[level-membership-for-emergency-medicine-category]
146 Insecticides, Herbicides, and Rodenticides
 Key Points
Key Points• Organophosphorus and carbamate poisonings cause excessive stimulation of muscarinic and nicotinic receptors by acetylcholine, which can potentially lead to life-threatening bronchorrhea and bronchospasm.
• Aggressive airway management and liberal use of atropine are important in the management of both organophosphorus and carbamate poisoning.
• Only a nondepolarizing neuromuscular blocker, such as vecuronium or rocuronium, should be used for intubation. Succinylcholine is metabolized by plasma cholinesterase, and prolonged paralysis may result if it is used in the setting of organophosphate poisoning.
• Timely administration of pralidoxime is key to the treatment of organophosphorus poisoning, but pralidoxime is not indicated for carbamate poisoning.
• Unintentional pediatric ingestion of 4-hydroxycoumarins (superwarfarins) accounts for the vast majority of rodenticide exposures and rarely results in toxicity.
• Ingestion of an anticoagulant rodenticide should be considered when a child younger than 6 years has an elevated prothrombin time or bleeding without another explanation.
• The prothrombin time should be measured at 24 and 48 hours after large ingestions of 4-hydroxycoumarins.
• Because no specific antidote or pharmacologic intervention has proved beneficial in treating paraquat or diquat poisoning, early decontamination is the most important step.
insecticides
Organophosphorus Compounds and Carbamates
Epidemiology
Organophosphate (OP) compounds and carbamates are used extensively worldwide for agricultural, industrial, and domestic pest control and, as a result, represent a significant public health issue in the developing world. An estimated 3 million poisonings and more than 200,000 deaths occur from OP compounds each year worldwide.1 In the United States in 2008, 4642 exposures to OP compounds and 2644 exposures to carbamates were reported to the National Poison Data System of the American Association of Poison Control Centers.2
Pathophysiology
Under normal circumstances, ACh is hydrolyzed by AChE to yield acetic acid and choline. In the presence of OP insecticides, AChE is phosphorylated, whereas in the presence of carbamate insecticides, the enzyme is carbamylated. As a result, the rate of regeneration of active AChE is slowed, and its function is inhibited. Within 24 to 72 hours of OP poisoning, an alkyl group may dissociate from the AChE-OP complex and thereby result in “aging” of the AChE. Once aging occurs, reactivation of AChE is no longer possible, and only synthesis of new enzyme can restore activity. In the case of carbamate poisoning, breakdown of the carbamate-AChE complex occurs much more rapidly and aging does not occur (Box 146.1).3
Box 146.1 Effects of Organophosphate and Carbamate on Acetylcholinesterase (AChE)
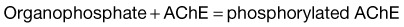
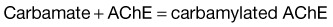
These complexes inactivate AChE and allow acetylcholine to sit on the nicotinic and muscarinic receptors and produce the symptoms of toxicity.
Three things can happen to the phosphorylated or carbamylated AChE:
ACh accumulates in the autonomic nervous system at postganglionic muscarinic (parasympathetic and sympathetic) receptors and preganglionic nicotinic (sympathetic) receptors. It also accumulates at the neuromuscular junction and in the central nervous system (CNS). Overstimulation of these receptors is responsible for the cholinergic toxidrome seen with OP and carbamate insecticide poisoning (Table 146.1).
See Table 146.1, Effects of Organophosphorus and Carbamate Insecticides, at www.expertconsult.com
Table 146.1 Effects of Organophosphorus and Carbamate Insecticides
| RECEPTOR | TARGET TISSUE | CLINICAL EFFECT |
|---|---|---|
Presenting Signs and Symptoms
The clinical effects are summarized in Table 146.1; only caveats in the clinical findings are emphasized here. Bronchorrhea occurs commonly with moderate to severe poisonings4 and can progress to pulmonary edema and respiratory failure. Miosis in the setting of cholinergic symptoms is fairly specific for OP and carbamate insecticide poisoning and may help make the diagnosis. Unfortunately, it is not consistently present.
Although the parasympathetic muscarinic effects are most often emphasized, certain sympathetic effects may predominate. Sinus tachycardia is more common than bradycardia,4,5 and mydriasis may even be seen.5 Nicotinic effects often predominate in mild cases and occur early in severe cases. Excessive nicotinic stimulation at the neuromuscular junction has effects that resemble the actions of a depolarizing neuromuscular blocking agent. Therefore, patients with OP or carbamate insecticide poisoning may exhibit muscle fasciculations and weakness. Paralysis occurs as the toxicity worsens, and the primary cause of death in acute poisonings is probably respiratory arrest secondary to paralysis and bronchorrhea.
One to 3 days after apparent resolution of the symptoms, patients may experience profound weakness and paralysis of the proximal muscles, neck flexor muscles, and cranial nerves. This development, termed the intermediate syndrome,6 is probably explained by ongoing AChE inhibition (Box 146.2).
Box 146.2 Paralysis Seen After Organophosphate Poisoning
Type II (Intermediate Syndrome)
• Develops 1 to 3 days after resolution of the acute organophosphate poisoning symptoms
• Manifested as paralysis and respiratory distress secondary to weakness of the proximal muscles, neck flexor muscles (with relative sparing of the distal muscle groups), and cranial nerve palsies
• Lasts for 4 to 18 days and may require mechanical ventilation
• Results from ongoing acetylcholinesterase inhibition or suboptimal treatment
Differential Diagnosis and Medical Decision Making
A detailed history in a patient with signs and symptoms of cholinergic excess often elucidates exposure to OP or carbamate insecticides. The diagnosis of OP or carbamate insecticide poisoning is therefore usually straightforward; however, certain clinical aspects may be mimicked by other entities. Table 146.2 is a partial list of other agents or diagnoses to consider.
