Cystic Fibrosis
After reading this chapter, you will be able to:
• List the anatomic alterations of the lungs associated with cystic fibrosis.
• Describe the causes and classifications of cystic fibrosis.
• List the cardiopulmonary clinical manifestations associated with cystic fibrosis.
• Describe the general management of cystic fibrosis.
• Describe the clinical strategies and rationales of the SOAPs presented in the case study.
• Define key terms and complete self-assessment questions at the end of the chapter and on Evolve.
Anatomic Alterations of the Lungs*
The abundance of stagnant mucus in the tracheobronchial tree also serves as an excellent culture medium for bacteria, particularly Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa. Some gram-negative bacteria are also commonly associated with cystic fibrosis, such as Stenotrophomonas maltophilia, Burkholderia cepacia, Burkholderia pickettii, and Burkholderia gladioli. The infection stimulates additional mucous production and further compromises the mucociliary transport system. This condition may lead to secondary bronchial smooth muscle constriction. Finally, as the disease progresses, the patient may develop signs and symptoms of recurrent pneumonia, chronic bronchitis, bronchiectasis, and lung abscesses (Figure 14-1).
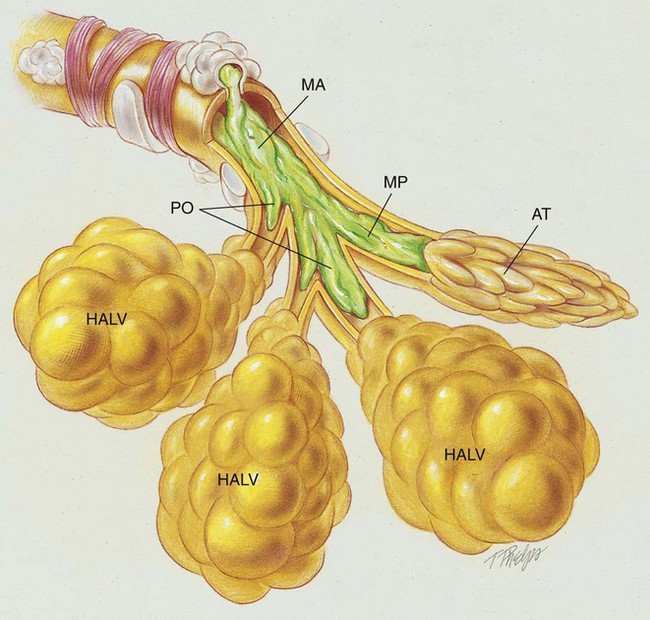
The major pathologic or structural changes associated with cystic fibrosis are as follows:
Etiology and Epidemiology
How the Cystic Fibrosis Gene Is Inherited
Because cystic fibrosis is a recessive gene disorder, the child must inherit two copies of the defective cystic fibrosis gene—one from each parent (cystic fibrosis carriers)—to have the disease. Even though the carrier of the cystic fibrosis gene may be identified through genetic testing, the carrier (heterozygote) does not demonstrate evidence of the disease. However, if both parents carry the cystic fibrosis gene, the possibility of their children having cystic fibrosis (regardless of gender) follows the standard Mendelian pattern: there is a 25% chance that each child will have cystic fibrosis, a 25% chance that each child will be completely normal (and not carry the gene), and a 50% chance that each child will be a carrier. Thus, when both patients have the cystic fibrosis gene, there is a one in four chance that the child will have cystic fibrosis (Figure 14-2). It is estimated that more than 10 million Americans are unknowing, symptomless carriers of the mutant cystic fibrosis gene.
