[level-membership-for-pulmolory-and-respiratory-category]
Interstitial Lung Diseases
After reading this chapter, you will be able to:
• List the anatomic alterations of the lungs associated with chronic interstitial lung disease.
• Describe the causes of chronic interstitial lung disease.
• List the cardiopulmonary clinical manifestations associated with chronic interstitial lung disease.
• Describe the general management of chronic interstitial lung disease.
• Describe the clinical strategies and rationales of the SOAPs presented in the case study.
• Define key terms and complete self-assessment questions at the end of the chapter and on Evolve.
Anatomic Alterations of the Lungs
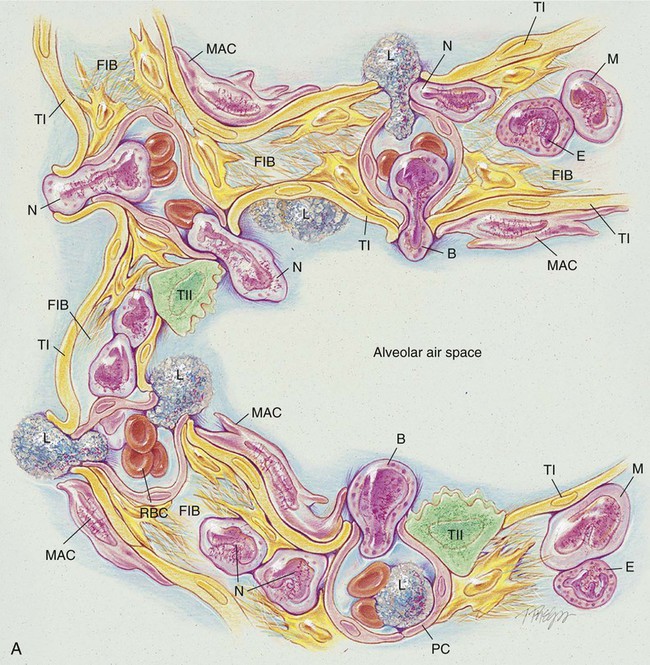
The anatomic alterations of ILD may involve the bronchi, alveolar walls, and adjacent alveolar spaces. In severe cases the extensive inflammation leads to pulmonary fibrosis, granulomas, honeycombing, and cavitation. During the acute stage of any ILD, the general inflammatory condition is characterized by edema and the infiltration of a variety of white blood cells (e.g., neutrophils, eosinophils, basophils, monocytes, macrophages, and lymphocytes) in the alveolar walls and interstitial spaces (see Figure 25-1, A). Bronchial inflammation and thickening and increasing airway secretions may be also present.
The major pathologic or structural changes associated with chronic ILDs are as follows:
• Destruction of the alveoli and adjacent pulmonary capillaries
• Fibrotic thickening of the respiratory bronchioles, alveolar ducts, and alveoli
• Honeycombing and cavity formation
• Fibrocalcific pleural plaques (particularly in asbestosis)
• Excessive bronchial secretions (caused by inflammation of airways)
Etiology and Epidemiology
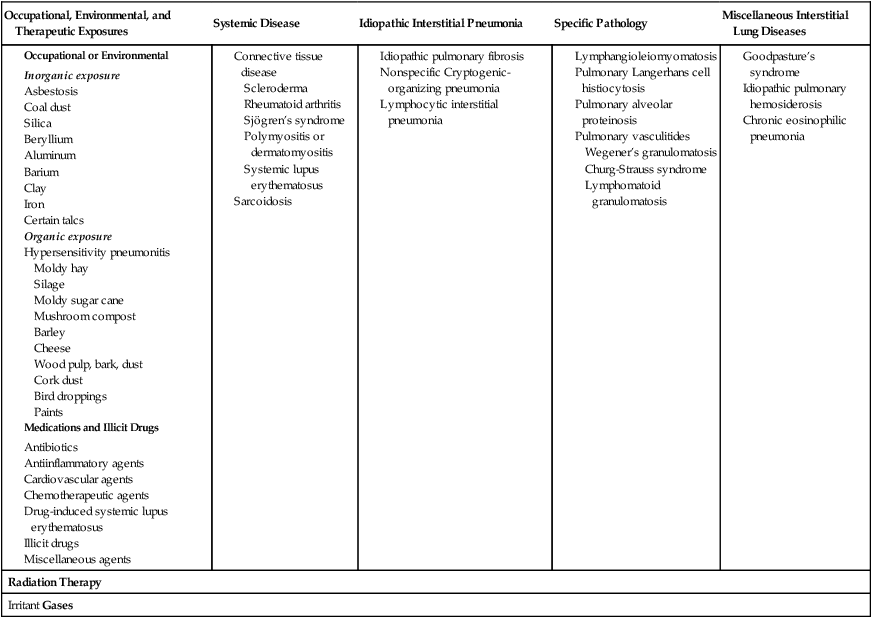
Because there are over 180 different pulmonary disorders classified as ILD, it is helpful to group them according to their occupational or environmental exposure, disease associations, and specific pathology. Table 25-1 provides an overview of common ILD groups. A discussion of the more common ILDs follows.
