called into question the effect of whole-body vibration on disc degeneration. The study, performed by Battié et al.,85,125 arguably the best-controlled study to date evaluating this variable, found no evidence to suggest that whole-body vibration led to accelerated disc degeneration.
Toxic Exposure
The most common toxic exposure suggested to contribute to early disc degeneration is smoking. Nicotine has both direct and indirect negative effects on the disc. Indirectly, nicotine has been shown in a rabbit model to reduce the density of the vascular buds as well as narrow the vascular lumina to the vertebrae, resulting in decreased nutrient diffusion and necrosis and hyalinization of the nucleus pulposus.126 Nicotine has also been shown in in vitro and animal studies directly to affect the cellular and matrix components of the disc by decreasing the production of collagen and proteoglycans and inhibiting cell proliferation.127,128 In addition, an increased level of IL-1 and other inflammatory cytokines has been shown in animal discs.129 Despite the convincing in vitro evidence to suggest the negative role that nicotine plays in the degenerating disc, monozygotic twin studies have demonstrated only a small effect of long-term cigarette smoking in disc degeneration. The total variance explained by long-term smoking in the lumbar spine was less than 2%.86
Disc Degeneration and Back Pain
It is well accepted that the degenerated disc can lead to secondarily painful states such as disc herniation, nerve root impingement, deformity, and spinal stenosis.49,130 What has been more controversial has been the notion of the disc itself as the primary source of pain. Multiple imaging studies support the concept that disc degeneration is asymptomatic in many individuals yet painful in others.7,49,131,132 This evidence suggests that the painful degenerative disc process differs from asymptomatic degeneration and may be more related to unique biochemical changes than the morphologic changes evident on imaging (Box 13-2; see Fig. 13-5).
BOX 13-2 Potential Protein Therapies for Intervertebral Disc Degeneration
From Fassett DR, Kurd MF, Vaccaro AR: Biologic solutions for degenerative disk disease. J Spinal Disord Tech 22:297, 2009.
The concept that a disc is capable of producing painful stimuli assumes that the disc is innervated. The healthy adult disc is only peripherally innervated by mechanoreceptors, which are thought to take part in proprioception. Innervation is limited to the outer anulus, where substance P-producing nociceptors have been found that are potentially involved with inflammatory pain.18,133,134 The bony vertebral end plate is also innervated and is likely also involved in primary disc pain.135 Evidence to support end plate pain perception has been provided by pain resolution after vertebroplasty as well as by pain provocation tests in humans.19,136 In degenerated discs in which anular tears attempt to heal by formation of granulation tissue, neovascularization as well as neoinnervation spread into the planes of tissue damage, thus providing the degenerated disc with additional innervation.14,137–141 Nerve growth factor is also up-regulated at this time and may actually lower the disc’s threshold to noxious stimuli.142–144 This increased density of nerve fibers along with a lower threshold to noxious stimuli may contribute to painful disc degeneration.
Growing evidence also suggests that the presence of inflammatory cytokines may be the distinguishing factor between nonpainful and painful disc degeneration. IL-1, IL-6, IL-9, and TNF-α have all been measured in degenerating human discs and have been associated with pain.48,49 The mechanism and reason for their elevation in the pathologic state are unknown. Animal models have shown, however, that after acute loading or injury a short-term inflammatory cascade is initiated as a part of the healing process. This proinflammatory status usually lasts approximately 1 to 3 weeks, depending on the severity of the injury. When chronic aberrant loading or a pathologic stimulus is present, however, a more persistent inflammatory state arises.48,52 This prolonged inflammation has been seen in animal tail stab models and has been shown to produce Modic changes in the adjacent vertebrae in the presence of a prolonged inflammatory cascade.48 In addition to its destructive modulation, the proinflammatory response may be responsible for pain sensitization through production of cytokines and mediators such as prostaglandin E2, IL-6, and IL-8.53,54,145
Future Treatment for Intervertebral Disc Degeneration
The potential advantages of these treatments are numerous, including the preservation of normal anatomy, maintenance of normal biomechanics, and conservation of normal motion segments.146 One of the key concerns with these therapies, however, remains the lack of vascularity and nutrition to an already degenerating disc.28,147 Molecular, genetic, and cellular therapies all require the presence of healthy nuclear cells or the ability to sustain these cells after implantation in order to produce matrix and prevent further degeneration.
The majority of the current molecular, genetic, and cellular studies do not seem to consider the avascular and austere physiologic status of the disc, which may lead to unfavorable results in vivo. One possible way of dealing with the lack of nutrition and low metabolic rate of disc cells would be to focus on decreasing the catabolic influences within the disc. This would require less energy and less cellular activity, yet still decrease disc degeneration.148
Molecular Therapies
In an effort to promote an anabolic state in the disc, various growth factors have been tested in animal studies and have shown the capability to increase cellular proliferation, up-regulate proteoglycan and collagen production, and increase disc height.146,149–153 The potential for a number of select growth factors to reverse degenerative disc changes has been reproducibly demonstrated in animal studies.146,154–156 The catabolic influences on the degenerating disc have also been addressed by the use cytokine inhibitors in an effort to down-regulate the proinflammatory cytokines TNF-α and IL-1 and shift the disc to a more anabolic state.146,157 Proteinase antagonists such as TIMPs also inhibit the breakdown of proteoglycans and may serve a role in future molecular therapy.146
Although early in vivo and in vitro studies have suggested a tremendous potential for these molecular therapies, the half-life of most of these molecules is relatively short.146 For these therapies to be clinically effective, repeated infusions or a long-term delivery device will likely be needed. For this reason, genetic therapy, with its ability to increase the translation of desired molecules, may prove more promising in the future (see Box 13-2).
Genetic Therapies
Genetic therapy transfers an exogenous gene into a cell along with a promoter in order to induce the translation of a desired target protein. Genetic therapy has the potential to overcome the major problem with molecular-based growth factor treatments by using existing disc cells to create a continuous, prolonged production of a desired protein.146 Genetic transfer has been performed most frequently using a viral vector, which for safety reasons has been the key limiting step in genetic therapy. Genes encoding for key anabolic growth factors and catabolic antagonists have been successfully transferred to disc cells using viral vectors, resulting in an improved anabolic state.146,158–161 Despite these promising results, the key challenge remains the safety of genetic transfer. Adenoviral vector transfer has been associated with death and life-threatening immune reactions.146,162–164 Alternative, nonviral methods of genetic transfer are under investigation and may play a key role in the future.148,165–168
Cellular Therapies
Cellular therapies attempt to address the primary disadvantage of molecular and genetic therapies, which is the common lack of viable cells in the disc because of its degenerative state. In the absence of viable, healthy disc cells, molecular and genetic therapies will not work. Although the transplantation of new cells into the disc may solve this dilemma, the continued nutrition and survival of these cells remains a significant challenge.28,147 Cell-based therapies have used both allograft and autograft nucleus pulposus cells, as well as a variety of stem cells, to regenerate discs. All three cell types have been successfully used in in vivo animal studies, resulting in improved disc biochemical profiles.146,169
In addition to these animal studies, one human clinical trial is underway in Europe investigating the role of autologous disc cell reimplantation 12 weeks after microdiscectomy and culture. Interim results were recently reported, indicating that 29 patients with 2 years of follow-up had improved clinical outcomes and visual analogue scores and less disc desiccation, but no difference in disc height. Although these results are encouraging, there is concern regarding the study because it is not blinded and a significant placebo effect may be detrimental to the study conclusions.146,170,171
Stem cell transplantation has also seen a surge in research interest. Multiple stem cell lineages have been studied, but mesenchymal stem cells have become the most popular owing to ethical concerns with fetal/embryonic stem cells, and their ready availability. Although good outcomes with in vivo animal studies using stem cells have been demonstrated, only one nonrandomized, uncontrolled study using autologous hematopoietic stem cells has been performed, which revealed no significant improvement in low back pain after treatment.146,172 One of the most significant challenges facing this technology is that the exact phenotype of the chondrocyte-like cells of the disc is not fully understood and, as such, it is currently impossible to reproduce these cells exactly.146,169
Summary
The internal processes and mechanics of disc degeneration may be partially understood, but the key initiating factors and the interactions of the various processes, including nutritional, molecular, cellular, genetic, inflammatory, and environmental influences, as well as why the disc begins degenerating almost immediately after birth, are poorly understood. Despite our relatively poor understanding of these interactions and the key factors promoting degeneration, research in molecular, genetic, and cellular therapies has rapidly advanced and provides a promising outlook for combating these degenerative changes in the future. Despite the significant challenges facing investigators in translating research into successful clinical treatments, the increasing amount of research in these fields as well as our improved understanding of disc physiology and pathology suggest that significant laboratory and clinical progress may be seen in the very near future.
