[level-membership-for-neurology-category]
Chapter 53A Infections of the Nervous System
Neurological Manifestations of Human Immunodeficiency Virus Infection
Epidemiology and Current Trends
HIV Genome, Replication, and Molecular Heterogeneity of HIV
Natural History of HIV Infection and Neuro-HIV Disease
Neuropathogenesis of HIV Disease
Antiretroviral Therapy and Its Effect on Neuro-HIV Disease
Clinical Spectrum of Neuro-HIV Disease
Epidemiology and Current Trends
Now, 3 decades after the first report of AIDS, this disease has become one of the greatest public health challenges globally. The HIV pandemic has resulted in an estimated 64 million infections worldwide, and it has claimed the lives of more than 30 million persons (www.unaids.org). In 2008, more than 1 million persons were living with HIV/AIDS in the United States, and based on new methods of determining incidence, approximately 56,000 new HIV infections and 25,000 deaths occurred (www.cdc.org). An estimated 44% of new HIV infections are in homosexual men, 17% are in IDUs, and in 35% the infection is transmitted by heterosexual contact. The demography of newly infected individuals has changed considerably since the 1990s in the United States; HIV infection is spreading rapidly in certain populations (racial and ethnic minority populations and women of color) and decreasing (IDUs) or fluctuating in others (homosexual men).
HIV Genome, Replication, and Molecular Heterogeneity of HIV
HIV is a ribonucleic acid (RNA) virus belonging to the family of human retroviruses (Retroviridae) and the subfamily of lentiviruses. Electron microscopy shows that the HIV virion is an icosahedral structure. It contains numerous external spikes formed by the two major envelope proteins (gp120 and gp41) and one major core protein (gp24). Like other retroviruses, HIV has genes that encode the structural and enzyme proteins of the virus: env encodes the surface glycoproteins, gag encodes the core protein, and pol encodes for the enzyme responsible for reverse transcription and viral integration into the host genome. HIV also contains at least six other genes (tat, rev, nef, vif, vpr, and vpu) that encode for proteins involved in gene regulation. The replication cycle of HIV begins with high-affinity binding of the gp120 envelope protein to its receptor, the CD4 molecule, on the host cell surface (Greene and Peterlin, 2002). After gp120 binds to the CD4 molecule, it undergoes a conformational change that facilitates its binding to a co-receptor (CCR5, CXCR4) (Xiao et al., 2000). These co-receptors belong to the family of transmembrane G protein–coupled cellular receptors that determine the cellular tropism of the virus strain. Following surface attachment and fusion, the HIV genomic RNA is uncoated and internalized into the host cell. The subsequent steps in viral replication include activation of reverse transcriptase enzyme, reverse transcription of the genomic RNA into double-stranded deoxyribonucleic acid (DNA), and integration of DNA into the host cell chromosome through the action of another virally encoded enzyme—integrase. Molecular analyses of various HIV isolates from a single person over time or from cohorts of patients reveal sequence variations over many parts of the viral genome. The degree of difference in coding sequences of the viral envelope protein, for example, can vary from a few percent to 50%. The immune pressure and functional constraints on viral proteins (e.g., by antibodies) and the use of retroviral therapy influence the level of variation in viral genes and protein products.
Natural History of HIV Infection and Neuro-HIV Disease
The current Centers for Disease Control and Prevention (CDC) classification system of HIV-infected adolescents and adults categorizes persons on the basis of clinical conditions associated with HIV infection and CD4+ T-cell counts. The system is based on three ranges of such lymphocyte counts and three clinical categories, making a total of nine mutually exclusive categories (Table 53A.1). HIV disease follows a progressive course; once individuals have had a clinical condition in category B (or C), their disease cannot again be classified into a lower category (A or B). AIDS-defining conditions were originally designed for surveillance purposes, and clinicians should not focus on whether AIDS is present but instead should view HIV disease as a spectrum ranging from primary infection through asymptomatic stage to advanced disease.
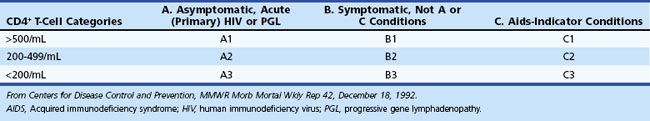
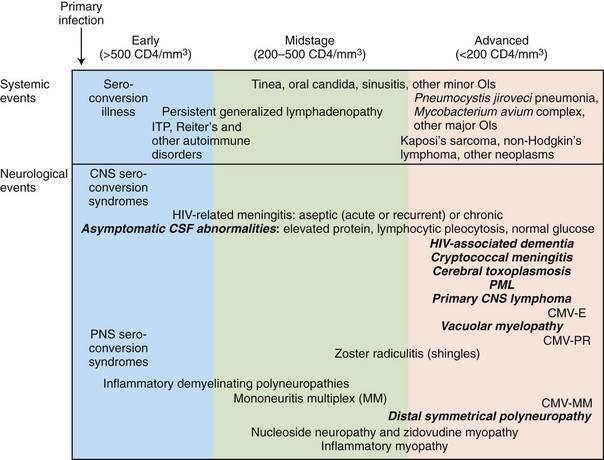
The stage of systemic HIV infection influences both the risk and the nature of neurological disease; hence CD4+ T-cell count provides critical information that helps guide the evaluation (Fig. 53A.1). In early infection (corresponding to CD4+ T-cell counts >500/mL) and excess immune stimulation, autoimmune disorders such as demyelinating neuropathies may develop. During midstage infection (CD4+ T-cell counts of 200-500/mL), primary HIV-related disorders such as HIV-associated neurocognitive dysfunction (HAND) and certain opportunistic infections such as varicella-zoster virus (VZV) radiculitis (shingles) may appear. In advanced HIV infection (CD4+ T-cell count <200/mL), the risk of dementia, myelopathy, and painful neuropathy increases further, and patients become increasingly vulnerable to major opportunistic infections (OIs) such as cerebral toxoplasmosis, progressive multifocal leukoencephalopathy (PML), and cryptococcal meningitis, as well as to neoplasms—in particular, primary central nervous system lymphoma (PCNSL).
