Chapter 53B Infections of the Nervous System
Viral Encephalitis and Meningitis
Hundreds of viruses exhibit tropism for the central (CNS) and/or peripheral (PNS) nervous systems. In the case of some viruses, involvement of the CNS or PNS is the predominant feature of illness, whereas in others, involvement of the nervous system is a rare complication of a more generalized illness. Viral infection of the nervous system can result in a myriad of clinical presentations occurring separately or in combinations including acute or chronic meningitis, encephalitis, myelitis, ganglionitis, and polyradiculitis. Viruses may also incite para- or postinfectious CNS inflammatory or autoimmune syndromes such as acute disseminated encephalomyelitis (ADEM) (see Chapter 54). The neurological complications of human immunodeficiency virus (HIV) and human T-cell lymphotropic virus (HTLV) infections are discussed separately (see Chapter 53A).
Table 53B.1 lists the most common viral causes of nervous system disease in North America and the relative propensity of each virus to cause meningitis, encephalitis, postinfectious encephalomyelitis, or myelitis. Infections that occur in residents or travelers to areas outside the United States are listed in Table 53B.2. In the United States, the most common viruses causing meningitis are enteroviruses, herpes simplex virus type 2 (HSV-2), and arboviruses. The most common identified viral causes of encephalitis are herpesviruses, notably HSV-1, and arboviruses. Even with the best diagnostic efforts, up to 70% of cases of suspected viral encephalitis remain of unknown etiology (Glaser et al., 2003, 2006; Kupila et al., 2006). Worldwide, there are tens of thousands of deaths from rabies each year (Warrell and Warrell, 2004), and in Asia, 30,000 to 50,000 cases and 10,000 deaths from Japanese encephalitis virus (Solomon, 2004). In the United States, nearly 12,000 cases of West Nile virus neuroinvasive disease, with an estimated 1150 deaths, have occurred since 1999 (Davis et al., 2006; DeBiasi and Tyler, 2006; see also: http://www.cdc.gov/ncidod/dvbid/westnile/index.htm).
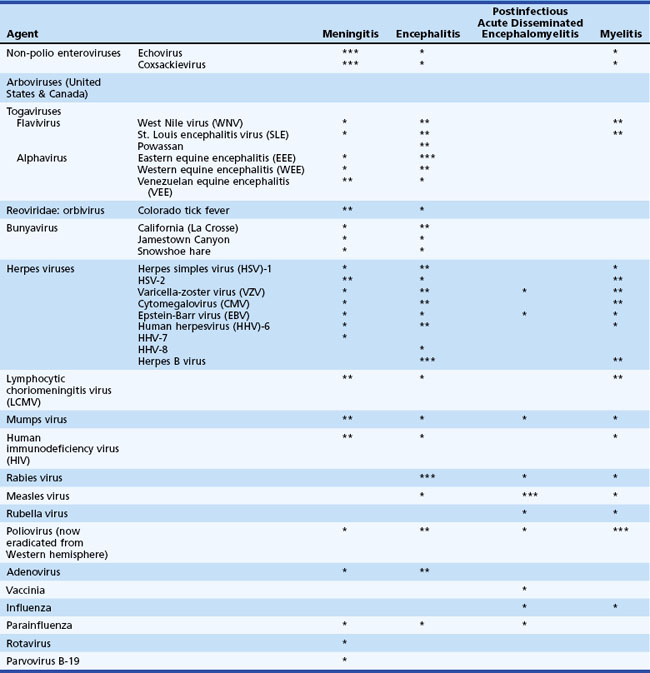
Table 53B.2 Additional Causes of Viral Nervous System Infection Resulting from Foreign Exposures
| Agent | Geographical Distribution | |
|---|---|---|
| Nipah virus | Indonesia, Singapore, India | |
| Filovirus | Ebola | Africa |
| Marburg | ||
| Arbovirus | ||
| Togavirus | ||
| Mosquito-borne | Eastern equine | Caribbean and South America (plus United States) |
| Venezuelan equine | Central and northern South America (plus United States) | |
| St. Louis | Caribbean, Central and northern South America (plus United States) | |
| Japanese B | Japan, China, Southeast Asia, India | |
| Kunjin | Australia | |
| Murray Valley | Australia and New Guinea | |
| West Nile | Africa and Middle East, parts of Europe (plus United States) | |
| Ilheus | South and Central America | |
| Rocio | Brazil | |
| Sand fly–borne | Toscana | Italy, Spain, Portugal, France |
| Tickborne complex | Far Eastern | Eastern Russia |
| Central European | Eastern and Central Europe, Scandinavia | |
| Kyasanur Forest | India | |
| Louping Ill | England, Scotland, and Northern Ireland | |
| Negishi | Japan | |
| Russian spring-summer | Eastern Europe, Asia | |
| Bunyavirus | Tahyna | Czechoslovakia, Yugoslavia, Italy, southern France |
| Inkoo | Finland | |
| Rift Valley | East Africa | |
| Rhabdovirus | Rabies | Many developing countries |
| Enterovirus | Poliovirus | Nigeria, Afghanistan, India (endemic) |
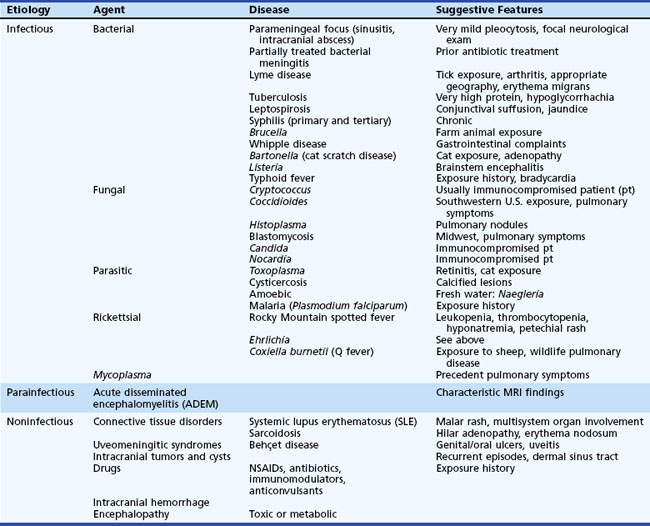
Although the basic clinical features of most types of viral meningitis and encephalitis are generally similar, specific physical examination findings may help narrow the possible viral etiologies of nervous system disease (Tables 53B.3 and 53B.4). It is important to recognize that several nonviral diseases can mimic the clinical features of viral CNS infection (Table 53B.5) (DeBiasi and Tyler, 2006). The treatment, prophylaxis, and immunotherapy of specific viral infections are summarized later in the chapter.
Table 53B.3 Skin/Mucous Membrane Findings Suggesting Specific Viral Central Nervous System Diseases
| Exanthem or Mucous Membrane Change | Viral Agent | Specific Changes |
|---|---|---|
| Vesicular eruption | Enterovirus | “Hand, foot, and mouth disease”: Macules/papules/vesicles on palms, soles, buttocks |
| Herpes simplex | Grouped small (3 mm) vesicles on an erythematous base | |
| Varicella-zoster virus | Zoster: Vesicles in dermatomal distribution Primary VZV: Multiple vesicles, papules, pustules in various stages of eruption |
|
| Maculopapular eruption | Epstein-Barr virus | Diffuse maculopapular eruption following ampicillin treatment |
| Measles | Diffuse maculopapular erythematous eruption beginning on face/chest and extending downward | |
| HHV-6 | Roseola: Diffuse maculopapular eruption following 4 days of high fever | |
| Colorado tick fever | Maculopapular rash in 50% | |
| LCMV | Occasionally occurs with lymphadenopathy | |
| WNV | Diffuse erythematous macular rash on chest and arms | |
| Erythema multiforme | (Mycoplasma) | Many types of rash |
| Confluent macular rash | Parvovirus | Confluent erythema over cheeks (“slapped cheeks”) followed by lacy reticular rash over extremities (late) |
| Purpura | Parvovirus | Rare “stocking glove” syndrome: Purpuric lesions on distal extremities |
| Pharyngitis | Enterovirus | Herpangina: Vesicles on soft palate |
| Adenovirus | Pharyngoconjunctivitis | |
| Conjunctivitis | St. Louis encephalitis | Conjunctivitis |
| Adenovirus | Conjunctivitis with pharyngitis (see above) |
HHV-6, Human herpesvirus type 6; LCMV, lymphocytic choriomeningitis virus; VZV, varicella-zoster virus; WNV, West Nile virus.
Table 53B.4 Other Specific Findings Associated with Viruses Causing Central Nervous System Disease
| Finding | Viruses |
|---|---|
| Alopecia | LCMV |
| Arthritis | LCMV, parvovirus |
| Biphasic illness | LCMV, Colorado tick fever |
| Lymphadenopathy | LCMV, mumps |
| Mastitis | Mumps |
| Mononucleosis | CMV, EBV |
| Myelitis | WNV, St. Louis encephalitis virus, VZV, herpes B virus, LCMV |
| Myocarditis/pericarditis | Enterovirus, (mumps, LCMV) |
| Orchitis/oophoritis | Mumps (LCMV, EBV) |
| Paresthesias | Colorado tick fever, LCMV, rabies |
| Parotitis | Mumps (LCMV) |
| Pneumonia | Influenza, parainfluenza |
| Retinitis | CMV |
| Tremors, myoclonus | Arbovirus (e.g., WNV) |
| Urinary problems | St. Louis encephalitis virus, VZV, herpes B virus, LCMV |
CMV, Cytomegalovirus; EBV, Epstein-Barr virus; LCMV, lymphocytic choriomeningitis virus; VZV, varicella-zoster virus; WNV, West Nile virus.
Deoxyribonucleic Acid Viruses
Herpesviruses
Herpes Simplex Viruses Type 1 and 2
Herpes Simplex Encephalitis
HSV-1 encephalitis is the most common identified cause of sporadic fatal encephalitis in the United States, accounting for approximately 10% of all cases of encephalitis and occurring with a frequency of about 1 case per 250,000 population per year (Whitley, 2006). Early recognition is important because the efficacy of the antiviral drug acyclovir (ACV) in reducing morbidity and mortality decreases as neurological disease progresses (Whitley et al., 1986). HSV-1 strains cause over 90% of cases of herpes simplex encephalitis (HSE) in adults, with the remainder due to HSV-2. Conversely, HSV-2 is a more common cause of meningitis and neonatal meningoencephalitis than HSV-1. Both HSV types 1 and 2 have been associated with myelitis.
Fever is present in about 90% and headache in about 80% of patients with biopsy or polymerase chain reaction (PCR) assay–proven HSE. Other common features include disorientation (70%), personality change (70%-85%), focal or generalized seizures (40%-67%), memory disturbance (25%-45%) motor deficit (30%-40%), and aphasia (33%) (Domingues et al., 1997; Whitley, 2006; Whitley et al., 1986).
It is important to recognize that no sets of signs or symptoms are pathognomonic of HSE, and multiple infectious and inflammatory processes can mimic HSE (Whitley, 2006; Whitley et al., 1986). Definitive diagnosis of HSE requires demonstration of viral deoxyribonucleic acid (DNA) in cerebral spinal fluid (CSF) using an HSV-specific PCR assay or isolation of virus, or detection of viral antigen or viral nucleic acid from brain tissue at biopsy or autopsy (see later discussion).
Examination of CSF is the single most important diagnostic test in suspected cases of HSE. CSF is usually under increased pressure, with a lymphocytic pleocytosis of 10 to 1000 white blood cells (WBC) per µL. Over 95% of PCR or biopsy-proven cases of HSE have a CSF pleocytosis, although rare cases with an initially normocellular CSF have been reported (Domingues et al., 1997; Whitley et al., 1986). HSE is often hemorrhagic, and both red blood cells and xanthochromia can be detected in CSF, although neither feature occurs with significantly greater frequency in HSE compared to other causes of focal encephalitis (Whitley et al., 1986). CSF protein is usually moderately elevated and glucose is normal in more than 90% of patients with HSE.
Virus cannot be cultured from CSF in over 95% of cases of HSE; however, HSV DNA can be detected in the CSF by PCR assay, and this is the diagnostic procedure of choice. Amplification of HSV DNA from CSF by PCR testing has a sensitivity of 98% and specificity of 94% for the diagnosis of HSE compared to brain biopsy (Lakeman and Whitley, 1995; Tebas et al., 1998) (Table 53B.6). HSV CSF PCR results must be interpreted in light of the pre-test probability that a patient has HSE (Bayesian decision analysis). For example, a negative test in a patient with a low prior probability of HSE (e.g., 5%) reduces the posttest likelihood of HSE to approximately 0.2%, whereas a negative PCR in a patient with a high pre-test likelihood of HSE (e.g., 60%) only reduces the posttest likelihood of HSE to around 6% (Tebas et al., 1998). CSF PCR remains a sensitive technique for the detection of HSE, even in patients who have received up to a week of antecedent antiviral therapy (Lakeman and Whitley, 1995). Despite the high overall sensitivity of CSF HSV PCR, false-negative results have been reported, most notably in patients in whom CSF was obtained within the first 72 hours of illness onset (Weil et al., 2002). For this reason, caution should be used in stopping ACV therapy in patients with suspected HSE on the sole basis of a single negative CSF PCR test obtained within 72 hours of symptom onset, unless a suitable alternative diagnosis has been established.
| Virus | Sensitivity | Specificity |
|---|---|---|
| Adenovirus | Unknown | |
| Dengue | Unknown | |
| Enterovirus | >95% | >95% |
| Herpesviruses | ||
| CMV | 100% in immunocompromised | |
| >60% in congenital CMV infection | ||
| EBV | 98.5% as tumor marker in HIV patients with CNS lymphoma | 100% |
| HSV-1 and -2 | >95% | >95% |
| HHV-6 | >95% | |
| VZV | >95% | >95% |
| HIV | HIV RNA present at all stages | Excellent |
| HTLV I and II | 75% | 98.5% |
| Influenza | Unknown but > culture | Unknown |
| Japanese encephalitis virus | Unknown | |
| JC virus | 50%-90% in PML | 98% |
| LCMV | Unknown | Unknown |
| Mumps | Unknown | Unknown |
| Measles | Unknown | Unknown |
| Parvovirus B-19 | Unknown | Unknown |
| Rabies | 100% | Unknown |
| WNV | 70% | Unknown |
CMV, Cytomegalovirus; EBV, Epstein-Barr virus; HHV, human herpesvirus; HSV, herpes simplex virus; HTLV, human t-cell lymphotropic virus; LCMV, lymphocytic choriomeningitis virus; PCR, polymerase chain reaction; PML, progressive multifocal leukoencephalopathy; VZV, varicella-zoster virus; WNV, West Nile virus.