Battié M.C., Videman T., Gibbons L.E., et al. 1995 Volvo Award in clinical sciences. Determinants of lumbar disc degeneration: a study relating lifetime exposures and magnetic resonance imaging findings in identical twins. Spine (Phila Pa 1976). 1995;20:2601.
Battié M.C., Videman T., Kaprio J., et al. The Twin Spine Study: contributions to a changing view of disc degeneration. Spine J. 2009;9:47.
Boos N., Weissbach S., Rohrbach H., et al. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine (Phila Pa 1976). 2002;27:2631.
Roughley P. Biology of intervertebral disc aging and degeneration: involvement of the extracellular matrix. Spine (Phila Pa 1976). 2004;29:2691.
Thompson J., Oegema T.J., Bradford D. Stimulation of mature canine intervertebral disc by growth factors. Spine (Phila Pa 1976). 1991;16:253.
Ulrich J., Liebenberg E., Thuillier D., et al. ISSLS prize winner: repeated disc injury causes persistent inflammation. Spine (Phila Pa 1976). 2007;32:2812.
1. Masuda K., Oegema T.J., An H.S. Growth factors and treatment of intervertebral disc degeneration. Spine (Phila Pa 1976). 2004;29:2757.
2. Praemer A., Furner S., Rice D.P. Musculoskeletal conditions in the United States. Rosemont, IL: American Academy of Orthopaedic Surgeons; 1999.
3. Buckwalter J. Aging and degeneration of the human intervertebral disc. Spine (Phila Pa 1976). 1995;20:1307.
4. Miller J.A., Schmatz C., Schultz A. Lumbar disc degeneration: correlation with age, sex, and spine level in 600 autopsy specimens. Spine (Phila Pa 1976). 1988;13:173.
5. Boos N., Weissbach S., Rohrbach H., et al. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine (Phila Pa 1976). 2002;27:2631.
6. Borenstein D., O’Mara J.J., Boden S., et al. The value of magnetic resonance imaging of the lumbar spine to predict low-back pain in asymptomatic subjects: a seven-year follow-up study. J Bone Joint Surg [Am]. 2001;83:1306.
7. Boden S., McCowin P., Davis D., et al. Abnormal magnetic-resonance scans of the cervical spine in asymptomatic subjects: a prospective investigation. J Bone Joint Surg [Am]. 1990;72:1178.
8. Haefeli M., Kalberer F., Saegesser D., et al. The course of macroscopic degeneration in the human lumbar intervertebral disc. Spine (Phila Pa 1976). 2006;31:1522.
9. Roughley P. Biology of intervertebral disc aging and degeneration: involvement of the extracellular matrix. Spine (Phila Pa 1976). 2004;29:2691.
10. Roberts S. Disc morphology in health and disease. Biochem Soc Trans. 2002;30:864.
11. Yu J. Elastic tissues of the intervertebral disc. Biochem Soc Trans. 2002;30:848.
12. Vernon-Roberts B., Moore R., Fraser R. The natural history of age-related disc degeneration: the pathology and sequelae of tears. Spine (Phila Pa 1976). 2007;32:2797.
13. Skaggs D., Weidenbaum M., Iatridis J., et al. Regional variation in tensile properties and biochemical composition of the human lumbar anulus fibrosus. Spine (Phila Pa 1976). 1994;19:1310.
14. Osti O., Vernon-Roberts B., Fraser R. 1990 Volvo Award in experimental studies. Anulus tears and intervertebral disc degeneration: an experimental study using an animal model. Spine (Phila Pa 1976). 1990;15:762.
15. Errington R., Puustjarvi K., White I., et al. Characterisation of cytoplasm-filled processes in cells of the intervertebral disc. J Anat. 1998;192:369.
16. Veres S., Robertson P., Broom N. ISSLS prize winner. Microstructure and mechanical disruption of the lumbar disc annulus: Part II. How the annulus fails under hydrostatic pressure. Spine (Phila Pa 1976). 2008;33:2711.
17. Bogduk N., Tynan W., Wilson A. The nerve supply to the human lumbar intervertebral discs. J Anat. 1981;132:39.
18. Palmgren T., Gronblad M., Virri J., et al. An immunohistochemical study of nerve structures in the anulus fibrosus of human normal lumbar intervertebral discs. Spine (Phila Pa 1976). 1999;24:2075.
19. Kuslich S., Ulstrom C., Michael C. The tissue origin of low back pain and sciatica: a report of pain response to tissue stimulation during operations on the lumbar spine using local anesthesia. Orthop Clin North Am. 1991;22:181.
20. Raj P. Intervertebral disc: anatomy-physiology-pathophysiology-treatment. Pain Pract. 2008;8:18.
21. Coventry M., Ghormley R., Kernohan J. The intervertebral disc: its microscopic anatomy and pathology. Part II: Changes in then intervertebral disc concomitant with age. J Bone Joint Surg. 1945;27:233.
22. Inoue H. Three-dimensional architecture of lumbar intervertebral discs. Spine (Phila Pa 1976). 1981;6:139.
23. Hadjipavlou A., Tzermiadianos M., Bogduk N., et al. The pathophysiology of disc degeneration: a critical review. J Bone Joint Surg [Br]. 2008;90:1261.
24. Urban J., Maroudas A. Swelling of the intervertebral disc in vitro. Connect Tissue Res. 1981;9:1.
25. Lyons G., Eisenstein S., Sweet M. Biochemical changes in intervertebral disc degeneration. Biochim Biophys Acta. 1981;673:443.
26. Eyre D.R., Matsui Y., Wu J. Collagen polymorphisms of the intervertebral disc. Biochem Soc Trans. 2002;30:844.
27. Johnstone B., Bayliss M. The large proteoglycans of the human intervertebral disc: changes in their biosynthesis and structure with age, topography, and pathology. Spine (Phila Pa 1976). 1995;20:674.
28. Urban J., Smith S., Fairbank J. Nutrition of the intervertebral disc. Spine (Phila Pa 1976). 2004;29:2700.
29. Oegema T.J. Biochemistry of the intervertebral disc. Clin Sports Med. 1993;12:419.
30. Nachemson A., Lewin T., Maroudas A., et al. In vitro diffusion of dye through the end-plates and the annulus fibrosus of human lumbar inter-vertebral discs. Acta Orthop Scand. 1970;41:589.
31. Rudert M., Tillmann B. Detection of lymph and blood vessels in the human intervertebral disc by histochemical and immunohistochemical methods. Ann Anat. 1993;175:237.
32. Hassler O. The human intervertebral disc: a micro-angiographical study on its vascular supply at various ages. Acta Orthop Scand. 1969;40:765.
33. Ratcliffe J. The arterial anatomy of the adult human lumbar vertebral body: a microarteriographic study. J Anat. 1980;131:57.
34. Crock H.V., Goldwasser M. Anatomic studies of the circulation in the region of the vertebral end-plate in adult greyhound dogs. Spine (Phila Pa 1976). 1984;9:702.