Primary Infection and Dissemination of Virus
HIV is transmitted by both homosexual and heterosexual contact, by sharing of contaminated needles in IDUs, by tainted blood and blood products, and from infected mothers to infants either intrapartum, perinatally, or via breast milk (Hansasuta and Rowland-Jones, 2001). Virus that enters directly into the bloodstream via infected blood or blood products is trapped rapidly by the spleen and lymphoid tissue, whereas entry of virus through a mucosal surface (sexual contact) requires mucosal dendritic cells to first carry the virus to the regional lymphoid tissue. In either case, virus replication in CD4+ T cells intensifies (more rapidly with bloodborne infection) prior to development of the HIV-specific immune response, leading to a burst of viremia and rapid dissemination of the virus to other lymphoid organs, the brain, and other tissues. The initial viremic burst generally results in “acute HIV syndrome.” Viral levels are measured in millions of HIV RNA virions per milliliter. Some 50% to 70% of individuals with acute HIV infection experience an “infectious mononucleosis”–type syndrome persisting 1 to 6 weeks and characterized by fever, erythematous or maculopapular rash, headache, nausea, anorexia, lethargy, arthralgia, sore throat, and lymphadenopathy.
Chronic Persistent HIV Infection
In HIV disease, a chronic infection develops and persists with varying degrees of virus replication and progressive immunological impairment for a median of 10 years before the patient becomes clinically ill. Chronic persistent infection is the biological hallmark of HIV disease. Establishment of chronic HIV infection requires that the virus must evade control and elimination by the immune system. The virus accomplishes this via a number of mechanisms, including high rates of genomic mutation and molecular heterogeneity (see earlier section on the HIV genome), sequestration of infected cells in immunologically privileged sites (e.g., brain), down-regulation of human leukocyte antigen (HLA) class I molecules on the surface of HIV-infected cells by viral proteins (e.g., nef), conformational masking of receptor-binding sites that fails to be neutralized by antibodies, and perhaps deletion of the initially expanded CD8+ T-cell clones by the overwhelming initial burst of viremia and antigenemia. Despite a vigorous immune response and the marked down-regulation of virus replication following primary HIV infection, HIV establishes a state of chronic low-grade infection. The half-life of a productively infected cell is approximately 1 day, and that of a circulating virion is 30 to 60 minutes. Given the relatively steady level of plasma viremia and of infected cells in an individual, it is estimated that extremely large amounts of virus (over a billion or so copies) are produced and cleared from the circulation each day. Thus, clinical latency should not be confused with microbial latency. Vigorous virus replication, though with low-level viremia, is present during the period of clinical latency. Even the term clinical latency is misleading because immunological progression of the HIV disease is generally relentless during this period. The CNS continues to harbor and mount a host reaction to the HIV throughout the asymptomatic or latent stage, yet without apparent immediate clinical sequelae (McArthur et al., 2010). The CSF in patients with latent HIV infection generally shows abnormalities including abnormal cell count, protein and immunoglobulin elevation, unique oligoclonal bands, and local synthesis of anti-HIV antibodies within the CNS compartment; the intact virus can be recovered from the CSF (Price and Spudich, 2008). Pathological studies have shown evidence of inflammatory reactions in the CNS, with perivascular mononuclear cell infiltrations, although the HIV RNA burden, as measured by polymerase chain reaction (PCR) from CSF and brain samples, appears to be low at this stage. Neither overt nor subclinical cognitive or motor dysfunction appears to be common in the early latent stage. From a practical standpoint, the risk of cognitive decline in asymptomatic individuals is sufficiently small as to provide no basis for disability or disqualification from work based simply on HIV-positive status.
The length of the asymptomatic stage is determined by viral and host factors (Kinter et al., 2000; McArthur et al., 2010; Price and Spudich, 2008; Xiao et al., 2000). Some patients who are termed long-term nonprogressors show little if any decline in CD4+ T-cell counts over many years. These patients generally have extremely low levels of HIV RNA. Other patients remain entirely asymptomatic despite progressive declines in the CD4+ count to extremely low levels. In these patients, the appearance of a systemic or CNS OI may be the first manifestation of HIV disease.
Neuropathogenesis of HIV Disease
Neurons lack the conventional surface receptor for HIV binding and fusion and therefore are not directly infected by the virus. The main cell types infected in the brain in vivo are those of the monocyte-macrophage lineage. These include monocytes that have migrated into the brain from the peripheral blood, perivascular macrophages, and resident microglial cells (Sabri et al., 2003; Yadav and Collman, 2009). Although there have been reports of infrequent HIV infection of astrocytes, there is no convincing evidence that brain cells other than those of monocyte-macrophage lineage can be productively infected in vivo (Price and Spudich, 2008). Nevertheless, in vitro infection of a neural cell line has been reported (Klein et al., 1999), and it appears that galactosylceramide on the neuronal surface is an essential component of the HIV gp120 receptor; antibodies to galactosylceramide inhibit HIV entry into the neural cell lines. Some studies have demonstrated that viral entry is due in part to the ability of virus-infected and immune-activated macrophages to induce adhesion molecules such as vascular adhesion molecule 1 (VCAM-1) on brain endothelium. Other studies have demonstrated that HIV gp120 enhances the expression of intercellular adhesion molecule 1 (ICAM-1) in glial cells; this effect may facilitate entry of HIV-infected cells into the CNS and may promote syncytia formation (Fig. 53A.2).
The R5 (CCR5 co-receptor) virus strains that are macrophage-tropic preferentially gain access into the brain rather than R4 (CXCR4 co-receptor) strains. HIV-infected individuals who are heterozygous for CCR5-del.32 (co-receptor gene with 32-base deletion) appear to be relatively protected against the development of HIV encephalopathy, compared to persons with homozygous wild-type CCR5 (Weiss et al., 1999). Distinct HIV envelope sequences are also linked with the clinical manifestation of HAND (McArthur et al., 2010).