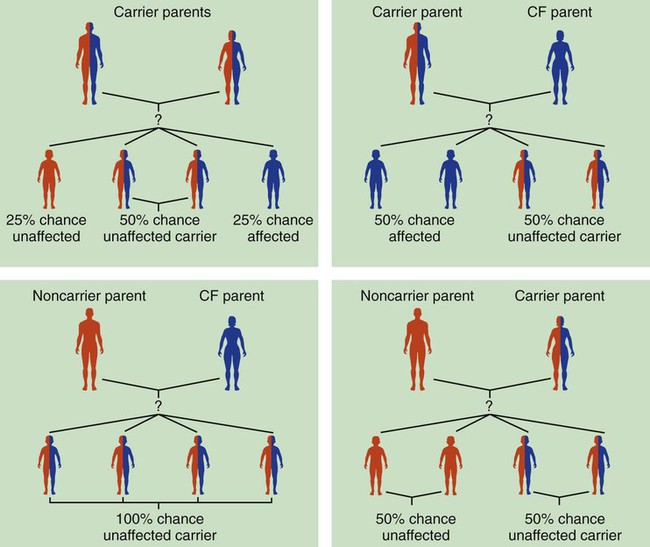
According to the Cystic Fibrosis Foundation, cystic fibrosis affects about 30,000 children and adults in the United States. About 1000 new cases of cystic fibrosis are diagnosed each year in the United States (70,000 worldwide). More than 70% of the patients are diagnosed by age 2. More than 40% of the cystic fibrosis patient population is age 18 or older.* Other researchers state that whites are most often affected (1 in 2500 to 3500). Cystic fibrosis is less common in Hispanics (1 in 9500) and African-Americans (1 in 17,000). Cystic fibrosis is rarely seen in Asians (1 in 31,000). The predicted median life expectancy is 37 years. Death usually is caused by pulmonary complications.
Screening and Diagnosis
The diagnosis of cystic fibrosis is based on clinical manifestations associated with cystic fibrosis, family history of cystic fibrosis, and laboratory findings. Box 14-1 provides common clinical indicators that justify evaluation for cystic fibrosis.
Sweat Test
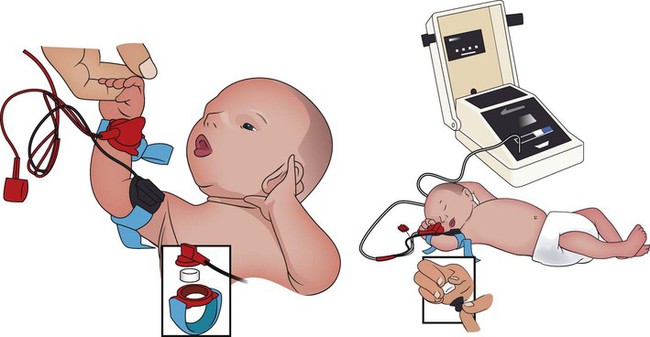
The sweat test (sometimes called the sweat chlorine test) is the gold standard diagnostic test for cystic fibrosis. The sweat test is a reliable test for the identification of approximately 98% of patients with cystic fibrosis. This test measures the amount of sodium and chloride in the patient’s sweat. During the procedure a small amount of a colorless, odorless sweat-producing chemical called pilocarpine is applied to the patient’s arm or leg—usually the forearm. An electrode is attached to the chemically prepared area, and a mild electric current is applied to stimulate sweat production (Figure 14-3). To collect the sweat, the area is covered with a gauze pad or filter paper and wrapped in plastic. After about 30 minutes the plastic is removed, and the sweat collected in the pad or paper is sent to the laboratory for analysis. The test is usually done twice.
Prenatal Testing
• Amniocentesis—the amniotic fluid is obtained and tested to determine if both of the CFTR genes from the fetus are normal. Amniocentesis can be used to diagnose a large number of genetic and chromosomal abnormalities, including cystic fibrosis.
• Chorionic villus biopsy—with the aid of an ultrasound examination, a thin tube is inserted into the uterus to obtain a piece of the placenta to biopsy. The cells of the placenta are then tested for a variety of genetic defects, including cystic fibrosis.
 OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Cystic Fibrosis
OVERVIEW of the Cardiopulmonary Clinical Manifestations Associated with Cystic Fibrosis
The following clinical manifestations result from the pathophysiologic mechanisms caused (or activated) by Atelectasis (see Figure 9-8), Bronchospasm (see Figure 9-11), and Excessive Bronchial Secretions (see Figure 9-12)—the major anatomic alterations of the lungs associated with cystic fibrosis (CF) (see Figure 14-1).