35. Oki S., Matsuda Y., Shibata T., et al. Morphologic differences of the vascular buds in the vertebral endplate: scanning electron microscopic study. Spine (Phila Pa 1976). 1996;21:174.
36. Mauck R., Hung C., Ateshian G. Modeling of neutral solute transport in a dynamically loaded porous permeable gel: implications for articular cartilage biosynthesis and tissue engineering. J Biomech Eng. 2003;125:602.
37. O’Hara B.P., Urban J., Maroudas A. Influence of cyclic loading on the nutrition of articular cartilage. Ann Rheum Dis. 1990;49:536.
38. Ferguson S., Ito K., Nolte L. Fluid flow and convective transport of solutes within the intervertebral disc. J Biomech. 2004;37:213.
39. Goupille P., Jayson M., Valat J., et al. Matrix metalloproteinases: the clue to intervertebral disc degeneration? Spine (Phila Pa 1976). 1998;23:1612.
40. Ohshima H., Urban J. The effect of lactate and pH on proteoglycan and protein synthesis rates in the intervertebral disc. Spine (Phila Pa 1976). 1992;17:1079.
41. Liu J., Roughley P., Mort J. Identification of human intervertebral disc stromelysin and its involvement in matrix degradation. J Orthop Res. 1991;9:568.
42. Kanemoto M., Hukuda S., Komiya Y., et al. Immunohistochemical study of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 human intervertebral discs. Spine (Phila Pa 1976). 1996;21:1.
43. Hsieh A.H., Lotz J. Prolonged spinal loading induces matrix metalloproteinase-2 activation in intervertebral discs. Spine (Phila Pa 1976). 2003;28:1781.
44. Crean J.K., Roberts S., Jaffray D., et al. Matrix metalloproteinases in the human intervertebral disc: role in disc degeneration and scoliosis. Spine (Phila Pa 1976). 1997;22:2877.
45. Roberts S., Caterson B., Menage J., et al. Matrix metalloproteinases and aggrecanase: their role in disorders of the human intervertebral disc. Spine (Phila Pa 1976). 2000;25:3005.
46. Tang B.L. ADAMTS: a novel family of extracellular matrix proteases. Int J Biochem Cell Biol. 2001;33:33.
47. Kobayashi M., Squires G., Mousa A., et al. Role of interleukin-1 and tumor necrosis factor alpha in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52:128.
48. Ulrich J.A., Liebenberg E., Thuillier D., et al. ISSLS prize winner: repeated disc injury causes persistent inflammation. Spine (Phila Pa 1976). 2007;32:2812.
49. Lotz J., Ulrich J. Innervation, inflammation, and hypermobility may characterize pathologic disc degeneration: review of animal model data. J Bone Joint Surg [Am]. 2006;88(Suppl 2):76.
50. Seguin C., Pilliar R., Roughley P., et al. Tumor necrosis factor-alpha modulates matrix production and catabolism in nucleus pulposus tissue. Spine (Phila Pa 1976). 2005;30:1940.
51. Shinmei M., Masuda K., Kikuchi T., et al. Interleukin 1, tumor necrosis factor, and interleukin 6 as mediators of cartilage destruction. Semin Arthritis Rheum. 1989;18:27.
52. Anderson D., Izzo M.W., Hall D.J., et al. Comparative gene expression profiling of normal and degenerative discs: analysis of a rabbit annular laceration model. Spine (Phila Pa 1976). 2002;27:1291.
53. Peng B., Wu W., Hou S., et al. The pathogenesis of discogenic low back pain. J Bone Joint Surg [Br]. 2005;87:62.
54. Burke J., Watson R., McCormack D., et al. Intervertebral discs which cause low back pain secrete high levels of proinflammatory mediators. J Bone Joint Surg [Br]. 2002;84:196.
55. Trout J.J., Buckwalter J., Moore K. Ultrastructure of the human intervertebral disc: II. Cells of the nucleus pulposus. Anat Rec. 1982;204:307.
56. Buckwalter J., Pedrini-Mille A., Pedrini V., et al. Proteoglycans of human infant intervertebral disc: electron microscopic and biochemical studies. J Bone Joint Surg [Am]. 1985;67:284.
57. Hollander A., Heathfield T., Liu J., et al. Enhanced denaturation of the alpha (II) chains of type-II collagen in normal adult human intervertebral discs compared with femoral articular cartilage. J Orthop Res. 1996;14:61.
58. Bushell G., Ghosh P., Taylor T., et al. Proteoglycan chemistry of the intervertebral disks. Clin Orthop Relat Res. 129:115, 1977.
59. Salisbury J., Watt F. Lack of keratan sulphate in the human notochord. J Anat. 1988;157:175.
60. Oegema T.J., Bradford D., Cooper K. Aggregated proteoglycan synthesis in organ cultures of human nucleus pulposus. J Biol Chem. 1979;254:10579.
61. Ghosh P., Melrose J., Cole T., et al. A comparison of the high buoyant density proteoglycans isolated from the intervertebral discs of chondrodystrophoid and non-chondrodystrophoid dogs. Matrix. 1992;12:148.
62. Inerot S., Axelsson I. Structure and composition of proteoglycans from human annulus fibrosus. Connect Tissue Res. 1991;26:47.
63. Antoniou J., Steffen T., Nelson F., et al. The human lumbar intervertebral disc: evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J Clin Invest. 1996;98:996.
64. Duance V.C., Crean J., Sims T., et al. Changes in collagen cross-linking in degenerative disc disease and scoliosis. Spine (Phila Pa 1976). 1998;23:2545.
65. Pokharna H., Phillips F. Collagen crosslinks in human lumbar intervertebral disc aging. Spine (Phila Pa 1976). 1998;23:1645.
66. Yokosuka K., Park J., Jimbo K., et al. Advanced glycation end-products downregulating intervertebral disc cell production of proteoglycans in vitro. J Neurosurg Spine. 2006;5:324.
67. Schleicher E., Wagner E., Nerlich A. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. 1997;99:457.
68. Pokharna H., Pottenger L. Nonenzymatic glycation of cartilage proteoglycans: an in vivo and in vitro study. Glycoconj J. 1997;14:917.
69. Verzijl N., Degroot J., Oldehinkel E., et al. Age-related accumulation of Maillard reaction products in human articular cartilage collagen. Biochem J. 2000;350:381.
70. Brownlee M., Cerami A., Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315.
71. Kilhovd B.K., Berg T., Birkeland K., et al. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care. 1999;22:1543.
72. Degroot J., Verzijl N., Jacobs K., et al. Accumulation of advanced glycation endproducts reduces chondrocyte-mediated extracellular matrix turnover in human articular cartilage. Osteoarthritis Cartilage. 2001;9:720.
73. Degroot J., Verzijl N., Bank R., et al. Age-related decrease in proteoglycan synthesis of human articular chondrocytes: the role of nonenzymatic glycation. Arthritis Rheum. 1999;42:1003.
74. Jhawar B., Fuchs C., Colditz G., et al. Cardiovascular risk factors for physician-diagnosed lumbar disc herniation. Spine J. 2006;6:684.
75. Wagner D.R., Reiser K., Lotz J. Glycation increases human annulus fibrosus stiffness in both experimental measurements and theoretical predictions. J Biomech. 2006;39:1021.
76. Yoshida T., Park J., Yokosuka K., et al. Up-regulation in receptor for advanced glycation end-products in inflammatory circumstances in bovine coccygeal intervertebral disc specimens in vitro. Spine (Phila Pa 1976). 2009;34:1544.
77. Iatridis J., Maclean J., Roughley P., et al. Effects of mechanical loading on intervertebral disc metabolism in vivo. J Bone Joint Surg [Am]. 2006;88(Suppl 2):41.
78. Stokes I., Iatridis J. Mechanical conditions that accelerate intervertebral disc degeneration: overload versus immobilization. Spine (Phila Pa 1976). 2004;29:2724.