Table 146.2 Differential Diagnosis of Organophosphorus and Carbamate Poisoning
| Other acetylcholinesterase inhibitors | Physostigmine, neostigmine, pyridostigmine |
| Other organophosphorus cholinesterase inhibitors (chemical weapon nerve agents) | Sarin, tabun, soman, Vx |
| Cholinomimetics | Pilocarpine, carbachol, methacholine, bethanechol, muscarine-containing mushrooms |
| Nicotinic alkaloids | Nicotine, coniine, lobeline |
| Other (symptom based) |
All patients with potential OP poisoning should undergo erythrocyte (red blood cell [RBC], or true) cholinesterase and plasma (pseudo) cholinesterase measurement from specimens obtained after arrival at the emergency department (ED). Though not often useful or necessary for making a diagnosis in the ED, the results of this measurement may help guide continued therapy. RBC cholinesterase hydrolyzes ACh and correlates with toxicity, whereas plasma cholinesterase is the first to decline and may be a more sensitive marker of exposure.7 Both substances should be measured because one may exhibit greater inhibition than the other, depending on the specific OP to which the patient was exposed. Box 146.3 summarizes the tests that may be helpful in evaluating a patient with moderate to severe toxicity.
Treatment
The treatment algorithm for OP and carbamate insecticide poisoning is summarized in Figure 146.1. The first step is adequate decontamination of the patient by removal of wet clothing and washing of contaminated skin with soap and water. ED personnel should wear gowns, gloves, and masks to prevent exposure to contaminated body fluids.8
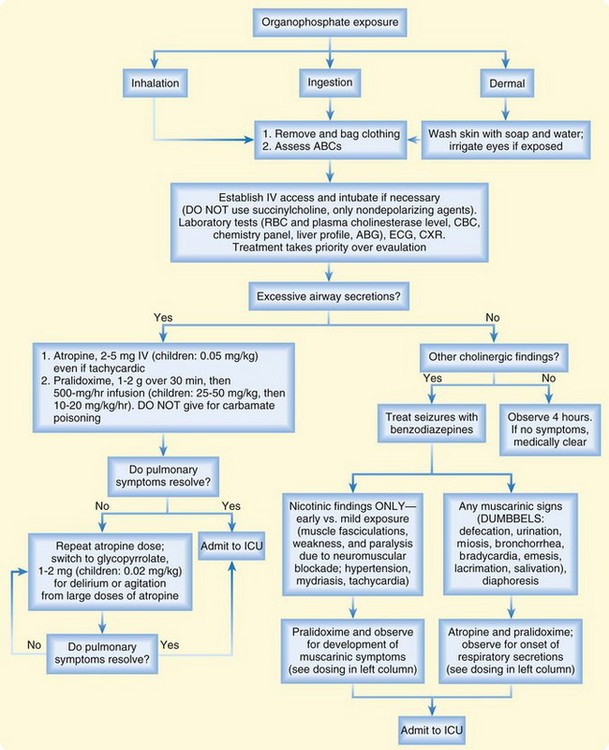
As the patient is being decontaminated, the emergency physician (EP) should focus on the ABCs (airway, breathing circulation), with particular attention paid to early airway, management for copious secretions, seizures, coma, severe weakness, and paralysis. If intubation is necessary, only a nondepolarizing neuromuscular blocking agent, such as vecuronium or rocuronium, should be used. Succinylcholine is metabolized by plasma cholinesterase, so prolonged paralysis may result if this agent is used a patient with OP poisoning.9
 Priority Actions
Priority Actions
Organophosphates
Treatment should next be directed at controlling muscarinic activity. Atropine is the drug of choice and should be administered intravenously at a dose of 2 to 5 mg (pediatric dose, 0.05 mg/kg) every 3 to 5 minutes, with the end point being control of respiratory secretions. Tachycardia is not a contraindication to atropine administration. Mild poisonings may resolve with just 1 to 2 mg of atropine, and severe poisonings may require more than 1000 mg.10 Large doses of atropine may lead to antimuscarinic CNS toxicity. If such toxicity occurs, glycopyrrolate (1 to 2 mg; pediatric dose, 0.025 mg/kg) can be used in place of atropine.
Organochlorines
Epidemiology
Organochlorines are heavily chlorinated aromatic compounds that are nonvolatile and poorly water soluble. They are divided into four classes on the basis of their structural characteristics, and they vary tremendously with respect to dermal absorption, lipid solubility, and toxic doses. The clinical toxicity, which is similar for each of the classes, is summarized in (Table 146.3).