Both white-matter changes and neuronal loss are observed in HIV infection. It is unlikely that direct infection of neurons or glial cells accounts for this cell loss. Rather, the brain pathology is thought to be due to a combination of direct effects, either toxic or function-inhibitory, of virus or viral antigens on neuronal cells and effects of a variety of neurotoxins released from the infiltrating monocytes, resident microglial cells, and astrocytes (McArthur et al., 2010; Price and Spudich, 2008; Scaravilli et al., 2007; Yadav and Collman, 2009). Neurotoxins can be released from monocytes as a consequence of other HIV-associated infections or immune activation. Activated monocyte-derived neurotoxic factors have been reported to injure neurons via the N-methyl-d-aspartate (NMDA) receptor. Additionally, HIV gp120 and tat shed by virus-infected monocytes and a variety of cytokines including tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, interferon alpha (IFN-α), and endothelin can contribute directly or indirectly to the neurotoxic effects in HIV infection. Activation of microglial cells, leading to increased production of eicosanoids, nitric oxide, and quinolinic acid, may also be neurotoxic.
Astrocytes may play diverse roles in HIV neuropathogenesis. Astrocytosis and reactive gliosis occur in brains of HIV-infected individuals; TNF-α and IL-6 have been shown to induce astrocyte proliferation. In addition, astrocyte-derived IL-6 can induce HIV expression in infected cells in vitro. Older HIV-infected individuals and those with the E4 allele for apolipoprotein E (APOE) appear to have an increased risk of HIV encephalitis and polyneuropathy (Corder et al., 1998). Neuropsychiatric abnormalities may rapidly improve following the initiation of antiretroviral therapy, indicating that HIV or products are involved in the neuropathogenesis of these disorders and suggesting that the dominant changes are either structural or functional at the synapses and dendritic spines (Yadav and Collman, 2009).
Antiretroviral Therapy and Its Effect on Neuro-HIV Disease
The dynamics of in vivo HIV production and turnover have been quantified using mathematical modeling in the setting of HAART. Treatment with effective HAART typically results in precipitous decline in the level of plasma viremia, often by well over 95% within a few weeks. The number of CD4+ T cells increases concurrently. Combination antiretroviral therapy is the cornerstone of management of HIV infection (Fauci and Lane, 2006). Suppression of HIV replication prolongs and improves the quality of life in patients with HIV infection.
Following the widespread use of HAART in the United States since 1996, dramatic declines have been noted in the incidence of most AIDS-defining conditions, including neurological diseases. Reconstitution of immune defenses with HAART has enabled some patients to discontinue secondary prophylaxis against CNS opportunistic pathogens. Primary neurological diseases can be prevented or delayed by successful HAART (Geraci and Simpson, 2001; Sacktor et al., 2006), although the evolution of drug-resistant viral strains may limit the sustained benefits of antiretroviral therapy (D’Aquila et al., 2003).
Once the decision has been made to initiate therapy, the physician must decide which drugs to use as the first therapeutic regimen. Initial choice of drugs will determine the immediate response to therapy, and it will have implications regarding options for future therapeutic regimens. The goal of treatment is to achieve a viral load of fewer than 50 copies/mL within 4 to 6 months of initiation (Gazzard et al., 2008). The two options for initial therapy most commonly in use today involve a three-drug regimen from two different antiretroviral classes (Table 53A.2). Recommendations regarding specific regimens are evolving. Combination therapy must be employed.
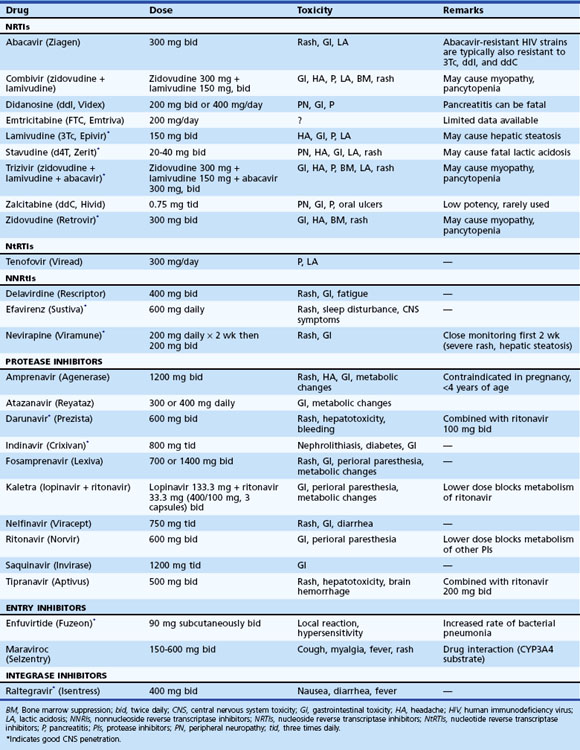
For HIV-associated brain disease, drugs that have good blood-brain barrier penetration should be preferred (see Table 53A.2). Following the initiation of therapy, one should monitor virological (HIV RNA levels) and immunological (CD4+ counts) responses periodically. The HIV RNA levels in serum generally reflect viral levels in CSF, at least until late stages of the HIV disease. In terminal stages, the CNS compartment may harbor slightly different and divergent HIV strains, with different degrees of drug susceptibility (Ellis et al., 2007; Liu et al., 2006; McArthur et al., 2010; Price and Spudich, 2008). In an attempt to determine an optimal therapeutic regimen, antiretroviral drug susceptibility through genotyping or phenotyping of HIV quasispecies has been attempted. Maximal suppression of viral replication is the goal of therapy, not just to prevent the disease progression but also to prevent the appearance of drug-resistant HIV quasispecies. The principles of current therapy for HIV infection are well articulated in publications of the U.S. Department of Health and Human Services (see updates at www.aidsinfo.nih.gov/guidelines/). As HAART has become widely available and has prolonged the life of patients with AIDS, an increasingly commonly observed manifestation was noted, characterized by a paradoxical clinical deterioration following therapy referred to as the immune reconstitution inflammatory syndrome (IRIS) and attributed to immunological recovery. Although typically observed with opportunistic infection, it may also occur as a response to noninfectious antigens. In the CNS, IRIS has been well described with Cryptococcus neoformans cytomegalovirus and PML. Patients with IRIS either present with clinical deterioration of a recognized infection following initiation of HAART, or the neurological disease may first manifest upon immunological recovery following the initiation of effective antiretroviral therapy. Neuroimaging generally shows worsening of previously observed lesions, frequently with contrast enhancement. Neuropathological findings at autopsy or from brain biopsy specimens reveal severe inflammatory reaction, with intraparenchymal and perivascular infiltration by lymphocytes (predominantly CD8+ T lymphocytes) and macrophages (Gray et al., 2005). The symptoms mostly regress with continued HAART, although a short course of high-dose parenteral corticosteroids (Riedel et al., 2006) has been suggested as effective in abrogating the response. Death may complicate IRIS (Gray et al., 2005).