79. Adams M., Bogduk N., Burton K., et al. The biomechanics of back pain, ed 2. Edinburgh: Churchill Livingstone; 2006.
80. Iatridis J., Setton L., Weidenbaum M., et al. Alterations in the mechanical behavior of the human lumbar nucleus pulposus with degeneration and aging. J Orthop Res. 1997;15:318.
81. Gu W., Mao X., Foster R., et al. The anisotropic hydraulic permeability of human lumbar anulus fibrosus: influence of age, degeneration, direction, and water content. Spine (Phila Pa 1976). 1999;24:2449.
82. Mimura M., Panjabi M., Oxland T., et al. Disc degeneration affects the multidirectional flexibility of the lumbar spine. Spine (Phila Pa 1976). 1994;19:1371.
83. Adams M., McNally D., Dolan P. ‘Stress’ distributions inside intervertebral discs: the effects of age and degeneration. J Bone Joint Surg [Br]. 1996;78:965.
84. Boxberger J., Sen S., Yerramalli C., et al. Nucleus pulposus glycosaminoglycan content is correlated with axial mechanics in rat lumbar motion segments. J Orthop Res. 2006;24:1906.
85. Battié M.C., Videman T., Parent E. Lumbar disc degeneration: epidemiology and genetic influences. Spine (Phila Pa 1976). 2004;29:2679.
86. Battié M.C., Videman T., Gibbons L.E., et al. 1995 Volvo Award in clinical sciences. Determinants of lumbar disc degeneration: a study relating lifetime exposures and magnetic resonance imaging findings in identical twins. Spine (Phila Pa 1976). 1995;20:2601.
87. Battié M.C., Videman T., Kaprio J., et al. The Twin Spine Study: contributions to a changing view of disc degeneration. Spine J. 2009;9:47.
88. Solovieva S., Lohiniva J., Leino-Arjas P., et al. COL9A3 gene polymorphism and obesity in intervertebral disc degeneration of the lumbar spine: evidence of gene-environment interaction. Spine (Phila Pa 1976). 2002;27:2691.
89. Zerba K.E., Sing C. The role of genome type-environment interaction and time in understanding the impact of genetic polymorphisms on lipid metabolism. Curr Opin Lipidol. 1993;4:186.
90. Loughlin J. Genetic epidemiology of primary osteoarthritis. Curr Opin Rheumatol. 2001;13:111.
91. Kalichman L., Hunter D. The genetics of intervertebral disc degeneration: associated genes. Joint Bone Spine. 2008;75:388.
92. Videman T., Levalahti E., Battié M.C. The effects of anthropometrics, lifting strength, and physical activities in disc degeneration. Spine (Phila Pa 1976). 2007;32:1406.
93. Videman T., Gibbons L.E., Kaprio J., et al. Challenging the cumulative injury model: positive effects of greater body mass on disc degeneration. Spine J. 2010;10:26.
94. Holm S., Maroudas A., Urban J., et al. Nutrition of the intervertebral disc: solute transport and metabolism. Connect Tissue Res. 1981;8:101.
95. Ogata K., Whiteside L. 1980 Volvo award winner in basic science. Nutritional pathways of the intervertebral disc: an experimental study using hydrogen washout technique. Spine (Phila Pa 1976). 1981;6:211.
96. Urban J., Holm S., Maroudas A., et al. Nutrition of the intervertebral disk: an in vivo study of solute transport. Clin Orthop Relat Res. 129:101, 1977.
97. Holm S., Selstam G., Nachemson A. Carbohydrate metabolism and concentration profiles of solutes in the canine lumbar intervertebral disc. Acta Physiol Scand. 1982;115:147.
98. Ishihara H., Urban J. Effects of low oxygen concentrations and metabolic inhibitors on proteoglycan and protein synthesis rates in the intervertebral disc. J Orthop Res. 1999;17:829.
99. Grunhagen T., Wilde G., Soukane D., et al. Nutrient supply and intervertebral disc metabolism. J Bone Joint Surg [Am]. 2006;88(Suppl 2):30.
100. Razaq S., Wilkins R., Urban J. The effect of extracellular pH on matrix turnover by cells of the bovine nucleus pulposus. Eur Spine J. 2003;12:341.
101. Kurunlahti M., Tervonen O., Vanharanta H., et al. Association of atherosclerosis with low back pain and the degree of disc degeneration. Spine (Phila Pa 1976). 1999;24:2080.
102. Kauppila L. Prevalence of stenotic changes in arteries supplying the lumbar spine: a postmortem angiographic study on 140 subjects. Ann Rheum Dis. 1997;56:591.
103. Biering-Sorensen F. Physical measurements as risk indicators for low-back trouble over a one-year period. Spine (Phila Pa 1976). 1984;9:106.
104. Caplan P., Freedman L., Connelly T. Degenerative joint disease of the lumbar spine in coal miners: a clinical and x-ray study. Arthritis Rheum. 1966;9:693.
105. Friberg S., Hirsch C. Anatomical and clinical studies on lumbar disc degeneration. Acta Orthop Scand. 1949;19:222.
106. Frymoyer J., Newberg A., Pope M., et al. Spine radiographs in patients with low-back pain: an epidemiological study in men. J Bone Joint Surg [Am]. 1984;66:1048.
107. Kellgren J., Lawrence J. Rheumatism in miners: II. X-ray study. Br J Ind Med. 1952;9:197.
108. Sairanen E., Brushaber L., Kaskinen M. Felling work, low-back pain and osteoarthritis. Scand J Work Environ Health. 1981;7:18.
109. Setton L., Chen J. Cell mechanics and mechanobiology in the intervertebral disc. Spine (Phila Pa 1976). 2004;29:2710.
110. Maclean J., Lee C., Alini M., et al. Anabolic and catabolic mRNA levels of the intervertebral disc vary with the magnitude and frequency of in vivo dynamic compression. J Orthop Res. 2004;22:1193.
111. Maclean J., Lee C., Grad S., et al. Effects of immobilization and dynamic compression on intervertebral disc cell gene expression in vivo. Spine (Phila Pa 1976). 2003;28:973.
112. Adams M., Hutton W. The effect of fatigue on the lumbar intervertebral disc. J Bone Joint Surg [Br]. 1983;65:199.
113. Adams M.A., Hutton W. The relevance of torsion to the mechanical derangement of the lumbar spine. Spine (Phila Pa 1976). 1981;6:241.
114. Adams M.A., Hutton W. 1981 Volvo Award in basic science. Prolapsed intervertebral disc: a hyperflexion injury. Spine (Phila Pa 1976). 1982;7:184.
115. Farfan H., Cossette J., Robertson G., et al. The effects of torsion on the lumbar intervertebral joints: the role of torsion in the production of disc degeneration. J Bone Joint Surg [Am]. 1970;52:468.
116. Farfan H.F. Mechanical disorders of the low back. Philadelphia: Lea & Febiger; 1973.
117. Porter R.W., Adams M., Hutton W. Physical activity and the strength of the lumbar spine. Spine (Phila Pa 1976). 1989;14:201.
118. Kelsey J.L., Hardy R. Driving of motor vehicles as a risk factor for acute herniated lumbar intervertebral disc. Am J Epidemiol. 1975;102:63.
119. Heliovaara M. Occupation and risk of herniated lumbar intervertebral disc or sciatica leading to hospitalization. J Chronic Dis. 1987;40:259.
120. Jensen M., Tuchsen F., Orhede E. Prolapsed cervical intervertebral disc in male professional drivers in Denmark, 1981-1990: a longitudinal study of hospitalizations. Spine (Phila Pa 1976). 1996;21:2352.
121. Frymoyer J., Pope M., Costanza M., et al. Epidemiologic studies of low-back pain. Spine (Phila Pa 1976). 1980;5:419.