When definitive diagnosis of HSE was dependent on brain biopsy, patients often had significantly depressed levels of consciousness and advanced neurological disease at the time of diagnosis. With routine use of HSV CSF PCR instead of brain biopsy for definitive diagnosis of HSE, patients are often identified at an earlier stage of illness, exhibit less depression of consciousness, and have improved response to therapy. Over 75% of cases with PCR-proven HSE had a Glasgow Coma Score (GCS) above 12 (Domingues et al., 1997), whereas only 30% of cases with biopsy-proven HSE had a GCS above 10 in the Collaborative Antiviral Study Group (CASG) trial of vidarabine versus ACV therapy (Whitley et al., 1986).
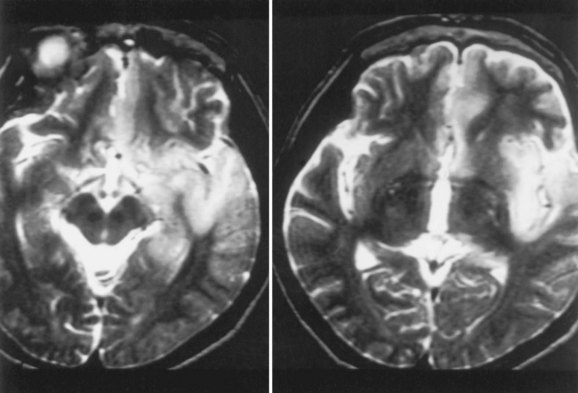
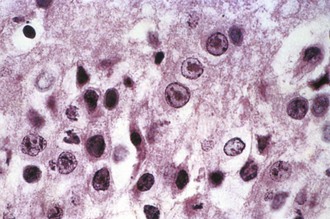
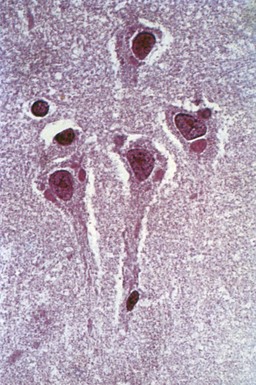
Magnetic resonance imaging (MRI) is significantly more sensitive than computed tomography (CT) and is the neuroimaging procedure of choice in patients with suspected HSE. Approximately 90% of patients with PCR-proven HSE will have MRI abnormalities involving the temporal lobes (Domingues et al., 1997; Raschilas et al., 2002) (Fig. 53B.1). The electroencephalogram (EEG) may be abnormal early in the course of disease, demonstrating diffuse slowing, focal abnormalities in the temporal regions, or periodic lateralizing epileptiform discharges (PLEDs). EEG abnormalities involving the temporal lobes are seen in approximately 75% of patients with PCR-proven HSE (Domingues et al., 1997). Brain biopsy is now only rarely performed for diagnosis of HSE. Biopsy is reserved for atypical cases in which the diagnosis remains in question for those who respond poorly to treatment. Biopsy specimens from patients with HSE show hemorrhagic necrosis, HSV antigen in infected cells, and accumulations of viral particles forming acidophilic intranuclear inclusion bodies in neurons (Cowdry type A inclusions) (Fig. 53B.2).
Because of its safety, empirical therapy with ACV should be started immediately in acute cases of focal encephalitis of suspected viral etiology (Table 53B.7). Mortality in untreated cases of HSE is around 70%, but this is reduced to 19% to 28% in patients treated with ACV (Whitley et al., 1986). Morbidity due to HSV-1 encephalitis remains high even in patients receiving ACV, with only 37.5% of all patients surviving with no or only mild deficits (Whitley et al., 1986). However, in specific subpopulations of ACV-treated HSE patients, prognosis can be considerably better. For example, 50% (12/24) of patients in whom treatment with ACV was initiated when their GCS exceeded 6 survived with no or only minor sequelae, and more than 60% who were younger than age 30 and had a GCS above 6 survived with no or only minor sequelae (Whitley et al., 1986). Outcomes have been generally similar in cases of PCR-proven HSE treated with ACV, with 37% to 56% of patients returning to normal functional status or having only mild disability or residual sequelae at 6 months follow-up (Domingues et al., 1997; Raschilas et al., 2002). Survival and prognosis are influenced by several factors including level of consciousness at initiation of therapy (e.g., GCS), patient age, and duration of disease before therapy (Marton et al., 1996; Raschilas et al., 2002; Whitley et al., 1986). In the CASG trial, all (9/9) patients treated with ACV within 4 days of onset of fever, headache, and focal neurological deficits survived, whereas the mortality was 35% in those in whom ACV treatment was initiated when disease was more than 4 days old (Whitley et al., 1986). In another study, the recovery rate was 50% in patients treated within 5 days of illness onset (Marton et al., 1996). In a third study of PCR-proven HSE, 75% of patients with an ultimately favorable outcome had received ACV therapy within 2 days of hospital admission compared to only 30% of those with a poor outcome (Raschilas et al., 2002).
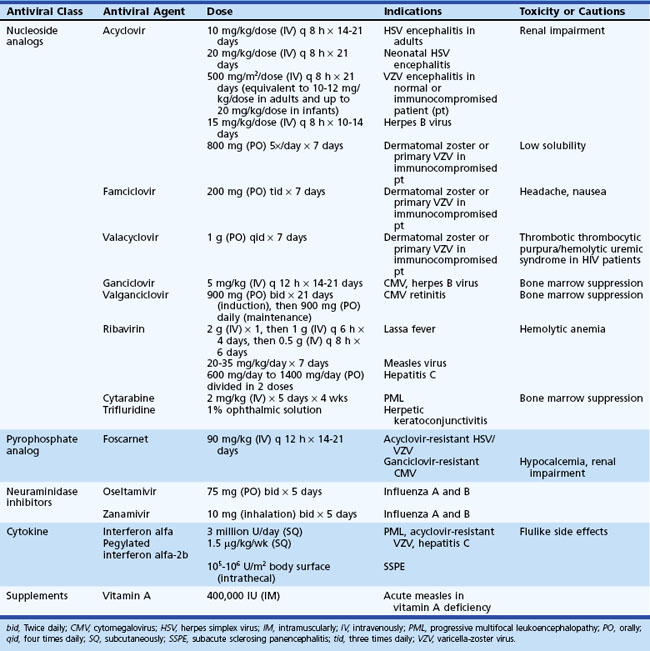
The standard adult dose of acyclovir for HSE is 10 mg/kg, given intravenously (IV) every 8 hours (20 mg/kg every 8 hours in neonates and children) for 14 to 21 days. Renal insufficiency is an infrequent, usually reversible, side effect of ACV therapy. ACV dosing should be adjusted appropriately in patients with renal insufficiency. There are no reliable data on the efficacy of oral antiviral drugs in treatment of HSE, so their use should be avoided. Retrospective studies have suggested that there may be some benefit in adding corticosteroids to ACV treatment (Kamei et al., 2006), although controlled clinical trials are needed before the role of steroid therapy as an adjunct to ACV can be definitively established. Foscarnet is an alternative therapy for patients with suspected or proven ACV-resistant strains or with allergy to ACV.
Neonatal Herpes Simplex Virus Meningoencephalitis
In contradistinction to adults, in whom HSE is usually caused by HSV-1, HSV-2 is the most common causal agent of meningoencephalitis in neonates (although HSV-1 disease may also occur). Neonates who acquire HSV from the birth canal develop infection of the CNS in 50% of cases (Kimberlin, 2005). CNS disease occurs as either a component of an overwhelming sepsis-like disseminated disease with multiorgan involvement (in the first week of life) or as isolated CNS disease that usually presents later (weeks 2-10 of life), with or without accompanying vesicular skin, mucous membrane, or conjunctival lesions (skin, eye, mouth disease). The presence of vesicular skin or mucosal lesions in an infant of this age, even in the absence of fever or systemic symptoms, warrants immediate evaluation of CSF for HSV infection, because up to 30% of infants with presumed isolated skin, eye, and mouth disease are subsequently identified as having CNS involvement. Neonates with possible HSV disease should be treated empirically with IV acyclovir, 20 mg/kg, every 8 hours. Treatment should be continued for 14 days in HSV-infected infants with isolated skin, eye, and mouth disease and 21 days in infants with sepsis or CNS involvement. Relapses of skin, eye, and mouth disease (with the potential for CNS involvement and subsequent neurological deficits) are common in the first year of life following neonatal HSV disease. For this reason, the use of oral ACV prophylaxis for 3 to 6 months following IV therapy is advised by some experts (Kimberlin, 2005).
Herpes Simplex Virus Meningitis
HSV meningitis accounts for approximately 10% of acute viral meningitis in adults. At the time of their first episode of genital herpes (generally due to HSV-2), approximately 36% of women and 11% of men have symptoms of meningitis including fever, headache, and nuchal rigidity. Of these patients, 20% will develop recurrent episodes of meningitis. Patients with HSV-2 meningitis only rarely have concomitant genital lesions, and in one study more than 80% had no history of genital lesions and no active genital lesions at presentation (O’Sullivan et al., 2003). CSF viral cultures are invariably negative during recurrent episodes of meningitis, although the virus may be isolated during the first (primary) episode. Other neurological complications such as paresthesias, urinary retention, and transverse myelitis have been described with HSV-2 infections.
CSF PCR has identified HSV-2 as the primary etiological agent in patients with benign recurrent lymphocytic meningitis (Mollaret meningitis) (Shalabi and Whitley, 2006; Tedder et al., 1994). This syndrome may be associated with the presence in the CSF of large cells of monocyte-macrophage lineage (Mollaret cells).
Varicella-Zoster Virus
Primary Infection
At least 95% of the adult population has been infected with VZV, and the lifetime risk of developing shingles is 3% to 5%. VZV can involve virtually every part of the CNS and PNS (Gilden, 2004; Gilden et al., 2000, 2005; Kleinschmidt-DeMasters and Gilden, 2001). Primary VZV may produce meningitis or encephalitis in immunocompromised patients. An acute self-limited cerebellar ataxia occurs in about 1 in 4000 children (<15 years of age) during or immediately following primary VZV infection (chickenpox). Postinfectious encephalomyelitis follows an estimated 1 in 2500 cases of primary VZV infection.
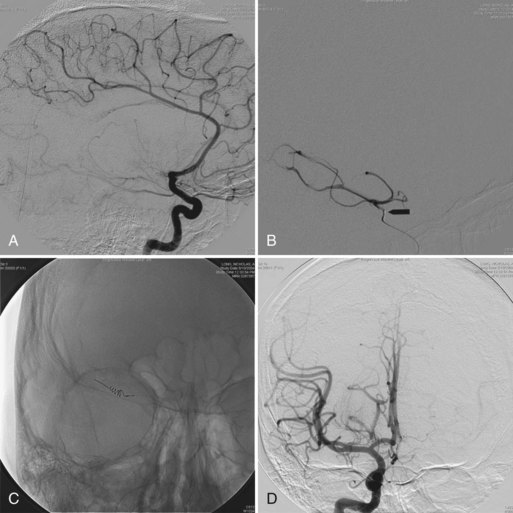
Reactivation of herpes zoster can produce meningitis, myelitis, and encephalitis in older children and adults. Zoster “encephalitis” can occur in both immunocompetent and immunocompromised hosts and typically results from reactivation of herpes zoster decades after an initial infection with varicella (chickenpox). VZV encephalitis is in fact a vasculopathy rather than a true encephalitis (Gilden, 2004; Gilden et al., 2000, 2005; Kleinschmidt-DeMasters and Gilden, 2001). In older (>60 years) immunocompetent adults, VZV vasculopathy (previously called granulomatous arteritis) typically presents with acute focal stroke-like deficits due to inflammatory involvement of large cerebral arteries, typically occurring in the trigeminal distribution. Most cases are monophasic, although rare cases follow a chronic or relapsing-remitting course (Gilden, 2004; Gilden et al., 2005). CSF shows a lymphocytic pleocytosis. MRI shows a large single infarct, most commonly in the carotid, middle, or anterior cerebral territory. At autopsy, viral particles, antigen, and DNA can be found in the involved artery. Diagnosis can be made by demonstration of VZV DNA in CSF by PCR or by demonstration of VZV immunoglobulin (IgM) or intrathecal synthesis of VZV IgG in CSF.
In immunocompromised individuals, VZV reactivation produces a multifocal vasculopathy predominantly involving small and medium-sized arteries, resulting in a clinical syndrome of mental status changes, focal deficits, and a CSF mononuclear pleocytosis (Gilden, 2004; Gilden et al., 2005). The typical rash of zoster may be absent. Neuroimaging shows multifocal hemorrhagic and ischemic cortical and subcortical infarcts. Diagnosis can be established by VZV CSF PCR or by demonstration of CSF VZV IgM or intrathecal VZV-specific IgG antibody synthesis.
VZV has been increasingly recognized as an important cause of viral meningitis in immunocompetent hosts even in the absence of a characteristic varicella (chickenpox) or zoster (shingles) rash (Kupila et al., 2006; Mogensen and Larsen, 2006). In one recent study, VZV accounted for 8% of viral meningitis cases, making it third in importance behind enteroviruses and HSV-2 (Kupila et al., 2006).
