[level-membership-for-pediatrics-category]
Chapter 399 Inherited Disorders of Surfactant Metabolism
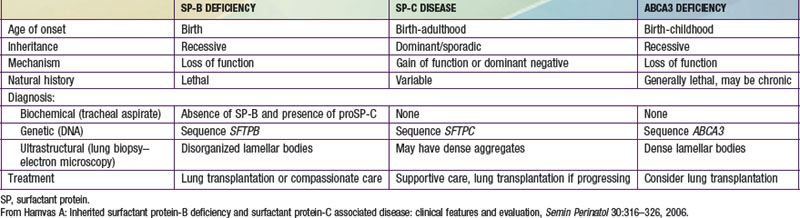
Pulmonary surfactant is a mixture of phospholipids and proteins synthesized, packaged, and secreted by type II pneumocytes that line the distal air spaces. This mixture forms a monolayer at the air-liquid interface that lowers surface tension at end-expiration of the respiratory cycle and thereby prevents atelectasis and ventilation-perfusion mismatch. Four surfactant-associated proteins have been described: surfactant proteins A and D (SP-A, SP-D) participate in host defense in the lung, whereas surfactant proteins B and C (SP-B, SP-C) contribute to the surface tension–lowering activity of the pulmonary surfactant. The ATP (adenosine triphosphate) binding cassette protein member A3, ABCA3, is a transporter located on the limiting membrane of lamellar bodies, the storage organelle for surfactant within alveolar type II cells, and has an essential role in surfactant phospholipids metabolism. Two genes for SP-A [SFTPA1, SFTPA2] and one gene for SP-D [SFTPD] are located on human chromosome 10, whereas single genes encode SP-B [SFTPB] and SP-C [SFTPC] and ABCA3 [ABCA3], which are located on human chromosomes 2, 8, and 16, respectively. Genetically engineered mice deficient in SP-A or SP-D are susceptible to viral and bacterial infections, and lineages deficient in SP-D accumulate lipids and foamy macrophages in their lungs and demonstrate emphysema as they age. Although inherited deficiencies of SP-D have not been identified in humans, mutations in the gene encoding SP-A2 may result in pulmonary fibrosis and lung cancer, although the disease did not manifest until adulthood in these cases. Inherited disorders of SP-B, SP-C, and ABCA3 have been identified in humans (see Table 399-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com).
Deficiency of Surfactant Protein B (Surfactant Metabolism Dysfunction, Pulmonary, 1; SMDP1; OMIM #265120)
Clinical Manifestations
Infants with an inherited deficiency of SP-B present in the immediate neonatal period with respiratory failure. This autosomal recessive disorder is clinically and radiographically similar to the respiratory distress syndrome (RDS) of premature infants (Chapter 95.3) but typically affects full-term infants. The initial degree of respiratory distress is variable, but the disease is progressive and is refractory to mechanical ventilation, surfactant replacement therapy, glucocorticoid administration, and extracorporeal membrane oxygenation. SP-B deficiency has been recognized in diverse racial and ethnic groups. Almost all affected patients have died without lung transplantation, but prolonged survival is possible in cases of partial deficiency of SP-B. Although murine lineages heterozygous for SP-B deficiency are susceptible to oxidant injury and pulmonary infection, humans heterozygous for loss-of-function mutations in SFTPB are clinically normal as adults and have normal pulmonary function.
Diagnosis
A rapid, definitive diagnosis can be established with sequence analysis of SFTPB, which is available through clinical laboratories (www.genetest.org). In families in which a mutation has been previously identified, antenatal diagnosis can be established by molecular assays of DNA from chorionic villus biopsy or amniocytes, which permits advanced planning of a therapeutic regimen. Other laboratory tests are investigational; they include analysis of tracheal effluent by enzyme-linked immunosorbent assay or Western blotting for the presence or absence of SP-B protein and for aberrantly processed precursor proSP-C peptides that have been found in SP-B–deficient human infants and animals. Immunostaining of lung biopsy tissue for the surfactant proteins can also support the diagnosis, although immunohistochemical assays for SP-B and SP-C are generally available only on a research basis. Staining for SP-B is usually absent, but robust extracellular staining for proSP-C due to aberrantly processed proSP-C peptides is observed and is diagnostic for SP-B deficiency. Such studies require a lung biopsy in a critically ill child but may be performed on lung blocks acquired at the time of autopsy, allowing for retrospective diagnosis.