122. Kelsey J. An epidemiological study of the relationship between occupations and acute herniated lumbar intervertebral discs. Int J Epidemiol. 1975;4:197.
123. Kelsey J. An epidemiological study of acute herniated lumbar intervertebral discs. Rheumatol Rehabil. 1975;14:144.
124. Bovenzi M., Zadini A. Self-reported low back symptoms in urban bus drivers exposed to whole-body vibration. Spine (Phila Pa 1976). 1992;17:1048.
125. Battié M., Videman T., Gibbons L.E., et al. Occupational driving and lumbar disc degeneration: a case-control study. Lancet. 2002;360:1369.
126. Iwahashi M., Matsuzaki H., Tokuhashi Y., et al. Mechanism of intervertebral disc degeneration caused by nicotine in rabbits to explicate intervertebral disc disorders caused by smoking. Spine (Phila Pa 1976). 2002;27:1396.
127. Uematsu Y., Matsuzaki H., Iwahashi M. Effects of nicotine on the intervertebral disc: an experimental study in rabbits. J Orthop Sci. 2001;6:177.
128. Akmal M., Kesani A., Anand B., et al. Effect of nicotine on spinal disc cells: a cellular mechanism for disc degeneration. Spine (Phila Pa 1976). 2004;29:568.
129. Oda H., Matsuzaki H., Tokuhashi Y., et al. Degeneration of intervertebral discs due to smoking: experimental assessment in a rat-smoking model. J Orthop Sci. 2004;9:135.
130. Brisby H. Pathology and possible mechanisms of nervous system response to disc degeneration. J Bone Joint Surg [Am]. 2006;88(Suppl 2):68.
131. Jensen M., Brant-Zawadzki M.N., Obuchowski N., et al. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med. 1994;331:69.
132. Carragee E., Paragioudakis S., Khurana S. 2000 Volvo Award winner in clinical studies. Lumbar high-intensity zone and discography in subjects without low back problems. Spine (Phila Pa 1976). 2000;25:2987.
133. Roberts S., Eisenstein S., Menage J., et al. Mechanoreceptors in intervertebral discs: morphology, distribution, and neuropeptides. Spine (Phila Pa 1976). 1995;20:2645.
134. Ozawa T., Aoki Y., Ohtori S., et al. The dorsal portion of the lumbar intervertebral disc is innervated primarily by small peptide-containing dorsal root ganglion neurons in rats. Neurosci Lett. 2003;344:65.
135. Fagan A., Moore R., Vernon Roberts B., et al. ISSLS prize winner. The innervation of the intervertebral disc: a quantitative analysis. Spine (Phila Pa 1976). 2003;28:2570.
136. Niv D., Gofeld M., Devor M. Causes of pain in degenerative bone and joint disease: a lesson from vertebroplasty. Pain. 2003;105:387.
137. Freeman B., Walters R., Moore R., et al. Does intradiscal electrothermal therapy denervate and repair experimentally induced posterolateral annular tears in an animal model? Spine (Phila Pa 1976). 2003;28:2602.
138. Kaapa E., Han X., Holm S., et al. Collagen synthesis and types I, III, IV, and VI collagens in an animal model of disc degeneration. Spine (Phila Pa 1976). 1995;20:59.
139. Kaigle A.M., Holm S., Hansson T. 1997 Volvo Award winner in biomechanical studies. Kinematic behavior of the porcine lumbar spine: a chronic lesion model. Spine (Phila Pa 1976). 1997;22:2796.
140. Lipson S., Muir H. 1980 Volvo award in basic science. Proteoglycans in experimental intervertebral disc degeneration. Spine (Phila Pa 1976). 1981;6:194.
141. Melrose J., Smith S., Little C., et al. Spatial and temporal localization of transforming growth factor-beta, fibroblast growth factor-2, and osteonectin, and identification of cells expressing alpha-smooth muscle actin in the injured anulus fibrosus: implications for extracellular matrix repair. Spine (Phila Pa 1976). 2002;27:1756.
142. Graiani G., Emanueli C., Desortes E., et al. Nerve growth factor promotes reparative angiogenesis and inhibits endothelial apoptosis in cutaneous wounds of type 1 diabetic mice. Diabetologia. 2004;47:1047.
143. Dyck P., Peroutka S., Rask C., et al. Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology. 1997;48:501.
144. Sullins J.S., Carnes D.L.Jr., Kaldestad R.N., et al. Time course of the increase in trk A expression in trigeminal neurons after tooth injury. J Endod. 2000;26:88.
145. Peng B., Hao J., Hou S., et al. Possible pathogenesis of painful intervertebral disc degeneration. Spine (Phila Pa 1976). 2006;31:560.
146. Fassett D., Kurd M., Vaccaro A. Biologic solutions for degenerative disk disease. J Spinal Disord Tech. 2009;22:297.
147. Kandel R., Roberts S., Urban J. Tissue engineering and the intervertebral disc: the challenges. Eur Spine J. 2008;17(Suppl 4):480.
148. Nishida K., Suzuki T., Kakutani K., et al. Gene therapy approach for disc degeneration and associated spinal disorders. Eur Spine J. 2008;17(Suppl 4):459.
149. Osada R., Ohshima H., Ishihara H., et al. Autocrine/paracrine mechanism of insulin-like growth factor-1 secretion, and the effect of insulin-like growth factor-1 on proteoglycan synthesis in bovine intervertebral discs. J Orthop Res. 1996;14:690.
150. Thompson J., Oegema T.J., Bradford D. Stimulation of mature canine intervertebral disc by growth factors. Spine (Phila Pa 1976). 1991;16:253.
151. Okuda S., Myoui A., Ariga K., et al. Mechanisms of age-related decline in insulin-like growth factor-I dependent proteoglycan synthesis in rat intervertebral disc cells. Spine (Phila Pa 1976). 2001;26:2421.
152. Yoon S.T., Patel N.M. Molecular therapy of the intervertebral disc. Eur Spine J. 2006;15(Suppl 3):S379.
153. Gruber H.E., Fisher E.J., Desai B., et al. Human intervertebral disc cells from the annulus: three-dimensional culture in agarose or alginate and responsiveness to TGF-beta1. Exp Cell Res. 1997;235:13.
154. Masuda K., Imai Y., Okuma M., et al. Osteogenic protein-1 injection into a degenerated disc induces the restoration of disc height and structural changes in the rabbit anular puncture model. Spine (Phila Pa 1976). 2006;31:742.
155. Imai Y., Okuma M., An H., et al. Restoration of disc height loss by recombinant human osteogenic protein-1 injection into intervertebral discs undergoing degeneration induced by an intradiscal injection of chondroitinase ABC. Spine (Phila Pa 1976). 2007;32:1197.
156. Chujo T., An H., Akeda K., et al. Effects of growth differentiation factor-5 on the intervertebral disc: in vitro bovine study and in vivo rabbit disc degeneration model study. Spine (Phila Pa 1976). 2006;31:2909.
157. Masuda K. Biological repair of the degenerated intervertebral disc by the injection of growth factors. Eur Spine J. 2008;17(Suppl 4):441.
158. Wehling P., Schulitz K., Robbins P., et al. Transfer of genes to chondrocytic cells of the lumbar spine: proposal for a treatment strategy of spinal disorders by local gene therapy. Spine (Phila Pa 1976). 1997;22:1092.
159. Nishida K., Kang J., Suh J., et al. Adenovirus-mediated gene transfer to nucleus pulposus cells: implications for the treatment of intervertebral disc degeneration. Spine (Phila Pa 1976). 1998;23:2437.
160. Paul R., Haydon R., Cheng H., et al. Potential use of sox9 gene therapy for intervertebral degenerative disc disease. Spine (Phila Pa 1976). 2003;28:755.
161. Zhang Y., An H., Thonar E., et al. Comparative effects of bone morphogenetic proteins and sox9 overexpression on extracellular matrix metabolism of bovine nucleus pulposus cells. Spine (Phila Pa 1976). 2006;31:2173.