There are no controlled randomized clinical trials of antiviral therapy of CNS VZV infection. Intravenous acyclovir (10 mg/kg every 8 hours) for 7 to 10 days is generally recommended for treatment of immunocompromised children and adults with chickenpox. Patients with VZV CNS disease including vasculopathy should receive the combination of IV acyclovir (10-20 mg/kg or 500 mg/m2 every 8 hrs) for a minimum of 7 (Gilden, 2004) to 14 (Steiner et al., 2005) days, combined with a steroid pulse for 3 to 5 days (e.g., prednisone 60-80 mg/day) (Gilden, 2004; Steiner et al., 2005). HIV-infected individuals with localized (nondisseminated) dermatomal zoster can be treated with oral famciclovir (500 mg 3 times daily) or valacyclovir (1000 mg 3 times daily) for 7 to 10 days.
Herpes Zoster
Herpes zoster typically begins with pain and paresthesias in one or two adjacent spinal or cranial dermatomes (Gilden et al., 2000). Pain is followed in 3 to 4 days by a painful pruritic vesicular eruption in the area supplied by the affected root. The eruption typically lasts 10 to 14 days. Eruption most commonly occurs in the lower thoracic dermatomes but also commonly involves the trigeminal distribution and the cervical or lumbosacral dermatomes. Involvement of the first division of the trigeminal ganglion produces ophthalmic zoster and may be associated with conjunctivitis, keratitis, anterior uveitis, or iridocyclitis. Fortunately, vision loss following herpes zoster ophthalmicus is rare. Involvement of the geniculate ganglion produces otic zoster, or the Ramsay Hunt syndrome—painful facial paresis accompanied by tympanic membrane and external auditory canal vesicular rash. Herpes zoster involving cervical and thoracic levels may be associated with myelitis, and in the lumbosacral region may be accompanied by bladder dysfunction or ileus. Complications of zoster include postherpetic neuralgia, segmental motor atrophy in the affected dermatome, meningitis, myelitis, large-vessel vasculitis (usually involving the carotid or its branches on the side of zoster ophthalmicus), and multifocal leukoencephalitis or encephalitis with generalized cerebral vasculopathy (Gilden, 2004; Gilden et al., 2000, 2005).
Risk factors for postherpetic neuralgia in patients with shingles include age older than 50 years and prodromal sensory symptoms. Treatments are directed toward lessening pain, reducing virus shedding, and shortening healing time. Acyclovir (800 mg orally, 5 times daily for 7 days), famciclovir (200 mg orally, 3 times daily for 7 days), or valacyclovir (1 g orally, 4 times daily for 7 days) accelerate cutaneous healing and decreases acute zoster pain if begun within 72 hours of onset of rash. Whether these agents significantly decrease the incidence, duration, or severity of postherpetic neuralgia is uncertain. In patients without contraindications, a short course of corticosteroids (e.g., 40 mg prednisolone/day, tapered over 3 weeks) may be added to antiviral therapy. Compared with ACV therapy alone, addition of corticosteroids has been shown to improve comfort levels (pain reduction during the acute phase) following herpes zoster, although its efficacy in reducing subsequent risk of postherpetic neuralgia remains uncertain. Intrathecal methylprednisolone and oral gabapentin have been studied as treatments for postherpetic neuralgia and have been efficacious in some studies. A live attenuated varicella vaccine (Oka/Merck) was evaluated in a multicenter placebo-controlled double-blind study in over 38,000 adults older than age 60 for its capacity to reduce the incidence of herpes zoster and secondary postherpetic neuralgia. The vaccine reduced the incidence of herpes zoster by 51% and the incidence of postherpetic neuralgia in those who still developed zoster by 67% (Oxman et al., 2005).
Cytomegalovirus
CMV causes both acute and latent or persistent infections in humans, and 40% to 100% of adults are seropositive, reflecting past infection with this virus. CMV-related neurological complications include retinitis, encephalitis, polyradiculomyelopathy, neuropathy, and Guillain-Barré syndrome (Griffiths, 2004). In immunocompetent hosts, CMV may cause inapparent infection, a mononucleosis syndrome, aseptic meningitis, or the Guillain-Barré syndrome. CMV encephalitis is rare in immunocompetent hosts beyond the neonatal period and usually presents as encephalopathy, with or without focal features, in the context of a subacute febrile illness. However, immunocompromised adults and developing fetuses are at high risk of developing CNS disease due to CMV. CMV infection of peripheral nerves, nerve roots, and spinal cord, particularly in patients with AIDS, causes ascending myeloradiculitis (Miller et al., 1996).
Congenital Cytomegalovirus
CMV infection is the most common human congenital infection and can cause severe injury to the infected fetus. It has been estimated that about 1% of newborns have CMV infection, of whom some 7% have symptomatic disease. The mortality for newborns with symptomatic disease is 10% to 30%, and up to 90% of survivors will have neurological sequelae. The rate of sequelae in newborns with asymptomatic primary infection has been estimated at roughly 15% (Griffiths and McLaughlin, 2004). In addition to congenital cases, there are cases that arise in the perinatal period as a consequence of passage through an infected birth canal or following breastfeeding. Persistent high levels of viral replication in the eye and brain of the developing fetus produce encephalitis, ependymitis, and retinitis, a pattern similar to that seen in patients with opportunistic CMV infection in the setting of HIV infection. Pathologically, encephalitis occurs in a periventricular pattern and may cause polymicrogyria and hydrocephalus (Fig. 53B.3). CT scans show characteristic periventricular calcifications in 20% to 30% of children with symptomatic CMV infection. Retinitis with optic atrophy is seen in about 15% of those affected and results in characteristic hyperpigmented retinal scars. As noted, 90% of survivors of congenital CMV infection have residual neurological sequelae including psychomotor retardation, learning delays, mental retardation, seizures, optic atrophy and retinitis, and hearing loss. Mild or subclinical congenital infections may also manifest later in childhood as sensorineural deafness or developmental delay. Congenital human CMV infection is the most common, nonheritable cause of hearing loss in the United States.
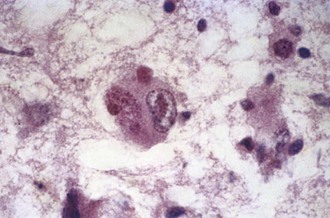
Diagnosis of congenital CMV is made by identification of CMV DNA in urine, saliva, or CSF during the immediate postnatal period. Because urinary excretion of CMV can often persist during the first year of life, isolation of virus or viral DNA from urine may be useful in later diagnosis. CMV inclusion-bearing cells are found in affected organs (Fig. 53B.4) and in stained preparations of urinary sediment and saliva. Serological studies may be difficult to interpret owing to transplacental transfer of antibody from the mother. Detection in the neonate of CSF IgM antibodies or CMV-specific intrathecal antibody synthesis is supportive of acute CNS infection.
In a study evaluating the efficacy of IV ganciclovir therapy (4-6 mg/kg every 12 hours for 6 weeks) in neonates with symptomatic congenital CMV infection, 69% of treated children, compared to only 39% of untreated children, showed improvement in brainstem auditory evoked potentials. More significantly, none of the ganciclovir-treated children, compared to 42% of those not treated, showed worsening hearing loss over the initial 6 months post infection (Kimberlin et al., 2003; Whitley et al., 1997;).
Cytomegalovirus in Immunocompromised Adults
CMV encephalitis in immunocompromised patients with acquired immunodeficiency syndrome (AIDS) or organ transplants (Griffiths, 2004; Tselis and Lavi, 2000) often presents as a nonspecific febrile encephalopathy, with or without focal features. CMV encephalitis typically occurs in AIDS patients with a CD4+ cell count of less than 50 cells/mm3, although patients with counts below 100/mm3 are at increased risk of developing CMV viremia (Griffiths, 2004). AIDS-associated CMV encephalitis presents either as microglial nodular encephalitis with acute onset of confusion and delirium, or as a more slowly progressive ventriculoencephalitis characterized by confusion and cranial nerve palsies. There is a broad pathological spectrum of CMV infection of the brain in AIDS patients, ranging from scattered microglial nodules to widespread necrotizing ependymitis and perivascular infiltrates to necrotizing leukoencephalopathy or focal necrosis deep in parenchyma.
CMV is one of the most important pathogens influencing the outcome of transplantation. In contrast to AIDS patients, allogeneic hematopoietic stem cell transplant patients develop encephalitis alone without retinitis. Risk factors for CNS disease in this group include T-cell depletion, umbilical cord blood transplantation, and a history of recurrent CMV viremia. Active graft-versus-host disease, a risk factor for a variety of late-onset infections after allogeneic stem cell transplantation, may be present (Reddy et al., 2010). Antiviral prophylaxis for prevention of primary infection or recurrence when either or both donor and recipient are seropositive is administered with oral or IV ganciclovir (1000 mg every 8 hours). In donor+/recipient− solid organ transplant, oral valganciclovir, 900 mg/day × 200 days is also used. Preemptive treatment is prevention of development of disease when reactivation has occurred. Induction is with IV ganciclovir 5 mg/kg, 2 or 3 times daily × 2 to 4 weeks, followed by a lower maintenance dose of 5 mg/kg/day × 4 weeks. CNS disease treatment is with IV ganciclovir, 5 to 7.5 mg/kg per dose 2 or 3 times daily, and the addition of IV foscarnet 90 mg/kg twice daily for refractory cases (Ljungman, 2008).
Diagnosis of CMV encephalitis may be difficult, particularly by serological testing, because CMV antibody titers can fluctuate spontaneously. Virus detection methods focus on quantitation of viral load (particularly in peripheral blood leukocytes; i.e., virus DNA in buffy coat) for patients with persistent infection, as well as to help predict which immunosuppressed patients might develop end-organ disease. CSF CMV PCR has a reported sensitivity of 82% and specificity of 99% in AIDS patients with CNS disease due to CMV (Cinque et al., 1997). In AIDS patients, in whom detection of CMV viremia by PCR may predict the development of retinitis, a wide spectrum of neuroimaging results has been reported, ranging from normal findings to detection of generalized atrophy, periventricular abnormalities, and focal discrete white-matter lesions. The most characteristic MRI finding is periventricular increased signal on T2-weighted images and ependymal enhancement following gadolinium administration on T1-weighted images. CMV polyradiculitis manifests as diffuse enhancement of cauda equina nerve roots and is often associated with concurrent CMV infection elsewhere in the body (Miller et al., 1996). Many patients with this syndrome have an almost pathognomonic CSF profile of neutrophilic pleocytosis with a low glucose concentration. The diagnosis is confirmed by PCR amplification of CMV DNA in CSF.
Ganciclovir, foscarnet, and cidofovir all have efficacy against CMV in vitro and in some clinical settings in vivo. In patients with AIDS, immune reconstitution following introduction of highly active antiretroviral therapy (HAART) may help control CMV replication and disease. General recommendations for immunocompromised patients, including those with AIDS, are initial antiviral therapy with IV ganciclovir (5 mg/kg every 12 hours for 2-3 weeks), followed by maintenance dosing with either IV ganciclovir (5 mg/kg/day, 5 days/week) or oral valganciclovir (900 mg daily) for at least an additional 4 weeks. Foscarnet should be reserved for treatment of ganciclovir-resistant CMV because of its nephrotoxicity and IV administration (Griffiths, 2004). Foscarnet dosage is 60 mg/kg IV, 3 times daily for 2-3 weeks for initial therapy, then 90 to 120 mg/kg IV daily for maintenance. Maintenance therapy should be continued in HIV patients until their CD4 count remains above 100 to 150 cells/mm3 for more than 6 months. There are limited clinical data on the use of cidofovir for treatment of CMV CNS disease (Table 53B.8).
Epstein-Barr Virus
Primary EBV infection may be asymptomatic, present as a nonspecific febrile illness, or classically, as the infectious mononucleosis syndrome with cervical lymphadenopathy, exudative pharyngitis, and splenomegaly. The pathogenesis of EBV-associated CNS disease remains uncertain because virus, viral antigen, or viral nucleic acid are only rarely isolated directly from CNS tissue in patients with encephalitis or myelitis, raising the possibility that at least some CNS manifestations may be post- or para-infectious immune-mediated phenomena. However, recent studies suggest that patients with EBV-associated neurological disease frequently have EBV DNA that can be amplified from CSF, and that this is frequently associated with amplifiable viral RNA consistent with lytic viral replication (Weinberg et al., 2002, 2005).
Significant nervous system disease occurs in less than 1% of EBV infectious mononucleosis cases and can manifest as meningitis, encephalitis, cerebellitis, transverse myelitis, optic neuritis, cranial neuropathy, Guillain-Barré syndrome, or as small-fiber sensory or autonomic neuropathy syndromes (Doja et al., 2006). The most common symptomatic EBV-associated CNS infection is meningitis. Cases of asymptomatic laboratory-defined meningitis probably vastly exceed symptomatic cases; up to 25% of patients with acute infectious mononucleosis may have a CSF pleocytosis despite the absence of signs or symptoms of meningitis. There are no unique features of EBV meningitis, although the presence of atypical lymphocytes in CSF may suggest the diagnosis. Diagnosis is typically made by amplification of EBV DNA by CSF PCR. In rare cases, EBV-specific IgM antibodies can be detected in CSF. Attempts to isolate virus from CSF are almost invariably negative. The presence of serum serologies indicative of acute infection supports the diagnosis (e.g., IgM antibodies against viral capsid antigen (VCA), presence of antibodies against early antigen [EA], but not Epstein-Barr nuclear antigen [EBNA]).
