Inherited Metabolic and Developmental Disorders of the Pediatric and Adult Liver
Angshumoy Roy
James M. Crawford
Milton J. Finegold
Introduction
Jaundice, which is observed in almost every newborn, is termed “physiologic” because it clears within a few days, after activation of bilirubin conjugation. This phenomenon reflects a unique feature of prenatal life: Many functions of the liver that are required after birth for nutrition, metabolic balance, and detoxification and excretion of endogenous chemicals are provided to the developing fetus by the placenta and the mother. “Pathologic” jaundice is the most frequent indication for liver biopsy in children, especially infants, because functional immaturity is not limited to glucuronidation, and intrinsic defects in many processes and structures lead to cholestasis. This is even more evident in premature infants. Not only do the first challenges to hepatobiliary function account for liver diseases that “adult” pathologists encounter very rarely, but maternal-fetal interactions are not always beneficial. Certain infections and immunologically mediated injuries are observed only in infants.
This chapter focuses on constitutional deficiencies of the liver that necessitate examination of tissue for diagnosis and treatment. Myriad chromosomal imbalances and heritable mutations that manifest with dysmorphism and multisystem disease, such as Down syndrome, may affect the liver but can be diagnosed clinically; they are included in this chapter only if they offer a challenge to diagnosis. Anatomic and synthetic defects, such as clotting factor deficiencies that do not lead to hepatobiliary dysfunction, are covered in other publications.1,2
We have incorporated a practical approach to liver biopsy that is derived from Jevon and Dimmick’s classification of the histologic pattern of pediatric liver biopsies, which identify six dominant patterns.3 Finally, the progress made during the past decade with regard to decoding the genetic basis of disease has resulted in improved therapeutics as well as reclassification and renaming of disease entities, genes, and proteins. In the preparation of this chapter, we have benefited from Online Mendelian Inheritance in Man (OMIM; www.omim.org), an online compendium of human genes and phenotypes maintained by the Johns Hopkins University and developed by the National Center for Biotechnology Information (NCBI). This searchable database provides a unique accession number for each entity and incorporates all alternative disease names and gene nomenclature. Throughout the text and in the tables, we have provided OMIM numbers for heritable conditions that may be beneficial to readers.
Pediatric Liver Biopsies
Indications for Liver Biopsy in Children
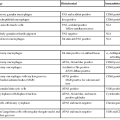
Other than prior liver or bone marrow transplantation (see Chapter 42), the most common indications for liver biopsy in the pediatric age group are conjugated hyperbilirubinemia in young infants (Table 54.1); tumor diagnosis (see Chapter 55); and assessment of liver injury, inflammation, and fibrosis. Metabolic diseases may manifest with fetal demise immediately after birth or at any age thereafter (Table 54.2).
Table 54.1
Liver Disease in Infants
Mitochondrial cytopathies, Reye syndrome
Hereditary disorders of bilirubin metabolism
Dubin-Johnson syndrome and Rotor syndrome
Hereditary disorders of bile formation and transport
Progressive familial intrahepatic cholestasis, types 1-3
Disorders of bile acid biosynthesis
Disorders of protein biosynthesis and targeting
Miscellaneous inherited disorders
Miscellaneous non-neoplastic conditions
Shock/hypoperfusion (as from congestive heart failure)
Multiple hemangiomas (with high-output cardiac failure)
Idiopathic neonatal hepatitis/syncytial giant cell hepatitis

Table 54.2
Presentation of Metabolic Diseases That Involve the Liver
| Age | Hepatic Failure Encephalopathy (±Bleeding) | Jaundice (Hepatitis) | Failure to Thrive and/or Hepatomegaly | Portal Hypertension (Ascites, Bleeding, Splenomegaly) | |
| (Hypoglycemia) | (Normal Sugar) | ||||
| Newborn | Galactosemia, mitochondriopathies, urea cycle defects, glutaric aciduria II | Crigler-Najjar syndrome type I | Leprechaunism, fructose 1.6 diphosphatase deficiency | ||
| First 2 mo | Wolman disease, tyrosinemia, perinatal hemochromatosis | α1-Antitrypsin deficiency, NPD type C | GSD 1a, Ib | Zellweger syndrome | GSD IV |
| First 6 mo | Hereditary fructose intolerance, LCAD deficiency, carnitine deficiency, propionic acidemia | Byler disease, Alagille syndrome, THCA, 3β-HSD, isomerase deficiency | GSD III | Lysinuric protein intolerance, MPS, other storage diseases | |
| First 2 yr | MCAD deficiency, mitochondriopathies | Cystic fibrosis, Rotor syndrome | GSD VI and IX, congenital disorder of glycosylation type Ib, glycoprotein | ||
| Up to 6 yr | Reye syndrome | Cholesterol ester storage, NPD types A and B, cystinosis, hereditary fructose intolerance | |||
| Puberty/and adolescence | Wilson disease, erythropoietic porphyria | Gilbert syndrome, Wilson disease, Dubin-Johnson syndrome | α1-Antitrypsin deficiency, Wilson disease, lipoatrophic diabetes | ||
| Adults | Gaucher disease, citrullinemia, hemochromatosis | ||||
GSD, Glycogen storage disorder; 3β-HSD, 3β-hydroxysteroid dehydrogenase; LCAD, long-chain acyl-coenzyme A dehydrogenase; MCAD, medium chain acyl-coenzyme A dehydrogenase; MPS, mucopolysaccharidoses; NPD, Niemann-Pick disease; THCA, trihydroxycholestanoic acid.
For young infants with conjugated hyperbilirubinemia, extrahepatic biliary atresia (EHBA) is the one condition that is amenable to surgical treatment. Choledochal cysts and other rare causes of duct obstruction that lead to jaundice shortly after birth are rare and are typically diagnosed by imaging studies rather than liver biopsy. Biliary atresia must be recognized quickly if surgical hepatic portoenterostomy is to be successful in reestablishing biliary drainage. Biopsy specimens from these patients are often obtained before the results of noninvasive studies, such as protease inhibitor typing, are available. Even in infants with probable biliary atresia, a liver biopsy may be performed to exclude other potential causes of jaundice. Therefore, clinical management decisions rely heavily on morphologic assessment of liver biopsy specimens (Table 54.3). After exclusion of biliary atresia and infections, consideration should be given to the possibility of an inherited disease as the cause of the patient’s illness.
Table 54.3
Pathologist’s Role in Infantile Cholestasis
| Action | Examples |
| Find treatable condition | Obstruction |
| Galactosemia | |
| Prevent inappropriate treatment | α1-Antitrypsin deficiency mimics EHBA |
| No surgery for Alagille syndrome | |
| Secure samples for diagnosis | Urine for FAB-GC/MS for bile acids |
| Frozen liver for enzymes | |
| PCR for virus | |
| Provide data for family | Hereditable conditions |
| Monitor course of treatment and prognosis | After Kasai procedure or transplantation, biopsies and serial α-fetoprotein levels |
| Elucidate pathogenesis (with a goal of prevention) | Role of fasting versus TPN |
EHBA, Extrahepatic biliary atresia; FAB-GC/MS, fast atom bombardment gas chromatography/mass spectrometry; PCR, polymerase chain reaction; TPN, total parenteral nutrition.
In older children, hepatomegaly, liver tumors, or chronic liver disease may prompt a liver biopsy. When liver disease appears after the neonatal period, clinical studies are typically used to identify the specific cause of the disease. Clinically diagnosed disorders include hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, Wilson disease, reticuloendothelial storage disorders, steatosis, drug-induced hepatitis, autoimmune hepatitis, and cholangiopathy. Unusual causes include Alagille syndrome and metabolic storage disorders. On occasion, liver tissue may be obtained from a child with portal hypertension in whom none of these conditions is suspected. In such cases, congenital vascular anomalies (see Chapter 51) and congenital hepatic fibrosis should be considered. Liver biopsies are used to assess the severity of disease and the response to treatment in all of these disorders.
Pediatric Liver Biopsy Specimen
Because of the broad range of diseases in children, evaluation of liver biopsies in these patients is distinct from that in adults. To perform all potentially necessary tests on a liver biopsy specimen, prior arrangements should be in place to ensure adequate specimen processing (Table 54.4).
Table 54.4
Bedside Processing of Pediatric Liver Biopsy Specimens
| Snap-freezing | Use liquid nitrogen; core tissue 2 cm in length; air-sealed specimen vial |
| Electron microscopy | Use electron microscopy fixative; core tissue 0.3 cm in length |
| Formalin fixation | Use neutral-buffered formalin; core tissue 1 cm in length |

Formalin-fixed specimens should be processed for routine light microscopy. Serial sections should be obtained. For example, a ribbon of 20 sections may be placed on 10 slides, with two tissue sections per slide. The first and last slides should be stained with hematoxylin and eosin (H&E). Periodic acid–Schiff (PAS) stain with and without diastase digestion, trichrome stain, Perls iron stain, and reticulin techniques may be used on intervening slides. The remaining slides may be held for possible future use.
Normal and Potentially Misleading Features of the Liver in Infants and Children
Some key features in the liver of infants and children differ from those in adult liver (Table 54.5). Variations occur in architecture, specific cell populations, content of hepatocytes, and response to injury.
Table 54.5
Normal and Potentially Misleading Features of Pediatric Liver Tissue
| Architecture | Physiologic hyperplasia: liver cell plates two cells thick |
| Residual ductal place architecture, particularly at periphery of liver | |
| Extramedullary hematopoiesis | Portal tracts: granulocytic extramedullary hematopoiesis |
| Parenchyma: erythropoietic extramedullary hematopoiesis | |
| Hepatocyte contents | Hemosiderin granules Copper deposits |
Architecture
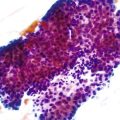
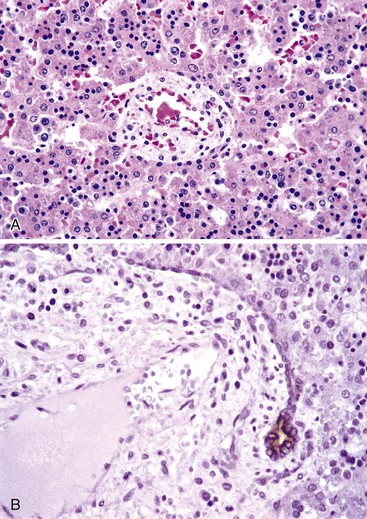
The liver undergoes substantial growth after birth. It normally doubles in weight within the first month of life, doubles again during the first year of life, and does not reach its mature size until late adolescence. The portal tract system grows in parallel with the liver. Therefore, the most peripheral aspects of the liver may exhibit developmental residua of fetal histology. For instance, residual bile duct plates may rim the portal tracts, and the latter contain a more cellular mesenchyme and a centrally placed portal vein (Fig. 54.1).4 The dimensions of hepatic lobules remain constant with growth. However, hepatocyte cords may remain two cells thick well into the fourth postnatal year. This should not be misinterpreted as regenerative hyperplasia in response to tissue injury.
Cell Populations
Hematopoietic elements are commonly present in liver biopsy specimens obtained during the postnatal months. Granulopoiesis predominates in portal tracts, whereas erythropoiesis is common in the parenchyma.
Hepatocyte Content
Until a postnatal age of approximately 3 months, hepatocytes normally contain copper-binding protein and copper (demonstrable by orcein and rhodanine techniques, respectively) and granules of hemosiderin, particularly in periportal hepatocytes. These deposits are considered to be physiologic and disperse with time. Conversely, hepatocyte alterations characteristic of various storage disorders may be inconspicuous in early infancy because of the time required to accumulate abnormal substances, such as α1-antitrypsin (A1AT). One dramatic exception to the concept of physiologic iron deposition occurs in newborns who exhibit liver failure at birth, which is usually attributable to severe liver injury in utero. In this scenario, marked iron deposits may be present in hepatocytes at birth, giving rise to the term neonatal iron storage disease, or perinatal hemochromatosis.5 A severe degree of necrosis and fibrosis is also present in patients with this condition. The extrahepatic reticuloendothelial system does not exhibit iron accumulation, a fact that highlights the primacy of the liver injury.6 The finding of severe perinatal hepatic siderosis is nonspecific and indicates the development of liver injury during gestation.7 A lesser degree of hemosiderosis, with reticuloendothelial system deposits, may be seen in cases of maternal-fetal blood group incompatibility with significant hemolysis.
Response to Injury
Giant, multinucleated hepatocytes, with or without bile pigment, are commonly present in infants with liver disease, regardless of the etiology. This change is considered nonspecific and reactive. Multinucleated hepatocytes are formed by syncytial breakdown of cell–cell borders but with partial preservation of the canalicular aspects of the cell membrane.8 The canalicular remnants, with retained bile, may be observed within the cytoplasm. Giant cells exhibit multiple nuclei, either scattered throughout the cytoplasm or clustered toward one pole of the cell. This reaction may persist well into childhood if the inciting disorder is not resolved. Multinucleated giant cell change is unusual in older children and adults, but it may occur in some disorders, such as autoimmune hepatitis and paramyxovirus hepatitis.9,10
The histologic spectrum of neonatal hepatitis includes giant cell change in hepatocytes, intralobular cholestasis, necrosis of hepatocytes, and intrahepatic hematopoiesis. All of these features are nonspecific events in infancy and can be observed in EHBA, A1AT storage disorder, and many other conditions (Table 54.6). With advances in biochemistry and molecular genetics, many conditions formerly grouped within the category of neonatal giant cell hepatitis can now be specifically diagnosed, including progressive familial cholestasis types 2 and 3 and various bile salt synthetic defects.
Table 54.6
Etiology and Differential Diagnosis of Neonatal Hepatitis
| Normal or Low GGT | Elevated GGT |

* Prompt medical intervention is possible and can be lifesaving, protect the central nervous system, and avert transplantation.
† Medical intervention is possible.
ARC, Arthrogryposis, renal dysfunction, and cholestasis; BSEP, bile salt export pump; GGT, γ-glutamyltransferase; PFIC, progressive familial intrahepatic cholestasis.
Approach to the Diagnosis of Pediatric Liver Disorders in Liver Biopsies
When evaluating pediatric liver biopsy specimens, a careful review of patient age at disease onset (see Table 54.2), clinical manifestations, and routine laboratory workup findings is essential (Table 54.7). If the presentation includes hepatomegaly, awareness of extrahepatic involvement is also helpful (Table 54.8). It is useful to initially classify the histologic pattern of disease into one of the six patterns of injury described initially by Jevon and Dimmick.3 Although these patterns often overlap, it is usually possible to define the predominant pattern in an individual case.11 This section describes an algorithmic approach to the diagnosis of liver disorders, beginning with the histologic patterns of tissue injury (see Boxes 54.1 through 54.6). A detailed description of the major entities is found later in the chapter.
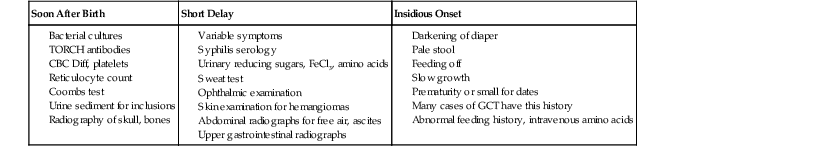
* Acute presentation: bleeding, seizures, and vomiting.
CBC Diff, Complete blood cell count with differential; GCT, giant cell transformation; TORCH, toxoplasmosis, other agents, rubella, cytomegalovirus, and herpes simplex.