Table 25-1
Interstitial Lung Diseases of Known Causes or Associations
Occupational, Environmental and Therapeutic Exposures
Inorganic particulate (dust) exposure
Asbestos
Exposure to asbestos may cause asbestosis—a common form of ILD. Asbestos fibers are a mixture of fibrous minerals composed of hydrous silicates of magnesium, sodium, and iron in various proportions. There are two primary types: the amphiboles (crocidolite, amosite, and anthophyllite) and chrysotile (most commonly used in industry). Asbestos fibers typically range from 50 to 100 µm in length and are about 0.5 µm in diameter. The chrysotiles have the longest and strongest fibers. Box 25-1 lists common sources associated with asbestos fibers.
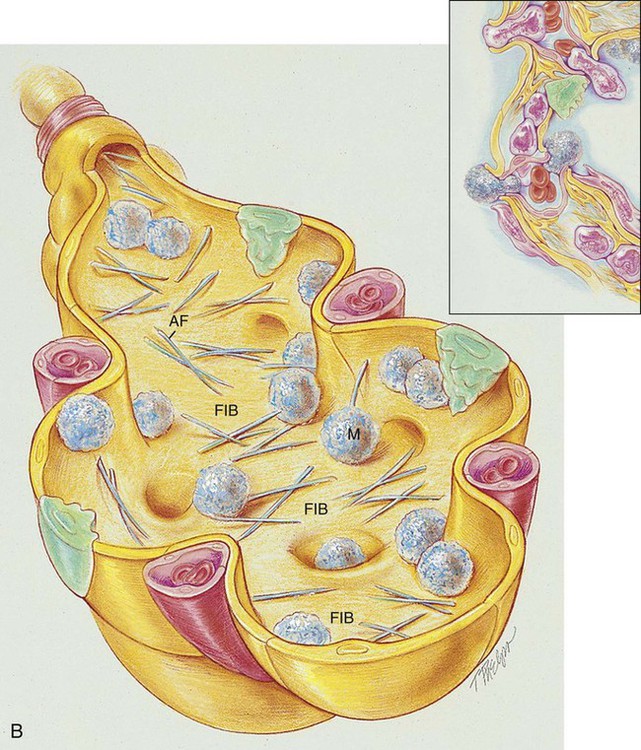
As shown in Figure 25-1, B, asbestos fibers can be seen by microscope within the thickened septa as brown or orange baton-like structures. The fibers characteristically stain for iron with Perls’ stain. The pathologic process may affect only one lung, a lobe, or a segment of a lobe. The lower lobes are most commonly affected. Pleural calcification is common and diagnostic in patients with an asbestos exposure history.
Silica
Complicated silicosis is characterized by nodules that coalesce and form large masses of fibrous tissue, usually in the upper lobes and perihilar regions. In severe cases the fibrotic regions may undergo tissue necrosis and cavitate. Box 25-2 lists common occupations associated with silica exposure.
Organic materials exposure
Hypersensitivity pneumonitis
Hypersensitivity pneumonitis (also called allergic alveolitis or extrinsic allergic alveolitis) is a cell-mediated immune response of the lungs caused by the inhalation of a variety of offending agents or antigens. Such antigens include grains, silage, bird droppings or feathers, wood dust (especially redwood and maple), cork dust, animal pelts, coffee beans, fish meal, mushroom compost, and molds that grow on sugar cane, barley, and straw. The immune response to these allergens causes production of antibody and an inflammatory response. The lung inflammation, or pneumonitis, develops after repeated and prolonged exposure to the allergen. The term hypersensitivity pneumonitis (or allergic alveolitis) is often renamed according to the type of exposure that caused the lung disorder. For example, the hypersensitivity pneumonitis caused by the inhalation of moldy hay is called farmer’s lung. Table 25-2 provides common causes, exposure sources, and disease syndromes associated with hypersensitivity pneumonitis.