In occasional patients, EBV is associated with frank encephalitis, presenting with altered consciousness including coma, seizures, and focal neurological signs and symptoms. EEG abnormalities such as diffuse slowing are common. Cases of EBV CNS disease may occur before, during, or after infectious mononucleosis or even in its absence. In one series of 21 cases of EBV encephalitis in children (Doja et al., 2006), only one patient had classic infectious mononucleosis; the remainder had a nonspecific prodrome that included fever (81%) and headache (66%). Seizures occurred in 48%, and 57% had EEGs with a diffusely slow background. CSF pleocytosis (81%) and MRI abnormalities (71%) were common. Mortality was 10%, with 80% neurologically normal at follow-up and an additional 10% having mild deficits. Some patients have a syndrome suggestive of involvement of the brain, spinal cord, and peripheral nerves or roots (encephalomyeloradiculitis). EBV encephalitis may mimic HSE, and in the CASG studies, EBV encephalitis accounted for about 8% of the HSV-negative cases of focal encephalitis in which an etiology was established. In a registry of childhood (ages 3-17) encephalitis cases at a large children’s hospital, EBV accounted for 6% of total cases (Doja et al., 2006).
EBV myelitis can occur as an isolated syndrome in association with meningoencephalitis. Most patients have CSF mononuclear pleocytosis. Diagnosis is made by amplification of EBV DNA from CSF or by appropriate serological testing (see earlier discussion). Myelitis typically follows mononucleosis, although in some patients the symptoms of the initial infection may be mild or even absent. A variety of clinical forms have been reported and include transverse myelitis, myeloradiculitis, and a poliomyelitis-like syndrome of acute flaccid paralysis. MRI may show increased T2-weighted intramedullary signal or evidence of cord swelling. Nerve root enhancement has been reported in patients with myeloradiculitis. No controlled treatment trials are available, although isolated cases have been treated with IV ACV and ganciclovir with or without the addition of corticosteroids (Tyler, 2004).
CSF PCR for EBV is positive during the acute phase of illness in children with infectious mononucleosis and neurological complications such as transverse myelitis, meningoencephalitis, and aseptic meningitis. CSF PCR is negative in EBV-seropositive individuals in the absence of CNS infection. However, positive EBV PCR may be seen in patients with evidence of other viral or nonviral CNS infection, raising the possibility that these infections may trigger viral reactivation. EBV has been one of the most frequent agents associated with dual-positive CSF PCR testing and may not always correlate clinically with the presence of CNS infection known to be caused by this virus (Weinberg et al., 2005).
Human Herpesvirus Type 6
HHV-6 was first isolated in 1986 from human peripheral blood mononuclear cells of patients with lymphoproliferative disorders. Two variants, HHV-6A and HHV-6B are known. Primary infection with HHV-6 usually occurs during infancy, producing exanthem subitum (or roseola) or a syndrome of generalized lymphadenopathy. Primary HHV-6 infection may cause febrile seizures or acute meningoencephalitis. The role of HHV-6 in other neurological diseases remains uncertain. Cases of focal encephalitis in immunocompetent patients have been attributed to HHV-6, as has a syndrome of acute limbic encephalitis occurring in transplant patients (posttransplant acute limbic encephalitis [PTALE]) (Seeley et al., 2007; Isaacson et al., 2005; McCullers et al., 1995) (Fig. 53B.5). The role, if any, for HHV-6 in inducing medial temporal sclerosis temporal lobe epilepsy (Fotheringham et al., 2007) and as a co-factor in other neurological disorders including multiple sclerosis remains unproven.
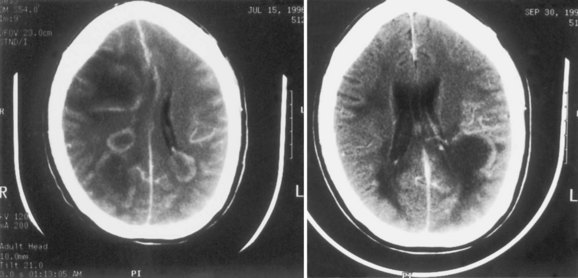
HHV-6 isolates generally resemble CMV in their susceptibility to antiviral drugs, with isolates being resistant to ACV and related drugs and sensitive to ganciclovir and foscarnet (Birnbaum et al., 2005; Seeley et al., 2007). Antiviral drugs do not inhibit the development of latency nor do they “clear” latent virus.
Herpes B Virus (Cercopithecine Herpesvirus 1)
Herpes B virus of Old World monkeys is highly pathogenic to humans, and untreated infection has an estimated 70% mortality. Ocular, oral, and genital secretions of monkeys, as well as CNS tissues and CSF, are potentially infectious. Disease is transmitted by direct contact with the virus, usually by animal bite or by virus-containing fomites. One reported fatal case of B virus infection was due to mucosal splash exposure (Cohen et al., 2002).
Polyomaviruses (JC) and Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML), a subacute demyelinating disease of the CNS, is a result of infection of oligodendrocytes by the polyomavirus, JC virus (JCV) (Koralnik et al., 2006). Seroepidemiological studies indicate that asymptomatic primary infection with JCV occurs in childhood, and that by adult life roughly 55% to 85% of the population is seropositive. Following primary infection, JCV becomes latent in sites including kidney, bone marrow, and tonsil. It is unclear whether CNS latency occurs. Reactivation in immunocompetent hosts usually takes the form of asymptomatic viruria. In the setting of impaired cell-mediated immunity, including immunosuppressive drug treatment, lymphoproliferative disorders, chronic infectious or inflammatory diseases such as tuberculosis and sarcoidosis, and most importantly AIDS, virus can reactivate to produce PML. In most modern series, over 80% of PML patients have underlying HIV infection. Treatment of patients with several immunomodulatory biologicals has been linked to increased risk of developing PML (Clifford et al., 2010; Major, 2010; Tan et al., 2010). The first reports involved patients treated for multiple sclerosis (2 cases) or Crohn disease (1 case) with natalizumab (Tysabri), a humanized monoclonal antibody that blocks lymphocyte binding to α4-integrin and inhibits trafficking into the CNS (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005). A total of about 150 additional cases of natalizumab-associated PML have now been reported (Clifford et al., 2010). The risk appears to increase with duration of therapy over the first 3 years (Clifford et al., 2010), increasing to approximately 1 in 1000 for patients treated for 25 to 36 months (Clifford et al., 2010), a rate identical to that originally estimated after the initial three cases were reported (Yousry et al., 2006). All cases reported to date for which sera is available and has been tested (n ≈ 25) were JCV antibody–positive prior to the development of disease. It remains to be determined whether assessment of JCV antibody testing prior to initiation of natalizumab therapy will aid in risk stratification. PML results from reactivation of latent JCV infection, and as such it would be expected that patients at risk for disease would be JCV seropositive, reflecting past exposure to the virus and serving as a presumptive marker for latency. Conversely, seronegative individuals may not have been infected with virus, would therefore not harbor latent virus, and would presumably not be at risk of PML.
The likely pathogenesis of PML includes reactivation from an extraneural primary site, dissemination of virus to the CNS through the bloodstream, and subsequent productive lytic infection of oligodendrocytes to induce demyelination (Koralnik et al., 2006). Infection of astrocytes is abortive, although resulting in striking and characteristic bizarre enlarged cells. Neuronal infection does not occur except in granule cell neurons of the cerebellum (Koralnik et al., 2006). Rearrangements in the regulatory region of the viral genome appear to play a key role in neuropathogenesis insofar as viruses lacking these regulatory region rearrangements have not been isolated from PML brain specimens.
CSF cell counts and protein levels are usually normal. Neuroimaging results help suggest the diagnosis. MRI studies show focal or multifocal lesions of subcortical white matter, sometimes involving the cerebellum, brainstem, and spinal cord, without mass effect or contrast enhancement. Fluid-attenuation inversion recovery (FLAIR) sequences, which remove CSF signals, are particularly good for demonstrating paraventricular disease (Fig. 53B.6). White-matter lesions are larger and more confluent than those of multifocal leukoencephalitis of VZV. Lack of CSF pleocytosis and an indolent course distinguish PML from acute disseminated encephalomyelitis. Brain biopsy establishes the diagnosis by showing characteristic changes, including oligodendrocytes with enlarged nuclei that contain inclusion bodies, as well as viral particles and antigen or viral DNA. JCV has never been cultured from CSF, but JCV DNA may be detected in CSF using PCR amplification. Finding JCV DNA in CSF by PCR in the appropriate clinical setting is diagnostic of PML and obviates the need for brain biopsy. Although the specificity of CSF PCR for JCV in the appropriate clinical setting approaches 100%, its sensitivity may be as low as 75%, reflecting variances in the amount of viral load in the CSF in different conditions. For example, 57% of Tysabri-associated PML cases were reported to have fewer than 500 copies of JCV DNA per mL, which is close to the sensitivity limit of some commercial assays (Clifford et al., 2010).
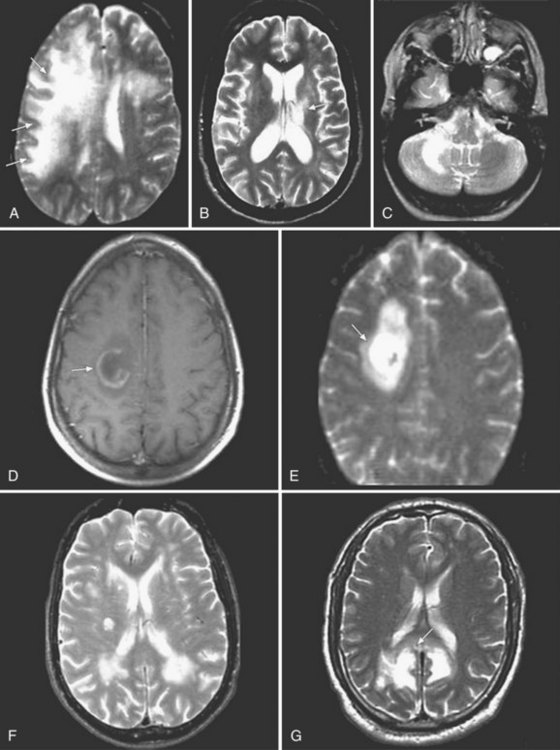
No specific therapy is available. Isolated reports of benefit from cytarabine were not confirmed in a randomized prospective clinical trial (Hall et al., 1998). Cidofovir was also found not to be of significant clinical benefit in HIV-associated PML in a prospective clinical trial (Marra et al., 2002). Interferon alfa has been reported to be of benefit in case reports but has not been tested in clinical trials. A study showing that JCV binds the 5HT receptor in cultured cells led to the use of mirtazapine, a serotonin receptor blocker, in patients with PML (Elphick et al., 2004). However, 12-month follow-up in patients with PML treated with mirtazapine showed no evidence of improved survival (Marzocchetti et al., 2009). A screen of chemical compounds found that mefloquine, a malarial drug, inhibits JCV replication. A multicenter clinical trial investigating the role of mefloquine treatment of JCV in PML patients did not show significant clinical benefit (clinicaltrials.gov identifier NCT00746941) (Brickelmaier et al., 2009). Improvement of the immune deficiency with HAART may lead to regression or stabilization of PML lesions.
Adenovirus
Adenoviruses cause acute respiratory disease in children and military recruits, conjunctivitis, hemorrhagic cystitis, and gastroenteritis. Meningoencephalitis or unilateral deafness coincident with nasopharyngeal infection are rare complications in normal hosts. Immunosuppression is a risk factor for severe encephalitis, with fatal meningoencephalitis reported in AIDS and bone marrow transplant patients. Clinical and histopathological features of adenovirus disease may resemble those of CMV disease. Adenovirus encephalitis and ependymitis was reported in a child with AIDS (Anders et al., 1990). Diagnosis is by isolation of virus from extraneural sites or by serology. Diagnosis of adenoviral infection is complicated by the existence of 51 viral serotypes; serotypes 2, 3, 5, 6, 7, 26, and 31 in mixture with 49 have been implicated in CNS infection.
Orthopoxviruses
Monkeypox
Monkeypox represents a newly emergent poxvirus zoonosis in North America. In June 2003, an outbreak of human monkeypox transmitted from infected exotic pets was identified in the midwestern United States. The source of the U.S. outbreak was traced to native prairie dogs housed with pets from Africa. The index animal source was suspected to be imported wild rodents from Africa, where the disease is endemic. Human monkeypox was first identified in Zaire, now the Democratic Republic of Congo, in 1970 toward the end of smallpox eradication efforts in Africa. In western and central African countries, it continues to produce sporadic cases clinically similar to smallpox. The 37 confirmed human infections in the June 2003 outbreak represented the first time human monkeypox infection was documented in the Western Hemisphere. Most cases were associated with a mild, self-limited febrile rash illness. However, one 6-year-old developed fever, rash, encephalitis, and seizures, with CSF pleocytosis, normal protein and glucose, edema, meningeal enhancement, and multifocal gray- and white-matter abnormalities on MRI. Monkeypox-specific IgM antibodies were present in serum and CSF. She made a full recovery. Diagnosis is by serum and CSF IgM and IgG, skin lesion viral culture, PCR, and immunohistochemistry (IHC) testing. Viral cultures and PCR in CSF were negative. Management is strict isolation and supportive care. Cidofovir, other selective antipoxvirus drugs, and vaccinia immune globulin are investigational treatments. Smallpox vaccine protects against monkeypox both by preventing disease and by decreasing disease severity (Sejvar et al., 2004). For both monkeypox and smallpox, vaccination within 3 days of exposure lessens the severity of symptoms in the majority of individuals.
Smallpox and Smallpox Vaccination
There are two forms of smallpox, variola major and variola minor, with differing outcomes. Mortality from variola major has been estimated at 30%, and from variola minor approximately 1%. Neurological complications of acute smallpox are uncommon but include stupor, coma, seizure, headache, hemiparesis, incontinence, or flaccid paralysis (Boos and Davis, 2003).