CLINICAL DATA OBTAINED AT THE PATIENT’S BEDSIDE
Peripheral Edema and Venous Distention
Because polycythemia and cor pulmonale are associated with CF, the following may be seen:
Spontaneous pneumothorax commonly is seen in patients with CF. The incidence is greater than 20% in adults with CF. When a patient with CF has a pneumothorax, there is about a 50% chance that it will recur. The respiratory care practitioner must be alert for the signs and symptoms of this complication (e.g., pleuritic pain, shoulder pain, sudden shortness of breath). Precipitating factors include excessive exertion, high altitude, and positive-pressure breathing (see Pneumothorax, Chapter 22.)
CLINICAL DATA OBTAINED FROM LABORATORY TESTS AND SPECIAL PROCEDURES
Pulmonary Function Test Findings
Moderate to Severe Cystic Fibrosis (Obstructive Lung Pathophysiology)*
FORCED EXPIRATORY FLOW RATE FINDINGS
| FVC | FEVT | FEV1/FVC ratio | FEF25%-75% |
| ↓ | ↓ | ↓ | ↓ |
| FEF50% | FEF200-1200 | PEFR | MVV |
| ↓ | ↓ | ↓ | ↓ |

LUNG VOLUME AND CAPACITY FINDINGS
| VT | IRV | ERV | RV | |
| N or ↑ | N or ↓ | N or ↓ | ↑ | |
| VC | IC | FRC | TLC | RV/TLC ratio |
| ↓ | N or ↓ | ↑ | N or ↑ | N or ↑ |
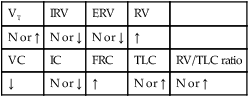
*Cystic fibrosis is primarily an obstructive lung disorder. However, when extensive total lung obstruction (from mucous plugging) and atelectasis is present throughout the lungs, restrictive pulmonary function testing values will likely appear.
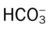

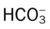

Oxygenation Indices*
Moderate to Severe Stages
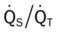 |
Do2† | 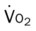 |
C(a-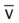 )o2 )o2 |
O2ER | 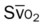 |
| ↑ | ↓ | N | N | ↑ | ↓ |

†The Do2 may be normal in patients who have compensated to the decreased oxygenation status with (1) an increased cardiac output, (2) an increased hemoglobin level, or (3) a combination of both. When the Do2 is normal, the O2ER is usually normal.
*C(a-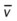 )O2, Arterial-venous oxygen difference; DO2, total oxygen delivery; O2ER, oxygen extraction ratio;
)O2, Arterial-venous oxygen difference; DO2, total oxygen delivery; O2ER, oxygen extraction ratio; 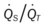 , pulmonary shunt fraction;
, pulmonary shunt fraction; 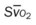 , mixed venous oxygen saturation; , oxygen consumption.
, mixed venous oxygen saturation; , oxygen consumption.
Hemodynamic Indices†
Moderate to Severe Stages
| CVP | RAP |  |
PCWP | CO | SV |
| ↑ | ↑ | ↑ | N | N | N |
| SVI | CI | RVSWI | LVSWI | PVR | SVR |
| N | N | ↑ | N | ↑ | N |
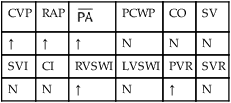
†CO, Cardiac output; CVP, central venous pressure; LVSWI, left ventricular stroke work index; 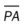 , mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; RVSWI, right ventricular stroke work index; SV, stroke volume; SVI, stroke volume index; SVR, systemic vascular resistance.
, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; RVSWI, right ventricular stroke work index; SV, stroke volume; SVI, stroke volume index; SVR, systemic vascular resistance.