See Table 146.3, Major Organophosphorus Insecticides, at www.expertconsult.com
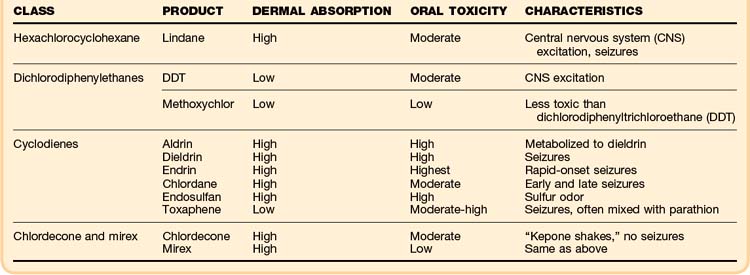
Most organochlorines have been banned in North America because of concern about their environmental persistence and bioconcentration. The only organochlorine still in common use in the United States is lindane (Kwell). It is used in agriculture as a seed treatment and medicinally as a topical scabicide in a 1% formulation. Toxicity from therapeutic lindane application is exceedingly rare, and most clinically relevant toxicity events occur as a result of inappropriate dermal application or ingestion.11–13
Pathophysiology
Lindane acts as an antagonist of γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the CNS.14 Toxicity results from loss of inhibitory tone and subsequent CNS hyperexcitability.
Presenting Signs and Symptoms
Symptoms, which can occur within 30 minutes of the ingestion of lindane,12 often include nausea and vomiting. With excessive or repeated topical applications, the onset of symptoms may be delayed from a few hours up to 4 to 5 days.11,14 CNS excitation is the hallmark of lindane toxicity. It is manifested by paresthesias, agitation, tremor, myoclonus, hallucinations, and most important, seizures. Seizures may occur suddenly and without prodrome. Complications of prolonged seizures may develop, including respiratory failure, metabolic acidosis, rhabdomyolysis, and hyperthermia.
Pyrethrins and Pyrethroids
Epidemiology
Pyrethrins are naturally occurring esters of chrysanthemum resin that possess insecticidal activity, whereas pyrethroids are synthetic derivatives of pyrethrins. Exposures to these agents are commonly reported to poison centers. Most are accidental, and serious clinical effects are rare.2
Pathophysiology
Pyrethrins and pyrethroids delay closure of sodium channels. The delay results in prolonged depolarization, repetitive firing, and eventually conduction blockade.15 Some pyrethroids may inhibit GABA chloride channels, but it is unlikely that such inhibition plays a significant role in toxicity.
Presenting Signs and Symptoms
Most cases of clinically relevant toxicity from pyrethrins result from pulmonary allergic reactions rather than from direct toxic effects. The signs and symptoms are similar to those of asthma exacerbations and consist of wheezing, cough, dyspnea, and chest pain. Most reactions are mild and easily treated. However, fatal status asthmaticus has been reported with exposure to pyrethrin-containing shampoo.16,17
Accidental or occupational exposure to pyrethroids usually produces minimal, if any, toxicity. The most common symptoms reported are facial paresthesias, dizziness, headache, nausea, anorexia, and fatigue.18 Massive exposures or large intentional ingestions may lead to more serious manifestations: seizures, altered mental status, coma, respiratory failure, and death.
Fipronil
Presenting Signs and Symptoms
The majority of human exposures are unintentional and most commonly result in neurologic symptoms such as dizziness and headache. Ocular and upper respiratory irritation has been reported commonly in addition to nausea and vomiting.19 More severe exposures or large intentional ingestions can cause CNS excitation and seizures.
Treatment
Tips and Tricks
Insecticides
Use glycopyrrolate in patients who need more atropine but show signs of central nervous system antimuscarinic toxicity, such as delirium and agitation.
Do not rely on the pupils to rule in or rule out the diagnosis.
The presence of tachycardia should not prevent administration of atropine to a patient with bronchorrhea or wheezing.
Base treatment on clinical signs and symptoms, not on acetylcholinesterase levels.
 Red Flags
Red Flags
Insecticides
Miosis in the setting of cholinergic symptoms, though not consistently present, is fairly specific for organophosphate and carbamate insecticides and may help make the diagnosis.
Although the parasympathetic muscarinic effects are most often emphasized, certain sympathetic effects may predominate (sinus tachycardia is more common than bradycardia, and mydriasis may be seen).
Nicotinic effects often predominate in mild cases and occur early in severe cases.
“Normal” cholinesterase levels do not necessarily rule out poisoning if the history and clinical picture are otherwise indicative.
Symptoms can occur within 30 minutes of the ingestion of lindane, but with excessive or repeated topical applications, the onset of symptoms may be delayed from a few hours to 4 to 5 days.
Herbicides
Paraquat and Diquat
Epidemiology
Paraquat and diquat account for only 4.9% of herbicide poisonings but are responsible for more than 50% of herbicide-related deaths.20 This fact points to the extremely toxic nature of these compounds. Most serious toxicity events and deaths are secondary to intentional ingestion.21
Pathophysiology
Paraquat is rapidly absorbed after ingestion and is concentrated in type I and type II alveolar epithelial cells. It is subsequently reduced to a free radical, which then reacts with oxygen to form a superoxide anion (O2−). This anion then may form H2O2, which in the presence of Fe2+, will generate highly reactive species such as the hydroxyl radical (OH). These reactive molecules cause lipid peroxidation and cellular destruction.22 Initially, acute alveolitis may occur. Later, proliferative changes and pulmonary fibrosis are seen. Although paraquat concentrates mostly in the lungs, it is also distributed throughout the entire body and causes cellular destruction in multiple organs.