Clinical Spectrum of Neuro-HIV Disease
The high prevalence and striking diversity of neurological disorders complicating AIDS were recognized early in the epidemic (Berger et al., 1987a). Clinical description of neurological OIs and malignancies predominated in early reports, but it also became clear that AIDS was associated with distinct neurological syndromes such as dementia and painful neuropathy that appeared to result solely from HIV. It also became recognized that the risk of neurological complications increased with the progression of HIV infection and decline of CD4+ counts. Clinically apparent neurological disease develops in approximately half of HIV-infected patients. Neuropathological abnormalities are nearly universal in patients dying with AIDS, suggesting subclinical disease, underdiagnosis, or both in many cases. Neurological disorders cause significant morbidity and mortality, and they may be the AIDS-defining illnesses in previously asymptomatic HIV disease or, occasionally, herald unrecognized HIV infection. Nervous system complications may directly threaten life as well as impair a patient’s ability to work or comply with complex HAART regimens necessary to manage HIV disease optimally. These disorders affect every level of the neuraxis, and a given patient may suffer more than one HIV-associated neurological disease.
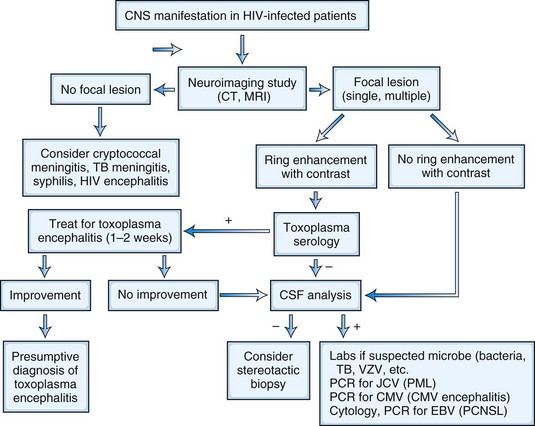
The neurological complications of HIV infection occur at all stages of the disease (Simpson and Berger, 1996). Disorders of both the CNS and peripheral nervous system (PNS) can complicate HIV infection from the period after initial infection through the end stages of the severe immunosuppression. These neurological complications can be classified in a number of ways. A classification based on the underlying pathophysiology and HIV disease stages is summarized in Fig. 53A.1 and Table 53A.3. Clinicians must be aware of the possibility that more than one nervous system site can be involved at the same time, and deficits from one site may be masked by other lesions elsewhere in the neuraxis. The clinician must also be vigilant for other common conditions that are not necessarily associated with HIV or AIDS. Experience in large HIV clinics indicates that the diagnosis of neurological complications of HIV infection and AIDS is far from an academic exercise. Rather, precise diagnosis is critical, and it frequently leads to specific therapy with resultant reduction in morbidity and mortality and in preservation of meaningful function and quality of life. Fig. 53A.3 summarizes a diagnostic algorithm for CNS manifestations in HIV-infected patients.
Table 53A.3 Major HIV-Associated CNS Disorders Classified by Neuroanatomical Localization
| MENINGES |
ALS, Amyotropic lateral sclerosis; CMV, cytomegalovirus; CNS, central nervous system; ddC, dideoxycytidine; d4T, didehydrodeoxythymidine; HIV, human immunodeficiency virus.
Major HIV-Associated Neurological Syndromes
Diffuse Disorders of the Meninges and Brain
HIV-Associated Neurocognitive Disorder
HAND, also known as AIDS dementia complex (ADC) or HIV encephalitis, is characterized by cognitive, behavioral, and motor dysfunction, usually developing later in the course of infection (McArthur et al., 2010; Price and Spudich, 2008; Yadav and Collman, 2009). HIV-associated dementia (HAD) and its less severe forms, minor neurocognitive disorder (MND) and asymptomatic neurocognitive impairment (ANI), are the most common complications of untreated HIV disease. An alternative terminology, HIV-associated cognitive/motor complex, has also been proposed to encompass the full constellation of this syndrome. Characteristically, HAD and MND forms of HAND manifest after patients have developed AIDS-defining systemic illnesses. However, a small number of patients present with HAD at a time when they do not yet fulfill formal diagnostic criteria of AIDS, although typically they show significant immunosuppression by laboratory criteria. Recognition of this early presentation was instrumental in the addition of HAD to the diagnostic criteria of AIDS.
Clinical Features
ANI is the mildest form of HAND and occurs in about 30% of individuals (McArthur et al., 2010). ANI is characterized by measurable neurocognitive impairment that is often unrecognized by the patient or fails to impact function. ANI can be considered as a presymptomatic form of HAND; individuals with ANI are likely to go on to develop symptomatic cognitive dysfunction.
One major component of HAD syndrome is psychomotor dysfunction. Although cognitive dysfunction usually appears earlier than motor symptoms and continues to predominate, motor manifestations in the form of poor balance and incoordination may be the initial presentation. Fine and skilled hand movements are affected early, resulting in deterioration of handwriting. Gait incoordination may result in frequent tripping or falling or a need for extra caution in walking. The latter may impart a slow and somewhat rigid character to the gait. Pivoting is slow and the posture often stooped. Concomitant VM is frequently observed at autopsy. Even if not symptomatic, motor abnormalities such as hyperreflexia and positive Babinski sign may be detected on examination early in the course of the disease. In HAART-treated individuals, the prevalence of HAD is low (approximately 5%); however, this disorder has dramatic effect on quality of life and survival (McArthur et al., 2010).