Compulsory vaccination was discontinued in U.S. civilians in 1973 and among military personnel in the late 1980s. The world was declared free of smallpox infection in 1980, but the United States reinstated vaccination against smallpox among military personnel in 2002 and among selected civilian groups in January 2003. Vaccination against smallpox is by inoculation with a preparation of vaccinia, a relatively benign orthopoxvirus. ACAM2000 is replacing Dryvax for use in the United States. Additional vaccines are in the Centers for Disease Control and Prevention (CDC) Strategic National Stockpile. Historically, routine smallpox vaccinations have been 90% effective in preventing smallpox and more than 99% effective in preventing fatalities (Sejvar et al., 2005).
A major complication of vaccination has been postvaccinal encephalomyelitis (PVEM). PVEM is more frequent after primary vaccination but has no predictor or marker. After primary vaccination, reported rates vary from 1 in 4000 to 1 in 80,000, and after revaccination, from 1 in 50,000 to 1 in 450,000. Neurological complications, particularly PVEM, accounted for the majority of vaccination-related deaths; PVEM fatality rates ranged from 10% to 50%. There are two forms of nervous system complications: an acute encephalopathy with brain swelling in infants and a perivenular demyelinating disease (ADEM) in vaccinees older than 2 years of age. Virus has been isolated from brain or CSF of some cases of PVEM. Other neurological syndromes temporally associated with smallpox vaccination include Guillain-Barré syndrome, Bell palsy or other cranial neuropathies, a poliomyelitis-like syndrome, and transverse myelitis. It is hoped that newer vaccines produced in cell culture will provide desired immunity with fewer complications (Johnson, 2003).
Administration of antivaccinia gamma globulin at the time of vaccination has been shown to reduce occurrence of PVEM, but there is no evidence that immunoglobulin treatment is effective in PVEM or other neurological complication after their onset. Evidence regarding safety of vaccination of individuals with known neurological illness has been limited (summarized in Abrahams and Kaufman, 2004).
Ribonucleic Acid Viruses
Poliovirus and Other Non-polio Enteroviruses
Poliovirus
Approximately one-quarter of polio patients develop an exacerbation of existing or new progressive lower motor neuron weakness 30 to 40 years after acute polio (“the post-polio syndrome”). Atrophy, fasciculations, and electromyographic evidence of active denervation are found in involved muscle groups. Diagnosis depends on the documentation of antecedent poliomyelitis and the exclusion of other new causes of lower motor neuron syndromes. Electrophysiology suggests that new weakness likely results from ongoing denervation that exceeds compensatory reinnervation. The exact cause of the syndrome remains uncertain; both immune-mediated processes and chronic persistent viral infection have been speculated to be potential triggers. No specific therapy has been shown to be of proven benefit, although patients may respond to a carefully designed program of physical therapy and the use of orthoses and assistive devices (Gonzalez et al., 2010).
Non-polio Enteroviruses
Meningitis
Non-polio enteroviruses are the most common cause of viral meningitis in adult and pediatric populations (Kupila et al., 2006), with over 75,000 cases of EV meningitis in the United States each year. Worldwide, the strains most commonly isolated in aseptic meningitis are coxsackie A9, B3-5, and echovirus 4, 6, 7, 8, 11, 18, and 30. Spread of infection is by fecal-oral and (rarely) respiratory routes. Outbreaks tend to cluster in the late summer and early fall and may be associated with pharyngitis and gastrointestinal symptoms such as anorexia, vomiting, or diarrhea. Other associated findings are the exanthem of herpangina or the rash of hand-foot-and-mouth disease (HFMD). Fortunately, EV meningitis occurring beyond the neonatal period in immunocompetent hosts is only rarely associated with severe disease or subsequent neurological deficits (Sawyer, 2002).
Meningitis caused by coxsackievirus produces CSF cell counts typically up to 250 WBC/µL with 10% to 50% polymorphonuclear cells. Echovirus infections are associated with CSF pleocytosis from several hundred to greater than 1000 WBC/µL, 90% of which may be polymorphonucleocytes in the first 24 hours of infection. Amplification of EV RNA from CSF by reverse transcriptase (RT)-PCR has now replaced viral culture as the diagnostic procedure of choice for enteroviral CNS infections (Ramers et al., 2000). PCR primers and probes are directed against the 5′ nontranslated region of the viral genome, which is highly conserved among almost all enteroviral strains. Currently available commercial enteroviral RT-PCR assays on CSF have better than 95% sensitivity and nearly 100% specificity for amplification of RNA from known strains of EV (Kost et al., 2007; Pillet et al., 2010).
Arboviruses
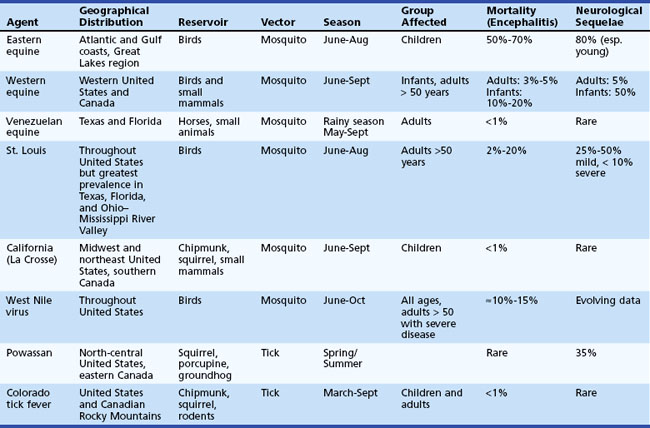
The term arbovirus (arthropod-borne virus) is a general term for viruses transmitted to humans by mosquito and tick (arthropod) vectors. Arboviruses exist in nature in complex cycles involving birds and mammals that serve as viral reservoirs and amplifying hosts. When transmitted to humans, arboviruses can cause fever, headache, meningitis, and encephalitis. Arboviruses comprise a group of over 500 RNA viruses, of which more than 100 are known to infect humans. The three taxonomic families into which arboviruses are divided are togaviruses (subdivided into flaviviruses and alphaviruses), reoviruses, and bunyaviruses. Considered together, arboviruses represent the leading cause of encephalitis worldwide. Key features of arboviral infections occurring in North America are summarized in Table 53B.9.
West Nile Virus (Flavivirus)
West Nile virus (WNV) was already one of the world’s most widely distributed arboviruses, present in many parts of Africa, West Asia, the Middle East, Eastern Europe, and Australia, when it emerged in North America in 1999. A reported 59 people were hospitalized with encephalitis or meningitis in late summer in the New York City metropolitan area, with 7 resultant fatalities (Nash et al., 2001). WNV has subsequently become the most important cause of arboviral meningitis, encephalitis, and acute flaccid paralysis in the continental United States (Bode et al., 2006; Davis et al., 2006; DeBiasi and Tyler, 2006).
Transmission occurs overwhelmingly as the result of the bite of an infected mosquito; however, person-to-person transmission through organ transplantation, blood and blood product transfusion, and intrauterine spread can occur. WNV is maintained and amplified in an enzootic transmission cycle with Culex mosquitoes and passeriform birds, with occasional spillover transmission to horses or humans who serve as dead-end hosts (Weaver et al., 2010). Since its emergence in 1999, the virus has rapidly spread across the United States and has emerged as a continuing and major cause of meningitis, encephalitis, and acute flaccid paralysis, resulting in over12,000 cases of neuroinvasive disease since 2001. After reaching a seasonal peak of nearly 3000 cases in 2003, the number of confirmed cases of neuroinvasive disease stabilized in the 900 to 1500 cases/yr range from 2004-7, but then declined dramatically to fewer than 700 cases in 2008 and fewer than 400 in 2009 before rebounding to over 600 cases in 2010. The incidence of disease in the United States remains greatest in the central part of the country, where it can exceed 100 cases per million population per year in specific areas. The epidemiology of WNV, including the case count and geographic distribution at county level resolution, can be followed in near real-time on the CDC’s WNV homepage (http://www.cdc.gov/ncidod/dvbid/westnile/index.htm).
Most human WNV infections are asymptomatic. Following an incubation period of 3 to 14 days, around 20% of infected individuals develop a nonspecific febrile illness (West Nile fever [WNF]). Approximately 1 in 150 infected persons will develop encephalitis and/or meningitis (Davis et al., 2006; DeBiasi and Tyler, 2006). Older individuals (>50 years of age) are at increased risk of developing encephalitis, although meningitis and acute flaccid paralysis occur in middle-aged and younger individuals. Neurological disease is rare in infants and children but does occasionally occur. Clinical features suggestive of WNV CNS infection include the presence of movement disorders (e.g., tremor, myoclonus, parkinsonism) and severe weakness, often of a lower motor neuron type (flaccid tone, reduced or absent reflexes) (Bode et al., 2006; Davis et al., 2006; DeBiasi and Tyler, 2006). In many cases, weakness appears to result from injury to spinal cord motor neurons (a true “poliomyelitis”). In patients with weakness, electromyography (EMG) and nerve conduction velocity (NCV) studies are often consistent with injury to motor neurons and anterior roots, including reduced amplitudes of compound motor action potentials (CMAPs) with relatively preserved sensorineural action potentials (SNAPs). Evidence of denervation may be present on EMG studies after an appropriate interval. The case fatality among hospitalized patients, most of whom have encephalitis, is approximately 12% to 14%.
Neuroimaging studies and CSF analysis may assist in the diagnosis. In contrast to HSE, in which MRI abnormalities in the temporal and frontal lobes are the rule, about 50% of patients with WNV encephalitis have initially negative or normal studies (Petropoulou et al., 2005). When abnormalities occur, they are best detected on FLAIR or diffusion-weighted images and typically involve the basal ganglia, thalamus, upper brainstem, and cerebellum. CSF invariably shows a pleocytosis (>95% of cases), with a mean of 225 cells/mm3; 45% of meningitis and 37% of encephalitis patients have a neutrophil-predominant CSF pleocytosis (Tyler et al., 2006). The protein is typically elevated, and the glucose is almost invariably normal.
There is no therapy with proven efficacy for WNV infection in humans, and treatment of WNV neuroinvasive disease is supportive. A clinical trial studying the efficacy of passive transfer of IVIG with high-titer WNV antibodies (clinicaltrials.gov identifier NCT00069316) has been completed, but results are not yet available (Agrawal and Petersen, 2003; Davis et al., 2006; DeBiasi and Tyler, 2006). A clinical trial studying the efficacy of a humanized monoclonal antibody (MGAWN1) for the treatment of WNV infection is currently enrolling subjects (clinicaltrials.gov identifier NCT00927953). Although the titer of WNV-specific antibody in U.S. lots of IVIG is increasing (Planitzer et al., 2007), its efficacy in treatment of WNV disease is unproven. Ribavirin did not show efficacy in uncontrolled clinical use. Although it is widely used, no controlled trials are yet available of the potential efficacy of interferon alfa preparations in treatment of WNV infection. Uncontrolled case reports of the use of interferon alfa have reported both positive and negative results (Chan-Tack and Forrest, 2005; Kalil et al., 2005). Three WNV vaccines have completed phase I human clinical trials for safety and immunogenicity. Two of these are chimeric vaccines in which genes encoding WNV structural protein envelopes are inserted into the backbone of attenuated yellow-fever or dengue virus strains (see NCT00094718), and the third is a recombinant plasmid DNA vaccine (NCT00300417).
St. Louis Encephalitis Virus (Flavivirus)
St. Louis encephalitis (SLE) virus is a cause of late summer encephalitis outbreaks in North America. Like WNV, SLE virus maintains an enzootic cycle between Culex species of mosquitoes and passeriform birds, with spillover transmission to dead-end hosts such as humans during late summer months. From 1999-2007, SLE virus caused 188 confirmed cases of neuroinvasive infection in the United States, with a peak in 2001-2003 paralleling that of the WNV epidemic (Reimann et al., 2008).
No specific antiviral treatment exists for St. Louis encephalitis. In an open-label study of interferon alfa-2b (Rahal et al., 2004), 13% of treated patients, compared to 65% of historical controls, had quadriplegia, quadriparesis, or respiratory insufficiency persist after the first week of hospitalization. At week 2, these endpoints were present in 7% of treated patients and 29% of historical controls. This study suggests that interferon alfa-2b might reduce the severity and duration of complications of St. Louis encephalitis, although this interpretation must be viewed with extreme caution because of significant limitations in study design and methodology.
Japanese Encephalitis Virus (Flavivirus)
Japanese encephalitis virus (JEV) is the most common cause of mosquito-borne encephalitis in the world, with an estimated 35,000 to 50,000 annual cases and 10,000 annual deaths (Solomon, 2004; Solomon et al., 2003). JEV is widely distributed in Asia throughout Japan, China, Taiwan, Korea, the Far Eastern former Soviet Union, Southeast Asia, and India. Worldwide, the virus cycles between Culex, Aedes, or Anopheles species of mosquitoes, pigs, and birds. Lineage I and II strains of JEV are now endemic in Northern Australia owing to the presence of competent mosquito vectors and susceptible vertebrate hosts (Weaver and Reisen, 2010).
No specific therapy is available for JEV encephalitis. In a randomized double blind trial, interferon alfa-2a (10 million units/m2 daily × 7 days) treatment failed to reduce either mortality (≈20%) or the incidence of severe neurological sequelae (≈15%) (Solomon et al., 2003). A formalin-inactivated vaccine with excellent efficacy in prevention of disease is available for travelers to and residents of endemic areas. The risk of developing encephalitis among travelers to endemic areas has been estimated to be between 1 in 5000 and 1 in 20,000 cases per week of travel. Primary immunization is with two doses separated by 7 to 14 days, and a single booster dose at 1 year. Revaccination is recommended at 3-year intervals. Because of the high seroprevalence in the adult population of endemic countries, the pediatric population endures the majority of neuroinvasive infections. Consequently, two clinical trials are evaluating the efficacy of vaccines to prevent disease in children (NCT01041573 and NCT01092507), and a randomized double-blind placebo-controlled trial comparing ribavirin therapy to placebo is enrolling patients in India (NCT00216268).