The pathophysiologic mechanism of diquat is similar to that of paraquat. Diquat does not concentrate in the lungs, however, and does not produce pulmonary fibrosis.23
Presenting Signs and Symptoms
Paraquat poisoning can be classified as mild, moderate, or severe according to the amount ingested.21 Physical examination findings are summarized in Table 146.4. Mild poisonings, which occur when small amounts of dilute preparations are ingested, are characterized by the development of gastrointestinal symptoms without other organ toxicity. As the amount of paraquat or diquat ion ingested rises, worsening gastrointestinal effects are seen, including severe oropharyngeal, esophageal, and gastric ulceration. Large ingestions produce renal and hepatic failure within a few days. Paraquat toxicity results in pulmonary fibrosis and refractory hypoxemia several days to weeks after ingestion, and death usually occurs within a few weeks. Massive ingestions cause multiorgan failure and death within a few days. Diquat toxicity does not produce pulmonary fibrosis. Diquat ingestion has been associated with brainstem infarction.23 Effects from dermal exposure to paraquat and diquat are usually mild, but ulcers and blistering can occur with highly concentrated formulations.
| DEGREE | AMOUNT INGESTED | CLINICAL FEATURES |
|---|---|---|
| Mild | <20 mg/kg paraquat ion |
Treatment
Supportive care should be provided, with airway protection and ventilation being paramount. Supplemental oxygen may worsen the toxicity by accelerating the damage caused by oxygen radicals. It is generally accepted that supplemental oxygen be withheld until the PaO2 value falls below 40 to 50 mm Hg. IV fluids should be given to ensure normal urine output and analgesics provided for the pain associated with mucosal ulcerations. Many other pharmacologic treatments of paraquat poisoning have been investigated, but none have proved useful.22 Hemoperfusion and hemodialysis are effective in removing paraquat from the blood, but neither improves the prognosis.
Chlorphenoxy Herbicides
Epidemiology
Chlorphenoxy herbicides are widely used to control the growth of broad-leaved weeds in pastures and crop fields and along public streets. Poisoning is uncommon, and most ED encounters consist of accidental dermal or inhalational exposure, for which serious systemic toxicity is rare. However, intentional ingestion of these compounds carries high morbidity and mortality. From 1962 to 2004, 69 cases of ingestion of chlorphenoxy herbicides alone (excluding other pesticides as coingestants) were reported. One third of the patients in these reports died.24
Pathophysiology
The pathophysiology of chlorphenoxy herbicide toxicity involves three mechanisms. First, a dose-dependent disruption of cell membranes is thought to be responsible for mediation of CNS toxicity through disruption of the blood-brain barrier. Second, these compounds may form analogues of acetyl coenzyme A (CoA) and thereby disrupt its role in cellular metabolism. Because acetyl CoA is involved in formation of the neurotransmitter ACh, false cholinergic transmitters may be formed. A third mechanism of toxicity results from uncoupling of oxidative phosphorylation, which leads to depletion of cellular adenosine triphosphate.25
Presenting Signs and Symptoms
Vomiting is common early after ingestion and may be accompanied by abdominal pain and diarrhea. Hypotension may occur secondary to volume loss, peripheral vasodilation, and direct myocardial toxicity. Severe ingestions are often associated with a rapid onset of coma. Other neurologic features that have been reported are hyperreflexia, hypertonia, seizures, hallucinations, clonus, and ataxia.24,25 Peripheral neuromuscular effects include weakness, loss of deep tendon reflexes, and fasciculations. Common metabolic effects are acidosis, hyperthermia, and rhabdomyolysis.
Treatment
Alkaline diuresis has been reported to reduce the half-life of 2,4-dichlorophenoxyacetic acid (2,4-D).26 Although hemodialysis and resin hemoperfusion enhance elimination of 2,4-D, no controlled trials have been conducted to assess whether these measures change the outcome. These modalities should be considered only for severe poisonings.
Glyphosate
Unintentional or small ingestions of glyphosate typically produce only mild gastrointestinal symptoms. An exception occurs with glyphosate-trimesium (Touchdown), which has produced rapid death after small ingestions.27 Most cases of significant toxicity result from intentional ingestion of the concentrated formulation of Roundup (41% glyphosate and 15% polyoxyethyleneamine surfactant). Common features are corrosive effects, such as oropharyngeal ulcers, dysphagia, abdominal pain, and vomiting. Significant laryngeal injury may lead to aspiration and lung injury. Metabolic acidosis is common with large ingestions of concentrated formulations. Hypovolemia and hypoperfusion may lead to secondary hepatic and renal insufficiency.28
Tips and Tricks
Herbicides
Withhold oxygen administration until a PaO2 value of less than 40 mm Hg occurs in patients with paraquat poisoning because it may worsen toxicity through acceleration of damage by oxygen radicals.
Consider urinary alkalinization with severe chlorphenoxy herbicide poisoning.
Early decontamination with activated charcoal takes priority in paraquat and diquat ingestions.
Glufosinate
Glufosinate is a nonselective herbicide used worldwide and marketed under the trade names BASTA, Ignite, Challenge, and Harvest. A glutamic acid analogue, glufosinate is combined with surfactants. As with glyphosate, ingestion of these products can lead to symptoms attributable to surfactants, such as corrosive injury, gastrointestinal symptoms, and acidosis. However, glufosinate is unique in that it may cause delayed onset of CNS toxicity. Ataxia, depressed level of consciousness, coma, and central apnea may develop 4 to 12 hours after ingestion.29,30 Delayed-onset seizures have been reported 29 hours after ingestion and may last for days.29
Rodenticides
Rodenticides vary greatly with respect to pathophysiology, signs and symptoms, degree of toxicity, and management. Because these poisonings are rarely encountered by EPs, a detailed discussion on each one is beyond the scope of this text. Some of the characteristics can be found in Table 146.5. Instead, attention is directed to the anticoagulant rodenticides warfarin and superwarfarin and the compound strychnine, which can be found in some rodenticides today.