Initially, formal mental status testing in ANI may be normal. As the disease progresses, however, patients begin to perform poorly on tasks requiring concentration and attention, such as digit and word reversals and serial sevens. With advancing disease, multiple domains of cognitive function are affected. However, mental slowing continues to remain a prominent feature, and afflicted individuals often appear apathetic with poor insight, and even indifferent to their illness. Parallel to cognitive deterioration, the gait disturbance may become disabling. Associated HIV VM causes worsening of leg weakness, and paraparesis limits walking. Postural tremors are common, and on occasion, patients may exhibit choreiform movements and myoclonic jerks (Cardoso, 2002). Bowel and bladder disturbances are late manifestations. Endstage patients are vegetative, lying in bed mute with a vacant stare, unable to ambulate, and incontinent. Characteristically, the course is notable for the absence of focal or lateralizing neurological deficit, such as hemiparesis or aphasia. Their presence suggests another etiology for cognitive impairment or a concomitant neurological disorder.
Diagnosis
Neuropsychological Testing
Formal neuropsychological testing can be useful in diagnosis, management, and clinical monitoring of individual patients and in clinical research studies. Appropriately chosen neuropsychological tests that target the same cardinal cognitive dysfunction delineated by the AIDS-directed clinical assessment provide a formal quantitative means of monitoring patients serially. These assessments focus chiefly on motor speed, concentration, and motor manipulation. Bedside neuropsychological tests (Power et al., 1995) have been developed to provide a quantitative scale of cognitive impairment in the same fashion as the Mini-Mental Status Examination for Alzheimer disease. Neuropsychological test findings are not disease specific and should always be interpreted in the clinical context. The neuropsychological tests do not substitute for the clinical neurological examination. Clinicians should be vigilant because some patients with MND perform within the population norms, and some without HAD/MND perform poorly on testing for other reasons.
Neuroimaging
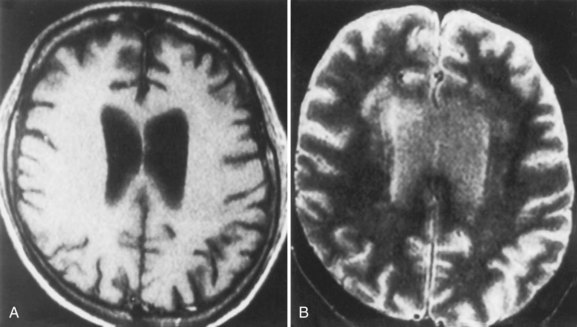
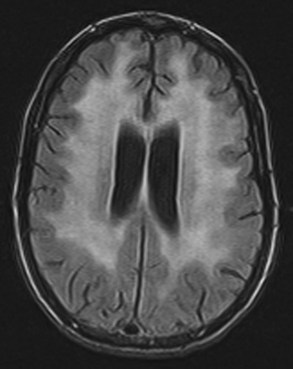
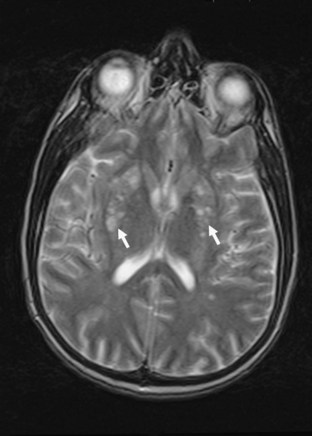
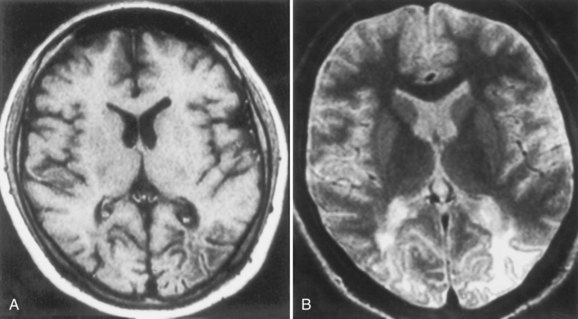
Neuroimaging procedures are often essential in evaluating the AIDS patient with cognitive dysfunction. First, they can be particularly helpful in ruling out other conditions such as primary CNS lymphoma. Second, they often reveal abnormalities that are characteristic, although not pathognomonic, of HAD. Diffuse cerebral atrophy, generally greater centrally than cortically, on either computed tomography (CT) or magnetic resonance imaging (MRI) is commonly observed in HAD (Fig. 53A.4). In some patients, MRI shows abnormalities in the hemispheric white matter (Fig. 53A.5) and less commonly in basal ganglia and thalamus, with patchy or diffusely abnormal signals most apparent on fluid-attenuated inversion recovery (FLAIR) images. Children with AIDS-related dementia may have basal ganglia calcification in addition to atrophy. Results of metabolic imaging, diffusion tensor imaging (DTI), single-photon emission computed tomography (SPECT), and MR spectroscopy have also been reported, although their utility in clinical diagnosis remains undefined.
Neuropathology
Histopathological abnormalities in HAD include white-matter pallor, perivascular mononuclear cellular infiltrates, multinucleated cell encephalitis (see Fig. 53A.2), and vacuolar myelopathy. Other findings include vacuolar changes in the brain, focal necrosis, and neuronal loss. These abnormalities are most common in the subcortical structures (i.e., hemispheric white matter, basal ganglia, thalamus, brainstem, spinal cord). The cerebral cortex is relatively spared. Findings on neuroimaging and neuropathology indicative of the subcortical nature of the disease correlate with the clinical findings of a predominantly subcortical dementia.