California Serogroup of Viruses (Bunyaviruses)
In the United States, the major causes of neuroinvasive disease in the California encephalitis serogroup include La Crosse virus, Jamestown Canyon virus, and California encephalitis virus. These viruses are maintained in an enzootic cycle between mosquito vectors and small mammals or deer hosts in a limited geographic range. Among this group, La Crosse virus is the most common cause of encephalitis and is found predominantly in the Midwestern United States (McJunkin et al., 2001). Jamestown Canyon virus (Michigan, New York) and Snowshoe Hare virus (Alaska, Canada, and the northern United States) are other bunyaviruses that can cause viral meningitis or encephalitis.
Me Tri Virus (Alphavirus)
Me Tri virus, a virus related to Semliki forest virus, was isolated from Culex tritaeniorhynchus mosquitoes collected in Hanoi province, Vietnam, in 1971. Serological surveys demonstrate wide distribution in both livestock and humans, with seroconversion to the virus associated with encephalitis in children. Since most cases of arboviral encephalitis in Vietnam and other Southeast Asian countries tend to be caused by JEV or occasionally by dengue, cases due to Me Tri virus might be underrecognized owing to lack of knowledge, reagents, or testing (Mackenzie, 2005).
Semliki Forest Virus (Alphavirus)
Semliki forest virus (SFV), first isolated in 1942 from Aedes abnormalis collected in the Semliki Forest of western Uganda, is widely distributed in Africa, with mosquito isolates found in Mozambique, Nigeria, and the Central African Republic. While serosurveys indicate that infection in man is relatively common in sub-Saharan Africa, SFV has been linked to human disease twice. In 1987, SFV was isolated from sera in patients with fever, headache, myalgias, and arthralgias in the Central African Republic. In Germany, a case of fatal SFV encephalitis was described in a laboratory worker (Willems et al., 1979).
Colorado Tick Fever Virus (Coltivirus) and Banna Virus (Seadornavirus)
Colorado tick fever virus is a member of the family Reoviridae and the genus Coltivirus (Attoui et al., 2005). Ticks (e.g., Dermacentor andersoni) are the principal vectors of disease transmission, although rare human cases have been associated with blood transfusion. The virus is found predominantly in the Rocky Mountain region of the United States and Canada. Human infection is characterized by abrupt onset of fever, chills, retroorbital pain, and myalgia (Attoui et al., 2005; Goodpasture et al., 1978). Up to 65% of cases have associated leukopenia and thrombocytopenia. Patients with encephalitis have a lymphocytic CSF pleocytosis. Diagnosis is usually based on isolation of virus from erythrocytes in blood, where it can persist for months, or on serology. RT-PCR testing is also available, but its sensitivity and specificity in CNS disease is unknown. A related virus, Banna virus, belonging to the genus Seadornavirus, is transmitted by mosquitoes and has been isolated in cases of encephalitis in humans and animals, predominantly in China and Indonesia.
Powassan Virus (Flavivirus)
Powassan encephalitis has been reported in Russia, Canada, and the United States. There have been less than 20 reported U.S. cases of Powassan encephalitis since 2000. The virus cycles between ticks and small mammals. Powassan virus has been called the most herpes-like of the arboviruses because of the temporal lobe involvement noted in several cases. Neurological sequelae occur in an estimated 35% of survivors (Outbreak of Powassan encephalitis, 2001).
Toscana Virus (Phlebovirus)
Toscana virus, a neurotropic arbovirus carried by sand flies, was first isolated in 1971. It has become the leading cause of summertime viral meningitis in Italy, accounting for 30% to 50% of cases in some series (Charrel et al., 2005; Valassina et al., 2003). Additional human cases of meningitis have subsequently been reported in many European countries including France, Spain, and Portugal (Charrel et al., 2005). The reservoir of Toscana virus is most likely the vector, Phlebotomus (sandfly) species. After an incubation period of a few days to 2 weeks, Toscana virus infection produces abrupt onset of either a self-limited febrile illness or various CNS syndromes: meningitis, encephalitis, or meningoencephalitis that may be severe and associated with coma and systemic disease (rash, adenopathy, hepatosplenomegaly, renal involvement, disseminated intravascular coagulation, and bleeding diathesis). Patients typically have headache (100%), fever (76%-97%), nausea and vomiting (67%-88%), and myalgia (18%). Nuchal rigidity occurs in 50% to 95%. Diagnosis is by immunoenzymatic serological testing and detection of Toscana viral sequences in CSF by PCR techniques.
Rabies
Rabies is a significant cause of encephalitis worldwide, with nearly 100% mortality. The disease is enzootic in all continents except Australia and Antarctica, as well as certain island states or nations: Great Britain, Ireland, Iceland, Japan, New Zealand, Hawaii, the Bahamas, and Bermuda. Reservoirs of infection are bats, wild carnivores (skunks, foxes, raccoons, coyotes, wolves), and unimmunized dogs. In the United States, the bulk of human rabies cases are frequently linked to bat exposures, often in the absence of a recognized or documented bite or scratch. Rare non-bite (aerosol) exposures have also been documented. In 2004, four recipients of kidneys, a liver, and an arterial segment from an organ donor died within a month of transplantation from encephalitis characterized by rapid neurological deterioration, seizures, respiratory failure, and agitated delirium progressing to coma. The cause of illness was subsequently identified as rabies, and the donor was retrospectively found to have a history of a possible bat bite before he died of unrelated causes (subarachnoid hemorrhage) (Srinivasan et al., 2005). There were three more cases in 2005 in recipients of solid organs. A liver recipient with a history of vaccination 20 years earlier and the two corneal recipients survived (Maier et al., 2010).
The incubation period of rabies is usually from 1 to 3 months but may vary from 1 week to several years. A prodrome of headache, fever, paresthesias, and pain at the site of inoculation is followed by an acute neurological phase, then coma. Cases in which hyperactivity dominates have been called furious rabies. Characteristic neuropathological intraneuronal inclusions (Negri bodies; Fig. 53B.7) and inflammatory changes are maximal in the brainstem and limbic system. Up to 80% of patients exhibit hydrophobia or aerophobia, spasms of pharyngeal and nuchal muscles lasting from 1 to 5 minutes, triggered by swallow attempts or tactile, auditory, visual, and olfactory stimuli. The spasms are thought to be an exaggerated respiratory tract protective reflex. As the disease progresses, attacks increase in frequency and are accompanied by agitation, hallucinations, autonomic hyperactivity, and seizures. Body temperature may reach 105°F to 107°F. Paralytic, myelitic, or “dumb” rabies, accounting for 20% of patients, is characterized by paresthesias, weakness, and flaccid paralysis in the bitten extremity, progressing to quadriplegia. Paralytic rabies is most often associated with bat rabies virus strains.
Examination of a skin biopsy from a hairy region of the posterior neck (nuchal biopsy) for the presence of rabies virus antigen by immunofluorescence, and RT-PCR testing of saliva are the most rapid methods of antemortem diagnosis. Corneal smears are rarely used because of low sensitivity. The presence of neutralizing antibodies in the serum and CSF of an unimmunized patient is diagnostic but are not highly sensitive methods. Active disease produces high titers (>1 : 5,000), which may be helpful for diagnosing acute rabies in previously immunized individuals. High titers also distinguish acute rabies from postvaccinal encephalomyelitis associated with vaccines derived from animal neural tissue. RT-PCR protocols for detection of viral sequences in saliva, skin biopsies, CSF, or brain specimens have been established (Wacharapluesadee and Hemachudha, 2001). The sensitivity for CSF is low, and this type of test is not usually needed for brain tissue.
Transdermal bites or scratches and mucous membrane contact with saliva from known reservoir animal species constitute exposure. In addition, any history of bat exposure, with or without recognized bite, should be considered a significant exposure. Superficial bat-inflicted wounds carry a high risk because of the ability of bat rabies variants to infect and multiply within epithelial cells and fibroblasts. Although CNS tissues are highly infectious, infectious rabies virus is only rarely isolated from CSF, and RT-PCR on CSF is not very sensitive. Saliva should be considered infectious, and appropriate precautions taken. Postexposure treatment and prophylaxis of rabies includes wound care and the early initiation of multiple doses of rabies vaccine and antirabies immunoglobulin to nonimmune individuals (Rupprecht and Gibbons, 2004). Wounds should be cleansed with soap and water followed by povidone-iodine. Human diploid cell rabies vaccine (HDCV) or purified chick-embryo vaccine (PCEC) should be administered intramuscularly on days 0, 3, 7, and 14 into the deltoid or anterolateral thigh muscles (Rupprecht and Gibbons, 2004). Previously immunized individuals should be given booster doses of vaccine on days 0 and 3. Modern cell-culture rabies vaccines, in use in many areas of the world, are improvements over the biological products derived from animal (usually sheep or suckling mice) neural tissue, which have caused Guillain-Barré syndrome or acute disseminated encephalomyelitis approximately once per 2000 vaccinations and other less-severe neurological complications as often as once in 120 vaccinations. Human rabies immunoglobulin, 20 IU/kg, should be administered once as soon as possible after exposure (up to 7 days after the first dose of vaccine) at the beginning of prophylaxis to patients who have not been previously vaccinated. As much of the entire dose as possible should be administered in the area of the wound, and the remainder should be administered intramuscularly at a site distant from the vaccine administration. Because human rabies immunoglobulin may partially suppress active production of antibody, the recommended dose should not be exceeded.
There has been one widely reported successful case of treatment of a patient with clinical rabies who did not receive antecedent postexposure prophylaxis (Willoughby et al., 2005). The patient was a 15-year-old girl who developed clinical rabies 1 month after a bat bite. She was treated with ketamine and induction of coma to a burst suppression on EEG using midazolam supplemented with phenobarbital. Antiviral therapy included IV ribavirin (33 mg/kg load, then 16 mg/kg every 6 hours) and enteral amantadine (200 mg/day). There have been many subsequent therapeutic failures using this approach, and it is unclear whether this patient’s survival was related to any of the specific therapies used (Jackson, 2009). Another patient who recently recovered from rabies virus infection without intensive care or use of specific therapies was classified abortive human rabies (CDC, 2010a).
Chandipura Virus “Encephalitis”
Chandipura virus, a member of the Rhabdoviridae family transmitted by phlebotomine sand flies, was isolated from humans in India with fever (1965) and encephalopathy (1980), and associated with childhood epidemic “encephalitis” during the monsoon season in Andhra Pradesh and Maharashtra in southern India in 2003 and Gujarat state, India, in 2004 (Rao et al., 2004). Whether these “epidemic brain attacks” were true Chandipura viral encephalitis with parenchymal brain invasion and infection has been questioned. Illnesses had features of Reye syndrome, with vomiting, cerebral edema, and bland spinal fluid but without liver abnormalities; in some, CT scans showed middle cerebral artery territory lesions consistent with arterial pathology (John, 2004; Sejvar, 2006). Still, these cases point to clusters of severe neurological disease at times of presumed heightened insect vector activity and the importance of surveillance, coordinated epidemiological and laboratory investigations, and public health activities for diagnosis, management, and prevention.
Measles
Despite the availability of an effective vaccine, measles, a highly contagious respiratory-borne disease, is still an important cause of childhood mortality and blindness in developing countries, as well as in sporadic outbreaks in industrialized nations. Endemic measles was eliminated from the United States in 2000, but outbreaks associated with imported measles continue to occur. In 2005, an epidemic was triggered in Indiana by a visiting unvaccinated Romanian girl incubating measles (Parker et al., 2006); 34 cases of measles were identified as a result of this exposure, 94% of which occurred in unvaccinated children. In 2007, a multistate measles outbreak was associated with an imported case occurring in a participant at an international youth sporting event. As long as measles remains endemic worldwide and subpopulations within the United States remain unvaccinated for religious or cultural reasons, there will be the risk of future outbreaks. Still, eradication of measles with the presently available vaccine is a rational objective.
Subacute Sclerosing Panencephalitis
SSPE has an annual incidence from under 0.1 cases to 5 or 6 cases per million in unimmunized populations. In areas of high early-life measles attack rates, SSPE accounts for a portion of childhood neurodegenerative conditions. Children infected in the first 2 years of life are at greater risk, and case series consistently show SSPE to be more frequent in boys. The median interval between acute measles infection and SSPE is 8 years, with a range from 2 to 12 years. The early stage is marked by behavioral or personality changes and declining school performance. Myoclonus, seizures, spasticity, choreoathetoid or ballistic movements, ataxia, and chorioretinitis follow in the second stage. Optic atrophy, quadriparesis, autonomic instability, akinetic mutism, and coma are seen in the final stage. The majority of cases follow a progressive downhill course to death within a few years; some temporarily plateau or improve, and possibly 5% remit spontaneously (Garg, 2002).
At the time neurological symptoms occur, neurons and glia contain nuclear and cytoplasmic viral inclusion bodies (Fig. 53B.8), and high titers of antimeasles antibody is found in both serum and CSF. The CSF/serum antibody ratio is consistent with high levels of intrathecal synthesis of measles antibody. CSF pleocytosis is absent, glucose is normal, and total protein is normal or elevated. Acute symptoms together with increased intracranial pressure are poor prognostic signs. The earliest MRI findings are high signal intensity on T2-weighted images of gray and subcortical white matter in posterior portions of the hemispheres. During the second stage of disease, the EEG shows a pattern of generalized slow-wave complexes with a regular periodicity (Fig. 53B.9). The complexes may last up to 3 seconds and occur at regular intervals, between 4 and 14 seconds, against a background of depressed activity.