Table 25-2
Causes of Hypersensitivity Pneumonitis
| Antigen | Exposure Source | Disease (Syndrome) |
| Bacteria, Thermophilic | ||
| Saccharopolyspora rectivirgula | Moldy hay, silage | Farmer’s lung |
| Thermoactinomyces vulgaris | Moldy sugarcane | Bagassosis |
| Thermoactinomyces sacchari | Mushroom compost | Mushroom worker’s lung |
| Thermoactinomyces candidus | Heated water reservoirs | Air conditioner lung |
| Bacteria, Nonthermophilic | ||
| Bacillus subtilis, Bacillus cereus | Water, detergent | Humidifier lung, washing powder lung |
| Fungi | ||
| Aspergillus species | Moldy hay | Farmer’s lung |
| Water | Ventilation pneumonitis | |
| Aspergillus clavatus | Barley | Malt worker’s lung |
| Penicillium casei, | Cheese | Cheese washer’s lung |
| Penicillium roqueforti | ||
| Alternaria species | Wood pulp | Woodworker’s lung |
| Cryptostroma corticale | Wood bark | Maple bark stripper’s lung |
| Graphium, Aureobasidium pullulans | Wood dust | Sequoiosis |
| Merulius lacrymans | Rotten wood | Dry root lung |
| Penicillium frequentans | Cork dust | Suberosis |
| Aureobasidium pullulans | Water | Humidifier lung |
| Cladosporium species | Hot tub mist | Hot tub HP* |
| Trichosporon cutaneum | Damp wood and mats | Japanese summer-type HP* |
| Amebae | ||
| Naegleria gruberi | Contaminated water | Humidifier lung |
| Acanthamoeba polyphaga | Contaminated water | Humidifier lung |
| Acanthamoeba castellani | Contaminated water | Humidifier lung |
| Animal Protein | ||
| Avian proteins | Bird droppings, feathers | Bird-breeder’s lung |
| Urine, serum, pelts | Rates, gerbils | Animal handler’s lung |
| Chemicals | ||
| Isocyanates, trimellitic anhydride | Paints, resins, plastics | Chemical worker’s lung |
| Copper sulfate | Bordeaux mixture | Vineyard sprayer’s lung |
| Phthalic anhydride | Heated epoxy resin | Epoxy resin lung |
| Sodium diazobenzene sulfate | Chromatography reagent | Pauli’s reagent alveolitis |
| Pyrethrum | Pesticide | Pyrethrum HP* |
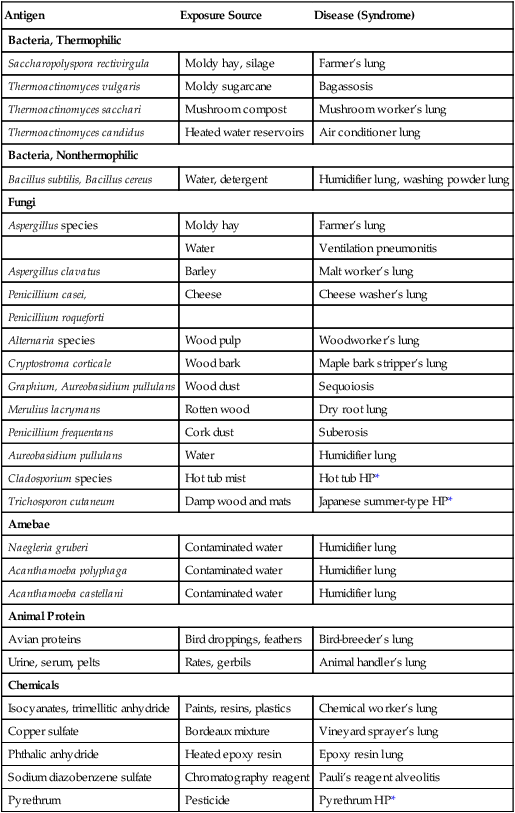
*HP, Hypersensitivity pneumonitis.
From Selman M: Hypersensitivity pneumonitis. In Schwarz MI, Kin TE, eds: Interstitial lung disease, ed 4, Hamilton, 2003, BC Decker.
Medications and illicit drugs
As the list of medications and illicit drugs continues to grow, so does the list of possible side effects (Box 25-4). Unfortunately, the lungs are major target organs affected by these side effects. Although it is impossible to discuss in detail the various lung-related side effects of every drug, it is possible to describe some of the general concerns related to drug-induced lung disease and to list some of the pharmacologic agents that may be responsible.
The chemotherapeutic (anticancer agents) are by far the largest group of agents associated with ILD. Bleomycin, mitomycin, busulfan, cyclophosphamide, methotrexate, and carmustine (BCNU) are the major offenders. Nitrofurantoin (an antibacterial drug used in the treatment of urinary tract infections) is also associated with ILD. Gold and penicillamine for the treatment of rheumatoid arthritis also have been shown to cause ILD. The excessive long-term administration of oxygen (oxygen toxicity) also is known to cause diffuse pulmonary injury and fibrosis (see Chapter 27). As a general rule, the risk of these drugs causing an interstitial lung disorder is directly related to the cumulative dosage. However, drug-induced interstitial disease may be seen as early as 1 month to as late as several years after exposure to these agents.
Irritant gases
The inhalation of irritant gases may cause an acute chemical pneumonitis and, in severe cases, ILD. Most exposures occur in an industrial setting. Table 25-3 lists some of the more common irritant gases and the industrial settings where they may be found.