See Table 146.5, Characteristics of Some Rodenticides, at www.expertconsult.com
| COMPOUND | CLINICAL CHARACTERISTICS | TREATMENT |
|---|---|---|
| Sodium monofluoroacetate, fluoroacetamide | Vomiting 2-20 hr after exposure, acidosis, coma, seizures, hypokalemia, hypocalcemia | Supportive; intravenous fluids (IVF), benzodiazepines for seizures, bicarbonate for refractory acidosis |
| Zinc phosphide | Gastrointestinal distress within 30 min, cough, dyspnea, acidosis, seizures, coma | Supportive; IVF, benzodiazepines for seizures, bicarbonate for refractory acidosis |
| Yellow phosphorus | Dermal burns, “smoking” vomitus, diarrhea, and cardiovascular collapse in severe cases | Supportive; gastric lavage with 0.1% potassium permanganate suggested |
| ANTU (α-naphthyl-thiourea) | Possible pulmonary edema | Supportive; observe for the development of pulmonary edema |
Anticoagulants
Epidemiology
In the 1980s, the 4-hydroxycoumarins and indanediones were developed (see Table 146.6). for a listing of brands and concentrations). These potent, long-acting superwarfarins are lethal to rodents and toxic to humans after a single acute ingestion. These compounds are now responsible for the majority of exposures to anticoagulant rodenticides. Of the 14,425 rodenticide exposures reported to poison control centers in 2008, 11,146 involved superwarfarins. Most were unintentional ingestions in children younger than 6 years.2
See Table 146.6, Anticoagulant Rodenticide (Superwarfarin) Brands and Concentrations, at www.expertconsult.com
Table 146.6 Anticoagulant Rodenticide (Superwarfarin) Brands and Concentrations
| RODENTICIDE | CONCENTRATIONS (%) | SELECTED BRAND NAMES |
|---|---|---|
| 4-Hydroxycoumarins | ||
| Brodifacoum | 0.005 | D-Con Mouse, Talon, Talon G, Havoc |
| Bromadiolone | 0.005 | Bromone, Super-Caid, Ratimus |
| Difenacoum | 0.005 | Endox, Endrocid, Racumin, Rodentin |
| Indanediones | ||
| Chlorophacinone | 0.005, 0.25, 2.5 | Caid, Drat, Liphadione, Microzul, Rozol |
| Diphacinone | 0.005-2.0 | Diphacin, Promar, Ramik |
| Pindone | 0.025-2.0 | Pival, Pivacin, Pivalyn |
Pathophysiology
The warfarins and superwarfarins inhibit the synthesis of vitamin K1–dependent clotting factors (II, VII, IX, X) by blocking conversion of inactive vitamin K to the active form. Bleeding may occur when factor levels fall to 25% of baseline. Because factor VII has the shortest half-life (about 5 hours), a rise in the prothrombin time may be seen in three to four half-lives (15 to 20 hours after ingestion) and certainly will be present within 48 hours.31
Presenting Signs and Symptoms
When a child is evaluated immediately after an unintentional ingestion, the child will be asymptomatic without signs of bleeding; 24 to 48 hours after a large ingestion, however, the child may have any manifestation of a coagulopathy, including, in order of decreasing frequency, ecchymosis, hematuria, uterine bleeding, gastrointestinal bleeding, epistaxis, spontaneous hematoma, gingival bleeding, hemoptysis, and hematemesis.32
Treatment
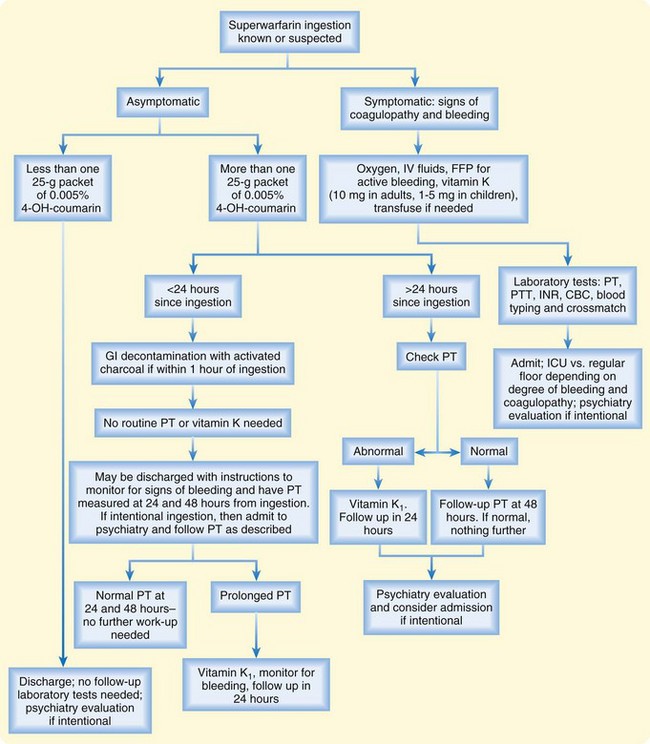
Figure 146.2 summarizes the management of warfarin or superwarfarin poisoning, which depends on the timing, amount ingested, and symptomatology. Accidental ingestions of less than one box of 4-hydroxycoumarin are unlikely to result in clinically significant toxicity and may be managed without gastric decontamination or laboratory evaluation unless signs of bleeding occur.33 Patients who ingest one or more boxes should be given activated charcoal if they are seen within 1 hour of ingestion. Acute hemorrhage is managed with oxygen and IV crystalloids to replace losses of volume. Fresh frozen plasma should be administered to patients with active bleeding and coagulopathy. Vitamin K1 is given at doses of 1 to 5 mg in children and 10 mg in adults. It may be administered intravenously at no more than 1 mg/min to reduce the likelihood of anaphylactoid reactions. Oral or subcutaneous administration is also acceptable.