• Translucent (dark) lung fields
• Depressed or flattened diaphragms
• Right ventricular enlargement
• Areas of atelectasis and fibrosis
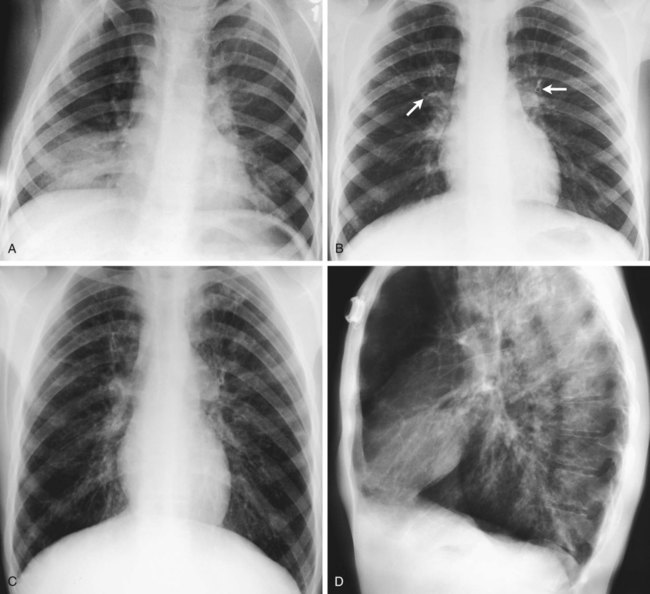
During the late stages of CF, the alveoli become hyperinflated, which causes the residual volume and functional residual capacity to increase. Because this condition decreases the density of the lungs and therefore reduces the resistance to x-ray penetration, the chest radiograph appears darker. As the patient’s residual volume and functional residual capacity increase, the diaphragm moves downward and appears flattened or depressed on the radiograph (Figure 14-4).
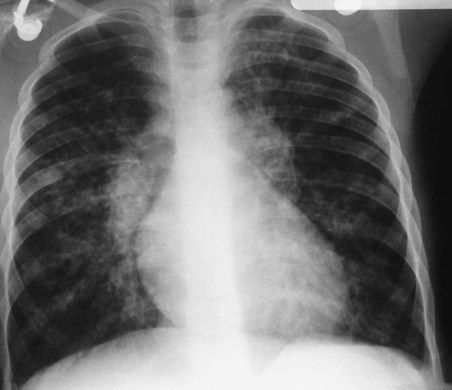
Figure 14-5 presents four serial chest radiographs of the progression of CF over 26 years. Because right ventricular enlargement and failure often develop as secondary problems during the advanced stages of CF, an enlarged heart may be identified on the radiograph. In some patients, areas of atelectasis, abscess formation, or a pneumothorax may be seen. Finally, computed tomography (CT) and positron emission tomography (PET) scans may be helpful when borderline radiographic findings are present.
General Management of Cystic Fibrosis
Respiratory Care Treatment Protocols
Oxygen Therapy Protocol
Oxygen therapy is used to treat hypoxemia, decrease the work of breathing, and decrease myocardial work. The hypoxemia that develops in cystic fibrosis is caused by the pulmonary shunting associated with the disorder. When the patient demonstrates chronic ventilatory failure during the advanced stages of cystic fibrosis, caution must be taken not to overoxygenate the patient (see Oxygen Therapy Protocol, Protocol 9-1).
Bronchopulmonary Hygiene Therapy Protocol
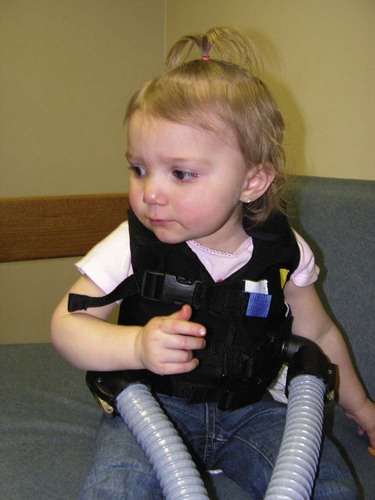
Because of the excessive mucous production and accumulation associated with cystic fibrosis, a number of respiratory therapy modalities are used to enhance the mobilization of bronchial secretions. Aggressive and vigorous bronchial hygiene—especially chest physical therapy and postural drainage—should be performed regularly on patients both in the hospital and at home. Because many patients with cystic fibrosis require bronchial hygiene therapy at least twice a day for 20 to 30 minutes, a mechanical percussor or a high-frequency chest compression vest can be especially helpful in moving thick bronchial secretions (Figure 14-6) (see Bronchopulmonary Hygiene Therapy Protocol, Protocol 9-2).