Table 25-3
Common Irritant Gases Associated with Interstitial Lung Disease
| Gas | Industrial Setting |
| Chlorine | Chemical and plastic industries; water disinfection |
| Ammonia | Commercial refrigeration; smelting of sulfide ores |
| Ozone | Welding |
| Nitrogen dioxide | May be liberated after exposure of nitric acid to air |
| Phosgene | Used in the production of aniline dyes |
Miscellaneous Diffuse Interstitial Lung Diseases
Goodpasture’s Syndrome
Chronic Eosinophilic Pneumonia
 OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Chronic Interstitial Lung Diseases
OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Chronic Interstitial Lung Diseases
The following clinical manifestations result from the pathophysiologic mechanisms caused (or activated) by an Increased Alveolar-Capillary Membrane Thickness (see Figure 9-10) and Excessive Bronchial Secretions (see Figure 9-12)—the major anatomic alterations of the lungs associated with chronic interstitial lung disease (see Figure 28-1).
CLINICAL DATA OBTAINED AT THE PATIENT’S BEDSIDE
CLINICAL DATA OBTAINED FROM LABORATORY TESTS AND SPECIAL PROCEDURES
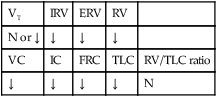
Pulmonary Function Test Findings Moderate to Severe Interstitial Lung Disease (Restrictive Lung Pathology)
FORCED EXPIRATORY FLOW RATE FINDINGS
| FVC | FEVT | FEV1/FVC ratio | FEF25%-75% |
| ↓ | N or ↓ | N or ↑ | N or ↓ |
| FEF50% | FEF200-1200 | PEFR | MVV |
| N or ↓ | N or ↓ | N or ↓ | N or ↓ |

Arterial Blood Gases
MILD TO MODERATE INTERSTITIAL LUNG DISEASE
Acute Alveolar Hyperventilation with Hypoxemia (Acute Respiratory Alkalosis)
| pH | Paco2 | 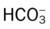 |
Pao2 |
| ↑ | ↓ | ↓ (slightly) | ↓ |

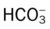

Oxygenation Indices*
Moderate to Severe Stage Interstitial Lung Disease
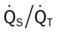 |
Do2† | 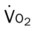 |
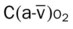 |
O2ER | 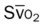 |
| ↑ | ↓ | N | N | ↑ | ↓ |

†The Do2 may be normal in patients who have compensated to the decreased oxygenation status with (1) an increased cardiac output, (2) an increased hemoglobin level, or (3) a combination of both. When the Do2 is normal, the O2ER is usually normal.
*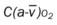 , Arterial-venous oxygen difference; DO2, total oxygen delivery; O2ER, oxygen extraction ratio;
, Arterial-venous oxygen difference; DO2, total oxygen delivery; O2ER, oxygen extraction ratio; 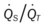 , pulmonary shunt fraction;
, pulmonary shunt fraction; 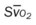 , mixed venous oxygen saturation;
, mixed venous oxygen saturation; 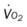 , oxygen consumption.
, oxygen consumption.
Hemodynamic Indices*
Severe Interstitial Lung Disease
| CVP | RAP |  |
PCWP | CO | SV |
| ↑ | ↑ | ↑ | N | N | N |
| SVI | CI | RVSWI | LVSWI | PVR | SVR |
| N | N | ↑ | N | ↑ | N |
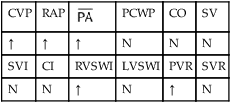
*CO, Cardiac output; CVP, central venous pressure; LVSWI, left ventricular stroke work index; 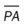 , mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; RVSWI, right ventricular stroke work index; SV, stroke volume; SVI, stroke volume index; SVR, systemic vascular resistance.
, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; RVSWI, right ventricular stroke work index; SV, stroke volume; SVI, stroke volume index; SVR, systemic vascular resistance.
Radiologic findings vary according to the cause.
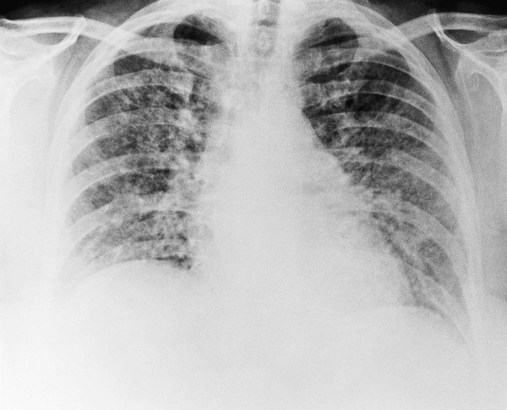
As shown in Figure 25-2, a patient with severe scleroderma, a bilateral reticulonodular pattern is commonly seen on the radiographs. In patients with asbestosis the opacity is often described as cloudy in appearance or as having a “ground-glass” appearance and is especially apparent in the lower lobes (Figure 25-3). Calcified pleural plaques may be seen on the superior border of the diaphragm or along the chest wall (Figure 25-4). The inflammatory response elicited by the asbestos fibers also may produce a fuzziness and irregularity of the cardiac and diaphragmatic borders.