Management
Accumulating evidence indicates that HAD can be treated and even prevented, at least to some extent, by antiretroviral therapy. Controlled studies have shown that HAART improves neuropsychological performance in patients with HAD (McArthur et al., 2010; Price and Spudich, 2008; Sacktor et al., 2006; Yadav and Collman, 2009). There has been at least a 50% decrease in HAD in developed countries since HAART introduction in the late 1990s. With a growing list of HIV therapies in several classes of drugs (described previously; see Table 53A.2), many combination regimens are potentially available.
For the successful management of HAD, HAART should be designed to enhance the probability of a patient’s adherence to the therapy. The very nature of the disorder can make adherence to complicated treatment schemes particularly difficult. Simpler regimens with the least number of drug side effects are important to enhance adherence to therapy. Selecting antiretroviral drugs that enter the CSF and brain in patients with HAD is optimal (Letendre et al., 2008). The relative CNS penetration of different antiretroviral drugs is summarized in Table 53A.2. Effective HAART outside the CNS compartment, with rapid reduction of plasma HIV RNA copies, also results in low viral copies in the CNS and clinical neurological improvement. This indicates a dynamic exchange of virus between these two compartments. In patients with effective HAART, HAD is now uncommon and has been replaced by milder forms of HAND. The possible neurotoxic role of viral antigens, cytokines, and other substances in HAND and strategies to counteract these neurotoxicities are currently under active research. Conventional antipsychotic medications are of help in some patients with psychobehavioral alteration. Patients should be encouraged to participate in clinical trial research that may provide the patient the best available therapy and also assist future development of therapy. Treatment trials and a listing of health providers with expertise in this area may be accessed through the Neurological AIDS Research Consortium website at http://narc.wustl.edu/narc/default.aspx.
Cryptococcal Meningitis
Cryptococcal meningitis is covered in Chapter 53C. In the pre-HAART era, approximately 10% of patients with AIDS developed cryptococcal meningitis, a neurological OI caused by the encapsulated yeast, Cryptococcus neoformans. Although the incidence has decreased with the widespread use of HAART in resource-rich countries (Bicanic and Harrison, 2005; Sacktor, 2002), cryptococcosis remains the most common cause of adult meningitis in Southern and Eastern Africa and a major cause of mortality (Park et al., 2009). Cryptococcal meningitis complicates advanced HIV infection (CD4+ counts generally <100/mL) and presents with headache, fever, stiff neck, and photophobia. However, meningeal symptoms and signs may be minimal or absent in over half of cases, and the rather broad clinical spectrum includes malaise, personality change, cognitive impairment, cranial neuropathy, altered mentation, and coma. Cranial CT or MRI may reveal hydrocephalus, gelatinous pseudocysts, infarction, or cryptococcoma (Fig. 53A.6). Not uncommonly, neuroimaging reveals only cerebral atrophy related to advanced HIV disease. Elevation of intracranial pressure (ICP) and hydrocephalus are common in cryptococcal meningitis. The CSF profile ranges from striking protein elevation, mononuclear pleocytosis, and hypoglycorrhachia to minimal abnormalities that overlap with those attributable to HIV infection alone. Fungal CSF culture is the gold standard, but incubation requires weeks. India ink smear is helpful when positive, but is too insensitive to exclude the diagnosis if negative. Fortunately, CSF cryptococcal antigen (CrAg) testing is a rapid, specific test with a sensitivity exceeding 90%. The rather diverse clinical presentations of cryptococcal meningitis may indicate that CrAg be performed routinely in patients with AIDS undergoing diagnostic CSF examination.
Similar to other CNS OIs in HIV-infected patients, the treatment for cryptococcal meningitis consists of an induction phase followed by maintenance therapy or secondary prophylaxis to prevent relapse (Table 53A.4). A typical acute (induction) regimen consists of amphotericin B (0.5 to 0.7 mg/kg/day) with or without flucytosine (75 to 150 mg/kg/day) for 2 to 3 weeks. Renal insufficiency, hypokalemia, and hypomagnesemia may complicate amphotericin B therapy, and the hematological toxicity of flucytosine sometimes precludes its use in patients with AIDS, in whom pancytopenia is common. Patients who are doing well can be switched to fluconazole, 200 mg twice a day for 8 to 10 weeks (Apisarnthanarak and Powderly, 2001) and then placed on maintenance therapy of 200 mg daily to prevent relapse. Although fluconazole therapy may be as effective as amphotericin B for acute therapy of cryptococcal meningitis, delayed CSF clearance of the fungus and a trend toward poorer outcomes among fluconazole-treated patients suggest this approach be reserved for patients with mild disease.
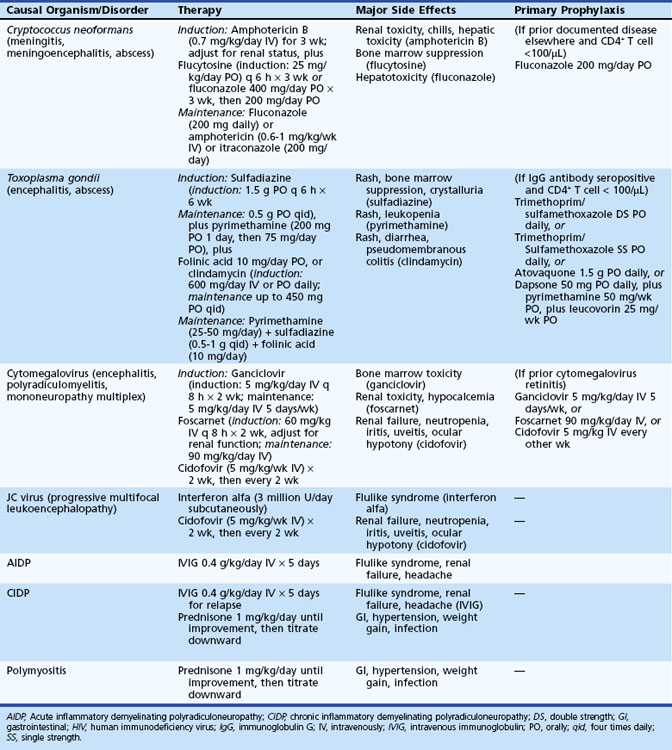
Poor prognostic features at presentation include impaired level of consciousness, CSF cell count less than 20 cells/µL, increased ICP, and CSF CrAg greater than 1 : 1024. Acute mortality in 10 weeks ranges from 10% to 25% in the developed world and up to 43% in resource-poor environments (Jarvis and Harrison, 2007). Medical management with corticosteroids or acetazolamide and CSF drainage with repeated lumbar punctures or ventriculostomy can be used to lower ICP, although none of these interventions has been subjected to clinical trial. Optic nerve sheath fenestration has been suggested as an adjunct to these measures when increased ICP threatens vision. Focal cerebral signs accompanying cryptococcal meningitis may suggest stroke from cryptococcoma or infectious vasculitis. Other complications include cranial neuropathies and obstructive or communicating hydrocephalus. Without chronic suppressive therapy, relapse rates exceed 50%. First-line maintenance therapy is fluconazole 200 mg daily; second-line agents include weekly amphotericin B or itraconazole. Secondary prophylaxis to prevent relapse after induction therapy should be continued at least until there is a sustained response in CD4+ T-cell counts (i.e., >200 cells/µL for at least 6 months). In patients who escape early complications, cryptococcal meningitis is compatible with long-term survival in patients who tolerate and adhere to HAART.