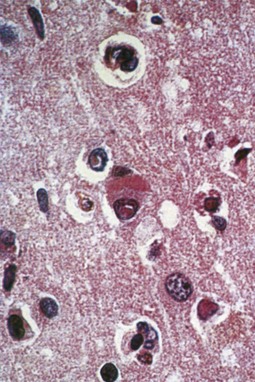
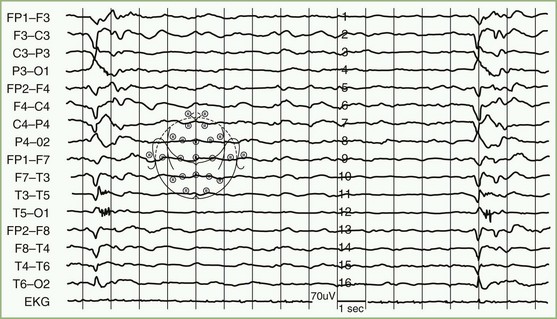
Some patients have improved or stabilized after one or several 6-week treatments with intraventricular interferon alfa through an Ommaya reservoir, starting at 10 U/m2 body surface area per day, with daily increments up to 106 U/m2/day on the fifth day, 5 days/week, combined with oral isoprinosine (inosiplex), 100 mg/kg/day. There have also been reports of response to IV ribavirin in combination with intrathecal interferon alfa (Tomoda et al., 2001) and symptomatic improvement in myoclonus and encephalopathy with levetiracetam. The laboratory endpoint of treatment is eradication of detectable measles antigen from the CSF. Systemic (subcutaneous) interferon alfa in daily doses of up to 5 million units has been used with intrathecal interferon alfa to simultaneously treat the peripheral reservoirs of measles virus, lymphoid, and glandular tissue. Prolonged or repeated treatments carry the risks of meningitis, interferon alfa–induced encephalopathy, and interferon alfa upper and lower motor neuron toxicity.
Immunization with attenuated live measles vaccine is recommended for infants between 12 and 15 months of age, with a second dose at age 4 to 6 years. Measles vaccine is one component of the combination quadrivalent measles, mumps, rubella, varicella (MMRV) vaccine and the trivalent measles, mumps, rubella (MMR) vaccine. Full recommendations of the Advisory Committee on Immunization Practices have been published (CDC, 2010b). In areas where measles circulates widely, immunization is performed early at 6 or 9 months of age. Fatal infection has followed measles vaccine in severely immunocompromised children, but there is no epidemiological evidence that vaccination causes SSPE.
Mumps
Mumps prevention is best achieved by vaccination with live attenuated virus at 12 to 15 months of age and an additional booster dose at 4 to 6 years of age; mumps vaccine is one component of the trivalent MMR or quadrivalent MMRV vaccine. Infrequent reports of mumps meningitis were reported following vaccination with strains of mumps vaccine (Urabe strain) that are no longer in use in the United States. Studies evaluating the use of the current MMR vaccine (containing Jeryl-Lynn strain) have not documented any association between vaccination and encephalitis, aseptic meningitis, or autism (Makela et al., 2002).
Rubella
Gestational rubella, especially infection acquired during the first trimester, has serious consequences for the fetus; 80% of children with a congenital rubella syndrome have some form of nervous system involvement. Signs in infancy include bulging fontanelle, lethargy, irritability, and abnormalities in muscle tone. CSF protein concentration is elevated, and the virus may be isolated from the CSF. Sequelae in survivors include mental retardation, sensorineural hearing loss, motor and posture abnormalities, cataracts, pigmentary retinopathy, and congenital heart disease. Congenital rubella syndrome has become rare in the United States since the institution of effective vaccination strategies in the 1960s (Reef et al., 2002).
Henipaviruses
Nipah Virus
NiV, closely related to HeV, was discovered in 1999 during the investigation of an outbreak of encephalitis in Malaysia. A major outbreak of disease in pigs and man from September 1998 to April 1999 resulted in 265 infected persons, 105 deaths, and the eventual destruction of 1.1 million pigs. Subsequent outbreaks have occurred in India. Clinical signs and symptoms include fever, headache, myalgia, drowsiness, stupor, coma, and stroke (Goh et al., 2000). Most patients in the initial outbreaks in Malaysia and Singapore had a history of direct contact with pigs. Like HeV, the natural reservoir is the fruit bat, found in regions extending from the western Pacific to the eastern coast of Africa.
Influenza
Influenza virus types and subtypes differ in the severity of respiratory and extra-respiratory disease they produce, and vary each year. Pandemic influenza A virus epidemics occurred in 1918 (H1N1), 1957 (H2N2), and 1968 (H3N2) and produced high morbidity in all age groups. In 1997 in Hong Kong, transmission of highly pathogenic avian influenza viruses (H5N1) from domestic poultry to humans resulted in 18 cases, with a 33% mortality. The unusual incidence of gastrointestinal, hepatic, renal and hematological disorders indicated that H5N1 viruses might have wider tissue tropism than human H3N2 viruses (Rowe et al., 2003). During the spring of 2009, a new pandemic influenza A (H1N1) virus emerged. Neurological complications of this pandemic H1N1 virus have been rare and reported mostly in children, typically but not exclusively in individuals with an underlying neurological disease. The usual presentation has been with encephalopathy and/or seizures. CSF pleocytosis is rare, and viral RNA has not been amplified from CSF, suggesting that CNS complications may be immune-mediated rather than the result of direct viral invasion (MMWR, 2009).
Influenza infections have been associated with neurological complications involving muscle (myositis, myopathy), the peripheral nervous system (GBS), and the CNS. CNS manifestations can take on a variety of forms including Reye syndrome, acute necrotizing encephalopathy, bilateral thalamic encephalitis, myelitis, and white-matter disease including ADEM and PRES (posterior reversible encephalopathy syndrome). Proinflammatory cytokines such as interferon alpha (IFN-α), and interleukin 6 (IL-6) are thought to have a role in pathogenesis of seizures or encephalopathy. Detection of influenza virus or nucleic acid in brain or CSF is very rare. The association of influenza A with childhood febrile seizures may be related in some cases to reactivation of HHV-6 or HHV-7 during influenza (Millichap and Millichap, 2006). The acute encephalopathy/encephalitis that has been reported to occur within 1 to 3 days of onset of respiratory symptoms (associated with both influenza A and B) has a high rate of mortality/morbidity (Morishima et al., 2002). On the other hand, the postencephalitis syndrome accompanied by an inflammatory spinal fluid is transient, with full recovery in most cases.
Antiviral treatment should be initiated as soon as possible for any hospitalized patient with neurological symptoms and suspected seasonal influenza or novel influenza (H1N1) infection. Treatment of 2009 H1N1 in adults is oral oseltamivir, 75 mg twice daily for 5 days; or zanamivir, 10 mg (2 5-mg inhalations) twice daily for 5 days (CDC, 2010c). Diagnosis is by detection of viral RNA by RT-PCR or by culture from nasopharyngeal specimens. The majority of 2009 H1N1 viruses and seasonal H3N2 viruses have been susceptible to the neuraminidase inhibitors, oseltamivir and zanamivir, but resistant to the adamantanes, amantadine and rimantadine.
An association between swine influenza vaccination and Guillain-Barré syndrome was reported in 1976-1977. When concerns were expressed again during the 1992-3 and 1993-4 flu seasons, no increase in risk for Guillain-Barré syndrome associated with influenza vaccination was found. Giant cell arteritis has been reported with influenza vaccination (Piyasirisilp and Hemachudha, 2002). Seasonal and 2009 H1N1 influenza vaccination are effective at preventing illness.
Other Hemorrhagic Fever Viruses
Arboviral Agents of Hemorrhagic Fevers
Dengue (Flavivirus)
Neurological complications including encephalopathy, encephalitis, stroke, mononeuritis multiplex of cranial and peripheral nerves, Guillain-Barré and Reye syndromes have been associated with both self-limited (classic) dengue fever and dengue hemorrhagic fever (Solomon et al., 2000). Diagnosis is by virus isolation from blood in the early stages or serological tests. The CSF may contain few (<30) or no WBCs. Care is supportive, and salicylates are contraindicated because of the risk of hemorrhagic exacerbation and Reye syndrome.
Hepatitis Viruses
The viral causes of hepatitis are hepatitis A, B, C, D (delta), and E viruses. Hepatitis C virus (HCV) and occasionally hepatitis B have been associated with a systemic vasculitic disease and mixed cryoglobulinemia, and hepatitis B has been associated with polyarteritis nodosa. Extrahepatic manifestations of HCV infection include cranial and peripheral neuropathies, mononeuritis multiplex, polymyositis, anterior spinal artery stroke, and encephalopathy. Several studies have detected HCV RNA or antigen in brain or CSF (Wilkinson et al., 2009). It has been suggested that CNS co-infection with HCV and HIV may result in additive cognitive disturbances, compared to chronic HIV infection alone (Winston et al., 2009). Ribavirin plus Interferon alfa treats liver and renal involvement but has variable effects on the neuropathy. Plasma exchange may be of benefit for patients whose neuropathy is worsened by interferon alfa. Intracerebral hemorrhage is a complication of combination therapy (standard or pegylated interferon alfa-2b and ribavirin), in patients with chronic hepatitis C who may or may not have additional risk factors such as hypertension and diabetes (Nishiofuku et al., 2006).
Abrahams B.C., Kaufman D.M. Anticipating smallpox and monkeypox outbreaks. Complications of the smallpox vaccine. Neurologist. 2004;10:265-274.
Agrawal A.G., Petersen L.R. Human immunoglobulin as a treatment for West Nile virus infection. J Infect Dis. 2003;188:1-4.
Anders K.H., Park C.S., Cornford M.E., et al. Adenovirus encephalitis and widespread ependymitis in a child with AIDS. Pediatr Neurosurg. 1990/1991;16:316-320.
Attoui H., Jaafar F.M., de Micco P., et al. Coltiviruses and seadornaviruses in North America, Europe, and Asia. Emerg Infect Dis. 2005;11:1673-1679.
Birnbaum T., Padovan C.S., Sporer B., et al. Severe meningoencephalitis caused by human herpesvirus 6 type B in an immunocompetent woman treated with ganciclovir. Clin Infect Dis. 2005;40:887-889.
Bode A.V., Sejvar J.J., Pape W.J., et al. West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clin Infect Dis. 2006;42:1234-1240.
Boos J., Davis L.E. Smallpox and smallpox vaccination. Neurology. 2003;60:1241-1245.
Brickelmaier M., Lugovsky A., Kartikeyan R., et al. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother. 2009;53:1840-1849.
CDC. Presumptive abortive human rabies-Texas, 2009. MMWR Morb Mortal Wkly Rep. 2010;59:185-190.
CDC. Use of combination Measles, Mumps, Rubella and Varicella Vaccine. MMWR Morb Mortal Wkly Rep. 2010;59:1-12.
CDC. Updated interim recommendations for the use of antiviral medications in the treatment and prevention of influenza for the 2009-2010 season. Available at http://www.cdc.gov/h1n1flu/recommendations.htm, 2010.
Chan-Tack K.M., Forrest G. Failure of interferon alpha-2b in a patient with West Nile virus meningoencephalitis and acute flaccid paralysis. Scand J Infect Dis. 2005;37:944-946.
Charrel R.N., Gallian P., Navarro-Mari J-M., et al. Emergence of Toscana virus in Europe. Emerg Infect Dis. 2005;11:1657-1663.
Cinque P., Scarpellini P., Vago L., et al. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS. 1997;11:1-17.
Clifford D.B., Deluca A., Simpson D.M., et al. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438-446.
Cohen J.I., Davenport D.S., Stewart J.A., et al. Recommendations for prevention of and therapy for exposure to B virus (cercopithecine herpesvirus 1). Clin Infect Dis. 2002;35:1191-1203.
Davis L.E., DeBiasi R., Goade D.E., et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286-300.
Debiasi R.L., Tyler K.L. West Nile virus meningoencephalitis. Nat Clin Pract Neurol. 2006;2:264-275.
DeBiasi R.L., Tyler K.L. Viral meningitis and encephalitis. Continuum: Infectious diseases. American Academy of Neurology Publications. 2006;12:58-94.
Doja A., Bitnun A., Jones E.L., et al. Pediatric Epstein-Barr virus-associated encephalitis: 10-year review. J Child Neurol. 2006;21:385-391.
Domingues R.B., Tsanaclis A.M., Pannuti C.S., et al. Evaluation of the range of clinical presentations of herpes simplex encephalitis by using polymerase chain reaction assay of cerebrospinal fluid samples. Clin Infect Dis. 1997;25:86-91.
Elphick G.H., Querbes W., Jordan J.A., et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380-1383.
Fotheringham J., Donati D., Akhyani N., et al. Association of human herpesvirus-6B with mesial temporal lobe epilepsy. PLoS Med. 2007;4:e180.
Garg R.K. Subacute sclerosing panencephalitis. Postgrad Med J. 2002;78:63-70.
Gilden D.H. Varicella zoster virus and central nervous system syndromes. Herpes. 2004;11(Suppl 2):89A-94A.
Gilden D.H., Cohrs R.J., Mahalingam R. VZV vasculopathy and postherpetic neuralgia: progress and perspective on antiviral therapy. Neurology. 2005;64:21-25.
Gilden D.H., Kleinschmidt-DeMasters B.K., et al. Neurologic complications of the reactivation of varicella-zoster virus. N Engl J Med. 2000;343:635-645.
Glaser C.A., Gilliam S., Schnurr D., et al. California Encephalitis Project, 1998-2000. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998-2000. Clin Infect Dis. 2003;36:731-742.
Glaser C.A., Honamand S., Anderson C.J., et al. Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin Infect Dis. 2006;43:1565-1577.
Goh K.J., Tan C.T., Chew N.K., et al. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med. 2000;342:1229-1235.
Gonzalez H., Olsson T., Berg K. Management of post-polio syndrome. Lancet Neurol. 2010;9:634-642.