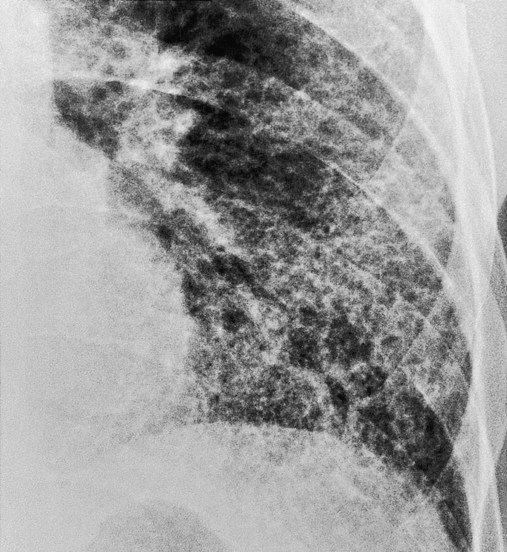
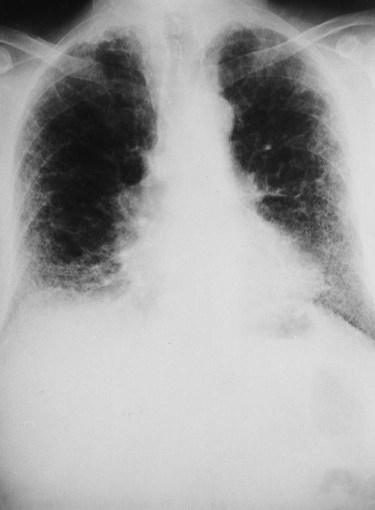
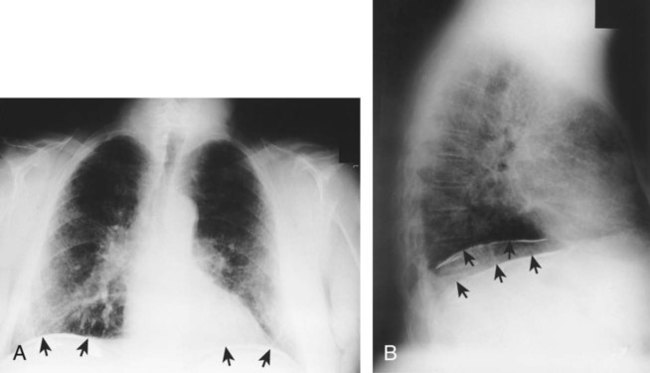
Figure 25-5 shows a diffuse parenchymal ground-glass pattern with some areas of consolidation in a patient with acute farmer’s lung. The severity of parenchymal opacification in this case is rare.
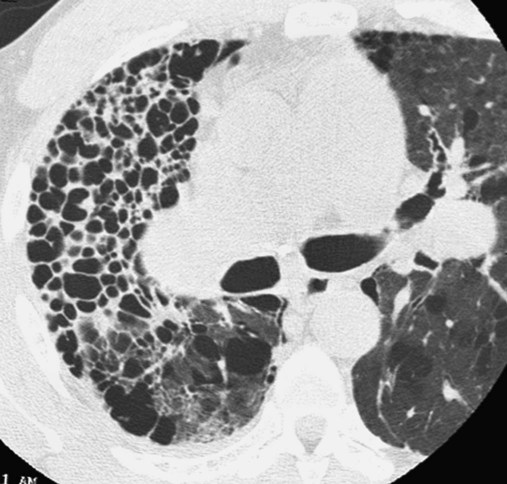
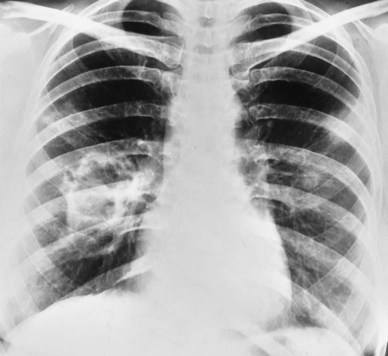
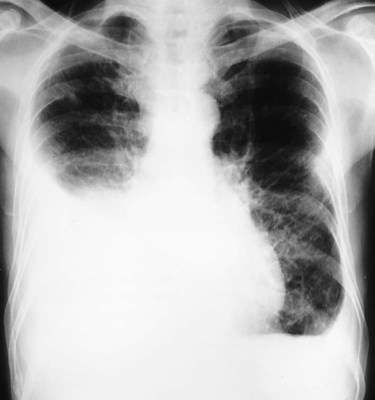
In Figure 25-6, the honeycomb appearance is nicely illustrated in a computed tomography (CT) scan of a patient with sarcoidosis. Figure 25-7 shows a patient with Wegener’s granulomatosis with numerous nodules with a large cavity lesion adjacent to the right hilus. Figure 25-8 shows a pleural effusion in a patient with rheumatoid disease.