Table 54.8
Extrahepatic Associations with Hepatomegaly
| Hepatomegaly | Splenomegaly | Mental Retardation | Neurodegeneration |
| Steatohepatitis (liver biopsy) | Biliary cirrhosis (liver biopsy) | Sly (fibroblast culture; β-galactosidase) | GSD type IV (liver; branching enzyme) |
| Budd-Chiari (MRI) | NPD type C (fibroblast culture; cholesterol esterification) | Wolman (fibroblast culture; acid lipase) | GSD type VIII (liver; phosphorylase B) |
| GSD type VI (liver; phosphorylase) | Cholesteryl-ester storage disease (fibroblast culture; acid lipase) | NPD type C (fibroblast culture; cholesterol esterification) | Sialidosis (fibroblast culture; neuraminidase) |
| GSD type IX (liver; phosphorylase kinase) | NPD type B (leukocyte; acid sphingomyelinase) | Mannosidosis (fibroblast culture; α-mannosidase) | |
| GSD type X (liver; complement C3 and C5, AMP-dependent kinase) | Fucosidosis (fibroblast culture; α-fucosidase) | ||
| Congenital disorder of glycosylation type Ib (serum transferrin) | NPD type A (leukocyte; acid sphingomyelinase) |
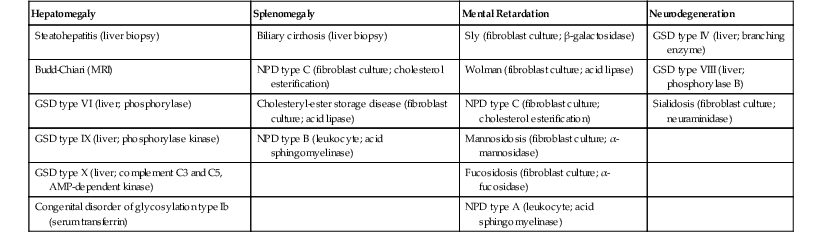
AMP, Adenosine monophosphate; GSD, glycogen storage disorder; NPD, Niemann-Pick disease.
Cholestatic Pattern
The differential diagnosis of cholestatic disease in childhood is extremely broad and includes extrahepatic biliary obstruction (EHBA, choledochal cyst), infections, immune regulatory defects such as Langerhans cell histiocytosis, genetic disorders, metabolic disorders, total parenteral nutrition (TPN) and toxin exposures (see Table 54.1). Liver biopsies to determine the cause of cholestasis should be performed only after completion of a thorough radiologic and laboratory workup, including ultrasonography, hepatobiliary scintigraphy, viral serology, Pi typing for A1AT deficiency, and sweat chloride testing to rule out the more common causes of cholestasis in this age group.12
When confronted with a predominantly cholestatic pattern of liver injury in a biopsy specimen, a useful starting point is the serum level of γ-glutamyltransferase (GGT) (Box 54.1). Serum levels of GGT, a canalicular membrane protein, are usually low in disorders of defective bile acid synthesis or bile salt secretion. These entities (Table 54.9) are discussed later in the chapter. Although the histologic features differ among some of these entities (e.g., lack of giant cells in progressive familial intrahepatic cholestasis type 1 [PFIC1] compared with PFIC2 and PFIC3), ultrastructural examination, specialized enzymatic assays, and genetic testing are crucial in diagnosing these disorders. Among the cholestatic disorders with normal or low serum GGT, congenital defects in bile acid synthesis are commonly diagnosed by urinary mass spectrometry. Peroxisomal biogenesis disorders, such as Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease, typically manifest with cholestasis, necrosis, and siderosis. These disorders are caused by mutations in multiple peroxin (PEX) genes13 and are usually diagnosed by measurement of very-long-chain fatty acids in plasma and erythrocyte plasmalogen.14
Table 54.9
Genetic Defects and Available Testing for Inherited Disorders That Manifest with a Cholestatic Pattern
| Disorder | Gene | Protein | Inheritance | Secondary Pattern | Confirmatory Testing |
| Progressive Familial Intrahepatic Cholestasis (PFIC) Syndrome | |||||
| PFIC1 (allelic disorder: BRIC) (OMIM 211600) |
ATP8B1 | ATPase, class I, type 8B, member 1 | AR | Cirrhotic | Gene sequencing |
| PFIC2 (OMIM 601847) |
ABCB11 | ATP-binding cassette, subfamily B, member 11 | AR | Hepatitic Cirrhotic |
Gene sequencing |
| PFIC3 (OMIM 602347) |
ABCB4 (MDR3) | ATP-binding cassette, subfamily B, member 4 | AR | Cirrhotic | Serum LPX, genotyping |
| Congenital Bile Acid Synthetic (CBAS) Defects | |||||
| CBAS1 (OMIM 607765) |
HSD3B7 | 3β-Hydroxy-Δ5-C27-steroid dehydrogenase | AR | Hepatitic | Blood spot ESI-MS or urine MS |
| CBAS2 (OMIM 235555) |
AKR1D1 | Δ4-3-Oxosteroid 5β-reductase | AR | Hepatitic Steatotic |
Blood spot ESI-MS or urine MS |
| North American Indian childhood cirrhosis (OMIM 604901) |
CIRH1A | Cirhin | AR | Cirrhotic | R565W (c.1741C→T) genotyping |
| Alagille syndrome (OMIM 118450; 610205) |
JAG1; NOTCH2 | Jagged1; Notch-2 | AD | Cirrhotic Hepatitic |
Gene sequencing |
| Niemann-Pick disease type C (OMIM 257220, 601015) |
NPC1; NPC2 | Niemann-Pick disease types C1 and C2 | AR | Hepatitic Storage |
Filipin staining in fibroblasts |
| Peroxisomal Biogenesis Disorders | |||||
| Zellweger syndrome (OMIM 214100) |
PEX genes | Peroxisomal biogenesis factors | AR | Hepatitic Cirrhotic Steatotic |
↑ Plasma VLCFA by GC |
| Neonatal adrenoleukodystrophy (OMIM 202370) |
PEX | Peroxisomal biogenesis factors | AR | — | ↑ Plasma VLCFA by GC |
| Infantile Refsum disease (OMIM 266510) |
PEX | Peroxisomal biogenesis factors | AR | — | ↑ Plasma VLCFA by GC |
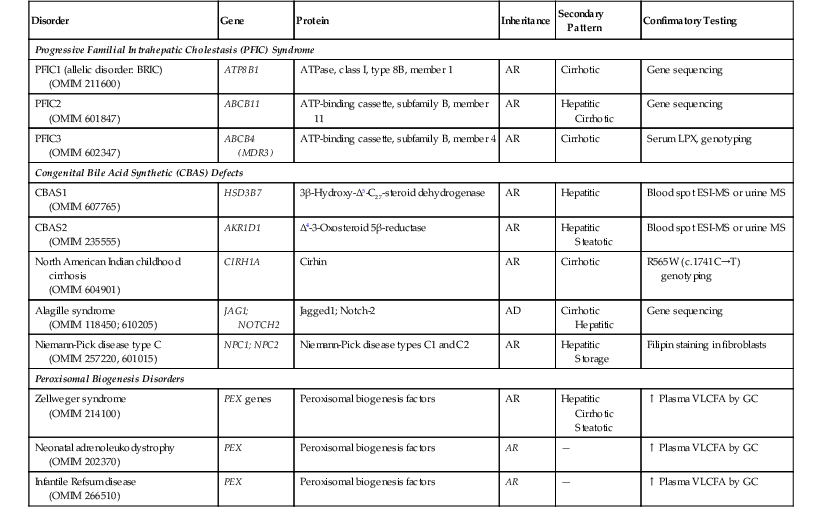
AD, Autosomal dominant; AR, autosomal recessive; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; BRIC, benign recurrent intrahepatic cholestasis; ESI, electrospray ionization; GC, gas chromatography; LPX, lipoprotein X; MS, mass spectrometry; OMIM, Online Mendelian Inheritance in Man (www.omim.org); VLCFA, very-long-chain fatty acids.
Two rare disorders of ductal plate malformation—congenital hepatic fibrosis and Caroli disease—deserve mention here. Both manifest with cholestasis and cholangitis but often with portal hypertension.15 Both are associated with autosomal recessive polycystic kidney disease (ARPKD), and both carry mutations in PKHD1 (fibrocystin), the that is gene defective in ARPKD,16 in approximately 30% of cases.
Steatotic Pattern
A steatotic pattern of injury is present when there is a prominent and diffuse distribution of fat vacuoles within hepatocytes. Steatosis is a common histopathologic finding in several types of inherited disorders that affect the liver; those in which other histologic features predominate are discussed separately. When one is considering the differential diagnosis of a primary steatotic pattern of liver injury, the most useful feature is the type of fat accumulation: microvesicular, macrovesicular, or mixed microvesicular and macrovesicular (Box 54.2).
Microvesicular steatosis results from perturbation of mitochondrial metabolism, fatty acid β-oxidation (FAO), or electron transport chain function, through either genetic defect or drug-induced17 inhibition of the pathways. The latter mechanism may result from a variety of drugs, including aspirin, ibuprofen, valproate, and zidovudine (see Chapter 48 for details on Reye syndrome). The diagnostic workup of genetic defects in FAO or electron transport chain function is often based on the clinical presentation and relies on specialized biochemical and metabolic testing of plasma, urine, and biopsied muscle tissue.3,18,19 Pathologic features in liver biopsy specimens, such as microvesicular steatosis, cholestasis, fibrosis, cirrhosis, abnormal mitochondrial ultrastructure, and immunohistochemical demonstration of mitochondrial enzyme deficiency, support the diagnosis.3 Fresh-frozen liver is essential for specialized biochemical and genetic assays, particularly for analysis of the ratio of mitochondrial DNA (mtDNA) to nuclear DNA by Southern blotting (used for diagnosis of mtDNA depletion syndrome). Also, histologic examination of the liver is highly relevant in the postmortem examination of patients with suspected mitochondrial disorders.
Diffuse macrovesicular steatosis or mixed microvesicular and macrovesicular steatosis can develop in several inherited and acquired conditions (Table 54.10). Two inborn errors of carbohydrate metabolism, galactosemia and hereditary fructose intolerance, are classically associated with steatosis in newborns and infants. Galactosemia is diagnosed through urine biochemical testing and red cell enzyme assays.20 A liver biopsy, if performed, shows macrovesicular steatosis with fibrosis and cirrhosis. A liver biopsy is indicated in hereditary fructose intolerance for confirmatory aldolase B enzyme assays on fresh-frozen tissue,20 although molecular assays to detect ALDOB mutations are now available.21 The pathologic features mimic those of galactosemia, except that cirrhosis is usually absent.22 Steatosis, with or without biliary cirrhosis, is common in cystic fibrosis (CF). Liver disease is relatively uncommon in young patients with CF but can lead to significant morbidity. In patients in whom the diagnosis was confirmed by positive sweat chloride testing and mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR), the diagnosis is suspected when eosinophilic material is present within bile ducts in liver biopsies.3
Table 54.10
Genetic Defects and Available Testing for Inherited Disorders That Manifest with a Steatotic Pattern
| Disorder | Gene(s) | Protein(s) | Inheritance | Secondary Pattern | Confirmatory Testing |
| Mitochondrial Disorders | |||||
| FAO disorders, ETC disorders, mtDNA depletion syndrome, etc. | Multiple ACADM, ACADL, etc. |
Multiple | Mostly AR | Plasma acylcarnitine and gene sequencing | |
| Inborn Errors of Carbohydrate Metabolism | |||||
| Hereditary fructose intolerance (OMIM 229600) |
ALDOB | Aldolase B | AR | Cholestatic | Liver enzyme activity and sequencing |
| Galactosemia (OMIM 230400) |
GALT | Galactose-1-phosphate uridyltransferase | AR | Cirrhotic | RBC GALT assay |
| Cystic fibrosis (OMIM 219700) |
CFTR | Cystic fibrosis transmembrane conductance regulator | AR | Hepatitic Cholestatic |
Sweat chloride, gene sequencing |
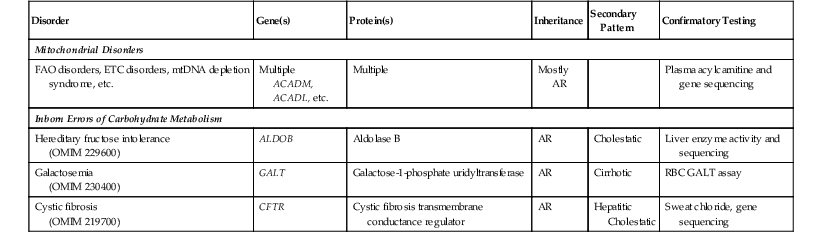
AR, Autosomal recessive; ETC, electron transport chain; FAO, fatty acid oxidation; mtDNA, mitochondrial DNA; RBC, red blood cell.
Organic acidurias and urea cycle disorders may occasionally demonstrate steatosis or focal glycogenosis,3 but these disorders are diagnosed through urine chromatographic analyses.23 In some cases, progression to fibrosis and even cirrhosis may occur. Other disorders may demonstrate steatosis as a secondary feature. Biopsies of glycogen storage disease (GSD) types I and III often reveal steatosis. The liver in Wilson disease may be steatotic, but the predominant pattern is hepatitic or cirrhotic or both.
Storage Pattern
A storage pattern is characterized by the presence of enlarged, swollen, and pale hepatocytes and/or reticuloendothelial cells, including Kupffer cells and portal macrophages. The stored material results from specific enzyme deficiencies in various metabolic pathways. A diverse group of disorders (>30) result in the development of a storage pattern within the liver, most of which cause hepatomegaly (see Table 54.8). Most, but not all (e.g., not the X-linked disorders, Fabry disease, or Hunter disease), are inherited in an autosomal recessive fashion. These disorders often demonstrate variable penetrance and expressivity, with different clinical and histologic manifestations among family members with the same genetic defect. For a comprehensive discussion of these disorders, readers are referred to several excellent review articles.3,20,24
For pathologists confronted with biopsy specimens revealing a storage pattern, the most efficient approach is one that involves a pediatric geneticist and a clinical biochemist, because the patient’s clinical presentation and laboratory findings often suggest the most likely diagnosis. The storage pattern can be subclassified as either lysosomal or cytoplasmic. Because reticuloendothelial cells are rich in lysosomes, Kupffer cells and histiocytes are typically more involved in lysosomal storage diseases than in disorders with cytoplasmic storage (e.g., GSD types I, III, and IV). However, this distinction is not absolute, because diffuse hepatic and extrahepatic involvement of the reticuloendothelial system is a well-documented feature in some GSDs, particularly GSD type IV.25 Distinguishing lysosomal (membrane-bound) from cytoplasmic storage disorders by electron microscopy is, therefore, quite useful.
Hepatic involvement in Pompe disease (GSD type II) is variable. The liver architecture is usually intact. PAS-positive diastase-sensitive inclusions are present within histiocytes and hepatocytes (Box 54.3). Diagnostic confirmation is usually established by measurement of acid α-glucosidase activity in muscle or fibroblasts. Additional confirmation can be done by GAA gene sequencing.
The differential diagnosis of cytoplasmic storage disorders includes GSD types I, III, IV, VI, and IX (Table 54.11). These disorders are discussed later in this chapter.
Table 54.11
Features of Important Glycogen Storage Disorders That Affect the Liver
| Disorder | Inheritance | Gene | Protein | Storage Material | Liver Histology | Other Features | Ultrastructure | Diagnostic Testing | |
| Parenchyma | RES | ||||||||
| GSD Ia (von Gierke disease) | AR | G6PC | Glucose-6-phosphate, catalytic subunit | Glycogen | PAS+, diastase-sensitive cytoplasmic glycogen | − | Steatosis, adenomas, HCC | − | Fresh liver enzyme activity, genetic testing |
| GSD Ib | AR | SLC37A4 | Glucose-6-phosphate transporter | ||||||
| GSD II (Pompe disease) | AR | GAA | Acid α-glucosidase | Glycogen | PAS+ vacuoles | − | − | Membrane-bound monoparticulate glycogen | Fibroblast and/or muscle enzyme activity, genetic testing |
| GSD III | AR | AGL | Glycogen debranching enzyme | Glycogen | PAS+, diastase-sensitive cytoplasmic glycogen | − | Steatosis, cirrhosis, HCC | − | Fresh liver or muscle enzyme activity, genetic testing |
| GSD IV (Andersen disease) | AR | GBE1 | Glycogen debranching enzyme | Glycogen | PAS+, diastase-sensitive cytoplasmic glycogen | Kupffer cell inclusions± | Cirrhosis, HCC | Amylopectin-like cytoplasmic glycogen | Fresh liver or fibroblast enzyme activity, genetic testing |
AR, Autosomal recessive; GSD, glycogen storage disorder; HCC, hepatocellular carcinoma; PAS, periodic acid–Schiff stain; RES, reticuloendothelial system.