162. Driesse M., Esandi M., Kros J., et al. Intra-CSF administered recombinant adenovirus causes an immune response-mediated toxicity. Gene Ther. 2000;7:1401.
163. Wood M., Charlton H., Wood K., et al. Immune responses to adenovirus vectors in the nervous system. Trends Neurosci. 1996;19:497.
164. Somia N., Verma I. Gene therapy: trials and tribulations. Nat Rev Genet. 2000;1:91.
165. Lawrie A., Brisken A., Francis S., et al. Ultrasound enhances reporter gene expression after transfection of vascular cells in vitro. Circulation. 1999;99:2617.
166. Lawrie A., Brisken A., Francis S., et al. Microbubble-enhanced ultrasound for vascular gene delivery. Gene Ther. 2000;7:2023.
167. Newman C., Lawrie A., Brisken A., et al. Ultrasound gene therapy: on the road from concept to reality. Echocardiography. 2001;18:339.
168. Nishida K., Doita M., Takada T., et al. Sustained transgene expression in intervertebral disc cells in vivo mediated by microbubble-enhanced ultrasound gene therapy. Spine (Phila Pa 1976). 2006;31:1415.
169. Sakai D. Future perspectives of cell-based therapy for intervertebral disc disease. Eur Spine J. 2008;17(Suppl 4):452.
170. Meisel H., Ganey T., Hutton W., et al. Clinical experience in cell-based therapeutics: intervention and outcome. Eur Spine J. 2006;15(Suppl 3):S397.
171. Meisel H., Siodla V., Ganey T., et al. Clinical experience in cell-based therapeutics: disc chondrocyte transplantation A treatment for degenerated or damaged intervertebral disc. Biomol Eng. 2007;24:5.
172. Haufe S., Mork A. Intradiscal injection of hematopoietic stem cells in an attempt to rejuvenate the intervertebral discs. Stem Cells Dev. 2006;15:136.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 13 Intervertebral Disc Process of Degeneration
Physiology and Pathophysiology
In the United States alone, nearly 5.7 million people are diagnosed with intervertebral disc disorders each year.1,2 These disorders are responsible for widespread disability and account for tremendous costs associated with their treatment and loss of productivity. The intervertebral disc is a unique multifunctional structure. It provides a major stabilizing effect for the spine and allows for the dissipation of axial load, and yet is still flexible enough to confer significant mobility. Despite these essential roles in spinal mechanics, the disc also begins to show signs of biochemical and structural change almost immediately after birth, and signs of degeneration are present at an extremely young age.3–5 Unfortunately, these degenerative changes are often progressive, rendering the disc biochemically and biomechanically incapable of absorbing the forces necessary for normal physiologic spinal function and potentially leading to rapid degeneration of the spine.
Understanding the anatomy and function of the normal disc serves as the framework for comprehension of the complex processes of aging and the pathologic degeneration that afflict the disc. One of the most significant challenges to a complete understanding of disc disease is the lack of consistency with which radiographically detectable signs of disc degeneration translate to clinical symptomatology.6 As such, there is abundant evidence to suggest that aberrant disc morphology does not necessarily equate with back pain.6,7 This poses a significant challenge to researchers in this field because individual variation in disc degeneration makes it difficult to distinguish between age-related alterations and pathologic premature degenerative changes.5,8 In the future, the ability to make this distinction will lead to improved clarity regarding the complex process of disc degeneration.
Normal Intervertebral Disc
The intervertebral disc is composed of water, proteoglycan, collagen, and a small amount of noncollagenous proteins. There are three major regions to the intervertebral disc with differing structural compositions: the nucleus pulposus, anulus fibrosus, and cartilaginous end plates. Each has different functions. Of significant note, the intervertebral disc is one of the most hypocellular structures in the body9 (Fig. 13-1).
Anulus Fibrosus
The anulus fibrosus is the outer circumferential portion of the disc and is often subdivided into the inner and outer anulus. The outer anulus is a collection of highly organized, densely packed collagen fibrils, creating a lamellar structure of up to 25 lamellae in the mature disc.9,10 These collagen fibrils insert into the adjacent vertebral end plates and join them together to stabilize the spine. In addition to its predominantly collagenous structure, a small amount of elastin is present in the anulus, which helps it to restore and recoil after stretching in response to compression and tensile forces.9,11 The strong anulus provides the ability to absorb significant hoop stresses and maintain stiffness in the presence of the tensile forces induced by bending and twisting of the spine.12,13 The importance of this ability to resist hoop stress has been shown in large animal studies, which demonstrated that even a partial-thickness laceration into the anulus rapidly produced advanced disc degeneration.12,14 The outer anulus is populated with a low density of fibroblast-like cells.15 It is the only tissue in the mature disc that remains innervated and is capable of producing pain when exposed to noxious stimuli.16–19
The inner anulus appears to serve as a transition zone to the vastly different biochemical composition of the nucleus. It is less dense and lacks the organized lamellar structure of the outer anulus. It is populated with rounder, chondrocyte-like cells, and the concentration of proteoglycans and water content is increased.20
Nucleus Pulposus
The nucleus pulposus, the inner, less-organized region of the disc, occupies the middle and posterior thirds of the intervertebral disc.21 It consists of a rich proteoglycan matrix with a lesser amount of disorganized collagen than in the anulus.20,22 There is a significantly higher content of water in the nucleus. The large amount of proteoglycan and water present in the nucleus make it ideal for developing the “swelling pressures” necessary to resist the compressive forces of the spine. At birth, the nucleus is populated almost exclusively with notochordal cells. These cells rapidly begin to necrose shortly after birth and by the age of 4 to 10 years they are completely replaced by rounder, chondrocyte-like cells of mesenchymal origin9 (Table 13-1).
| Component | Anulus Fibrosus | Nucleus Pulposus |
|---|---|---|
| Collagen (mainly types I and II; others present include types III, V, VI, IX, XI, XII, and XIV) | 70% of dry weight 80% type I collagen |
20% of dry weight 80% type II collagen |
| Proteoglycan | 25% of dry weight | 50%–70% of dry weight |
| Water | Lowest content | Highest content |
Cartilaginous End Plate
The cartilaginous end plate is a hyaline cartilage layer separating the disc from the adjacent bony vertebral end plate. It is usually less than 1 mm thick in the mature disc and lies immediately adjacent to the perforated bony end plate. It is loosely cemented to the underlying bony end plate by a thin layer of calcium that is absent near the perforated areas of the bony plate, which allows for nutrient diffusion from the vertebra to the disc.21
Matrix of the Intervertebral Disc
The matrix content of the intervertebral disc is critical to disc function. The uniquely hydrophilic nature of proteoglycans provides the swelling pressure required to increase the intradiscal water content necessary to resist compressive strains on the spine.9,23,24 The matrix composition changes with age, and the quantitative loss and qualitative deterioration of the proteoglycans is thought to represent the single largest biochemical contribution to disc degeneration.20,25
Proteoglycans are highly negatively charged molecules consisting of a core protein and chains of glycosaminoglycans radiating from the core. The chains are composed mainly of keratin and chondroitin sulfate. Multiple proteoglycans then are linked to hyaluronic acid to form large proteoglycan aggregates capable of producing even larger swelling pressures. The aggregates are held together by type II collagen and are cross-linked by type IX collagen.23,26
Aggrecan is the most abundant proteoglycan in the disc.20 It is responsible for 70% of the dry weight of the nucleus pulposus and 25% of the anulus fibrosus. Other proteoglycans present at lower levels include decorin, biglycan, fibromodulin, lumican, versican, and perlican.20,27
Nutrition of the Intervertebral Disc
The intervertebral disc is the largest avascular tissue in the human body. Nutritional support for cell survival, matrix production, and disc homeostasis occurs through diffusion of nutrients through the vertebral end plates.28–30 In the newborn and young child, small blood vessels penetrate the cartilaginous end plates and enter the disc anulus and nucleus.21 These vessels rapidly become less efficient, yet remain partially functional until about 8 years of age. They then become completely nonfunctional over the next two decades of life.5,21,28,31,32
The vertebral body is supplied by an arterial network that drains into a subchondral venous plexus within the bone.28,33 Capillaries from this region penetrate into the subchondral plate and terminate in loops at the bony end plate–cartilaginous end plate junction.28,34,35 Through these capillaries, the nutrients necessary for disc metabolism are delivered and diffuse through the cartilaginous end plates to reach the center of the disc.28 The end plate acts as a semipermeable membrane, letting smaller neutral charges (water, amino acids, oxygen) through more easily than larger, positively charged molecules28,36–38 (Fig. 13-2).