Neurosyphilis
Neurosyphilis is covered in Chapter 53C. Diagnosis and management of neurosyphilis in HIV-infection poses complex challenges. Syphilis and HIV infection often coexist, as the disorders share risk factors. Moreover, both infections are characterized by diverse neurological syndromes affecting brain, meninges, spinal cord, and nerve roots. CNS invasion in early syphilis appears to occur at similar rates in patients with and without HIV infection (Pao et al., 2002). Individuals without HIV infection frequently clear Treponema pallidum from CSF even without antibiotic therapy. Whether that is true in HIV-infected patients is less certain. Meningeal disorders characteristic of early neurosyphilis dominate case reports of HIV infection and neurosyphilis, perhaps implicating impaired CSF clearance of T. pallidum in HIV-infected patients, even with adequate therapy for early syphilis. Other clinical syndromes described in association with HIV infection include syphilitic eye disease, gumma, and myelopathy. Clinical experience indicates that co-infection with HIV and syphilis accelerates the development of meningovascular complications of the latter, often with early strokes.
Several factors complicate the diagnosis of neurosyphilis, particularly asymptomatic forms, in the setting of HIV infection. Individuals co-infected with HIV and T. pallidum may demonstrate unusual serological responses. When clinical suspicion of neurosyphilis is high and syphilis serology results are negative, repeat testing to exclude the prozone effect and darkfield examination or immunofluorescence staining of the lesion obtained by biopsy may be necessary (Berger, 1991; Berger and Levy, 1997). Assuming an atraumatic lumbar puncture, the CSF Venereal Disease Research Laboratory (VDRL) test is quite specific but not particularly sensitive for diagnosing neurosyphilis (see Chapter 53C). Moreover, relying on CSF pleocytosis and protein elevation to make the diagnosis, as is done for HIV-negative individuals, is complicated by the high frequency of these CSF abnormalities caused by HIV infection alone. A CSF pleocytosis is less common in HIV infection when CD4 counts are low (Marra et al., 2007). Treponeme-specific tests in CSF are quite sensitive but not specific for neurosyphilis. Hence, negative CSF fluorescent treponemal antibody or microhemagglutination-T. pallidum excludes the diagnosis of neurosyphilis, but a positive result does not establish the diagnosis. HIV-infected patients with late latent syphilis or syphilis of unknown duration should undergo CSF examination before therapy, regardless of whether there are associated ophthalmic, vestibular, or neurological symptoms.
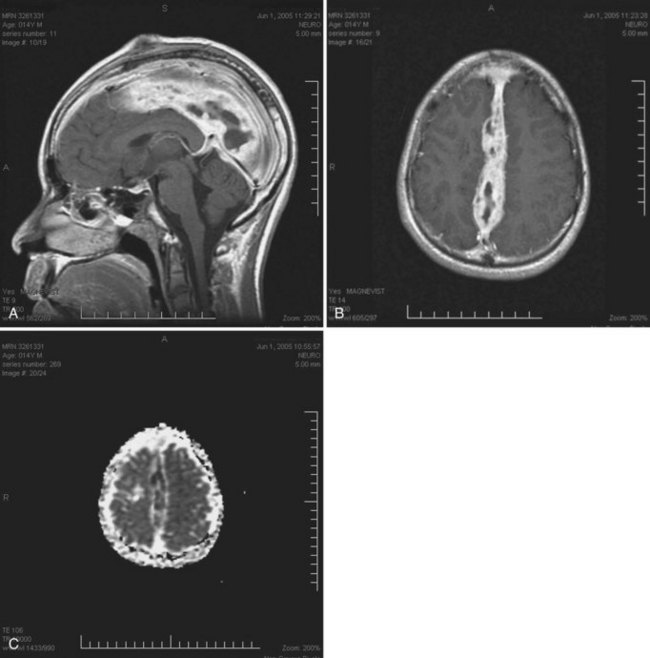
Cytomegalovirus Encephalitis and Ventriculoencephalitis
Cytomegalovirus is a B herpesvirus with worldwide distribution; most adults have serological evidence of latent CMV infection. Clinically evident CMV encephalitis represented about 2% of all neurological complications in HIV patients in the pre-HAART era. The incidence of CMV disease, including CNS infection, has decreased substantially in the HAART era. Now an uncommon cause of global cerebral dysfunction in advanced AIDS (CD4+ count <50/mL), CMV encephalitis and ventriculoencephalitis often cause death within weeks to months (Williams, 1999). Prior or active disseminated CMV disease, such as retinitis, esophagitis, or colitis, may provide important clues to the neurological diagnosis. CMV encephalitis typically presents as a confusional state evolving over weeks and can resemble HAD. In addition to a course that is more subacute than chronic, focal cerebral signs, hyponatremia, and cranial MRI showing periventricular enhancement are other factors that favor a diagnosis of CMV encephalitis over HAD. CSF abnormalities are typically nonspecific, and CMV PCR is positive in fewer than half of cases. Pathological findings include microglial nodules and cytomegalic cells in cortical and subcortical gray matter, thought to be consistent with hematogenous spread of CMV to brain. By contrast, CMV ventriculoencephalitis may reflect dissemination from CSF and presents more acutely than CMV encephalitis, often on a background of CMV retinitis or concurrently with polyradiculomyelopathy. Brainstem signs and neuroimaging studies revealing dilated ventricles also suggest the diagnosis. CSF abnormalities tend to be more striking than with CMV encephalitis, revealing elevated protein, polymorphonuclear or lymphocytic pleocytosis, and normal or low glucose levels. Identifying CMV DNA in CSF by PCR helps confirm the diagnosis (see Fig. 53A.3). Pathological features in CMV ventriculitis include ependymal necrosis and subependymal gliosis with cytomegalic cells but generally no microglial nodules. CMV viremia is quite common in late-stage AIDS, so viral detection in this setting by culture, antigen testing, or PCR from blood does not help establish the diagnosis of CMV-related neurological syndromes. Prospective data are needed to confirm the impression that CSF PCR for CMV DNA is specific and reasonably sensitive for these and other CMV-related neurological disorders.