The differential diagnosis of lysosomal storage disorders with foamy histiocytes includes lipidoses (Table 54.12), chiefly Gaucher disease, Farber disease, Niemann-Pick disease (NPD) types A and B, GSD type II, mucopolysaccharidoses, and GM1 gangliosidosis. In Gaucher disease, the most common lysosomal storage disorder, there is a characteristic diffuse infiltration by engorged histiocytes (Gaucher cells) containing PAS-negative cytoplasmic (“crinkled-paper”) inclusions of glucosylceramide.3 Hepatocytes are typically spared. On demonstration of tubular structures by electron microscopy, one should, for confirmation, measure the level of leukocytic or fibroblastic acid β-glucosidase activity and sequence the GBA gene for mutations.24
Table 54.12
Major Disorders of Lipid Metabolism That Affect the Liver
| Disorder | Inheritance | Gene | Protein | Storage Material | Liver Histology | Other Features |
Ultrastructure | Diagnostic Testing | |
| Parenchyma | RES | ||||||||
| Gaucher disease | AR | GBA | Acid β-glucosidase | Glycosylceramide | − | Gaucher cells (20-100 µm): eosinophilic corrugated “crinkled-paper” cytoplasm | Fibrosis, rarely cirrhosis | Spindled tubular structures | Enzymes activity (l, f) ± GBA mutations |
| Niemann-Pick disease types A and B | AR | SMPD1 | Acid sphingomyelinase | Sphingomyelin | Vacuolated hepatocytes | Niemann-Pick cells (25-75 µm): foamy histiocytes, lipofuscin+ | ORO+, LXB+, PAS− | Laminated myelin-like figures | Enzymes activity (l, f) |
| Niemann-Pick disease type C | AR | NPC1 NPC2 |
NPC1, NPC2 | Cholesterol | Cholestasis, giant cell transformation | Sea-blue histiocytes | Cirrhosis | Whorled aggregates | Filipin staining (f) + NPC1, NPC2 mutations |
| Farber disease | AR | ASAH1 | Acid ceramidase | Ceramide | − | Lipogranulomas, foamy histiocytes | Fibrosis | Curvilinear Farber bodies | Enzymes activity (l, f, p) |
| GM1 gangliosidosis | AR | GLB1 | β-galactosidase 1 | Glycosphingolipids | Vacuolated hepatocytes | Finely vacuolated | Fibrillogranular material | Enzymes activity (l, f, p) | |
| Wolman disease CESD |
AR | LIPA | Acid liposomal lipase | Cholesterol esters | Microvesicular droplets | Enlarged vacuolated, periportal foamy histiocytes | ORO+, cirrhosis, (Wolman) |
Cholesterol crystal profiles | Enzymes activity (l, f) + LIPA mutations |
AR, Autosomal recessive; CESD, cholesteryl-ester storage disease; f, fibroblast enzyme activity; l, leukocyte enzyme activity; LXB, luxol-fast blue; ORO, oil red O; p, plasma enzyme activity; PAS, periodic acid–Schiff; RES, reticuloendothelial system.
Hepatitic Pattern
Hepatitic pattern, in infants, refers to a histologic pattern that reveals hepatocyte unrest with or without necrosis, diffuse giant cell transformation of hepatocytes, extramedullary hematopoiesis, and prominent cholestasis3 (see Table 54.6). In older children, portal, interface, or lobular inflammation (or some combination of these) is typically present, but usually without cholestasis. Chief among the inherited disorders that manifest with a hepatitic pattern are A1AT deficiency and Wilson disease. A number of other inherited disorders with characteristic histologic patterns of injury (e.g., NPD type C, Alagille syndrome with cholestasis) can also manifest with a superimposed hepatitic pattern on liver biopsies. These entities are discussed later in this chapter.
When investigating biopsy specimens with a hepatitic pattern for a suspected inherited disorder, it is crucial to rule out acquired causes of hepatitis (e.g., infection, toxin or drug exposure, TPN) that are far more common in this age group. It is also important to evaluate the clinical presentation and laboratory results. For example, A1AT deficiency liver disease can manifest either in the neonatal period with hepatitis or later in childhood with cirrhosis, but clinical manifestations of Wilson disease are rare before the age of 5 years26 (see Table 54.2). On biopsy, the presence of PAS-positive, diastase-resistant inclusions within zone 1 hepatocytes is characteristic of A1AT deficiency, but immunostaining is more sensitive and more specific. Serum A1AT phenotyping by isoelectric focusing (Pi typing) is the confirmatory assay for diagnosis of A1AT deficiency in cases of suspected neonatal hepatitis, and it is particularly valuable in cases that mimic EHBA (i.e., bile duct proliferation, no biliary excretion on scintigraphy).27 Diagnosis of Wilson disease is less straightforward; screening for decreased serum ceruloplasmin levels (<20 mg/dL) can be problematic, because both decreased (hypoplasminemia or aceruloplasminemia) and elevated levels (acute phase reaction) may be seen in other conditions (Box 54.4). A low serum alkaline phosphatase level may be a useful clue to the etiology, as well as increased unconjugated bilirubin, which reflects hemolysis in children with acute liver decompensation.
Cirrhotic Pattern
Cirrhosis, an end-stage response to chronic liver injury, is common to several different types of inherited disorders. Therefore, the differential diagnosis of liver biopsies with a cirrhotic pattern rests largely on the presence or absence of other characteristic microscopic findings (Box 54.5). Metabolic disorders that lead to cirrhosis also carry an increased risk of neoplasia. Advanced stages of congenital hepatic fibrosis (discussed earlier) may be confused with cirrhosis; however, bile duct ectasia is a characteristic feature of the former.28 Although mitochondrial hepatopathies may lead to several different patterns of liver injury, it is most often steatosis. Less often, these disorders may manifest with hepatitis and cirrhosis.
Tyrosinemia, caused by a deficiency of fumarylacetoacetate hydrolase,29 can lead to cirrhosis in early life (infancy), hepatic failure, hepatocellular necrosis, or giant cell hepatitis.
Hereditary hemochromatosis (HH) is caused by mutations in HFE, the hemochromatosis gene,30 and leads to cirrhosis and hepatocellular carcinoma (HCC). Although the defect in iron metabolism is present at birth, the clinical manifestations of HH are rarely apparent before adulthood, when long-term effects of chronic iron overload typically manifest31 (see later discussion).
Neoplastic Pattern
Several types of inherited disorders predispose the child to the development of focal nodular hyperplasia, hepatic adenoma, and HCC.32 In contrast, hepatoblastoma, the most common malignancy in the liver of children, is not usually associated with inherited metabolic disorders, although trisomy 18, neurofibromatosis, and congenital hepatic fibrosis have been linked to hepatoblastoma. The increased risk for HCC is chiefly caused by the development of cirrhosis, but HCC can arise in noncirrhotic patients with A1AT deficiency, hemochromatosis, or GSD type I.2,33 The histologic features and biologic behavior of HCC and adenomas arising in patients with a metabolic disorder are similar to those of neoplasms that arise in cirrhosis resulting from other causes. Cirrhosis and neoplasms that arise in inherited metabolic disorders are indicative of advanced disease (not necessarily advanced age), and the diagnostic features of the underlying disorder can usually be identified in non-neoplastic areas of the liver tissue (Box 54.6).
Most patients with GSD type I have hepatic adenomas by 15 years of age, although adenomas may be present in early childhood. Dysplastic changes and HCC within individual adenomatous nodules have also been reported in this condition.34,35
Among all of the metabolic disorders of the liver, hereditary tyrosinemia (discussed later) carries the highest risk for development of HCC (13% to 15% incidence).29,36 Typically, hepatocellular dysplasia and foci of HCC develop in a background of mixed micronodular and macronodular cirrhosis, but treatment should begin in infancy to abort the mutagenic process. Not all infants manifest overt liver dysfunction with time.29
The incidence of HCC in patients with HH (discussed later) is approximately 10%. Most cases develop in a background of cirrhosis.33 Because of the widespread availability of testing for the HFE gene mutations C282Y and H63D and sensitive transferrin-iron screening tests, a biopsy diagnosis is required only in cases with a negative genotype or high ferritin levels.
Other inherited disorders that are less commonly associated with HCC include A1AT deficiency, PFIC2, Wilson disease, Alagille syndrome, and GSD types I, III, and IV. The characteristic biopsy features were described earlier. A1AT deficiency is a precursor to HCC.37 In some cases, PiZ heterozygotes were found to have HCC (and cholangiocarcinoma) in noncirrhotic livers.38 Fanconi anemia, Familial adenomatous polyposis and Beckwith-Weidemann syndrome are other syndromes that predispose to cancer. Three percent of patients with Fanconi anemia develop adenomas or HCC,39 and hepatoblastoma has a well-known association with both familial adenomatous polyposis and Beckwith-Weidemann syndrome.40–42 Finally, cholangiocarcinoma is a rare complication of congenital hepatic fibrosis.43 This tumor was recently described in two children with PFIC2.44
Liver Diseases of Infancy
Sudden Death
Although sudden death within 2 to 3 days after birth is usually caused by nonmetabolic conditions such as sepsis or congenital heart disease,45 some inborn errors of metabolism are also associated with acute life-threatening illness (Box 54.7). Defects of FAO lead to cardiac arrhythmias and can cause sudden death (see Mitochondrial Cytopathies).46 At autopsy, excess droplets of fat may be present in the liver and heart of patients with a defect of FAO. If a disorder of FAO is suspected, tissue specimens should be obtained as soon as possible after death, before autopsy, for metabolic testing47,48 (Table 54.13). Both liver and muscle tissue should be obtained for analysis. In addition, urine and cerebrospinal fluid should be snap-frozen and stored for further analysis. Blood spots should be obtained for analysis of acylcarnitines. Whole blood specimens should be placed in an ethylenediaminetetraacetic acid (EDTA) tube for DNA extraction and in a lithium heparin tube (spun and separated within 20 minutes of collection) for metabolite analysis. A full-thickness skin biopsy should be performed under sterile conditions within 12 hours of death for fibroblast culture and archiving.
Table 54.13
Investigation of Acute Life-Threatening Disease and Sudden Death
| Collect | Blood, urine, CSF, vitreous humor (dodecanoic acid = MCAD deficiency) Bile (carnitine and acylcarnitines = FAO disorders) |
| Freeze | Liver, skeletal, cardiac muscle |
| Sample for cell culture | Skin fibroblasts (DNA and enzyme analyses) |
CSF, Cerebrospinal fluid; FAO, fatty acid oxidation; MCAD, medium chain acyl-coenzyme A dehydrogenase.
Sudden and unexplained death in an infant or young child is often the first manifestation of medium chain acyl-coenzyme A dehydrogenase (MCAD) deficiency, the most common FAO disorder.49 MCAD deficiency manifests with hepatomegaly and steatosis and may be confused with Reye syndrome. A particular Lys304Glu mutation in the ACADM gene is highly prevalent in some populations. Since the institution of newborn screening, early diagnosis has led to prospective management of acute episodes of hypoketotic hypoglycemia.
Neonatal Hemochromatosis
Clinical Features
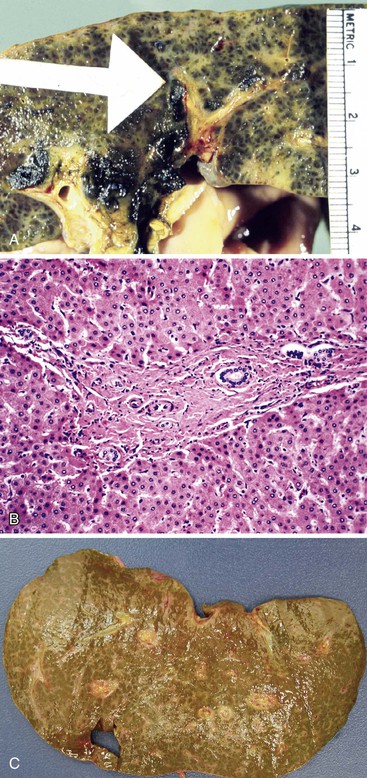
Neonatal hemochromatosis, also termed neonatal iron storage disease, is a rare syndrome that is characterized by the presence of congenital cirrhosis and fulminant liver failure. This condition exhibits abundant iron deposition in the liver and in other organs, but not in the spleen or bone marrow.6 Clinically, patients with neonatal hemochromatosis, either before birth or shortly thereafter, exhibit liver failure, including hypoglycemia, coagulopathy, hypoalbuminemia, ascites, and hyperbilirubinemia. Although some infants recover with exchange transfusion and some survive to transplantation, most die of their disease.
Pathogenesis
Neonatal hemochromatosis has been described in association with various conditions, including metabolic disorders (tyrosinemia, Δ4-3-oxosteroid 5β-reductase deficiency, mtDNA mutations, and Zellweger syndrome), infections (echovirus 9, cytomegalovirus [CMV], herpes simplex virus, rubella, and parvovirus B19), and karyotypic disorders (Down syndrome).50,51 As a result, some authors regard neonatal hemochromatosis as a final common phenotype of any gestational insult that culminates in abnormal iron metabolism.52,53 At least three patterns of disease transmission have been described: transmission of maternal alloantibodies, autosomal recessive inheritance, and matrilineal inheritance.54 In the last instance, several reports have documented women with more than one affected child, but the children were fathered by different men. Although gonadal mosaicism in these mothers has not formally been excluded, the possibility of mitochondrial inheritance also has not been excluded. Neonatal hemochromatosis is not associated with mutations in the HFE, the gene involved in most cases of HH (see later discussion).
In 2004, Whitington and Hibbard first reported giving high-dose intravenous γ-globulin to pregnant women with a history of a previously affected infant, and this treatment prevented recurrence in a small series.55 Their findings have since been confirmed in many centers, and it is now imperative to determine the etiology of all perinatal forms of acute liver injury. Mimicry of hemochromatosis has been seen in cases of mtDNA depletion50 and other circumstances, which presumably do not benefit from intravenous immunoglobulin therapy.
The antibody responsible for neonatal hemochromatosis has yet to be determined. Complement fixation induced by immunoglobulin G of maternal origin is detected by immunofluorescence for the membrane attack complex on hepatocytes. One patient with cirrhosis at birth did not have hemosiderosis and survived without specific therapy.56 However, the usual scenario is fatal necrosis with extensive parenchymal siderosis without reticuloendothelial iron, suggesting dysfunction of macrophages. This has been observed even prenatally: Three of eight stillborn or very premature infants had no extrahepatic siderosis.57,58 Recently, Whitington and colleagues have provided insight into the severe degree of parenchymal siderosis that accompanies the hepatocellular injury.58 The injured livers have significantly reduced hepcidin, hemojuvelin, and transferrin gene expression compared with normal livers.
Antibodies to mitochondrial proteins are responsible for primary biliary cirrhosis, a disease that affects women disproportionately. Two patients transmitted this disease transplacentally to their infants, producing transient liver injury. One infant was born with ascites and conjugated hyperbilirubinemia. In the other, the presentation was more insidious. His biopsy, at 5 weeks, showed portal inflammation involving bile ducts and ductules and mild portal fibrosis, as well as multinucleated giant hepatocytes typical of neonatal hepatitis. Immunofluorescence was able to detect immunoglobulin G deposits surrounding hepatocytes. Within 3 months, both infants showed no evidence of liver dysfunction, and the antibodies were undetectable.59
Neonatal hemochromatosis resembles neonatal hepatitis in infants with systemic lupus erythematosus, in which the mother has high titers of antinuclear anti Ro (SSA) and anti La antibodies.60
Pathologic Features
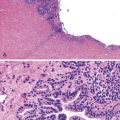
The liver in patients with neonatal hemochromatosis (usually seen at postmortem examination) reveals cirrhosis and cholestasis (Fig. 54.2). Confluent areas of hepatocellular loss and a variable degree of hepatocyte regeneration are typical. Residual hepatocytes may demonstrate giant cell or pseudoacinar transformation. Iron deposition is typically coarsely granular and located predominantly within hepatocytes, sparing Kupffer cells. Extrahepatic sites of siderosis include the parenchymal cells of the heart, thyroid, pancreas, adrenal glands, kidneys, and the submucosal glands of the gastrointestinal and upper respiratory tracts.