Strychnine
Pathophysiology
Strychnine blocks the postsynaptic binding of glycine in the spinal cord and brainstem. Because glycine is the major inhibitory neurotransmitter in these areas, disinhibition results in excessive stimulation of motor neurons.34
Presenting Signs and Symptoms
Symptoms usually begin within 15 to 30 minutes of ingestion. Initial symptoms include a heightened sense of awareness and muscle spasms. As the toxicity progresses, the muscular hyperexcitability worsens. Minimal stimuli can produce severe muscle spasms, opisthotonos, and trismus, which can be indistinguishable from seizures. Patients generally maintain a clear sensorium before and after these episodes, an effect unique to strychnine ingestion.34 The complications of strychnine poisoning are secondary to muscle spasms and include hyperthermia, metabolic acidosis, and rhabdomyolysis. Death is usually the result of respiratory failure from spasm of the respiratory muscles.
Treatment
 Documentation
Documentation
Patient Instructions
Document discussion with the patient regarding diagnosis, warning signs, what to do, follow-up, and when to return
With pediatric accidental ingestions, document poison prevention counseling for parents
For superwarfarin poisoning, instructions on where to return for measurements of the prothrombin time and who will monitor the results
1 Jeyaraatnam J. Acute pesticide poisoning: a major global health problem. World Health Stat Q. 1990;43:139–144.
2 Bronstein AC, Spyker DA, Cantilena LR, et al. 2008 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 26th Annual Report. Clin Toxicol. 2009;47:911–1084.
3 Kwong TC. Organophosphate pesticides: biochemistry and clinical toxicology. Ther Drug Monit. 2002;24:144–149.
4 Lee P, Tai DY. Clinical features of patients with acute organophosphate poisoning requiring intensive care. Intensive Care Med. 2001;27:694–699.
5 Sungur M, Guven M. Intensive care management of organophosphate insecticide poisoning. Crit Care. 2001;5:211–215.
6 Senanayake N, Karalliedde L. Neurotoxic effects of organophosphorus insecticides. An intermediate syndrome. N Engl J Med. 1987;316:761–763.
7 Lotti M. Cholinesterase inhibition: complexities in interpretation. Clin Chem. 1995;41:1814–1818.
8 Geller RJ, Singleton KL, Tarantino ML, et al. Nosocomial poisoning associated with emergency department treatment of organophosphate toxicity—Georgia, 2000. J Toxicol Clin Toxicol. 2001;39:109–111.
9 Selden BS, Curry SC. Prolonged succinylcholine-induced paralysis in organophosphate insecticide poisoning. Ann Emerg Med. 1987;16:215–217.
10 Du Toit PW, Muller FO, Van Tonder WM, et al. Experience with intensive care management of organophosphate insecticide poisoning. S Afr Med J. 1981;60:227–229.
11 Fischer TF. Lindane toxicity in a 24-year old woman. Ann Emerg Med. 1994;24:972–974.
12 Aks SE, Krantz A, Hryhorczuk DO, et al. Acute accidental lindane ingestion in toddlers. Ann Emerg Med. 1995;26:647–651.
13 Centers for Disease Control and Prevention (CDC). Unintentional topical lindane ingestions—United States, 1998-2003. MMWR Morb Mortal Wkly Rep. 2005;54(21):533–535.
14 Narahashi T, Frey JM, Ginsbury KS, et al. Sodium and GABA-activated channels as the targets of pyrethroids and cyclodienes. Toxicol Lett. 1992;64/65:429–436.
15 Tenenbein M. Seizures after lindane therapy. J Am Geriatr Soc. 1991;39:394–395.
16 Wax PM, Hoffman RS. Fatality associated with inhalation of a pyrethrin shampoo. Clin Toxicol. 1994;32:457–460.
17 Wagner SL. Fatal asthma in a child after use of an animal shampoo containing pyrethrin. West J Med. 2000;173:86–87.
18 He F, Wang S, Liu L, et al. Clinical manifestations and diagnosis of acute pyrethroid poisoning. Arch Toxicol. 1989;63:54–58.
19 Lee S, Mulay P, Diebolt-Brown B, et al. Acute illnesses associated with exposure to fipronil—surveillance from 11 states in the United States, 2001-2007. Clin Toxicol. 2010;48:737–744.
20 Klein Schwartz W, Smith GS. Agricultural and horticultural chemical poisonings: mortality and morbidity in the United States. Ann Emerg Med. 1997;29:232–238.
21 Vale JA, Merideth TJ, Buckley BM. Paraquat poisoning: clinical features and immediate general management. Hum Toxicol. 1987;6:41–47.
22 Bismuth C, Garnier R, Baud FJ, et al. Paraquat poisoning: an overview of the current status. Drug Saf. 1990;5:243–251.