Aerosolized Medication Protocol
Various sympathomimetic, parasympatholytic, and mucolytic agents are commonly used to induce bronchial smooth muscle relaxation and mucous thinning. Dornase alpha (Pulmozyme—also known as rhDNase or DNase) has been shown to be especially helpful in the management of cystic fibrosis. This aerosolized agent is an enzyme that breaks down the DNA of the thick bronchial mucus associated with cystic fibrosis. Dornase alpha has shown good results in improving the lung function of patients with cystic fibrosis while reducing the frequency and severity of respiratory infections (see Aerosolized Medication Protocol, Protocol 9-4 and Appendix II).
Mechanical Ventilation Protocol
Because acute ventilatory failure superimposed on chronic ventilatory failure often is seen in patients with severe cystic fibrosis, mechanical ventilation may be required to maintain an adequate ventilatory status. Continuous mechanical ventilation is justified when the acute ventilatory failure is thought to be reversible—for example, when pneumonia complicates the condition (see Mechanical Ventilation Protocols, Protocol 9-5, Protocol 9-6, and Protocol 9-7).
Medications and Special Procedures Prescribed by the Physician
Xanthines
Xanthines are occasionally used to enhance bronchial smooth muscle relaxation (see Appendix II, Xanthine Bronchodilators).
Antibiotics
Antibiotics are commonly administered to prevent or combat secondary respiratory tract infections. For example, an antibiotic treatment widely used to treat P. aeruginosa in the cystic fibrosis patient is inhaled tobramycin (TOBI). Unfortunately, a major drawback of long-term use of antibiotics is the development of bacteria that become resistant to drug therapy. In addition, the long-term use of antibiotics often leads to fungal infections of the mouth, oral pharynx, and tracheobronchial tree (see Appendix II, Antibiotic Agents).
Lung or Heart-Lung Transplantation
Several large organ transplant centers currently are performing lung or heart-lung transplantations in selected patients with cystic fibrosis whose general body condition is good. According to the Cystic Fibrosis Foundation, approximately 900 lung transplants are performed each year in the United States. Since 1991, about 1600 cystic fibrosis patients have received lung transplants—120 to 150 patients per year. In 2003, 368 patients were accepted for the lung transplant procedure. The success of lung transplantation in patients with cystic fibrosis is as good as or better than in patients with other lung diseases (e.g., emphysema). As many as 90% of the patients with cystic fibrosis are alive 1 year after transplantation, and 50% are alive after 5 years.*
Physical Examination
His chest x-ray examination on this admission revealed hyperlucent lung fields, depressed hemidiaphragms, and mild cardiac enlargement (Figure 14-7). His arterial blood gas values (ABGs) on 1.5 L/min oxygen by nasal cannula were as follows: pH 7.51, Paco2 58 mm Hg, 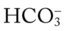 43, and Pao2 66 mm Hg. His hemoglobin oxygen saturation measured by pulse oximetry (Spo2) was 94%. On the basis of these clinical data, the following SOAP was documented.
43, and Pao2 66 mm Hg. His hemoglobin oxygen saturation measured by pulse oximetry (Spo2) was 94%. On the basis of these clinical data, the following SOAP was documented.
Respiratory Assessment and Plan
S “I’ve not been this short of breath in a long time.”