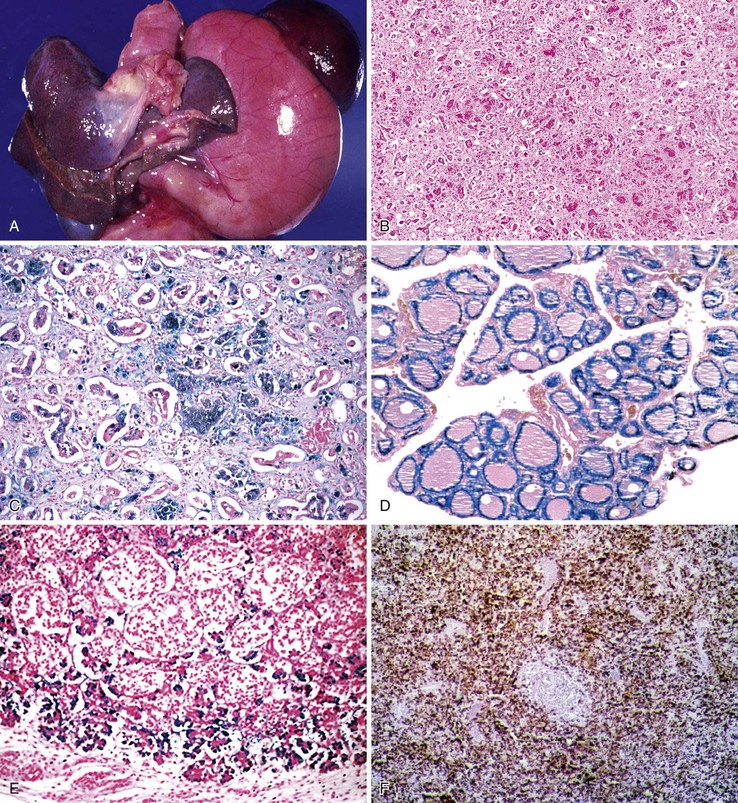
Natural History
Although neonatal hemochromatosis carries a high risk of death, it remains in the differential diagnosis of severely ill neonates who survive beyond the first days of life. Demonstration of siderosis in biopsy samples from oral submucosal glands is a rapid diagnostic method. T2-weighted magnetic resonance imaging can be used to assess the presence of iron in various tissues. Siderosis of the liver and extrahepatic organs may also accompany other conditions, such as erythropoietic disorders, sickle cell disease, thalassemia, and erythroblastosis from blood group incompatibility. However, hemosiderosis affects the reticuloendothelial system primarily. These other diseases need to be excluded by other clinical and laboratory studies.
Neonatal Jaundice
Heritable disorders of bilirubin conjugation can rarely produce liver dysfunction (see later discussion). Liver diseases of infancy most often manifest with jaundice owing to conjugated hyperbilirubinemia. This occurs because of the relative immaturity of hepatic secretory and excretory functions in early life. Some of the disorders of infancy that cause neonatal jaundice are listed in Table 54.1. In all cases of neonatal jaundice, the possibility of EHBA or hepatic damage due to drug exposure (including inadvertent drug overdose) should be excluded. Infants requiring TPN are at risk for cholestatic liver disease (see Chapter 48). Infectious causes of liver disease in infancy include enterovirus, parvovirus B19, and adenovirus, which cause direct cytopathic cell death, as well as bacterial sepsis, urinary tract infection, and intrauterine exposure to maternal infections (of the “TORCH” acronym), all of which cause liver damage to neonates and infants (see Chapter 46). Acquired syphilis is an exceedingly uncommon cause of perinatal liver disease.
Inherited metabolic disorders of carbohydrate, amino acid, and lipid and glycolipid metabolism, along with disorders of the biosynthetic and secretory pathways for bile acids, should always be considered in the differential diagnosis of neonatal jaundice (see Table 54.6). Storage of abnormal A1AT and abnormal function of the CFTR compromise hepatic formation of bile. Defects in the transporters responsible for bile formation give rise to progressive familial intrahepatic cholestasis. Inherited defects of peroxisomal and mitochondrial function can cause neonatal jaundice. Shock may cause cholestatic liver damage in neonates. Alagille syndrome may manifest in early infancy as “giant cell hepatitis”; it can mimic biliary atresia61 and may lead to Kasai portoenterostomy (KPE). The nonspecific designation of “neonatal hepatitis/giant cell hepatitis” applies to the 25% of patients in which a specific etiology for neonatal jaundice remains undetermined.
Extrahepatic Biliary Atresia
Clinical Features
EHBA accounts for more than 30% of all cases of cholestasis in a neonate. This disorder has an incidence of 1 in 8000 to 18,000 live births. Most cases are sporadic and do not reveal a positive family history of neonatal cholestasis. There is one report of a 20-year-old mother who underwent a portoenterostomy for EHBA at 64 days of age and subsequently gave birth to a daughter with EHBA.62 In contrast, in only 2 of 30 sets of twins were both infants affected, and both pairs were dizygotic, whereas none of the 7 monozygotic pairs displayed concordance.63 However, there are rare exceptions.64 Variation in epigenetic modifications of genomic DNA has been suggested for this developmental difference.65 Infants with biliary atresia are typically of normal gestational age and birth weight. However, the increasing detection of biliary cysts in midgestation, via ultrasonography, indicates that as many as 10% of patients with EHBA have very early onset.66,67 The pathology in those cases is not notably different that in other patients with EHBA.
Cholestatic jaundice is the main clinical presentation. It typically develops in the first few weeks of life and does not remit, unlike the mild physiologic jaundice of early infancy. Furthermore, in physiologic jaundice, the mildly elevated serum bilirubin is primarily unconjugated, and the serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are normal. The progressive biliary obstruction that characterizes biliary atresia leads to a progressive rise in serum bilirubin levels in which conjugated bilirubin represents 50% to 80% of the total. A recent study of conjugated or direct-reacting bilirubin in the first 48 hours of life revealed significant elevations in all infants with EHBA compared with aged-matched controls, even though the total bilirubin level was not increased.68 Serum levels of GGT are increased several times above normal. Liver biosynthetic function, as indicated by serum albumin levels and prothrombin time, is usually normal at initial presentation.
In all suspected cases, ultrasonography should be performed to exclude the presence of an extrahepatic biliary tract anomaly, such as a choledochal cyst. Hepatobiliary scintigraphy is also useful to assess the status of biliary tract function, but in many hepatocellular diseases of infants, there is also an absence of excretion of the labeled molecule into the intestine. Percutaneous liver biopsy is used to determine whether there is histologic evidence of large bile duct obstruction. Biopsy features of atresia may also occur with other extrahepatic forms of biliary obstruction (Box 54.8).
The overall accuracy rate of percutaneous liver biopsy for diagnosis of EHBA is 79% to 95%.69 The decision to proceed with a biliary tract exploratory surgical procedure does not rest solely on the pathologic findings in a liver biopsy specimen. For instance, the presence of acholic stools, an undetectable or irregular contour of the gallbladder on sonographic studies, and the findings of retrograde endoscopic choledochography are also used to diagnose biliary atresia clinically.
Pathogenesis
Potential mechanisms of biliary atresia include a defect in hepatic morphogenesis, a defect in prenatal hepatic circulation, immunologic dysregulation in the perinatal period, intrauterine or perinatal viral infection, and toxin exposure (Table 54.14). These hypotheses have been put forth on the basis of epidemiologic studies and molecular analyses of tissue specimens from human patients. Mouse models of biliary atresia have provided some additional clues.70,71 A growing consensus is that the pathogenesis in most cases probably involves an infectious or immune-mediated fibro-obliteration of the extrahepatic and intrahepatic biliary tract. Defective morphogenesis of the biliary system may be the cause in the remainder of cases.72
Table 54.14
Biliary Atresia: Pathogenesis
| Proposed Mechanisms | Classic versus Syndromic Presentations | Associated Malformations |

In favor of the first hypothesis are the finding of an association between biliary atresia and several viral agents (CMV, reovirus, rotavirus) and the ability of viruses introduced into neonatal mice to cause inflammatory destruction of the bile ducts.73 Studies of human liver specimens at different phases of disease progression point to the presence of a proinflammatory commitment of lymphocytes with a predominant type 1 helper T cell (Th1) phenotype. Prevention of experimental inflammatory destruction of bile ducts occurs in mice that are deficient in interferon-γ.72 Neonatal systemic lupus can mimic biliary atresia.74,75 Molecular profiling of liver tissue from children with biliary atresia has revealed a unique proinflammatory footprint related to activation of lymphocytes, particularly natural killer cells.65
The initial biopsies of 47 infants with biliary atresia showed overexpression of genes associated with inflammation or fibrosis or both.76 Not surprisingly, the patients with a fibrosis signature were older and had decreased duration of transplant-free survival. When serum samples collected at the same time from 19 infants with EHBA and 19 with other forms of neonatal cholestasis were subjected to gel electrophoresis and tandem mass spectrometry, a combination of 11 proteins was able to discriminate between the two groups.77 Among them were apolipoproteins CII and E, whose genes on chromosome 19 are regulated by farnesoid X receptor, which in turn is responsive to bile acids. Other clues to the pathogenesis of EHBA from this study were upregulation of proinflammatory “positive” acute phase reactants, such as complement C3, and a relatively reduced level of “negative” acute phase proteins such as prealbumin.
Prompted by the similarities between biliary duct inflammation and graft-versus-host disease, Suskind and colleagues were the first to describe maternal “microchimerism” in the livers of infants with biliary atresia.78 Fluorescence in situ studies found two to four lymphocytes per tissue section to be XX in seven of eight affected boys. By quantitative polymerase chain reaction, between 1 and 142 copies of maternal DNA were detected for every 25,000 copies of neonate DNA. This was further documented in 14 more boys with EHBA by Hayashida79 and Muraji80 and their associates, although control subjects without biliary atresia also had a few XX lymphocytes in these studies. CD8+ T cells were especially more abundant in the EHBA biopsies in the latter study. Human leukocyte antigen (HLA) class I matching was significantly more prevalent in 57 maternal-child pairs with biliary atresia than in 50 control pairs (odds ratio, 2.46), possibly providing for greater survival of the chimeric lymphocytes.81
In favor of the second hypothesis are data that show a systemic dysregulation of morphogenesis in patients with biliary atresia,72 such as the coexistence of nonhepatic embryologic abnormalities, evidence for abnormal development of the ductal plate of the maturing intrahepatic biliary tract, and overexpression of certain regulatory genes in children who have an early form of biliary atresia.82,83 Associated developmental abnormalities include polysplenia or asplenia, cardiovascular defects, abdominal situs inversus, intestinal malrotation, and anomalies of the portal vein and hepatic artery in 25% of patients (see Table 54.14).
Mutations in the laterality genes CFC1 and ZIC3 lead to laterality defects and biliary atresia in some patients. The Inv mouse that develops situs inversus also develops biliary obstruction. The JAG1 gene has also been implicated in the pathogenesis of biliary atresia, because affected patients with a poor outcome also show a high frequency of JAG1 single nucleotide polymorphisms.84 The persistent expression of HES1 protein in the nuclei of biliary epithelial cells specimens of biliary atresia obtained up to 3 months after birth offers more evidence for disorderly Notch signaling in this disease. In normal development, such expression is silenced by 16 weeks’ gestation.85
The cystic bile ducts detected by prenatal ultrasonography may or may not be associated with biliary atresia. Among 88 patients with atresia, Okada and co-workers86 found a better prognosis for those with common duct atresia, as expected, but no association with other malformations despite early fetal onset. Therefore, the concept of a “fetal” (i.e., developmental) versus an acquired variant of biliary atresia was challenged. Sequential imaging starting at 16 weeks’ gestation revealed disappearance of a cyst before birth and extreme narrowings of the common hepatic duct at 30 days after birth.87
The ciliopathies responsible for polycystic disease and congenital hepatic fibrosis (discussed later) may have a new and potentially meaningful relationship to biliary atresia. Other ciliary dysfunction contributes to laterality defects in embryonic development, including syndromic biliary atresia. Having observed a twofold greater incidence of renal cysts developing in children after transplantation for EHBA versus other liver diseases, Hartley and colleagues88 found two other examples in long-term survivors. Using immunohistochemistry, they found that expression of fibrocystin was missing in the biliary epithelium of the EHBA patients, suggesting a role for PKHD1 or genes involved in primary ciliogenesis in this disease. Chu and associates89 showed, for the first time, that the cilia in biliary epithelium of five affected children were fewer, shorter, and abnormally oriented. Two other patients with biliary atresia who had no other malformations displayed alterations similar to those in epithelium of patients with other cholestatic diseases. Therefore, the defects may be secondary and nonspecific.
An all-encompassing hypothesis is that a genetically susceptible individual undergoes inflammatory destruction of the extrahepatic biliary system in response to as yet undetermined environmental factors.
Pathologic Features
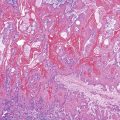
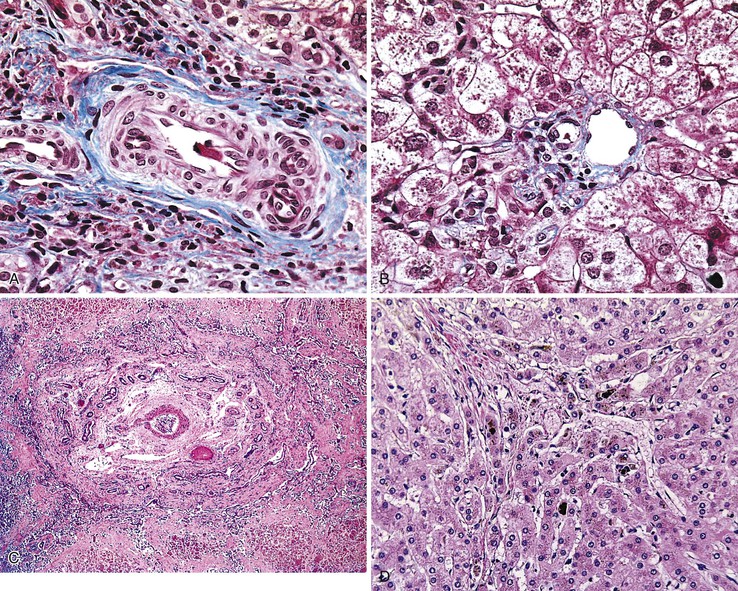
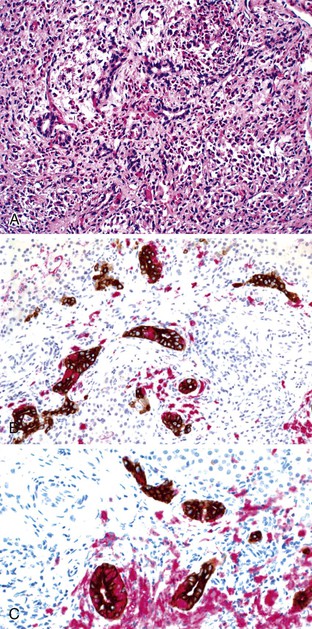
A diagnosis of EHBA is favored if a liver biopsy specimen exhibits bile ductular proliferation (neocholangioles), portal edema, and fibrosis but lacks sinusoidal fibrosis.69 Neutrophilic cholangitis and pericholangitis may or may not be present. The presence of bile plugs within bile duct lumina (which are distinct from the lumina of neocholangioles at the margins of portal tracts) and increased numbers of bile duct profiles are helpful diagnostic findings (Fig. 54.3). Fifteen percent of cases show giant cell transformation of hepatocytes (Fig. 54.4). The ductular reactions seen in cases of obstruction, as opposed to those that accompany hepatocellular necrosis due to toxins or ischemia, have been extensively analyzed by Desmet for many years.90 He hypothesized that the proliferation of duct epithelium with elongation and branching is controlled by molecular interactions with regional fibroblasts and represents an adaptive response to enhanced recycling of bile acids. The ductular “metaplasia” observed in other circumstances is attributed to the capacity of preexisting liver precursor cells in the canal of Hering to assume a hepatocellular or ductular phenotype and to the newly appreciated capacity of mature cells to “differentiate” and “redifferentiate” in vivo.91 Evidence for epithelial-mesenchymal transition as a mechanism for progressive fibrosis in EHBA has also been provided.92
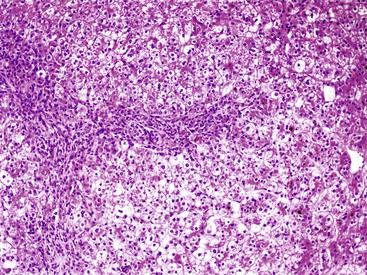
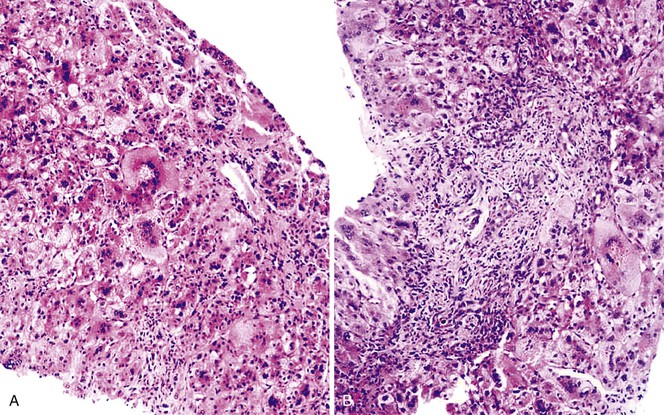
The classic “obstructive” findings of bile duct proliferation and inspissated bile plugs are not always the result of atresia (see Box 54.8). Some cases may show nonclassic features such as prominent portal tract arteries and an absence of bile duct proliferation (Fig. 54.5, A and B). In keeping with the developmental theory of biliary atresia, a subset of affected patients exhibit ductal plate malformation (see Fig. 54.5, C). Recapitulating immature fetal portal tracts, this latter entity consists of portal tracts without an interlobular bile duct but with a centrally located hepatic artery and a peripheral rim of bile ductular structures. However, this phenomenon is not limited to the 20% to 25% of patients with a laterality defect (biliary atresia splenic malformation). As a result, EHBA can be difficult to diagnose, even for the most experienced pediatric hepatopathologist (see Fig. 54.5, D).