Modulation and Regulation of Disc Matrix
Anabolic Influences
The intervertebral disc matrix is in a constant state of dynamic flux. Anabolic and catabolic influences on the disc matrix continuously interact with the disc to alter the physical composition of the matrix.23 Metalloproteinases (MMPs) modulate the matrix composition by breaking down aged proteoglycans under catabolic influences while newly synthesized matrix takes the place of the degraded proteoglycans.23,39 A very complex, multifactorial, poorly understood interaction of mechanical and biochemical factors defines whether a disc is capable of remodeling in response to loading conditions in order to maintain disc homeostasis.1 If the disc is unable to remodel and maintain a balance of anabolic and catabolic influences, then damage accumulates in a process that may eventually self-perpetuate into a state of continued degeneration.
The key anabolic modulating growth factors work by stimulating the chondrocyte-like cells to produce matrix while inhibiting the MMPs present in the disc.1 In addition to various growth factors, tissue inhibitors of MMPs (TIMPs) are present to suppress the activation of the MMPs23,40 (Table 13-2).
TABLE 13-2 Key Anabolic Modulators (Growth Factors) of the Intervertebral Disc
| Growth Factor | Role |
|---|---|
| Insulin-like growth factor-1 (IGF-1) | Stimulates proteoglycan synthesis Reduces cellular apoptosis |
| Epidermal growth factor (EGF) | Stimulates matrix synthesis Cellular proliferation |
| Basic fibroblast growth factor (bFGF) | Stimulates proteoglycan synthesis Cellular proliferation |
| Platelet-derived growth factor (PDGF) | Decreases cellular apoptosis |
| Transforming growth factor beta (TGF-β) | Greatly increased proteoglycan synthesis Cellular proliferation |
| Bone morphogenetic protein-2 (BMP-2) | Increased proteoglycan synthesis Cellular proliferation Increased mRNA for type II collagen, aggrecan, sox9, and osteocalcin Up-regulates gene expression of BMP-6, BMP-7, and TGF-β |
| Bone morphogenetic protein-7 (BMP-7; osteogenic protein-1 [OP-1]) | Increased proteoglycan synthesis Increased collagen synthesis Cellular proliferation |
| Link protein N-terminal peptide | In concentrations of 10–100 μg/mL stimulates matrix assembly |
From Masuda K, Oegema TJ, An H: Growth factors and treatment of intervertebral disc degeneration. Spine (Phila Pa 1976) 29:2757, 2004.
Catabolic Influences
Catabolic modulation in the disc occurs mainly through cytokine-induced and metalloproteinase degradation of matrix components.23 MMPs 1, 2, 3, 8, 9, and 13 are all present in the disc and serve to degrade collagen, aggrecan, versican, link protein, and the other proteoglycans.9,41–45 The aggrecanases ADAM-TS-4 and ADAM-TS-5, from the ADAM (a disintegrin and metalloproteinase) protein family, are also present in the disc and mainly function to degrade aggrecan and versican.9,46 These MMPs and aggrecanases allow for the large proteoglycans to be degraded into smaller components that may be more easily leached from the intervertebral disc by diffusion.20 In a pathologic catabolic state when the MMPs are working with little opposing matrix production, a significant net loss of proteoglycan and water content occurs, rendering the disc incapable of producing the swelling pressure necessary to resist compressive forces on the disc.20 Overproduction of MMPs can thus lead to increased forces on other spinal structures and contribute to disc and overall spinal degeneration.
In addition to the catabolic effect of MMPs, growing evidence suggests that inflammatory cytokines may have a significant catabolic influence on the disc.23,47,48 Animal studies have indicated that in the presence of injury to the intervertebral disc there are increased levels of interleukin (IL)-1, tumor necrosis factor alpha (TNF-α), and IL-8 near the injured regions of the disc.49 These cytokines have negative catabolic effects on the disc and also stimulate the disc cells to produce chemokines, which are responsible for macrophage attraction and stimulation, especially in the presence of disc herniation.48,50 Macrophages in the disc usually reflect an attempt to break up or remove herniated disc fragments, but their presence also leads to further inflammation and cytokine release, which may prove damaging to the disc.23,51 These inflammatory substances are also responsible for the activation of multiple cascades within the disc cell nuclei that lead to the production of metalloproteinases, which in turn contribute to matrix breakdown.48,52 In addition, these inflammatory cytokines are thought to contribute to the pain-producing effects of disc degeneration48,49,53,54 (Table 13-3).
TABLE 13-3 Key Catabolic Modulators of the Intervertebral Disc
| Catabolic Modulators | Actions |
|---|---|
| Interleukin-1 (IL-1) | Inhibits synthesis of matrix Promotes metalloproteinase production Stimulates nitric oxide synthetase to produce nitric oxide Enhances caseinase activity Triggers the translocation of transcription factors to cell nuclei that target genes that include collagenases and COX-2 |
| Tumor necrosis factor alpha (TNF-α) | Inhibits synthesis of matrix Promotes metalloproteinase production Stimulates nitric oxide synthetase to produce nitric oxide Triggers the translocation of transcription factors to cell nuclei that target genes that include collagenases and COX-2 |
| Nitric oxide | Produces free radicals that damage cell membranes and induce degenerative pathways Inhibits tissue inhibitors of metalloproteinases (TIMPs) |
| Interleukin-6 and interleukin-9 (IL-6, IL-9) fibronectin superoxide | Proinflammatory and pro-nociceptive effect stimulates proteoglycan degradation Degrades hyaluronic acid, causing aggregate breakdown |
COX-2, cyclooxygenase-2.
Data from references 1, 9, 48; and Anderson DG, Tannoury C: Molecular pathogenic factors in symptomatic disc degeneration. Spine J 5(6 Suppl):260S, 2005.
Degeneration
It is very difficult to differentiate the normal degenerative changes of an aging disc from those of pathologic disc degeneration.5,8 It is also crucial to understand that even pathologic disc degeneration is not always accompanied by back pain, and therefore degeneration as discussed here does not necessarily translate to clinical symptomatology. The biochemical and cellular structure of the intervertebral disc begins changing almost immediately after birth, and the process progresses through life.3–5 It is useful to divide disc changes into the following two categories:
Gross and Histologic Changes
The hallmark histologic finding in intervertebral disc degeneration is the progressively diminishing vascular supply, which begins shortly after birth and leads to tissue breakdown as early as the second decade of life.5 Changes in the nucleus pulposus also begin immediately after birth. Within the first two decades of life the nucleus begins to undergo mucoid degeneration, increasing cleft formation, and granular changes consistent with granulation tissue formation at sites of disc injury. These clefts and granular changes eventually lead to frank tears that communicate with anular rim lesions.5,21
In the anulus, peripheral rim lesions appear late in the second decade of life and eventually progress to frank tears that communicate with the nuclear clefts. The end plate also shows degenerative changes that begin with the loss of the end plate vessels early in childhood. After the loss of these vessels, cartilage disorganization increases and cracks in the cartilaginous end plate begin to appear. Microfractures in the adjacent subchondral bone follow and new subchondral bone formation and eventual sclerosis of the plate occur. All of these changes eventually lead to a “burned-out” state,5 apparent in the seventh and later decades of life. At this time, all components of the disc begin to resemble scar tissue and there is no longer a distinction between the different regions of the disc5,21 (Table 13-4 and Figs. 13-3 to 13-6).