General Management of Interstitial Lung Disease
Medications and Procedures Commonly Prescribed by the Physician
Respiratory Care Treatment Protocols
Oxygen Therapy Protocol
Oxygen therapy is used to treat hypoxemia, decrease the work of breathing, and decrease myocardial work. Because of the hypoxemia associated with ILDs, supplemental oxygen often is required. The hypoxemia that develops in an interstitial lung disorder most commonly is caused by the alveolar thickening, fibrosis, and capillary shunting associated with the disorder. In addition, because the patient may demonstrate chronic ventilatory failure during the advanced stages of an ILD, caution must be taken not to overoxygenate the patient (see Oxygen Therapy Protocol, Protocol 9-1).
Mechanical Ventilation Protocol
Mechanical ventilation may be needed to provide and support alveolar gas exchange and eventually return the patient to spontaneous breathing. Because acute ventilatory failure superimposed on chronic ventilatory failure often is seen in patients with severe ILD, continuous mechanical ventilation may be required. Continuous mechanical ventilation is justified when the acute ventilatory failure is thought to be reversible (see Mechanical Ventilation Protocols, Protocol 9-5, Protocol 9-6, and Protocol 9-7).
CASE STUDY
Interstitial Lung Disease
Admitting History
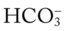 46, and Pa
46, and Pa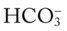 42, and Pa
42, and PaRespiratory Assessment and Plan
S “This is the worst my breathing has ever been.”
O Vital signs: BP 180/96, HR 108, RR 32, T 38.3° C (100.8° F); weak appearance; skin: cyanotic, damp, and clammy; distended neck veins and digital clubbing; cough: frequent, weak, moderate amount of thick, whitish yellow secretions; peripheral edema 3+ of ankles and feet. Bilateral dull percussion notes in lung bases. Over both lungs: rhonchi and crackles; pleural friction rub over right middle lobe between sixth and seventh ribs, between anterior axillary line and midaxillary line; CXR: ground-glass appearance in lower lobes; calcified pleural plaques in right and left lower pleural spaces; consolidation in right middle lung lobe; right heart enlargement; ABGs (1.5 L/min O2 by nasal cannula): pH 7.56, Paco2 51, 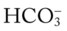 42, Pao2 47.
42, Pao2 47.
• Respiratory distress (general appearance, vital signs, ABGs)
• Pulmonary fibrosis (history, diagnosis of asbestosis, CXR)
• Alveolar consolidation in right middle lobe (CXR)
• Pleurisy (asbestosis or pneumonitis) in area of right middle lobe (pleural friction rub)
• Excessive bronchial secretions (rhonchi, sputum production)
• Chest infection likely (yellow sputum)
• Acute alveolar hyperventilation superimposed on chronic ventilatory failure with moderate-to-severe hypoxemia (history, ABGs)
P Begin Oxygen Therapy Protocol (HAFOE at Fio2 0.28). Bronchopulmonary Hygiene Therapy Protocol (C&DB q4h; obtaining sputum for Gram stain and culture). Initiate Lung Expansion Therapy Protocol (incentive spirometry followed by C&DB). Monitor with alarming pulse oximeter set at 85% Spo2.
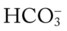 36, and Pa
36, and PaRespiratory Assessment and Plan
S N/A (patient too dyspneic to reply)
O Condition unstable; cough: frequent, weak, productive of thick, white and yellow secretions; skin: cyanotic, pale, cool, and damp; distended neck veins and peripheral edema, but improving; vital signs: BP 192/108, HR 113, RR 34, T 38° C (100.4° F); dull percussion notes over both lung bases; rhonchi and crackles throughout both lungs; pleural friction rub over right middle lobe between sixth and seventh ribs, between anterior axillary line and midaxillary line; ABGs (Fio2 = 0.28): pH 7.57, Paco2 47, 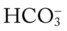 36, Pao2 40; Spo2 77%
36, Pao2 40; Spo2 77%
• Continued respiratory distress (general appearance, vital signs, ABGs)
• Pulmonary fibrosis in lower lobes (history, diagnosis of asbestosis, recent CXR)
• Alveolar consolidation in right middle lobe (CXR, pneumonia)
• Pleurisy or pneumonia that has extended into pleural space over right middle lobe (pleural friction rub)
• Excessive bronchial secretions (rhonchi, sputum production)
• Infection likely (yellow sputum)
• Acute alveolar hyperventilation superimposed on chronic ventilatory failure with severe hypoxemia, worsening (history, ABGs)
• Impending ventilatory failure (ABGs: increased alveolar hyperventilation and worsening Pao2)
P Up-regulate Oxygen Therapy Protocol (HAFOE to Fio2 0.40). Bronchopulmonary Hygiene Therapy Protocol (adding intensive nasotracheal suctioning q2h). Start Aerosolized Medication Protocal (nebulize 2 mL acetylcysteine to the premix albuterol). Continue Lung Expansion Therapy Protocol (continuing to coach and monitor incentive spirometry; if FVC falls below 15 mL/kg, administer CPAP mask at +10 cm H2O for 20 minutes qid while patient is awake). Continue to monitor closely.