 weeks) did not suggest the disease; exploratory laparotomy and cholangiography were required for definitive diagnosis.
weeks) did not suggest the disease; exploratory laparotomy and cholangiography were required for definitive diagnosis.On performance of a hepatoportoenterostomy, a bile duct remnant is usually submitted for pathologic examination. Typical findings in the bile duct remnant include fibrosis and obstruction of the lumen, a variable degree of periductal inflammation, and apoptotic degeneration of residual bile duct epithelium (Fig. 54.6). These findings do not differ in syndromic cases of biliary atresia and those discovered by prenatal ultrasonography, supporting the concept of a shared pathogenetic pathway.86,93
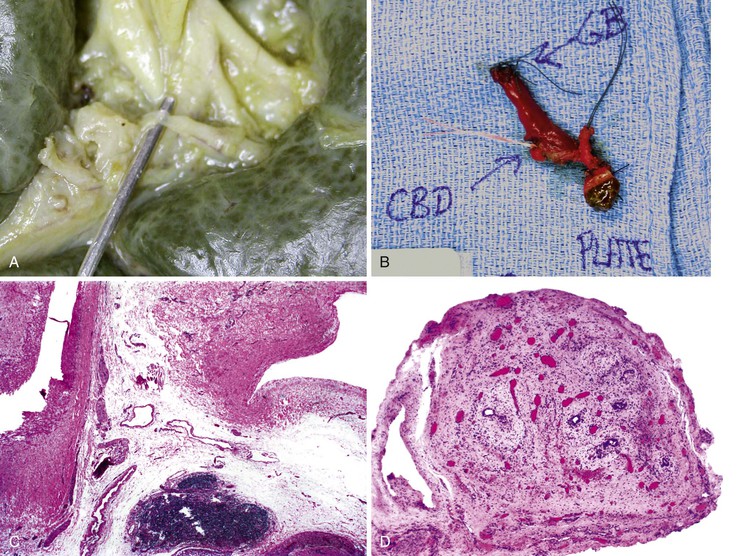
Natural History and Treatment
Before 1959, when the KPE procedure was introduced, biliary atresia was considered to be uniformly fatal by 2 years of age, with a median age at death of 10 months. After performance of a Kasai procedure, about 20% of affected infants show complete reversal of their jaundice and can lead relatively normal lives.86 The best survival rate in KPE patients with a native liver is 53% at 10 years, with jaundice clearing in 75% postoperatively. In the first report of the U.S. Biliary Atresia Research Consortium in 2006, a decline in total bilirubin at 3 months after KPE to less than 2 mg/dL was seen in 36.5% of children, and a good response (to <6 mg/dL) was seen in another 29%. Most patients in the excellent response group had a much better outcome. The 25% of children with a laterality defect fared more poorly, even if treated earlier.94 The surgical team in the study found a favorable drop in total bilirubin in 45% of patients in 2011, with a survival rate for those with a native liver of 54% at 1 year and 47% at 2 years. Atresia of the common hepatic duct or ducts at the liver hilum (Ohi94a types 2 and 3) had a worse prognosis than an atretic common bile duct (Ohi type 1), similar to the prognosis in patients with other malformations, ascites at surgery, or delay in surgery beyond day 75. Children operated on within the first 30 days had a 74% survival rate, with their native liver, at 2 years.95 At King’s College Hospital in London, 56% of infants with EHBA cleared jaundice, and the 2-year survival rate in patients with their native liver was 65%. The 51 patients with biliary atresia splenic malformation or biliary cysts tended to be operated on earlier but fared worse if the procedure was delayed.96 Nevertheless, the same group reported that even when surgery was performed on day 100 or even later, 45% of patients were alive with their native liver at 5 years, and 40% at 10 years, suggesting that patients may postpone transplantation by the KPE.97
Infants who do not respond to the Kasai procedure suffer from recurrent episodes of ascending cholangitis or sepsis due to the immediate proximity of a bowel segment to the porta hepatis of the residual biliary tree. Regardless of the presence or absence of ascending cholangitis, the liver may progress to cirrhosis with portal hypertension. Large bile lakes may form at the hilum, indicating a persistent effort at bile secretion by hepatocytes (Fig. 54.7). The pathogenesis of progressive fibrosis in chronic cholestatic diseases, including EHBA, PFIC1, PFIC2, and Alagille syndrome, has been linked to persistence of Hedgehog (Hh) signaling, which normally promotes embryonal duct differentiation and then shuts off as the liver matures.98 Immunohistochemical colocalization of the Hh transcription factor Gli2 and the mesenchymal markers vimentin and FSP1 in some of the reactive ductular epithelial cells in these diseases is evidence of reversal of mesenchymal-epithelial transition. Similar colocalization of FSP1 and cytokeratin 7 was shown in ductular epithelium, and this ductal cell derangement may lead to fibroplasia.98 Growth failure, malnutrition, deficiencies of lipid-soluble vitamins, and altered protein–energy and nutrient utilization are also potential complications.
A failed Kasai procedure in a patient with biliary atresia is the most frequent indication for liver transplantation in infants and young children. Patient survival after orthotopic transplantation for EHBA in the U.S. United Network for Organ Sharing from 1988 to 2003 was 85.8% at 10 years, with graft survival of 72.7%. Cadaveric split-liver grafts were associated with reduced survival, as was the need for life support at the time of surgery.99 Pathologic examination of explanted livers for occult malignancy is crucial for all forms of chronic liver disease in children.
Neonatal Sclerosing Cholangitis
In addition to neonatal sclerosing cholangitis associated with ichthyosis (OMIM 607626) due to CLDN1 mutations, several infants of consanguineous matings have been described with progressive sclerosing cholangitis from infancy.100,101 An 8-month-old Arab boy reported by Bar Meir and associates102 had elevated immunoglobulins and anti–smooth muscle titers, but there were no such abnormalities in his mother. These few patients serve to highlight the concept of an intrahepatic form of biliary “atresia,” but they also manifest extrahepatic ductal lesions by imaging, similar to older patients with progressive sclerosing cholangitis.103,104 Langerhans cell histiocytosis can lead to sclerosing cholangitis as part of a systemic process in young children (see Paucity of Intrahepatic Bile Ducts) (Fig. 54.8). Therefore, it may be most useful to consider that there is a continuum of inflammatory and sclerosing processes that may have a heritable basis in susceptibility105 and that show a variable degree of ductal obliteration.106
Neonatal Hepatitis Syndrome
A neonatal hepatitis syndrome resembling biliary atresia was first described by Burns in 1817. By the mid-1950s, two distinct disease categories of cholestasis of infancy were established: biliary atresia, an inflammatory sclerosing syndrome of the extrahepatic biliary tract with hepatic features of bile duct obstruction, and neonatal hepatitis, a nonobstructive type of neonatal cholestatic syndrome with characteristic hepatitic features. At that time, neonatal cholestasis due to disorders of bile formation and transport was not yet recognized. In the subsequent decades, enormous progress was made in the molecular characterization of neonatal cholestatic disorders. This entity is now best considered a clinicopathologic syndrome in which there are many potential etiologies, including infections, anatomic or structural defects, metabolic and inherited disorders, hormonal insufficiency, and vascular, toxic, and immune causes (see Table 54.6).
A review of all biopsies designated neonatal giant cell hepatitis at the Johns Hopkins and University of Chicago tertiary hospitals from 1984 to 2007 found 62 cases (75% boys; average age, 2 months).107 Of those cases with clinical follow-up, half proved to be idiopathic. Ultimately, 8% of patients were found to have biliary atresia, 6% Alagille syndrome, 6% bile salt synthetic defects, and 16% hypopituitarism. At the Texas Children’s Hospital in Houston, 151 infants up to 3 months old were biopsied because of persistent conjugated hyperbilirubinemia. Fifty-nine percent were boys. However, among the 80 patients (53%) who were found to have extrahepatic biliary obstruction, 60% were girls. Forty-eight patients had neonatal hepatitis, with or without giant cells, one of whom proved to have CMV infection. One infant had A1AT deficiency, one hypopituitarism, one McCune-Albright syndrome, and one Langerhans cell histiocytosis. Sixteen had paucity of intrahepatic bile ducts, but none was syndromic (i.e., Alagille syndrome) at this early age. Disorders without a specific identifiable etiology now account for about 25% of the total.12
Clinical Features
Persistent unconjugated hyperbilirubinemia raises the possibility of an inherited defect in bilirubin conjugation or hemolysis. Diagnostic assessment and therapeutic intervention is then critically important to avoid kernicterus and permanent neurologic damage. In contrast, predominantly conjugated hyperbilirubinemia in newborns should prompt a rigorous diagnostic investigation for biliary atresia. An approach to the workup of neonatal hepatitis was presented earlier (see Cholestatic Pattern and Table 54.7).
Pathologic Features
The hallmark histologic finding of many neonatal liver disorders is the formation of large multinucleated hepatocytes (giant cells), which are part of the broader spectrum of lobular disarray in neonatal hepatitis (Fig. 54.9). Nuclear inclusions in hepatocytes may suggest DNA viruses (Figs. 54.10 and 54.11); those in red cell precursors may suggest parvovirus B19. In some cases, most of the liver parenchyma is transformed into giant cells, and this is referred to as “syncytial giant cell hepatitis.” When this histologic picture predominates, electron microscopy can be extremely helpful, and even pathognomonic, if NPD type C is the cause. Of 40 infants evaluated for neonatal giant cell hepatitis in a Denver Children’s Series, 27% had NPD type C. Splenomegaly was a useful clue.108
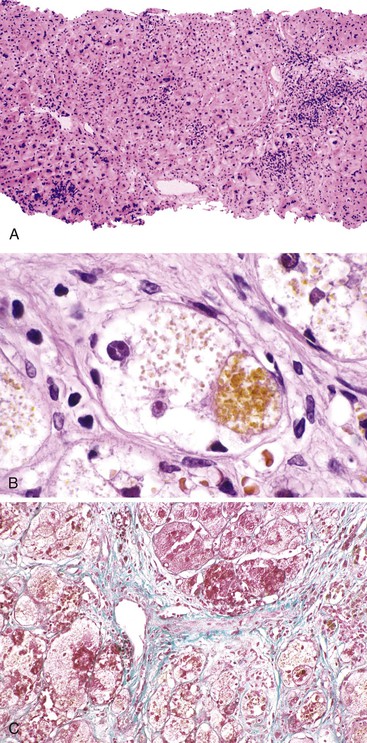
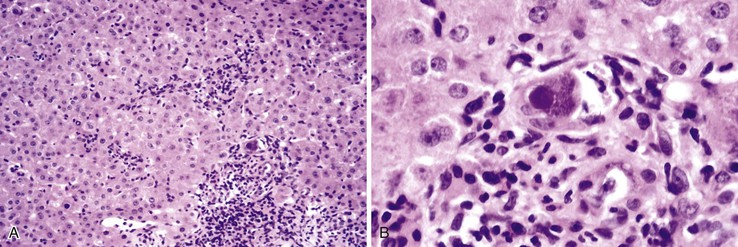
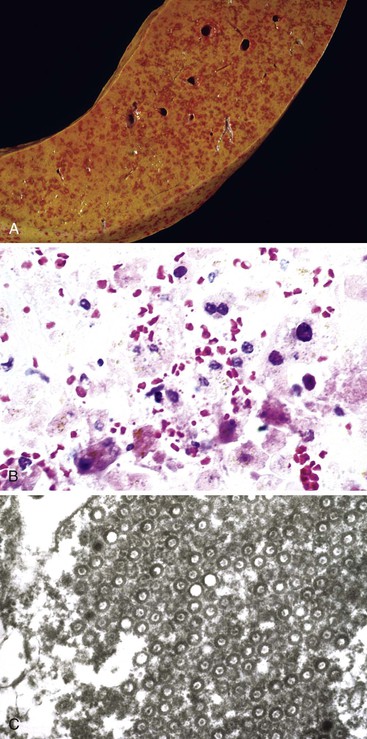
In cases of neonatal hepatitis, the relative proportion of inflammatory cells is not considered informative, because ongoing apoptosis may lead to extensive hepatocyte destruction and accumulation of residual lymphocytes and macrophages. In some cases, parenchymal inflammation is minimal or absent. in addition, parenchymal neutrophils are an uncommon finding. Occasional islands of erythropoiesis are a normal finding. In all instances, the biopsy specimen should be examined carefully for evidence of viral cytopathic change.
Portal tracts may be inconspicuous, whether normal or abnormal. In general, at least five to seven portal tracts are required to consider the tissue sample adequate for histologic evaluation. Ten or more portal tracts are preferred. Bile duct hypoplasia was more common among infants with hypopituitarism, compared with other conditions, in the Johns Hopkins/University of Chicago series.107 No other biopsy finding was helpful in distinguishing possible etiologies in that series. Exclusion of biliary atresia or A1AT storage disorder requires verification that most, if not all, portal tracts contain a terminal bile duct, a companion hepatic artery, and a portal vein and that bile ductular proliferation, edema, neutrophilic inflammation, and fibrosis are absent. Although the portal tracts in patients with neonatal hepatitis may be expanded by a mixed inflammatory infiltrate, this should not be confused with normal granulocytic hematopoiesis occasionally found within portal tracts.
Occasionally, massive hepatic necrosis, with severe accumulation of parenchymal iron, may be observed in patients with Down syndrome. One infant in the Texas Children’s Hospital series had an exceptional degree of hepatocellular necrosis, and the cause was pyruvate kinase deficiency. The observation of massive necrosis should also prompt consideration of neonatal hemochromatosis (discussed earlier; see Table 54.6).
Differential Diagnosis
Once EHBA and A1AT mutations have been excluded on the basis of both clinical findings and percutaneous liver biopsy, then the differential diagnosis of neonatal hepatitis syndrome must be pursued. Infections of the newborn have been mentioned, and Chapter 46 addresses some of these disorders. Extensive lobular disease, with or without giant cells, may be seen in patients with EHBA (see Fig. 54.4). Therefore, lobular changes should not divert attention from the features in the portal tracts. Second, nonclassic features of EHBA, in which intrahepatic bile ducts have yet to proliferate, may be observed in biopsy specimens from patients with biliary atresia (see Fig. 54.5, B). Sometimes, alert detection of acholic stools prompts an early biopsy, but the time of onset and rate of progression can vary greatly.109 (For a detailed discussion of the differential diagnosis, see Cholestatic Pattern and Table 54.6.)