Surfactant Protein C Gene Abnormalities (Surfactant Metabolism Dysfunction, Pulmonary, 2; SMDP2; OMIM #610913)
Disease Due to Mutations in ABCA3 (Surfactant Metabolism Dysfunction, Pulmonary, 3; SMDP3; OMIM #610920)
Brasch F, Schimanski S, Muhlfeld C, et al. Alteration of the pulmonary surfactant system in full-term infants with hereditary ABCA3 deficiency. Am J Respir Crit Care Med. 2006;174:571-580.
Bullard JE, Wert SE, Whitsett JA, et al. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. 2005;172:1026-1031.
Cameron HS, Somaschini M, Carrera P, et al. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. 2005;146:370-375.
Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120-1128.
Dunbar AE, Wert SE, Hamvas A, et al. Prolonged survival in hereditary surfactant protein B (SP-B) deficiency associated with a novel splicing mutation. Pediatr Res. 2000;48:275-282.
Garmany TH, Wambach JA, Heins HB, et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008;63:645-649.
Hamvas A, Nogee LM, Wegner DJ, et al. Inherited surfactant deficiency caused by uniparental disomy of rare mutations in the surfactant protein-B and ATP binding cassette, subfamily A, member 3 genes. J Pediatr. 2009;155:854-859.
Matsumura Y, Ban N, Inagaki N. Aberrant catalytic cycle and impaired lipid transport into intracellular vesicles in ABCA3 mutants associated with nonfatal pediatric interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2008;295:698-707.
Nogee LM, Dunbar AEIII, Wert SE, et al. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573-579.
Nogee LM, Wert SE, Proffit SA, et al. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am J Respir Crit Care Med. 2000;161:973-981.
Palomar LM, Nogee LM, Sweet SC, et al. Long-term outcomes after infant lung transplantation for surfactant protein B deficiency related to other causes of respiratory failure. J Pediatr. 2006;149:548-553.
Shulenin S, Nogee LM, Annilo T, et al. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296-1303.
Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52-59.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 399 Inherited Disorders of Surfactant Metabolism
Pulmonary surfactant is a mixture of phospholipids and proteins synthesized, packaged, and secreted by type II pneumocytes that line the distal air spaces. This mixture forms a monolayer at the air-liquid interface that lowers surface tension at end-expiration of the respiratory cycle and thereby prevents atelectasis and ventilation-perfusion mismatch. Four surfactant-associated proteins have been described: surfactant proteins A and D (SP-A, SP-D) participate in host defense in the lung, whereas surfactant proteins B and C (SP-B, SP-C) contribute to the surface tension–lowering activity of the pulmonary surfactant. The ATP (adenosine triphosphate) binding cassette protein member A3, ABCA3, is a transporter located on the limiting membrane of lamellar bodies, the storage organelle for surfactant within alveolar type II cells, and has an essential role in surfactant phospholipids metabolism. Two genes for SP-A [SFTPA1, SFTPA2] and one gene for SP-D [SFTPD] are located on human chromosome 10, whereas single genes encode SP-B [SFTPB] and SP-C [SFTPC] and ABCA3 [ABCA3], which are located on human chromosomes 2, 8, and 16, respectively. Genetically engineered mice deficient in SP-A or SP-D are susceptible to viral and bacterial infections, and lineages deficient in SP-D accumulate lipids and foamy macrophages in their lungs and demonstrate emphysema as they age. Although inherited deficiencies of SP-D have not been identified in humans, mutations in the gene encoding SP-A2 may result in pulmonary fibrosis and lung cancer, although the disease did not manifest until adulthood in these cases. Inherited disorders of SP-B, SP-C, and ABCA3 have been identified in humans (see Table 399-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com).
Deficiency of Surfactant Protein B (Surfactant Metabolism Dysfunction, Pulmonary, 1; SMDP1; OMIM #265120)
Clinical Manifestations
Infants with an inherited deficiency of SP-B present in the immediate neonatal period with respiratory failure. This autosomal recessive disorder is clinically and radiographically similar to the respiratory distress syndrome (RDS) of premature infants (Chapter 95.3) but typically affects full-term infants. The initial degree of respiratory distress is variable, but the disease is progressive and is refractory to mechanical ventilation, surfactant replacement therapy, glucocorticoid administration, and extracorporeal membrane oxygenation. SP-B deficiency has been recognized in diverse racial and ethnic groups. Almost all affected patients have died without lung transplantation, but prolonged survival is possible in cases of partial deficiency of SP-B. Although murine lineages heterozygous for SP-B deficiency are susceptible to oxidant injury and pulmonary infection, humans heterozygous for loss-of-function mutations in SFTPB are clinically normal as adults and have normal pulmonary function.