Goodpasture H.C., Poland J.D., Francy D.B., et al. Colorado tick fever: clinical epidemiologic, and laboratory aspects of 228 cases in Colorado in 1973-1974. Ann Intern Med. 1978;88:303-310.
Griffiths P. Cytomegalovirus infection of the central nervous system. Herpes. 2004;11(Suppl 2):95A-104A.
Griffiths P.D., McLaughlin J.E. Cytomegalovirus. In: Scheld W.M., Whitley R.J., Marra C.M. Infections of the Central Nervous System. third ed. Philadelphia: Lippincott Williams & Wilkins; 2004:159-173.
Hall C.D., Dafni U., Simpson D., et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. AIDS Clinical Trials Group 243 Team. N Engl J Med. 1998;338:1345-1351.
Isaacson E., Glaser C.A., Forghani B., et al. Evidence of human herpesvirus 6 infection in 4 immunocompetent patients with encephalitis. Clin Infect Dis. 2005;40:890-893.
Jackson A.C. Update on rabies diagnosis and treatment. Curr Infect Dis Rep. 2009;11:296-301.
John T.J. Chandipura virus, encephalitis, and epidemic brain attack in India. Lancet. 2004;364:2175.
Johnson R.T. Smallpox. The threat of bioterrorism and the risk of vaccine. Neurology. 2003;60:1228-1229.
Kalil A.C., Devetten M.P., Singh S., et al. Use of interferon-alpha in patients with West Nile encephalitis: report of 2cases. Clin Infect Dis. 2005;40:764-766.
Kamei S., Sekizawa T., Shiota H., et al. Evaluation of combination therapy using acyclovir and corticosteroid in adult patients with herpes simplex virus encephalitis. J Neurol Neurosurg Psychiatry. 2006;76:1544-1549.
Kimberlin D.W. Herpes simplex virus infections in neonates and early childhood. Semin Pediatr Infect Dis. 2005;16:271-281.
Kimberlin D.W., Lin C.Y., Sanchez P., et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease including the central nervous system: a randomized controlled trial. J Pediatr. 2003;143:16-25.
Kleinschmidt-DeMasters B.K., Gilden D.H. Varicella-zoster virus infections of the nervous system: clinical and pathologic correlates. Arch Path Lab Med. 2001;125:770-780.
Kleinschmidt-DeMasters B.K., Tyler K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369-374.
Koralnik I.J. Progressive multifocal leukoencephalopathy revisited: has the disease outgrown its name? Ann Neurology. 2006;60:162-173.
Kost C.B., Rogers B., Oberste M.S., et al. Multicenter beta trial of the GeneXpert enterovirus assay. J Clin Microbiol. 2007;45:1081-1086.
Kupila L., Vuorinen T., Vainionpää R., et al. Etiology of aseptic meningitis and encephalitis in an adult population. Neurology. 2006;66:75-80.
Lakeman F.D., Whitley R.J. Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. J Infect Dis. 1995;171:857-863.
Langer-Gould A., Atlas S.W., Green A.J., et al. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;35:375-381.
Ljungman P. CMV infections after hematopoietic stem cell transplantation. Bone Marrow Transplantation. 2008;42:S70-S72.
Mackenzie J.S. Emerging zoonotic encephalitis viruses: lessons from Southeast Asia and Oceania. J Neurovirol. 2005;11:434-440.
Maier T., Schwarting A., Mauer D., et al. Management and outcomes after multiple corneal and solid organ transplantations from a donor infected with rabies virus. Clin Infect Dis. 2010;50:1112-1129.
Major E.O. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35-47.
Makela A., Nuorti J.P., Peltola H. Neurologic disorders after measles-mumps-rubella vaccination. Pediatrics. 2002;110(No. 5):957-963.
Marra C.M., Rajicic N., Barker D.E., et al. A pilot study of cidofovir for progressive multifocal leukoencephalopathy in AIDS. AIDS. 2002;16(No. 13):1791-1797.
Marzocchetti A., Tompkins T., Clifford D.B., et al. Determinants of survival in progressive multifocal leukoencephalopathy. Neurology. 2009;73:1551-1558.
Marton R., Gotlieb-Steimatsky T., Klein C., et al. Acute herpes simplex encephalitis: clinical assessment and prognostic data. Acta Neurol Scand. 1996;93:149-155.
McCullers J.A., Lakeman F.D., Whitley R.J. Human herpesvirus 6 is associated with focal encephalitis. Clin Infect Dis. 1995;21:571-576.
McJunkin J.E., de los Reyes E.C., Irazuzta J.E., et al. La Crosse encephalitis in children. N Engl J Med. 2001;344:801-807.
Miller R.F., Fox J.D., Thomas P., et al. Acute lumbosacral polyradiculopathy due to CMV in advanced HIV disease: CSF findings in 17 patients. J Neurol Neurosurg Psychiatry. 1996;61:456-460.
Millichap J.G., Millichap J.J. Role of viral infections in the etiology of febrile seizures. Ped Neurol. 2006;35(No. 3):165-172.
MMWR. Neurologic complications associated with novel influenza A (H1N1) virus infection in children-Dallas, Texas, May 2009. MMWR Morb Mortal Wkly Rep. 2009;58:773-778.
Mogensen T.H., Larsen C.S. Aseptic meningitis caused by reactivation of varicella-zoster virus in immunocompetent adults. Scand J Infect Dis. 2006;38:815-818.
Morishima T., Togashi T., Yokota S., et al. Encephalitis and encephalopathy associated with an influenza epidemic in Japan. Clin Infect Dis. 2002;35(No. 5):512-517.
Nash D., Mostashari F., Fine A., et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
Nishiofuku M., Tsujimoto T., Matsumura Y., et al. Intracerebral hemorrhage in a patient receiving combination therapy of pegylated interferon alpha-2b and ribavirin for chronic hepatitis C. Intern Med. 2006;45(No. 7):483-484.
O’Sullivan C.E., Aksamit A.J., Harrington J.R., et al. Clinical spectrum and laboratory characteristics associated with detection of herpes simplex virus DNA in cerebrospinal fluid. Mayo Clin Proc. 2003;78:1347-1352.
2001 Outbreak of Powassan encephalitis—Maine and Vermont, 1999-2001. MMWR—Morb Mortali Wkly Rep. 2001;50(No. 35):761-764.
Oxman M.N., Levin M.J., Johnson G.R., et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
Parker A.A., Staggs W., Dayan G.H., et al. Implications of a 2005 measles outbreak in Indiana for sustained elimination of measles in the United States. N Engl J Med. 2006;355:447-455.
Petropoulou K.A., Gordon S.M., Prayson R.A., et al. West Nile virus meningoencephalitis: MR imaging findings. AJNR Am J Neuroradiol. 2005;26:1986-1995.
Pillet S., Billaud G., Omar S., et al. Multicenter evaluation of the ENTEROVIRUS R-gene real-time RT-PCR assay for the detection of enteroviruses in clinical specimens. J Clin Virol. 2010;47:54-59.
Piyasirisilp S., Hemachudha T. Neurological adverse events associated with vaccination. Curr Opin Neurol. 2002;15:333-338.
Planitzer C.B., Modrof J., Kreil T.R. West Nile virus neutralization by US plasma- derived immunoglobulin products. J Infect Dis. 2007;196(No. 3):435-440.
Rahal J.J., Anderson J., Rosenberg C., et al. Effect of interferon-alpha2b therapy on St. Louis viral meningoencephalitis: clinical and laboratory results of a pilot study. J Infect Dis. 2004;190:1084-1087.
Ramers C., Billman G., Hartin M., et al. Impact of a diagnostic cerebrospinal fluid enterovirus polymerase chain reaction test on patient management. JAMA. 2000;283:2680-2685.
Rao B.L., Basu A., Wairagkar N.T., et al. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet. 2004;364:869-874.
Raschilas F., Wolff M., Delatour F., et al. Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin Infect Dis. 2002;35:254-260.
Reddy S.M., Winston D.J., Territo M.C., et al. CMV central nervous system disease in stem-cell transplant recipients: an increasing complication of drug-resistant CMV infection and protracted immunodeficiency. Bone Marrow Transplantation. 2010. doi:10.1038/bmt.2010.35
Reef S.E., Frey T.K., Theall K., et al. The changing epidemiology of rubella in the 1990s: on the verge of elimination and new challenges for control and prevention. JAMA. 2002;287(No. 4):464-472.
Reimann C.A., Hayes E.B., Diguiseppi C., et al. Epidemiology of neuroinvasive arboviral disease in the United States: 1999-2008. Am J Trop Med Hyg. 2008;79:974-979.
Rowe T., Cho D.S., Bright R.A., et al. Neurological manifestations of avian influenza viruses in mammals. Avian Dis. 2003;47:1122-1126.
Rupprecht C.E., Gibbons R.V. Clinical practice. Prophylaxis against rabies. N Engl J Med. 2004;351:2626-2635.
Sawyer M.H. Enterovirus infections: diagnosis and treatment. Semin Pediatr Infect Dis. 2002;13:40-47.
Seeley W.W., Marty F.M., Holmes T.M., et al. Post-transplant acute limbic encephalitis: clinical features and relationship to HHV6. Neurology. 2007;69:156-165.
Sejvar J.J. The evolving epidemiology of viral encephalitis. Curr Opin Neurol. 2006;19:350-357.
Sejvar J.J., Chowdary Y., Schomogyi M., et al. Human monkeypox infection: a family cluster in the midwestern United States. J Infect Dis. 2004;190:1833-1840.
Sejvar J.J., Labutta R.J., Chapman L.E., et al. Neurologic adverse events associated with smallpox vaccination in the United States, 2002-2004. JAMA. 2005;294(No. 21):2744-2750.
Shalabi M., Whitley R.J. Recurrent benign lymphocytic meningitis. Clin Infect Dis. 2006;43:1194-1197.
Solomon T. Flavivirus encephalitis. N Engl J Med. 2004;351:370-378.
Solomon T., Dung N.M., Vaughn D.W., et al. Neurological manifestations of dengue infection. Lancet. 2000;355:1053-1059.
Solomon T., Dung N.M., Wills B., et al. Interferon alfa-2a in Japanese encephalitis: a randomised double-blind placebo-controlled trial. Lancet. 2003;361:821-826.
Srinivasan A., Burton E.C., Kuehnert M.J., et al. Transmission of rabies virus from an organ donor to four transplant recipients. N Engl J Med. 2005;352:1103-1111.
Steiner I., Budka H., Chaudhuri A., et al. Viral encephalitis: a review of diagnostic methods and guidelines for management. Eur J Neurol. 2005;12:331-343.
Tan C.S., Koralnik I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9:425-437.
Tebas P., Nease R.F., Storch G.A. Use of the polymerase chain reaction in the diagnosis of herpes simplex encephalitis: a decision analysis model. Am J Med. 1998;105:287-295.
Tedder D.G., Ashley R., Tyler K.L., et al. Herpes simplex virus infection as a cause of benign recurrent lymphocytic meningitis. Ann Intern Med. 1994;121:334-338.
Tomoda A., Shiraishi S., Hosoya M., et al. Combined treatment with interferon-alpha and ribavirin for subacute sclerosing panencephalitis. Pediatr Neurol. 2001;24:54-59.
Tselis A., Lavi E. Cytomegalovirus infection of the adult nervous system. In: Davis L., editor. Infectious Diseases of the Nervous System. London: Butterworth-Heinemann, 2000.
Tyler K.L. Acute viral myelitis. In: Scheld W.M., Whitley R.J., Marra C.M. Infections of the Central Nervous System. third ed. Philadelphia: Lippincott William & Wilkins; 2004:305-322.
Tyler K.L., Pape J., Goody R.J., et al. CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology. 2006;66:361-365.
Valassina M., Cusi M.G., Valensin P.E. A Mediterranean arbovirus: the Toscana virus. J Neurovirol. 2003;9:577-583.
Wacharapluesadee S., Hemachudha T. Nucleic-acid sequence based amplification in the rapid diagnosis of rabies. Lancet. 2001;385:892-893.
Warrell M.J., Warrell D.A. Rabies and other lyssavirus diseases. Lancet. 2004;363:959-969.
Weaver S.C., Reisen W.K. Present and future arbovirus threats. Antiviral Res. 2010;85:328-345.
Weil A.A., Glaser C.A., Amad Z., et al. Patients with suspected herpes simplex encephalitis: rethinking an initial negative polymerase chain reaction result. Clin Infect Dis. 2002;34:1154-1157.
Weinberg A., Bloch K.C., Li S., et al. Dual infections of the central nervous system with Epstein-Barr virus. J Infect Dis. 2005;191:234-237.
Weinberg A., Li S., Palmer M., et al. Quantitative CSF PCR in Epstein-Barr virus infections of the central nervous system. Ann Neurol. 2002;52:543-548.
Whitley R.J., Alford C.A., Hirsch M.S., et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314:144-149.
Whitley R.J. Herpes simplex encephalitis: adolescents and adults. Antiviral Res. 2006;71:141-148.
Whitley R.J., Cloud G., Gruber W., et al. Ganciclovir treatment of symptomatic congenital cytomegalovirus infection: results of a phase II study. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis. 1997;165:1080-1086.
Wilkinson J., Radowski M., Laskus T. Hepatitis C virus neuroinvasion: identification of infected cells. J Virol. 2009;83:1312-1319.
Willems W.R., Kaluza G., Boschek C.B., et al. Semliki forest virus: cause of a fatal case of human encephalitis. Science. 1979;203(No. 4385):1127-1129.
Willoughby R.E.Jr., Tieves K.S., Hoffman G.M., et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005;352:2508-2514.
Winston A., Garvey L., Scotney E., et al. Does acute hepatitis C infection affect the central nervous system in HIV-1 infected individuals? J Viral Hepatol. 2009. [Epub ahead of print] PMID 19780944
Yousry T.A., Major E.O., Ryschkewitsch C., et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354:924-933.