Natural History and Treatment
The natural history of neonatal hepatitis depends heavily on the specific etiology (Table 54.15). For instance, pharmacologic intervention is imperative for many of the infectious disorders. Many of the other disorders, including hypopituitarism, tyrosinemia, and bile salt synthetic defects, benefit dramatically from prompt intervention.110 In about one quarter of all infants, no cause is found.111 The prognosis for “idiopathic” neonatal hepatitis syndrome is considered favorable; the mortality rate is 13% to 25%.112 Predictors of a poor clinical outcome include severe or prolonged (>6 months) jaundice, alcholic stools, familial occurrence, and persistent hepatomegaly. Peak bilirubin level is not predictive of outcome. Sepsis is a devastating complication and is associated with a poor outcome. For infants whose liver disease resolves, the prognosis is quite good; most have no residual liver dysfunction.
Table 54.15
Natural History of Common Causes of Neonatal Jaundice
| Etiology | Natural History |
| Biliary atresia | Death by 1 to 2 yr of age without Kasai procedure or liver transplantation |
| α1-Antitrypsin storage disorder | Progressive chronic liver disease with risk of cirrhosis |
| Neonatal infection | Resolution of infection required for survival |
| Disorders of carbohydrate metabolism | |
| Glycogen storage disease | Maintenance of blood sugar levels enables survival Risk of chronic renal disease High probability of hepatic adenomatosis Liver transplantation is curative in some forms |
| Galactosemia | Dietary galactose restriction enables survival Neurodevelopmental complications may persist |
| Fructosemia | Avoidance of dietary fructose enables survival Lifelong risk of metabolic crises on fructose exposure |
| Disorders of amino acid metabolism | |
| Tyrosinemia | Pharmacologic treatment enables survival High lifetime risk of hepatocellular carcinoma |
| Disorders of glycolipid and lipid metabolism | |
| Niemann-Pick disease types A and C | Type A: fatal outcome of progressive neurologic disease Type C: survival into adulthood possible, neurologic compromise may occur |
| Hereditary disorders of bile formation | |
| Progressive familial intrahepatic cholestasis | Biliary diversion may ameliorate; liver transplantation for cirrhosis; persistent diarrhea in type 1 |
| Disorders of bile acid biosynthesis | Dietary treatment with bile acids can ameliorate effects of disease |
| Idiopathic neonatal hepatitis | Highly variable, most likely representing undiscovered genetic abnormalities Cholestatic liver disease may persist into later childhood |
The management of idiopathic neonatal hepatitis syndrome is supportive and includes adequate nutrition. Dietary measures, such as use of lactose-free, low-protein formulas, may mitigate further liver damage until the results of tests for galactosemia and hereditary tyrosinemia type I become known. Elemental formulas containing medium-chain triglycerides help maintain caloric intake in severely ill patients. Infants with chronic cholestasis require fat-soluble vitamin supplementation. Pruritus associated with chronic cholestasis is difficult to treat in many patients.2
Syncytial Giant Cell Hepatitis
In 1991, a putative novel form of hepatitis was first described in patients aged 5 months to 41 years.113 Clinical features of a severe type of hepatitis required liver transplantation in five patients; five others died of their liver disease. Liver histology revealed replacement of the parenchyma with abundant large syncytial multinucleated (giant) hepatocytes that contained up to 30 nuclei per cell. Electron microscopy revealed intracytoplasmic structures consistent with paramyxovirus nucleocapsids (Fig. 54.12). Injection of liver homogenate from an affected patient into two chimpanzees led to an increase in the titer of paramyxovirus antibodies in one.
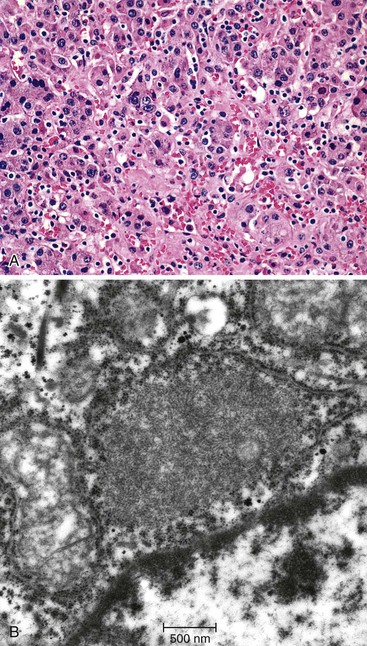
One year later, a separate report concluded that syncytial giant cell hepatitis may have a variety of causes, such as autoimmune hepatitis, hepatitis A virus (HAV) or HBV infection, Epstein-Barr virus infection, or, potentially, HCV infection.114 Three of the patients had fulminant hepatic failure; two others developed severe chronic hepatitis. However, a further seven patients fared quite well. Therefore, syncytial giant cell hepatitis represents a tissue reaction pattern that may develop as a result of multiple causes and does not always imply an ominous prognosis. This interpretation has been supported by subsequent reports in which generalized bacterial sepsis, viral infection, toxoplasmosis, syphilis, listeriosis, and even tuberculosis were shown to induce a similar histologic pattern of injury. Viral causes include CMV, rubella, herpes simplex virus, human herpesvirus 6, varicella, coxsackievirus, echovirus, reovirus 3, parvovirus B19, human immunodeficiency virus (HIV), enteroviruses, paramyxovirus, HAV, HBV, or (rarely) HCV; some adult patients were coinfected with HIV and HCV.115
The Paramyxoviridae are divided into the subfamily Paramyxovirinae, containing Paramyxovirus (Sendai virus, parainfluenza virus type 3), rubella, mumps, parainfluenza virus type 2, and Morbillivirus (measles), and the subfamily Pneumovirinae, containing the Pneumovirus genus (respiratory syncytial virus). Therefore, even by ultrastructural examination, the differential diagnosis for syncytial giant cell hepatitis is quite broad.
Paucity of Intrahepatic Bile Ducts
Neonates with conjugated hyperbilirubinemia may exhibit paucity of small intrahepatic (interlobular) bile ducts. A variety of disorders can cause nonsyndromic duct paucity (Table 54.16), including A1AT storage disorder and hypopituitarism. Langerhans cell histiocytosis may cause severe cholangitis with a dense infiltrate of eosinophils, as well as CD207-positive and even CD1a-positive histiocytes (see Fig. 54.8). In patients who survive infancy, duct loss can be severe. Sclerosing cholangitis due to Langerhans cell histiocytosis has been reported in an adult.116 Some cases of paucity are idiopathic.111 Among congenital infections, CMV infection is the most important; viral inclusions may be found within bile duct epithelial cells (see Fig. 54.10). In severe neonatal hepatitis, paucity of bile ducts may be evident on liver biopsy specimens. Clinical presentation of severe cholestasis, either early in life or later in childhood, should always raise the possibility of syndromic paucity of bile ducts (Alagille syndrome).
Table 54.16
Causes of Nonsyndromic Paucity of Bile Ducts
| Infants | Adolescents and Adults |
| Prematurity | Immune-mediated causes |
| Infection | Primary biliary cirrhosis |
| Cytomegalovirus | Primary sclerosing cholangitis |
| Rubella | Liver allograft rejection |
| Syphilis | Graft-versus-host disease |
| Hepatitis B virus | Metabolic |
| Metabolic disorders | Cystic fibrosis |
| α1-Antitrypsin storage disorder | Toxic insults |
| Niemann-Pick disease type C | |
| Cystic fibrosis | |
| Zellweger syndrome | |
| Mitochondrial cytopathies | |
| Byler syndrome | |
| Prune-belly syndrome | |
| Hypopituitarism | |
| Genetic: chromosomal disorders | |
| Trisomy 18, trisomy 21 | |
| Partial trisomy 11 | |
| Monosomy X | |
| Neonatal sclerosing cholangitis-ichthyosis | |
| Immune-mediated causes | |
| Liver allograft rejection | |
| Graft-versus-host disease | |
| Severe idiopathic neonatal hepatitis | |
| End-stage extrahepatic atresia |
Adapted from Roberts EA. Neonatal hepatitis syndrome. Semin Neonatol. 2003;8:357-374.
α1-Antitrypsin Deficiency
α1-Antitrypsin is a hepatic storage disorder that mostly manifests later in life (see later discussion). However, 11% of individuals with PiZZ mutations in SERPINA1, the A1AT gene, may develop conjugated hyperbilirubinemia in infancy.
A1AT storage disorder can mimic the histologic appearance of biliary atresia, with portal tract changes that include edema, scant acute inflammation, proliferating bile ductules, and bile plugs within bile duct lumina (Fig. 54.13). Characteristic globular inclusions of staining material (PAS) with diastase digestion [d-PAS] in periportal hepatocytes are typically not present for weeks or months, but immunohistochemistry for the protein is more sensitive, as is electron microscopy for demonstration of the protein within endoplasmic reticulum. Bile pigment, hemosiderin, and copper-binding protein may be present within periportal hepatocytes, all of which are highlighted as fine granules in tissue sections stained with d-PAS. Protease inhibitor typing by serum immunoelectrophoresis is required for diagnosis.
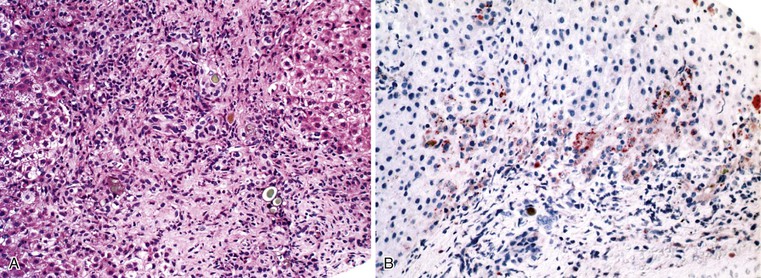
Syndromic Paucity of Bile Ducts: Alagille Syndrome
Clinical Features
Some infants and young children with conjugated hyperbilirubinemia have associated facial dysmorphism and cardiovascular, vertebral, and ocular malformations. The combination of extrahepatic abnormalities and deficiency of interlobular bile ducts is termed Alagille syndrome.117 Alagille syndrome is an autosomal dominant disorder with an estimated frequency of 1 in 70,000 live births. Sporadic cases account for 45% to 50%. The mortality rate is 15% to 20%. Deaths are mostly caused by hepatic or cardiac complications.
Before the advent of genetic testing, a diagnosis of Alagille syndrome required demonstration of paucity of interlobular bile ducts and clinical evidence of at least three major of the following abnormalities: characteristic facies, posterior embryotoxon, butterfly vertebrae, renal disease, and cardiac anomalies. However, bile duct paucity is evident in only 60% of infants biopsied before 6 months of age.118 Furthermore, there is wide phenotypic variability in patients with this disorder, ranging from mild subclinical findings to complete liver failure and complex congenital heart disease.119,120 Most symptomatic patients with hepatic involvement are seen clinically within the first 6 months of life with jaundice, hepatomegaly, pruritus, and failure to thrive. Laboratory studies reveal conjugated hyperbilirubinemia and increased serum levels of GGT, alkaline phosphatase, bile acids, and cholesterol. Serum ALT and AST levels may be normal or mildly elevated. Currently, genetic testing is almost always conclusive.
Pathogenesis
Mutations in Jagged1 (JAG1), a ligand in the Notch signaling pathway, have been identified in 94% of patients with Alagille syndrome.121–123 More recently, mutations in the NOTCH2 gene, which encodes a receptor for Jagged1, have been identified in some JAG1 mutation–negative patients.124 It is hypothesized that mutations in JAG1 result in an arrest of branching and elongation of bile ducts during postnatal liver growth.118 In support of this theory, Libbrecht and associates reported a case in which an explanted liver from a 16-year-old patient with Alagille syndrome demonstrated paucity of bile ducts in peripheral liver parenchyma but normally developed bile ducts in the central areas.125 The interaction of the NOTCH and WNT pathways in biliary epithelial development is of great interest for aberrant organogenesis and response to injury.126,127
Pathologic Features
To diagnose paucity of bile ducts, the liver biopsy specimen should contain at least 7 portal tracts, and preferably at least 10. Normally, on routinely stained tissue sections (H&E or Masson trichrome) of mature liver tissue, approximately 90% of portal tracts contain an interlobular bile duct, either as a single duct or as multiple duct profiles,4 in close apposition to the branch of the hepatic artery. The presence of multiple portal tracts containing arteries and veins but not bile ducts should prompt consideration of bile duct paucity, which is defined by an absence of, or marked reduction in, the number of interlobular bile ducts within portal tracts. In pediatric patients, a bile duct-to-portal tract ratio of less than 0.8 is often used as a cutoff point between normal and abnormal, although the number of bile ducts may be normally (physiologically) low in preterm infants. However, in any age group, a ratio of less than 0.4 is strongly suggestive of bile duct paucity. When making this assessment, large portal tracts should not be considered in the equation. Furthermore, incomplete portal tracts that are partially transected by the biopsy should be excluded from the analysis, because absence of a bile duct may simply represent a sampling artifact. Specimens from patients with Alagille syndrome are conspicuous for their lack of portal tracts and parenchymal inflammation, but they may have abundant giant cells, as in neonatal hepatitis (Fig. 54.14).
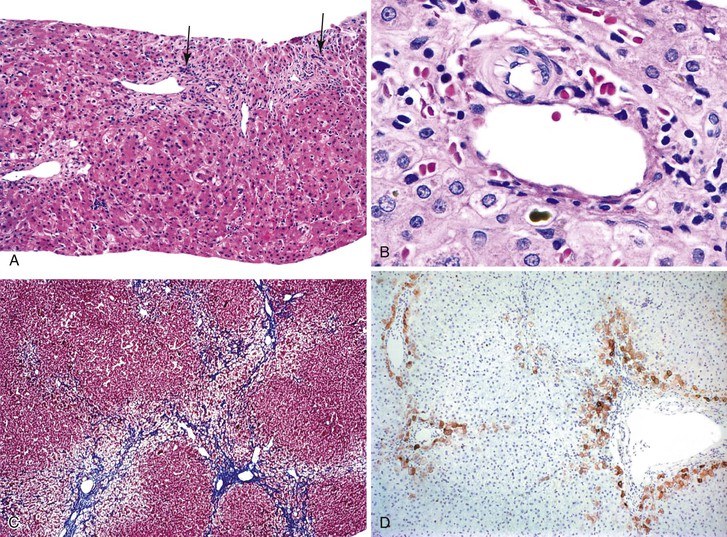
Immunohistochemical staining for cytokeratin 7 or 19 is useful to highlight bile duct epithelium. Because hepatocytes in cholestatic livers often acquire cytokeratin 7 immunoreactivity (see Fig. 54.14, D), cytokeratin 19 is preferred for quantitating bile ducts.128 Immunoreactive ductular profiles at the margins of portal tracts should not be confused with true interlobular bile ducts (see Fig. 54.1). It has been shown that a persistent lack of canalicular CD10 immunopositivity, although physiologic in infants younger than 2 years of age, is a characteristic finding in Alagille syndrome.129 The Masson trichrome stain is also helpful in identifying small portal tracts because of its ability to detect delicate amounts of connective tissue. These portal tracts should be included in the portal tract count when one is assessing bile ducts.
In the evaluation of a liver biopsy specimen for potential paucity of bile ducts, the pathology report should indicate how many portal tracts and bile ducts were identified. The staining method should also be reported. Paucity of interlobular bile ducts is not a specific disorder, instead, it represents a pathologic feature that may have a variety of causes.
Differential Diagnosis
When Alagille syndrome cannot be distinguished from biliary obstruction on liver biopsy and the clinical and radiologic features fail to detect the extrahepatic manifestations in an infant with Alagille syndrome, patients have at times been treated by KPE. Even at a major center with extensive experience with these diseases, 4.4% of patients were mistakenly operated on, and afterward their liver disease progressed more rapidly to end-stage cirrhosis.61 Because disease progression is highly variable, every effort must be made to avoid such surgery. No genotype–phenotype correlation with severity for JAGGED1 mutations was found among 33 patients with Alagille syndrome who were older than 10 years of age.130 Those children with elevated total bilirubin (>6.5 mg/dL), conjugated bilirubin (>4.5 mg/dL), and cholesterol (>520 mg/dL) before age 5 years had significantly worse liver disease later.