O Skin: pale, cyanotic; barrel chest and use of accessory muscles of respiration; digital clubbing; cough frequent and productive; sputum: sweet-smelling, thick, yellow-green; distended neck veins and peripheral edema; vital signs: BP 142/90, HR 108, RR 28, T normal; bilateral hyperresonant percussion notes; diminished breath sounds; crackles and rhonchi; CXR: hyperlucency, flattened diaphragms, and mild cardiac enlargement; ABGs (1.5 L/min O2 by nasal cannula): pH 7.51, Paco2 58, 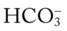 43, Pao2 66; Spo2 94%
43, Pao2 66; Spo2 94%
• Respiratory distress (general appearance, vital signs)
• Excessive tracheobronchial tree secretions (productive cough)
• Infection likely (yellow-green sputum)
• Hyperinflated alveoli (barrel chest, use of accessory muscles, CXR)
• Acute alveolar hyperventilation superimposed on chronic ventilatory failure with mild hypoxemia (history, ABGs)
• Possible impending acute ventilatory failure
• Cor pulmonale (distended neck veins, peripheral edema, CXR)
P Bronchopulmonary Hygiene Therapy Protocol (cough and deep breathe Tx q4h), sputum culture). Oxygen Therapy Protocol (2 L/min by nasal cannula). Monitor possible impending ventilatory failure closely (pulse oximetry, vital signs, ABGs).
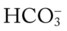 45, and Pa
45, and PaRespiratory Assessment and Plan
S “I can’t get enough air to sleep 10 minutes!”
O Cyanosis and use of accessory muscles of respiration; vital signs: BP 147/95, HR 117, RR 32, T 37° C (98.6° F); cough: frequent, weak, and productive of large amounts of thick, green sputum; Pseudomonas aeruginosa cultured; bilateral hyperresonant notes and diminished breath sounds; crackles, rhonchi, and wheezes; Spo2 92%; ABGs: pH 7.55, Paco2 54, 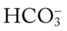 45, Pao2 57
45, Pao2 57
• Continued respiratory distress (general appearance, vital signs, use of accessory muscles)
• Excessive bronchial secretions (cough, sputum, breath sounds)
• Poor ability to mobilize secretions (weak cough)
• Acute alveolar hyperventilation superimposed on chronic ventilatory failure with mild- to-moderate hypoxemia (ABGs)
P Start Aerosolized Medication Protocol (0.5 cc albuterol in 2 cc rhDNase q4h). Up-regulate Bronchopulmonary Hygiene Therapy per protocol (CPT and PEP therapy q2h). Up-regulate Oxygen Therapy Protocol (HAFOE mask at Fio2 0.35). Continue to monitor possible impending ventilatory failure closely.
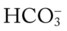 41, and Pa
41, and PaRespiratory Assessment and Plan
S “I can’t get into a comfortable position to breathe.”
O Cyanosis; pursed-lip breathing and use of accessory muscles of respiration; vital signs: BP 145/90, HR 120, RR 22, T 38° C (100.5° F); bilateral hyperresonant percussion notes, crackles, rhonchi, and wheezing; Spo2 65%; ABGs: pH 7.33, Paco2 79, 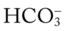 41, PaO2 37
41, PaO2 37
• Continued respiratory distress (general appearance, vital signs, use of accessory muscles, pursed-lip breathing)
• Excessive bronchial secretions (cough, sputum, breath sounds)
• Acute ventilatory failure superimposed on chronic ventilatory failure with severe hypoxemia (ABGs, vital signs)
P Contact physician stat. Consider intubation and implementation of the Mechanical Ventilation Protocol. Continue Aerosolized Medication and Bronchial Hygiene Therapy Protocol (after patient has been placed on ventilator). Up-regulate Oxygen Therapy Protocol (initially, Fio2 0.50 on ventilator). Monitor closely.
Discussion
1. Vigorous use of chest physical therapy (percussion and postural drainage)
2. Intermittent treatment of secretions with antibiotics and mucolytics, such as Pulmozyme
Note that on the initial physical examination, the patient demonstrated excessive bronchial secretions and a productive cough; no baseline arterial blood gases existed with which to compare his current values (see Bronchospasm, Figure 9-11). Thus the observation of an elevated Paco2 should be taken very, very seriously, because (at least initially) whether this value is a “chronic” arterial blood gas value is unclear.
*Cystic fibrosis does not affect the lungs exclusively. It also affects the function of exocrine glands in other parts of the body. In addition to being characterized by abnormally viscid secretions in the lungs, the disease is clinically manifested by pancreatic insufficiency and high sodium concentrations in the sweat.