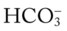 38, and Pa
38, and PaRespiratory Assessment and Plan
S N/A (patient intubated on ventilator)
O Vital signs: BP 135/90 on vasopressors, HR 84, T 38.3° C (100.8° F); frequent premature ventricular contractions; skin: pale, cyanotic, and clammy; distended neck veins; peripheral edema of ankles and feet; dull percussion notes over lung bases; rhonchi and crackles throughout both lungs; thick, greenish-yellow sputum frequently suctioned; pleural friction rub over right middle lung lobe between sixth and seventh ribs and between anterior axillary line and midaxillary line; hemodynamic indices: elevated CVP, RAP, PA, RVSWI, and PVR; ABGs (Fio2 = 1.0): pH 7.53, Paco2 56, 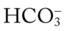 38, Pao2 246, Spo2 98%
38, Pao2 246, Spo2 98%
• Pulmonary fibrosis, lower lung lobes (history, diagnosis of asbestosis, recent CXR)
• Alveolar consolidation, right middle lobe (recent CXR showing pneumonia)
• Pneumonia possibly extended into pleural space over right middle lobe (pleural friction rub)
• Excessive bronchial secretions (rhonchi, sputum production)
• Infection likely (fever, greenish-yellow sputum, possible new organism)
• Acute alveolar hyperventilation superimposed on chronic ventilatory failure and overly corrected hypoxemia (ABGs)
• Overventilation and overoxygenation caused by mechanical ventilator
P Down-regulate Oxygen Therapy Protocol (reduction of Fio2 to 0.50). Down-regulate Mechanical Ventilation Protocol (decreasing tidal volume to increase Paco2 to patient’s baseline—80 to 90 mm Hg). Continue Bronchopulmonary Hygiene Therapy Protocol and Aerosolized Medication Protocol. Continue Lung Expansion Therapy Protocol (depending on mean airway pressure). Continue to closely monitor and reevaluate.
Discussion
In the initial assessment the patient’s severe hypertension and his fever were noted. Both deserved vigorous therapy if his pulmonary function were to improve at all. The patient’s severe hypoxemia reflected common clinical indicators caused by Alveolar-Capillary Membrane Thickening (see Figure 9-10) and Excessive Bronchial Secretions (see Figure 9-12). Although there is no therapy to reverse the increased alveolar membrane thickening, the excessive bronchial secretions can be effectively treated in most cases.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Interstitial Lung Diseases
After reading this chapter, you will be able to:
• List the anatomic alterations of the lungs associated with chronic interstitial lung disease.
• Describe the causes of chronic interstitial lung disease.
• List the cardiopulmonary clinical manifestations associated with chronic interstitial lung disease.
• Describe the general management of chronic interstitial lung disease.
• Describe the clinical strategies and rationales of the SOAPs presented in the case study.
• Define key terms and complete self-assessment questions at the end of the chapter and on Evolve.
Anatomic Alterations of the Lungs
The anatomic alterations of ILD may involve the bronchi, alveolar walls, and adjacent alveolar spaces. In severe cases the extensive inflammation leads to pulmonary fibrosis, granulomas, honeycombing, and cavitation. During the acute stage of any ILD, the general inflammatory condition is characterized by edema and the infiltration of a variety of white blood cells (e.g., neutrophils, eosinophils, basophils, monocytes, macrophages, and lymphocytes) in the alveolar walls and interstitial spaces (see Figure 25-1, A). Bronchial inflammation and thickening and increasing airway secretions may be also present.
The major pathologic or structural changes associated with chronic ILDs are as follows:
• Destruction of the alveoli and adjacent pulmonary capillaries
• Fibrotic thickening of the respiratory bronchioles, alveolar ducts, and alveoli
• Honeycombing and cavity formation
• Fibrocalcific pleural plaques (particularly in asbestosis)
• Excessive bronchial secretions (caused by inflammation of airways)
Etiology and Epidemiology
Because there are over 180 different pulmonary disorders classified as ILD, it is helpful to group them according to their occupational or environmental exposure, disease associations, and specific pathology. Table 25-1 provides an overview of common ILD groups. A discussion of the more common ILDs follows.
Table 25-1
Interstitial Lung Diseases of Known Causes or Associations
Occupational, Environmental and Therapeutic Exposures
Inorganic particulate (dust) exposure
Asbestos
Exposure to asbestos may cause asbestosis—a common form of ILD. Asbestos fibers are a mixture of fibrous minerals composed of hydrous silicates of magnesium, sodium, and iron in various proportions. There are two primary types: the amphiboles (crocidolite, amosite, and anthophyllite) and chrysotile (most commonly used in industry). Asbestos fibers typically range from 50 to 100 µm in length and are about 0.5 µm in diameter. The chrysotiles have the longest and strongest fibers. Box 25-1 lists common sources associated with asbestos fibers.