Caution is also required in the interpretation of hepatobiliary scintigraphy and cholangiographic studies. Typically, both extrahepatic and intrahepatic bile ducts in patients with Alagille syndrome are markedly hypoplastic; therefore, on hepatobiliary scanning, one may not see excretion of radioisotope into the intestines (mimicking EHBA).118 Similarly, intraoperative cholangiography may not necessarily demonstrate opacification of the proximal extrahepatic ducts. These diagnostic pitfalls underscore the importance of a careful and full physical examination and radiologic evaluation in patients with suspected Alagille syndrome. Sequencing of the entire coding region of JAG1 is now available and should be used in patients in whom there is a strong clinical suspicion and in family members of probands with mutations.
Natural History
Alagille syndrome is a benign illness in many children. However, young children with protracted severe jaundice usually have a poorer prognosis. Between 10% and 50% of patients with Alagille syndrome eventually progress to cirrhosis and liver failure.131 Cases of HCC in Alagille syndrome have been described, as in every chronic cholestatic disease of infants.132 The overall mortality rate from Alagille syndrome is estimated to be 20% to 25%; death occurs mainly from cardiac disease, intercurrent infection, or progressive liver disease. Liver transplantation is warranted for patients with severe hepatic disease. Because of the presence of multiple organ defects in Alagille syndrome, the outcomes of liver transplantation are poorer than for patients with EHBA, especially with regard to death during the first 30 postoperative days. No pretransplant factors were identified among the 87% of patients who survived to 1 year, compared with those who did not.133
Hereditary Disorders of Bilirubin Metabolism
Bilirubin is the end product of heme degradation, which is derived predominantly from the breakdown of senescent erythrocytes by the mononuclear phagocytic system. Heme oxygenase within reticuloendothelial cells oxidizes heme to biliverdin, which is then reduced to bilirubin by biliverdin reductase. Bilirubin is released into the circulation and binds to serum albumin. Uptake of bilirubin by hepatocytes occurs via a carrier-mediated system at the level of the sinusoidal membrane. Bilirubin is then conjugated with either one or two molecules of glucuronic acid by bilirubin uridine diphosphate (UDP)-glucuronosyltransferase (i.e., UGT1A1) within the endoplasmic reticulum. It is then excreted as water-soluble, nontoxic bilirubin glucuronides into bile.
Crigler-Najjar Syndrome and Gilbert Syndrome
Classification and Pathogenesis
The hepatic conjugating enzyme UGT1A1 is a product of the UGT1A1 gene, which is located on chromosome 2q37. It is a member of a family of UDP-glucuronosyltransferases (UGTs) that catalyze the glucuronidation of an array of substrates such as steroid hormones, carcinogens, and drugs. The various types of UGTs are distributed within a wide range of tissues, including liver, kidney, intestines, skin, lung, olfactory epithelium, and testis. UGT1A1 is located primarily within the smooth and rough endoplasmic reticulum of hepatocytes as a single isoform that catalyzes the glucuronidation of bilirubin to form its monoglucuronidated and diglucuronidated forms. In humans, two members of the UGT1 family possess the capability to glucuronidate bilirubin in vitro, but only one isoform is physiologically relevant in vivo. The bilirubin glucuronidating isoform is termed UGT1A1, because it is generated from the exon 1A of the UGT1 gene locus. Multiple mutations in UGT1A1 can cause hereditary unconjugated hyperbilirubinemia: Crigler-Najjar syndrome types I and II and Gilbert syndrome.134
Clinical and Pathologic Features
In Crigler-Najjar syndrome type I, the liver UGT1A1 enzyme is completely absent. Colorless bile contains only trace amounts of unconjugated bilirubin. Serum unconjugated bilirubin reaches very high levels, leading to severe jaundice and icterus. Without liver transplantation, this condition is invariably fatal (because of kernicterus), usually within 18 months of birth.
Crigler-Najjar syndrome type II is a less severe, nonfatal disorder in which the hepatic level of UGT1A1 enzyme activity is greatly reduced but not absent, and the enzyme is capable of forming only monoglucuronidated bilirubin. In contrast to the type I syndrome, the only major clinical consequence is the presence of extraordinarily yellow skin caused by moderate to high levels of circulating unconjugated bilirubin. Phenobarbital treatment may improve bilirubin glucuronidation by inducing hypertrophy of the hepatocellular endoplasmic reticulum.
Gilbert syndrome is a relatively common, benign, inherited condition. Affected patients have mild, fluctuating hyperbilirubinemia in the absence of hemolysis or liver disease. Typically, hepatic bilirubin glucuronidating activity is approximately 30% of normal levels. In most patients, the genetic defect consists of two extra bases (TA) in the TATAA element (TATA box) in the promoter region of the UGT1A1 gene. This creates an A(TA)7TAA element, rather than the normal A(TA)6TAA of the UGT1A1 gene, and results in reduced expression of UGT1A1. In some cases, patients are heterozygous for missense mutations in the UGT1A1 gene. Gilbert syndrome affects 3% to 10% of the population. Mild hyperbilirubinemia may go unrecognized for many years and is not associated with functional derangements of the liver. When detected in adolescents or adults, it is typically in association with an unrelated stress, such as intercurrent illness, strenuous exercise, or fasting, that reduces hepatic levels of the obligate cofactor for UGT1A1, UDP-glucuronic acid. Gilbert syndrome has no clinical consequence except for anxiety related to persistent jaundice. A 2007 study suggested that diabetic patients with coexistent Gilbert syndrome have a lower incidence of vascular complications and a reduction in serum markers of oxidase stress and inflammation, possibly because of the antioxidant effect of bilirubin.135
In all disorders of bilirubin conjugation, the liver is morphologically normal by both light and electron microscopy.
Dubin-Johnson Syndrome
Dubin-Johnson syndrome is an autosomal recessive disorder characterized by the presence of conjugated hyperbilirubinemia due to mutations in the ABCC2 gene. ABCC2 encodes the hepatocyte canalicular multispecific organic anion transporter (cMOAT), the transport protein responsible for secretion of bilirubin conjugates into bile.136,137 There are many different mutations in the protein that cause functional defects in protein maturation and localization to the canalicular plasma membrane.138,139 The key consequence is profound conjugated hyperbilirubinemia. Patients affected with this disorder show profound jaundice but are otherwise normal. Presentation may be at any age until the sixth decade of life. Rarely, neonatal hepatitis has been reported in patients with Dubin-Johnson syndrome.140
Affected livers reveal deep discoloration, not from retained bilirubin pigment but from accumulation of a chemical polymer in lysosomes that resembles epinephrine.141 On liver biopsy, the liver tissue core appears dark. By light microscopy, prominent globular pigmented inclusions are present within hepatocytes (Fig. 54.15). Pigmented inclusions may be inconspicuous in infants but become more prominent during childhood.
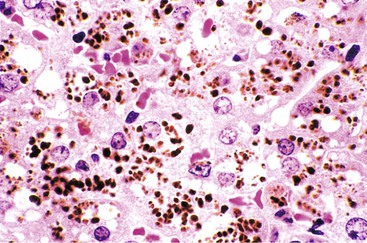
The conjugated hyperbilirubinemia of Dubin-Johnson syndrome is not toxic to the central nervous system or other tissues. Treatment with ursodeoxycholic acid may be beneficial for the rare infants who develop neonatal hepatitis.140
Rotor Syndrome
Rotor syndrome resembles Dubin-Johnson syndrome in that it produces a benign type of conjugated hyperbilirubinemia. The jaundice is more often evident in infancy. It is a rare autosomal recessive disorder (frequency, 1 in 106) that is caused by the simultaneous deficiency of two hepatocyte sinusoidal membrane organic anion transporters (OATP1B1 and OATP1B3), which are encoded by SLCO1B1 and SLCO1B3 on chromosome 12p.142 In Rotor syndrome, loss of these two proteins impairs hepatocyte uptake of conjugated bilirubin. In contrast to Dubin-Johnson syndrome, no pigment accumulates in hepatocytes, but urinary coproporphyrin 1 excretion is similarly very abundant. Although there is no clinical dysfunction, the defect may contribute to hepatotoxicity from drugs that require excretion of conjugates.
Hereditary Disorders of Bile Acid Metabolism
Bile is a complex mixture of bile salts, phosphatidylcholine, cholesterol, bilirubin, glutathione, and electrolytes combined with pigments and metabolites of drugs. Approximately 50% of the dry weight of bile consists of bile salts—conjugates of bile acids and glycine or taurine. The two primary bile acids in humans, cholic acid and chenodeoxycholic acid, are synthesized within hepatocytes, where they get conjugated to the amido groups of glycine or taurine to form bile salts. Bile salts are exported through the hepatocyte canalicular membranes into the canaliculus, where they arc admixed with other components of bile, and flow through the biliary tree. Defects in the synthesis of bile acids or in the export of bile salts to the canaliculus lead to cholestatic disorders in humans, as well as deficiency of fat-soluble vitamins and steatorrhea in the liver (see Table 54.9).
Inborn Errors of Bile Acid Synthesis
Inborn errors of bile acid synthesis are rare inherited disorders in which the normal bile acid biosynthetic pathway are severely impaired. This condition results in buildup of toxic biosynthetic intermediates and manifests clinically as neonatal hepatitis syndrome or in childhood as chronic cholestasis. These diseases are extremely rare. Patients can be screened for these disorders by mass spectrometric analyses of urinary bile acids and bile alcohols.143
3β-Hydroxy-Δ5-C27-Steroid Oxidoreductase Deficiency
3β-Hydroxy-Δ5-C27-steroid oxidoreductase, the second enzyme in bile acid biosynthesis, is a microsomal oxidoreductase that removes hydrogen atoms from the steroid nucleus. Mutations in the HSD3B7 gene that encodes the enzyme have been reported in 15 patients in 13 families studied.144 In another 18 patients,145 the majority had neonatal conjugated hyperbilirubinemia, rickets, and hepatomegaly. Liver biopsy revealed giant cell transformation and hepatocyte disarray with cholestasis, bridging fibrosis, and, rarely, cirrhosis. These infants typically have normal serum levels of GGT and low serum total bile acid concentrations. Dietary administration of cholic acid, with or without ursodeoxycholic acid, inhibits endogenous biosynthesis of bile acids and markedly reduces accumulation of aberrant bile acid precursors. With this regimen, infants can fare well and develop normally.
Δ4-3-Oxosteroid 5β-Reductase Deficiency
Δ4-3-Oxosteroid 5β-reductase is a cytosolic enzyme that responsible for 5β reduction of the nucleus of the steroid molecule. Infants affected by an autosomal recessive disorder in which the enzyme is inactivated produce excessive amounts of Δ4-3-oxo bile acids, which are toxic at high levels. Mutations in the AKRID1 gene,146 which encodes the enzyme, have been reported in patients with severe neonatal cholestasis and coagulopathy. Serum GGT levels are usually normal in this disorder. Liver biopsy may reveal giant cell hepatitis and hepatic scarring resembling neonatal hemochromatosis.147 Treatment with cholic acid, with or without ursodeoxycholic acid, is beneficial.
Other Bile Acid Biosynthetic Disorders
Neonatal jaundice with hepatosplenomegaly develops in patients who have a deficiency in the 24,25-dihydroxycholanoic cleavage enzyme. Because this enzyme is important for the alternative pathway of bile acid biosynthesis, which produces chenodeoxycholic acid, dietary treatment with chenodeoxycholic acid plus cholic acid is beneficial.
Other inborn errors of bile acid metabolism have been described in patients with neonatal liver disease.148,149
Defects in Bile Secretion or Transport
Several disorders manifest in neonates or children with cholestasis resulting from defective bile secretion or transport. Chief among them are the progressive familial intrahepatic cholestatic disorders.
Progressive Familial Intrahepatic Cholestasis
Progressive familial intrahepatic cholestasis (PFIC) comprises three autosomal recessive disorders (PFIC1, PFIC2, and PFIC3; see Table 54.9), which are caused, respectively, by mutations in three canalicular proteins: familial intrahepatic cholestasis type 1 (FIC1), encoded by ATP8B1; bile salt export pump (BSEP), encoded by ABCB11; and multidrug resistance protein 3 (MDR3), encoded by ABCB4.150 All three disorders typically manifest in early childhood with conjugated hyperbilirubinemia and are characterized by the presence of elevated plasma bile acids and defective bile secretion. Although PFIC2 and PFIC3 are caused by defective transport of bile components, PFIC1 results from a failure to maintain canalicular membrane lipid asymmetry and fluidity.150
When PFIC is suspected, measurements of serum GGT, another canalicular protein, can be helpful in providing a clue. PFIC3 is characterized by high GGT levels, whereas PFIC1 and PFIC2, along with a few other disorders, are referred to as low (normal) GGT cholestatic disorders (see Table 54.6). Liver biopsy plays a critical role in the diagnosis of PFIC disorders. Molecular genetic testing for the three genes is available and, ultimately, helpful for confirmation.151
Progressive Familial Intrahepatic Cholestasis 1
PFIC1 (also known as ATP8B1 disease, FIC1 disease, or Byler disease) is a multisystemic disease caused by mutations in the ATP8B1 gene located on chromosome 18q21.152 The ATP8B1 gene product, FIC1, is a P-type adenosinetriphosphatase that is believed to act as a lipid “flippase,” internalizing phosphatidylserine from the outer to the inner leaflet of the canalicular membrane. This activity has been found to be crucial in maintaining lipid asymmetry of the membrane. Before identification of the causative gene and its mutations, PFIC1 was referred to as Byler disease, named after the Amish kindred in whom the disease was first described. Patients with PFIC1 typically develop jaundice, extreme pruritus, coagulopathy, and growth failure within the first several months of life. Laboratory studies usually reveal elevated levels of ALT, AST, alkaline phosphatase, and bile acids. However, the serum level of GGT is normal or low, despite the presence of cholestasis. Liver disease in patients with PFIC1 is progressive and leads to the development of cirrhosis by 20 years of age.
Other entities caused by mutations in ATP8B1 include the allelic disorder benign recurrent intrahepatic cholestasis 1 (BRIC1; OMIM 243300),152,153 which is a milder, nonprogressive form that manifests with recurrent bouts of cholestasis and pruritus, and Greenland familial cholestasis, which is a form of PFIC1 seen in the Inuit population.
Histologically, PFIC1 is characterized by the presence of bland intracanalicular cholestasis, ductular bile plugs, and minimal or no inflammation (Fig. 54.16, A). Ultimately, a micronodular pattern of fibrosis and cirrhosis develops with disease progression. Transmission electron microscopy is useful in distinguishing PFIC1 from other causes of cholestasis. In PFIC1, the canalicular bile is coarse and granular, commonly referred to as “Byler bile”154 (see Fig. 54.16, B).
The ATP8B1 gene is expressed in several nonhepatic tissues (i.e., acinar cells of the pancreas and ileal and colonic enterocytes). Consequently, patients with PFIC1 may also show a variety of extrahepatic manifestations, such as chronic diarrhea, pancreatic insufficiency, pancreatitis, and elevated sweat chloride levels.155 Chronic diarrhea and pancreatitis persist, and are even exacerbated, after liver transplantation because of restoration of normal hepatic secretion of bile salts into bile and an impaired ability of intestinal epithelium to reabsorb bile salts for return to the enterohepatic circulation. Therefore, surgical interruption of the enterohepatic circulation of bile acids, by either partial external biliary diversion or partial ileal exclusion, is the recommended treatment of choice for affected patients.156
Progressive Familial Intrahepatic Cholestasis 2
Progressive familial intrahepatic cholestasis 2 (also called PFIC2, ABCB11 disease, or BSEP disease) is caused by mutations in the ABCB11 gene,157 in which more than 100 different mutations have been described.158 The ABCB11 gene encodes for the major canalicular bile salt transporter, BSEP. Like PFIC1, PFIC2 manifests in infancy as severe cholestasis, pruritus, jaundice, malabsorption of fats and fat-soluble vitamins, and failure to thrive. Laboratory studies show elevated serum levels of ALT and AST and normal or low serum GGT levels. Liver disease in patients with PFIC2 is more severe than in patients with PFIC1. Rapid progression to end-stage cirrhosis typically occurs within the first decade of life. Liver biopsies from patients with PFIC2 reveal neonatal hepatitis with hepatocellular disarray, edema, inflammation, giant cell transformation, and, ultimately, portal fibrosis (Fig. 54.17).159 Several cases of HCC and cholangiocarcinoma have been reported as well.44 Ultrastructurally, the bile in PFIC2 is amorphous or filamentous.
van Mil and colleagues160 identified eight distinct mutations in ABCB11, in eight different families whose members had recurrent bouts of cholestasis similar to BRIC1 but without mutations in ATP8B1. This disorder has now been termed BRIC2 (OMIM 605479) (Table 54.17). It is more often associated with cholelithiasis, compared with BRIC1. Liver biopsies typically show cholestasis with hepatocyte swelling.