FIGURE 13-3 Sagittal section through a normal intervertebral disc.
(From Thalgott J, Albert T, Vaccaro A, et al: A new classification system for degenerative disc disease of the lumbar spine based on magnetic resonance imaging, provocative discography, plain radiographs and anatomic considerations. Spine J 4[6 Suppl]:167S, 2004.)
FIGURE 13-4 Sagittal section through a disc with mild degeneration.
(From Thalgott J, Albert T, Vaccaro A, et al: A new classification system for degenerative disc disease of the lumbar spine based on magnetic resonance imaging, provocative discography, plain radiographs and anatomic considerations. Spine J 4[6 Suppl]:167S, 2004.)
The cellular content of the disc also changes over time. The earliest changes occur shortly after birth and include replacement of the rapidly decaying notochordal cells with mesenchymal chondrocyte-like cells.9 Cell density in both the nucleus and the anulus also rapidly declines after birth. This occurs as the matrix grows along with the expanding volume of the intervertebral disc. The cell density in the nucleus pulposus shortly after birth is approximately 4 × 106 cells/cm3 and in the anulus fibrosus, 9 × 106 cells/cm3. This is significantly lower then the cellular density of even relatively acellular hyaline cartilage,9 which has about 1.4 × 107 cells/cm3. As the disc continues to age, cell death accelerates and large numbers of decaying cells can be found in clusters. By adulthood, more than 50% of the disc’s cells are necrotic55 (Fig. 13-7 and Table 13-5).
FIGURE 13-7 Midsagittal view on T2 density-weighted MRI of discs graded according to a modified Pfirrmann classification (see Table 13-5).
(From Hangai M, Kaneoka K, Kuno S, et al: Factors associated with lumbar intervertebral disc degeneration in the elderly. Spine J 8:732, 2008.)
TABLE 13-5 Modified Pfirrmann Classification as Evaluated on Midsagittal T2-Density-Weighted MRI
| Grade | Signal Intensity of Nucleus Pulposus | Distinction between Nucleus and Anulus |
| I | Hyperintense or isointense to CSF (bright white) and homogeneous | Clear |
| II | Hyperintense of isointense to CSF (white) and inhomogeneous | Clear |
| III | Intermediate to CSF (light gray) and inhomogeneous | |
| IV | Hypointense to CSF (dark gray) and inhomogeneous | |
| V | Low intense to CSF (black) and inhomogeneous |
CSF, cerebrospinal fluid.
Adapted from Hanagai M, Kaneoka K, Kuno S, et al: Factors associated with lumbar intervertebral disc degeneration in the elderly. Spine J 8:732, 2008.
Biochemical Changes
The most significant biochemical change in the degenerating disc is the net loss of proteoglycan.20,25 In addition to this quantitative loss of proteoglycan, the quality of the remaining proteoglycan is compromised.9,27,56 With the loss of large quantities of aggregated proteoglycan, the disc’s ability to create the swelling pressure necessary to absorb the forces imposed on it decreases. These changes also begin shortly after birth, when the rich proteoglycan matrix composition is altered. At birth, chondroitin sulfate is the predominant glycosaminoglycan in the disc, but as the blood supply is progressively lost and the disc volume increases, the disc contents become increasingly anaerobic. Because of the lack of oxygen, chondroitin sulfate is progressively replaced by keratan sulfate chains, which are created in the absence of oxygen.9,57–59
In addition to the changing glycosaminoglycan composition in the disc, the overall structure of the large proteoglycans changes. In the young disc, aggrecan is the predominant proteoglycan. Multiple aggrecan molecules bind to hyaluronan to form an even larger structure called an aggregate. In the fetal and newborn disc, aggregating proteoglycans predominate and are extremely effective in creating large swelling pressures.9,60,61 The amount of aggregating proteoglycan is rapidly diminished with age by proteolytic degradation of the large aggregates. By 6 months of age, only 50% of the anular and 30% of the nuclear proteoglycan content is in aggregated form. In the mature adult disc, only 10% of the proteoglycan is aggregated and these aggregates are smaller and have a higher keratan sulfate composition compared with their younger equivalents.27,62 These degenerative features of disc proteoglycans may make them less effective in creating the swelling pressure necessary to resist compressive forces9 (Fig. 13-8).
FIGURE 13-8 The large aggrecan molecule forms an aggregate with hyaluronan and link protein.
(From Esko JD, Kimata K, Lindahl U: Proteoglycans and sulfated glycosaminoglycans. In Varki A, Cummings RD, Esko JD, et al., editors: Essentials of glycobiology, ed 2, Cold Spring Harbor, NY, 2009, Cold Spring Harbor Laboratory Press. Online text version available at www.ncbi.nlm.nih.gov/books/NBK1900/.)
The collagen content of the disc is not immune to change, also exhibiting signs of degeneration. The disc collagen demonstrates evidence of proteolytic collagenase action, which accumulates and eventually contributes to the weakening mechanical properties of the disc.9,57,63 It is significant that the triple helix structure of disc collagen is more denatured and damaged than that of the collagen in articular cartilage, which may indicate less effective repair mechanisms in the disc.57 In addition, the collagen present in the disc appears to be nonenzymatically cross-linked by advanced glycation end products (AGEs).9,64–66
AGEs are protein modifiers formed through the Maillard reaction when an amino acid and a reducing sugar react.66 With age, AGEs accumulate in various tissues in the body such as the skin, myocardial cells, renal cells, chondrocytes, and intervertebral discs.66–70 AGEs originally were given substantial attention because of their increased presence in diabetes.71 After research elucidated the presence and negative effect of AGEs on articular cartilage, their presence and potential role in intervertebral disc degeneration were sought.66,72,73 in vitro and animal studies suggest that AGEs play a role in disc degeneration. Their increased concentration in diabetes may contribute to the increased relative risk for disc herniation seen in at least one study of patients with diabetes.74
AGEs likely influence disc degeneration through negative biologic and biomechanical contributions. Biomechanical studies have demonstrated that the accumulation of AGEs leads to alterations in the disc’s physical properties, the most significant of which is increased mechanical stiffness.75 This increased stiffness of the disc also likely leads to a decreased ability to produce the swelling pressure necessary for normal disc function.9,69 Biologic in vitro studies have also demonstrated that the accumulation of AGEs in intervertebral disc cells leads to a down-regulation in the production of aggrecan.66 In addition, a recent bovine study documented the increased expression of the receptor for AGEs (RAGE) in intervertebral discs in the presence of an inflammatory state with increased IL-1β. All of these studies noted a decrease in aggrecan production, which may at least partially explain the increased disc degeneration seen in the presence of chronic inflammation.48,76
Disc Material Properties and Disc Degeneration
The gross structural and biochemical changes associated with early degeneration lead to changes in the intervertebral disc’s mechanical properties, which make it less capable of performing its necessary functions.77 Cadaveric studies conducted on discs of various ages and degrees of degeneration have demonstrated an increased shear modulus in aged and degenerated discs, indicating a decreased ability of the disc to absorb energy.77–79 In essence, the discs become increasingly stiff and appear to undergo a transition from “fluid-like” behavior to “solid-like” behavior with increasing degeneration.80 Alterations in disc viscoelasticity have also been seen, including both increased creep and creep rate. In addition to these changes, which more drastically affect the nucleus, alterations in the anulus result in a decreased Poisson’s ratio as well as decreased radial permeability, which transforms the anisotropic hydraulic permeability of the normal disc to a more isotropic permeability in degenerated discs81 (Box 13-1).