As shown in Figure 25-1, B, asbestos fibers can be seen by microscope within the thickened septa as brown or orange baton-like structures. The fibers characteristically stain for iron with Perls’ stain. The pathologic process may affect only one lung, a lobe, or a segment of a lobe. The lower lobes are most commonly affected. Pleural calcification is common and diagnostic in patients with an asbestos exposure history.
Silica
Complicated silicosis is characterized by nodules that coalesce and form large masses of fibrous tissue, usually in the upper lobes and perihilar regions. In severe cases the fibrotic regions may undergo tissue necrosis and cavitate. Box 25-2 lists common occupations associated with silica exposure.
Organic materials exposure
Hypersensitivity pneumonitis
Hypersensitivity pneumonitis (also called allergic alveolitis or extrinsic allergic alveolitis) is a cell-mediated immune response of the lungs caused by the inhalation of a variety of offending agents or antigens. Such antigens include grains, silage, bird droppings or feathers, wood dust (especially redwood and maple), cork dust, animal pelts, coffee beans, fish meal, mushroom compost, and molds that grow on sugar cane, barley, and straw. The immune response to these allergens causes production of antibody and an inflammatory response. The lung inflammation, or pneumonitis, develops after repeated and prolonged exposure to the allergen. The term hypersensitivity pneumonitis (or allergic alveolitis) is often renamed according to the type of exposure that caused the lung disorder. For example, the hypersensitivity pneumonitis caused by the inhalation of moldy hay is called farmer’s lung. Table 25-2 provides common causes, exposure sources, and disease syndromes associated with hypersensitivity pneumonitis.
Table 25-2
Causes of Hypersensitivity Pneumonitis
| Antigen | Exposure Source | Disease (Syndrome) |
| Bacteria, Thermophilic | ||
| Saccharopolyspora rectivirgula | Moldy hay, silage | Farmer’s lung |
| Thermoactinomyces vulgaris | Moldy sugarcane | Bagassosis |
| Thermoactinomyces sacchari | Mushroom compost | Mushroom worker’s lung |
| Thermoactinomyces candidus | Heated water reservoirs | Air conditioner lung |
| Bacteria, Nonthermophilic | ||
| Bacillus subtilis, Bacillus cereus | Water, detergent | Humidifier lung, washing powder lung |
| Fungi | ||
| Aspergillus species | Moldy hay | Farmer’s lung |
| Water | Ventilation pneumonitis | |
| Aspergillus clavatus | Barley | Malt worker’s lung |
| Penicillium casei, | Cheese | Cheese washer’s lung |
| Penicillium roqueforti | ||
| Alternaria species | Wood pulp | Woodworker’s lung |
| Cryptostroma corticale | Wood bark | Maple bark stripper’s lung |
| Graphium, Aureobasidium pullulans | Wood dust | Sequoiosis |
| Merulius lacrymans | Rotten wood | Dry root lung |
| Penicillium frequentans | Cork dust | Suberosis |
| Aureobasidium pullulans | Water | Humidifier lung |
| Cladosporium species | Hot tub mist | Hot tub HP* |
| Trichosporon cutaneum | Damp wood and mats | Japanese summer-type HP* |
| Amebae | ||
| Naegleria gruberi | Contaminated water | Humidifier lung |
| Acanthamoeba polyphaga | Contaminated water | Humidifier lung |
| Acanthamoeba castellani | Contaminated water | Humidifier lung |
| Animal Protein | ||
| Avian proteins | Bird droppings, feathers | Bird-breeder’s lung |
| Urine, serum, pelts | Rates, gerbils | Animal handler’s lung |
| Chemicals | ||
| Isocyanates, trimellitic anhydride | Paints, resins, plastics | Chemical worker’s lung |
| Copper sulfate | Bordeaux mixture | Vineyard sprayer’s lung |
| Phthalic anhydride | Heated epoxy resin | Epoxy resin lung |
| Sodium diazobenzene sulfate | Chromatography reagent | Pauli’s reagent alveolitis |
| Pyrethrum | Pesticide | Pyrethrum HP* |
*HP, Hypersensitivity pneumonitis.
From Selman M: Hypersensitivity pneumonitis. In Schwarz MI, Kin TE, eds: Interstitial lung disease, ed 4, Hamilton, 2003, BC Decker.
Medications and illicit drugs
As the list of medications and illicit drugs continues to grow, so does the list of possible side effects (Box 25-4). Unfortunately, the lungs are major target organs affected by these side effects. Although it is impossible to discuss in detail the various lung-related side effects of every drug, it is possible to describe some of the general concerns related to drug-induced lung disease and to list some of the pharmacologic agents that may be responsible.
[/not-level-membership-for-pulmolory-and-respiratory-category]