Table 54.17
Inherited Disorders of Bile Acid Synthesis and Transport
| Common Names | Gene | Chromosome | Function | Location | Diseases |
| FIC1 | ATP8B1 | 18q21.3 | Phosphatidylserine “flippase” | Hepatocyte canalicular (apical) membrane; cholangiocytes, enterocytes | PFIC1, BRIC1, Byler disease, Byler syndrome |
| BSEP | ABCB11 | 2q31 | Bile salts exporter | Hepatocyte canalicular (apical) membrane | PFIC2, BRIC2 |
| MDR3 | ABCB4 | 7q21 | Phosphatidylcholine “floppase” | Hepatocyte canalicular (apical) membrane | PFIC3, ICP |
| MRP2 (cMOAT) | ABCC2 | 10q24 | Anionic transporter | Hepatocyte, enterocyte, kidney tubular epithelium: apical membranes | Dubin-Johnson syndrome |
| OATP1B1 and OATP1B3 | SLCO1B1 and SLCO1B3 | 12p | Organic anion (bilirubin glucuronide) transporter | Hepatocyte sinusoidal membrane | Rotor syndrome |
| CFTR | CFTR | 7q31.2 | Chloride ion transporter | Bronchial epithelium, enterocyte, bile duct epithelium, pancreatic ductal epithelium: apical membranes | Cystic fibrosis |
| IBST | SLC10A2 | 13q33 | Bile salt/sodium transporter | Cholangiocytes, intestine: apical membranes | PBAM |
| ATP7B | ATP7B | 13q14.3 | Copper transporter | Hepatocyte endoplasmic reticulum | Wilson disease |
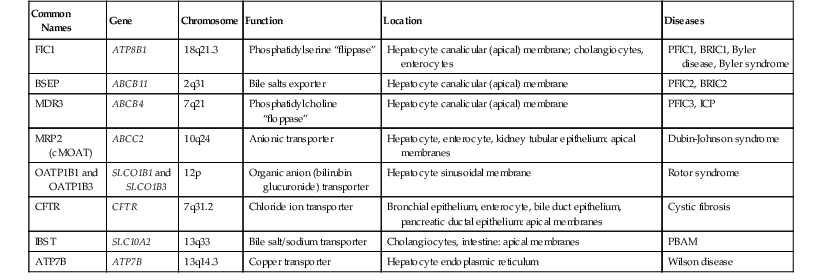
ATP7B, ATPase, Cu++ transporting, beta polypeptide; BRIC, benign recurrent intrahepatic cholestasis; BSEP, bile salt export pump; CFTR, cystic fibrosis transmembrane conductance regulator; cMOAT, canalicular multiple organic anion transporter; FIC, familial intrahepatic cholestasis; IBST, intestinal bile salt transporter; ICP, intrahepatic cholestasis of pregnancy; MRP, multidrug resistance protein; OATP, organic anion transport protein, B; PBAM, primary bile acid malabsorption; PFIC, progressive familial intrahepatic cholestasis.
Progressive Familial Intrahepatic Cholestasis 3
Progressive familial intrahepatic cholestasis 3 (also called PFIC3, ABCB4 disease, or MDR3 disease) is caused by mutations in the ABCB4 gene located on chromosome 7q21, which codes for MDR3.161 Mutations in ABCB4 have also been linked to intrahepatic cholestasis of pregnancy.162 MDR3 is a “floppase” in the canalicular membrane of hepatocytes that translocates phosphatidylcholine to the outer leaflet of the membrane, from which it is extruded by bile salts. Under normal conditions, extruded phosphatidylcholine complexes with bile salts to form mixed micelles, thereby protecting the biliary epithelium from the detergent action of secreted bile salts. In the absence of secreted phosphatidylcholine, the biliary epithelium becomes damaged and GGT is released into the systemic circulation. Markedly elevated serum GGT levels distinguish PFIC3 from PFIC1 and PFIC2, both of which exhibit normal serum levels of GGT.163
Similar to PFIC1 and PFIC2, PFIC3 manifests clinically as jaundice, pruritus, hepatomegaly, and elevated serum liver enzymes. Unlike PFIC1 and PFIC2, the age at onset is variable, ranging from 1 month to 20 years. The liver disease is progressive and eventually leads to the development of fibrosis and cirrhosis.164
The liver in PFIC3 exhibits extensive bile ductular proliferation, portal inflammation, and fibrosis165 (Fig. 54.18). These findings are nonspecific and may be confused with extrahepatic biliary obstruction.
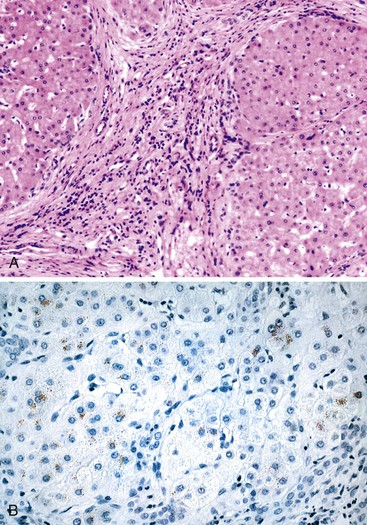
Differential Diagnosis
Clinically, PFIC1 or PFIC2 should be suspected in any neonate or infant who develops progressive symptomatic cholestasis, elevated serum bile acid levels, and low to normal serum GGT levels develops. On liver biopsy, bland intracanalicular cholestasis is more often seen in PFIC1 than in PFIC2; the latter more commonly reveals a neonatal hepatitis pattern. Ultrastructural analysis may show Byler bile in PFIC1. Several rare disorders, including inborn errors of bile acid synthesis (described earlier), familial hypercholanemia, and ARC syndrome (discussed in the next section) also manifest with low-GGT cholestasis and should be considered in the differential diagnosis. PFIC3 is characterized by elevated serum levels of GGT.
Natural History
Patients with PFIC1 or PFIC2 develop cholestasis as neonates or infants. Liver damage is progressive and leads to cirrhosis during the course of several years. Death caused by liver failure is common within the first or second decade of life. PFIC3 has a more variable time of presentation but eventually results in cirrhosis. The development of HCC and cholangiocarcinoma has been reported in patients with PFIC2. It appears that protein-truncating mutations have a greater risk of leading to malignancy than missense mutations.158
Liver transplantation resolves the cholestatic phenotype in these disorders, but extrahepatic symptoms, such as diarrhea and failure to thrive, remain.166 At least four patients with BSEP deficiency have been reported to develop autoantibodies to BSEP after liver transplantation, presumably because of lack of immune tolerance mediated by self-antigen exposure during development. Such antibodies have caused recurrence of cholestasis by targeting the graft BSEP protein. Although several such patients with recurrent BSEP disease after transplantation have undergone repeat transplantation, it appears that the antibody response may be effectively controlled by altering the immunosuppressive regimen.167,168
Patients with BRIC usually have a milder course of disease characterized by intervening periods of normal liver function. However, some patients have developed permanent cholestasis later in life.160
Arthrogryposis, Renal Dysfunction, and Cholestasis Syndrome
Arthrogryposis, renal dysfunction, and cholestasis (ARC) syndrome is a rare, autosomal recessive, multisystemic disorder characterized by germline mutations in two endosomal vesicular trafficking proteins, VPS33B and VIPAS39 (also called VIPAR).169,170 Affected individuals have low-GGT cholestatic jaundice in the newborn period. Liver biopsy shows bland canalicular and hepatocyte cholestasis with multinucleated hepatocytes. Biopsy in these individuals is associated with a significant risk of hemorrhage, presumably because of an associated platelet storage pool defect.169 Therefore, molecular analyses are extremely important to confirm the diagnosis. Recognition of this syndrome in the absence of arthrogryposis is challenging.171
Familial Hypercholanemia
Familial hypercholanemia (FHCA) is a low-GGT cholestatic disorder that is characterized by elevated serum bile acids and fat malabsorption. FHCA is caused by mutations in three genes: TJP2, BAAT, and EPHX1.172 Mutations in BAAT lead to failure of conjugation of bile acids to glycine or taurine. The inheritance pattern can be oligogenic, with genotypes of both BAAT and TJP2 influencing the phenotype.173 Liver biopsy findings may reveal only mild cholestasis. Patients usually respond to treatment with ursodeoxycholic acid.
Other extremely rare disorders that lead to defective bile acid transport and secretion and cause neonatal cholestasis include microvillus inclusion disease and the syndrome of ichthyosis, leukocyte vacuoles, alopecia, and sclerosing cholangitis (ILVASC; OMIM 607626), which is caused by mutations in the claudin1 gene, CLDN1.174 Readers are referred to other publications for details on these disorders.2
Cystic Fibrosis
CF is the most common lethal genetic disease in Caucasians, with an incidence of 1 in 2500 live births.175 This autosomal recessive disease is caused by mutations in CFTR, a protein that is important in the maintenance of fluid balance across epithelial cells.176 The basic defect lies in the production of thickened secretions in the airways and glandular ducts, which results from abnormal electrolyte transport. Organs affected in CF include the lungs, pancreas, salivary and sweat glands, biliary tree, liver, and intestines.
Clinical Features
Phenotypic expression of CF is extremely heterogeneous. Meconium ileus and failure to thrive are key features in infants. The diagnosis relies on a combination of clinical features, biochemical testing, and radiologic assessment. Chronic liver disease is one of the major complications of CF. In infants, the most common hepatic features are cholestasis and hepatomegaly. Laboratory studies in infants with liver involvement reveal elevated serum levels of AST, ALT, alkaline phosphatase, and GGT.
Pathogenesis
The genetic defect in CF resides in the gene responsible for the CFTR protein, which maintains fluid balance across epithelial cells. This cyclic adenosine monophosphate (AMP)-dependent chloride channel is responsible for transmembrane efflux of chloride ions (Cl−). Under physiologic conditions, cyclic AMP-stimulated chloride secretion through the low-conductance CFTR channels imposes a negative luminal potential and an osmotic gradient that triggers passive secretion of sodium ions (Na+) and water. The apical chloride gradient also facilitates extrusion of bicarbonate (HCO3−) into the lumen, via Cl−/HCO3− exchange. In the biliary tract, this causes biliary alkalinization, which is required for proper digestive function.
Because chloride exchange is critical for transepithelial fluid transport, all epithelia involved in the movement of water are potentially affected by defective CFTR function. In particular, the epithelia of the bronchopulmonary tree, the pancreatic ducts, the hepatobiliary duct system, and the intestines are unable to maintain proper fluid secretion. Hence, a tenacious, mucus-rich substance accumulates in the respiratory passages and in the ducts of the pancreas and liver. This results from a failure to secrete the fluid that is needed to mobilize proteinaceous secretions away from these passageways.
The CFTR protein undergoes a “life cycle”177 with several stages:
2. Endocytic retrieval from the plasma membrane to early or sorting endosomes
More than 900 mutations of the CFTR gene on chromosome 7 have been described.177 The predominant, but by no means the only, clinically significant mutation is ΔF508, which is found in 70% of patients with CF. The mutation in ΔF508 causes deletion of a phenylalanine at position 508, which results in the development of an immature protein that is not fully glycosylated. Instead, the translated peptide misfolds within the endoplasmic reticulum and is recognized by the quality control machinery of the endoplasmic reticulum. It is ubiquitinated and targeted for proteasomal degradation. Although CFTR is not the only cellular mechanism for chloride transport, failed delivery of ΔF508-CFTR to the plasma membrane leads to impaired epithelial fluid transport and, thus, to the clinical manifestations of CF. A newly described mutation in CFTR (c.3871G > T) was discovered in a 2-month-old infant in whom all signs pointed to EHBA.178
Pathologic Features
Liver involvement occurs in 20% to 50% of patients with CF, and the prevalence increases with age. Rarely, infants develop giant cell hepatitis.179 More often (in one third of infants with CF), a chronic form of biliary damage develops because of failure of adequate secretion of biliary fluid. Viscous, glycoprotein-rich bile plugs the intrahepatic biliary tree; this is the source of the historical term mucoviscidosis (Fig. 54.19). In neonates and infants, the histologic findings are typically nonspecific and include portal edema, bile ductular proliferation, inflammation, and early fibrosis. Neonatal hepatitis, paucity of intrahepatic bile ducts, and steatosis can also be present.
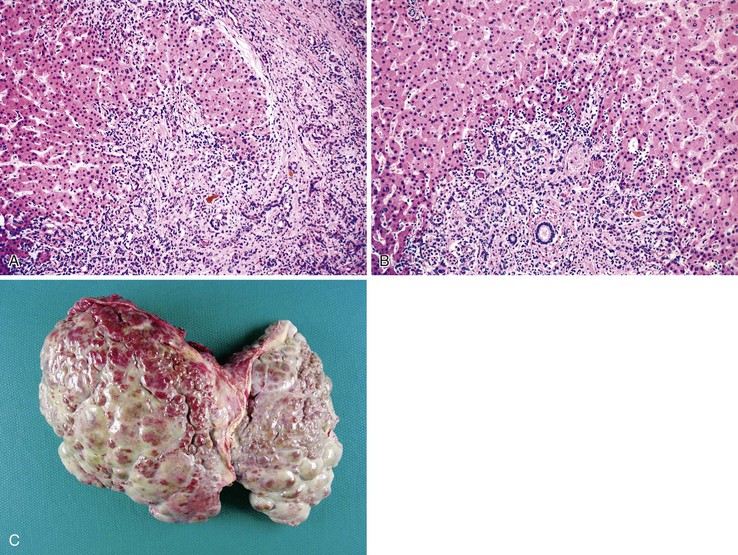
Hypoplasia of the extrahepatic bile ducts has been described in CF, which, together with the presence of bile ductular proliferation, may be confused with EHBA. With time, chronic biliary plugging leads to inflammation and portal tract scarring. Expanded portal tracts contain bile ducts filled with inspissated granular eosinophilic material, bile ductular proliferation, cholangitis, fibrosis, and chronic inflammation. Focal biliary fibrosis, the classic lesion of CF, may develop by adolescence.175
Natural History
Median survival time in patients with CF now exceeds 30 years. With improvement in therapy and long-term care of these patients, liver complications have emerged as a significant medical issue.180,181 Liver disease is now considered the third most common cause of death, after cardiorespiratory and lung transplantation complications. It accounts for approximately 2% of the overall mortality rate in CF. Oral administration of ursodeoxycholic acid stimulates secretion of bile and is free of serious side effects. Gene therapy and exogenous administration of nucleotide triphosphate are therapeutic approaches currently in the experimental phase.
Aagenaes Syndrome
Aagenaes syndrome, also referred to as cholestasis-lymphedema syndrome, is a rare autosomal recessive disorder that clinically manifests as intermittent cholestasis and lower limb edema.182 The genetic locus has been mapped to chromosome 15q, but the molecular defect is unknown.183 It was reported initially in a Norwegian kindred but has also been described in other ethnic groups. A neonatal hepatitis syndrome that evolves slowly into a chronic cholestatic condition is characteristic. There may be localized lower limb lymphedema or more subtle generalized edema despite normal serum albumin levels. These lymphatic abnormalities manifest clinically later than in neonatal jaundice. This cholestatic condition also shows clinical features of pruritus and deficiencies in fat-soluble vitamins.