23 Jones GM, Vale JA. Mechanisms of toxicity, clinical features, and management of diquat poisoning: a review. Clin Toxicol. 2000;38:123–128.
24 Bradberry SM, Watt BE, Proudfoot AT, et al. Mechanism of toxicity, clinical features, and management of acute chlorophenoxy herbicide poisoning: a review. Clin Toxicol. 2000;38:111–122.
25 Bradberry SM, Proudfoot AT, Vale JA. Poisoning due to chlorphenoxy herbicides. Toxicol Rev. 2004;23:65–73.
26 Prescott LF, Park J, Darrien I. Treatment of severe 2,4-D and mecoprop intoxication with alkaline diuresis. Br J Clin Pharmacol. 1979;7:111–116.
27 Sorensen FW, Gregersen M. Rapid lethal intoxication caused by the herbicide glyphosate-trimesium (Touchdown). Hum Exp Toxicol. 1999;18:735–737.
28 Bradberry SM, Proudfoot AT, Vale JA. Glyphosate poisoning. Toxicol Rev. 2004;23:159–167.
29 Koyama K, Andou Y, Saruki K, et al. Delayed and severe toxicities of a herbicide glufosinate and a surfactant. Vet Hum Toxicol. 1994;36:17–18.
30 Tanaka J, Yamashita M, Yamashita M, et al. Two cases of glufosinate poisoning with late onset convulsions. Vet Hum Toxicol. 1998;40:219–222.
31 Smolinske SC, Scherger DL, Kearns PS, et al. Superwarfarin poisoning in children: a prospective study. Pediatrics. 1989;84:490–494.
32 Katona B, Wason S. Superwarfarin poisoning. J Emerg Med. 1989;7:627–631.
33 Ingels M, Lai C, Manning BH, et al. A prospective study of acute, unintentional pediatric superwarfarin ingestions managed without decontamination. Ann Emerg Med. 2002;40:73–78.
34 Smith BA. Strychnine poisoning. The J Emerg Med. 1990;8:321–325.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
146 Insecticides, Herbicides, and Rodenticides
• Organophosphorus and carbamate poisonings cause excessive stimulation of muscarinic and nicotinic receptors by acetylcholine, which can potentially lead to life-threatening bronchorrhea and bronchospasm.
• Aggressive airway management and liberal use of atropine are important in the management of both organophosphorus and carbamate poisoning.
• Only a nondepolarizing neuromuscular blocker, such as vecuronium or rocuronium, should be used for intubation. Succinylcholine is metabolized by plasma cholinesterase, and prolonged paralysis may result if it is used in the setting of organophosphate poisoning.
• Timely administration of pralidoxime is key to the treatment of organophosphorus poisoning, but pralidoxime is not indicated for carbamate poisoning.
• Unintentional pediatric ingestion of 4-hydroxycoumarins (superwarfarins) accounts for the vast majority of rodenticide exposures and rarely results in toxicity.
• Ingestion of an anticoagulant rodenticide should be considered when a child younger than 6 years has an elevated prothrombin time or bleeding without another explanation.
• The prothrombin time should be measured at 24 and 48 hours after large ingestions of 4-hydroxycoumarins.
• Because no specific antidote or pharmacologic intervention has proved beneficial in treating paraquat or diquat poisoning, early decontamination is the most important step.
insecticides
Organophosphorus Compounds and Carbamates
Epidemiology
Organophosphate (OP) compounds and carbamates are used extensively worldwide for agricultural, industrial, and domestic pest control and, as a result, represent a significant public health issue in the developing world. An estimated 3 million poisonings and more than 200,000 deaths occur from OP compounds each year worldwide.1 In the United States in 2008, 4642 exposures to OP compounds and 2644 exposures to carbamates were reported to the National Poison Data System of the American Association of Poison Control Centers.2
Pathophysiology
Under normal circumstances, ACh is hydrolyzed by AChE to yield acetic acid and choline. In the presence of OP insecticides, AChE is phosphorylated, whereas in the presence of carbamate insecticides, the enzyme is carbamylated. As a result, the rate of regeneration of active AChE is slowed, and its function is inhibited. Within 24 to 72 hours of OP poisoning, an alkyl group may dissociate from the AChE-OP complex and thereby result in “aging” of the AChE. Once aging occurs, reactivation of AChE is no longer possible, and only synthesis of new enzyme can restore activity. In the case of carbamate poisoning, breakdown of the carbamate-AChE complex occurs much more rapidly and aging does not occur (Box 146.1).3
Box 146.1 Effects of Organophosphate and Carbamate on Acetylcholinesterase (AChE)
These complexes inactivate AChE and allow acetylcholine to sit on the nicotinic and muscarinic receptors and produce the symptoms of toxicity.
Three things can happen to the phosphorylated or carbamylated AChE:
ACh accumulates in the autonomic nervous system at postganglionic muscarinic (parasympathetic and sympathetic) receptors and preganglionic nicotinic (sympathetic) receptors. It also accumulates at the neuromuscular junction and in the central nervous system (CNS). Overstimulation of these receptors is responsible for the cholinergic toxidrome seen with OP and carbamate insecticide poisoning (Table 146.1).
See Table 146.1, Effects of Organophosphorus and Carbamate Insecticides, at www.expertconsult.com
Table 146.1 Effects of Organophosphorus and Carbamate Insecticides
| RECEPTOR | TARGET TISSUE | CLINICAL EFFECT |
|---|---|---|
Presenting Signs and Symptoms
The clinical effects are summarized in Table 146.1; only caveats in the clinical findings are emphasized here. Bronchorrhea occurs commonly with moderate to severe poisonings4 and can progress to pulmonary edema and respiratory failure. Miosis in the setting of cholinergic symptoms is fairly specific for OP and carbamate insecticide poisoning and may help make the diagnosis. Unfortunately, it is not consistently present.
Although the parasympathetic muscarinic effects are most often emphasized, certain sympathetic effects may predominate. Sinus tachycardia is more common than bradycardia,4,5 and mydriasis may even be seen.5 Nicotinic effects often predominate in mild cases and occur early in severe cases. Excessive nicotinic stimulation at the neuromuscular junction has effects that resemble the actions of a depolarizing neuromuscular blocking agent. Therefore, patients with OP or carbamate insecticide poisoning may exhibit muscle fasciculations and weakness. Paralysis occurs as the toxicity worsens, and the primary cause of death in acute poisonings is probably respiratory arrest secondary to paralysis and bronchorrhea.
One to 3 days after apparent resolution of the symptoms, patients may experience profound weakness and paralysis of the proximal muscles, neck flexor muscles, and cranial nerves. This development, termed the intermediate syndrome,6 is probably explained by ongoing AChE inhibition (Box 146.2).
Box 146.2 Paralysis Seen After Organophosphate Poisoning
Type II (Intermediate Syndrome)
• Develops 1 to 3 days after resolution of the acute organophosphate poisoning symptoms
• Manifested as paralysis and respiratory distress secondary to weakness of the proximal muscles, neck flexor muscles (with relative sparing of the distal muscle groups), and cranial nerve palsies
• Lasts for 4 to 18 days and may require mechanical ventilation
• Results from ongoing acetylcholinesterase inhibition or suboptimal treatment
Differential Diagnosis and Medical Decision Making
A detailed history in a patient with signs and symptoms of cholinergic excess often elucidates exposure to OP or carbamate insecticides. The diagnosis of OP or carbamate insecticide poisoning is therefore usually straightforward; however, certain clinical aspects may be mimicked by other entities. Table 146.2 is a partial list of other agents or diagnoses to consider.
Table 146.2 Differential Diagnosis of Organophosphorus and Carbamate Poisoning
| Other acetylcholinesterase inhibitors | Physostigmine, neostigmine, pyridostigmine |
| Other organophosphorus cholinesterase inhibitors (chemical weapon nerve agents) | Sarin, tabun, soman, Vx |
| Cholinomimetics | Pilocarpine, carbachol, methacholine, bethanechol, muscarine-containing mushrooms |
| Nicotinic alkaloids | Nicotine, coniine, lobeline |
| Other (symptom based) |
All patients with potential OP poisoning should undergo erythrocyte (red blood cell [RBC], or true) cholinesterase and plasma (pseudo) cholinesterase measurement from specimens obtained after arrival at the emergency department (ED). Though not often useful or necessary for making a diagnosis in the ED, the results of this measurement may help guide continued therapy. RBC cholinesterase hydrolyzes ACh and correlates with toxicity, whereas plasma cholinesterase is the first to decline and may be a more sensitive marker of exposure.7 Both substances should be measured because one may exhibit greater inhibition than the other, depending on the specific OP to which the patient was exposed. Box 146.3 summarizes the tests that may be helpful in evaluating a patient with moderate to severe toxicity.
Treatment
The treatment algorithm for OP and carbamate insecticide poisoning is summarized in Figure 146.1. The first step is adequate decontamination of the patient by removal of wet clothing and washing of contaminated skin with soap and water. ED personnel should wear gowns, gloves, and masks to prevent exposure to contaminated body fluids.8
As the patient is being decontaminated, the emergency physician (EP) should focus on the ABCs (airway, breathing circulation), with particular attention paid to early airway, management for copious secretions, seizures, coma, severe weakness, and paralysis. If intubation is necessary, only a nondepolarizing neuromuscular blocking agent, such as vecuronium or rocuronium, should be used. Succinylcholine is metabolized by plasma cholinesterase, so prolonged paralysis may result if this agent is used a patient with OP poisoning.9
Priority Actions
Organophosphates
Treatment should next be directed at controlling muscarinic activity. Atropine is the drug of choice and should be administered intravenously at a dose of 2 to 5 mg (pediatric dose, 0.05 mg/kg) every 3 to 5 minutes, with the end point being control of respiratory secretions. Tachycardia is not a contraindication to atropine administration. Mild poisonings may resolve with just 1 to 2 mg of atropine, and severe poisonings may require more than 1000 mg.10 Large doses of atropine may lead to antimuscarinic CNS toxicity. If such toxicity occurs, glycopyrrolate (1 to 2 mg; pediatric dose, 0.025 mg/kg) can be used in place of atropine.
Organochlorines
Epidemiology
Organochlorines are heavily chlorinated aromatic compounds that are nonvolatile and poorly water soluble. They are divided into four classes on the basis of their structural characteristics, and they vary tremendously with respect to dermal absorption, lipid solubility, and toxic doses. The clinical toxicity, which is similar for each of the classes, is summarized in (Table 146.3).
See Table 146.3, Major Organophosphorus Insecticides, at www.expertconsult.com
[/not-level-membership-for-emergency-medicine-category]