Three drugs are currently available for treating systemic CMV infection: ganciclovir, a guanosine nucleoside analog; foscarnet, a pyrophosphate analog; and cidofovir, a cytosine nucleotide analog. All have major limitations in AIDS patients because they are only virostatic, their administration is inconvenient, and they have major side effects. Clinical trial data for the efficacy of ganciclovir, foscarnet, cidofovir, or combination therapy for CMV encephalitis and ventriculoencephalitis are lacking, though a trial of empirical therapy is probably appropriate given the poor prognosis (see Table 53A.4). Effective HAART and thereby successful restoration of the immune response against CMV is the most efficacious therapy to improve the dismal outcome in these patients.
Other Meningitis and Meningoencephalitis Syndromes
Other causes of meningitis complicating HIV infection include other rare infections, neoplasms, and medications. Additional infectious causes of meningitis include bacteria (Salmonella typhi, Pneumococcus pneumoniae, Mycobacterium tuberculosis, T. pallidum, Nocardia asteroides, Listeria monocytogenes, Bartonella henselae) and fungi (Histoplasma capsulatum and Coccidioides immitis in individuals who have lived in endemic regions, and Candida albicans, Blastomyces dermatitidis, and Sporothrix schenckii). Pyogenic meningitis can be complicated by subdural empyema (Fig. 53A.7) that may require surgical drainage. Patients with AIDS are at increased risk for systemic lymphoma, which can cause lymphomatous meningitis. Medications are often overlooked as a cause of meningitis; implicated agents in common use for HIV infection include nonsteroidal antiinflammatory drugs (NSAIDs), trimethoprim/sulfamethoxazole (TMP/SMX), and intravenous immunoglobulin (IVIG). Causes of meningoencephalitic syndromes include VZV and the parasites Toxoplasma gondii, Trypanosoma cruzi, and Acanthamoeba spp. (Ambroise-Thomas, 2001).
Focal Central Nervous System Disorders
Three disorders account for most HIV-related focal CNS dysfunction: cerebral toxoplasmosis, PCNSL, and PML. The epidemiology of these CNS OIs changed dramatically following the availability of HAART in 1996. All complicate advanced HIV infection. While the clinical features may overlap, neuroimaging studies, CSF analysis, response to therapeutic trial, and brain biopsy permit distinction (see Fig. 53A.3).
Cerebral Toxoplasmosis
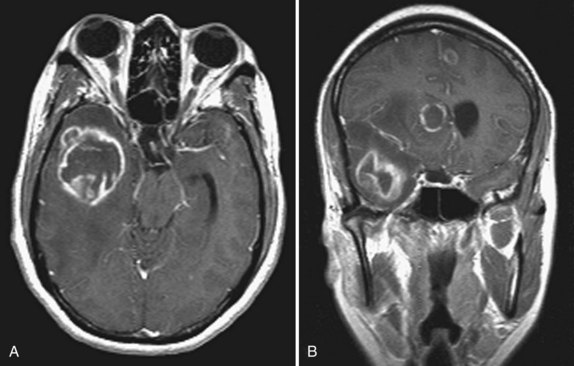
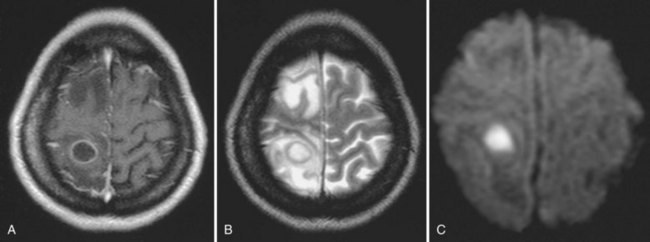
The most common clinical presentation in TE is headache and focal neurological deficit, with or without fever. Patients may present with seizure, hemiparesis, or aphasia as a manifestation of these focal lesions or with a picture more influenced by the accompanying cerebral edema, characterized by confusion, mental torpor, and lethargy, which can progress to coma. Rarely, toxoplasmosis manifests as acute meningoencephalitis or with predominantly muscle involvement. Diagnosis is usually suspected on the basis of CT or MRI findings of multiple lesions in multiple locations, although in some cases, only a single lesion is seen. The lesions have a predilection for the basal ganglia. Pathologically, these lesions generally exhibit inflammation and central necrosis and, as a result, demonstrate ring enhancement on contrast CT or MRI (Fig. 53A.8). There is usually surrounding edema. In addition to toxoplasmosis, the differential diagnosis of multiple mass lesions in the HIV-infected patient include PCNSL (see the following discussion) and, less commonly, tubercular, fungal, or bacterial abscesses. Resolution of lesions on antitoxoplasma therapy confirms the clinical diagnosis. The definitive diagnostic procedure is brain biopsy, but it carries morbidity and therefore is reserved for the patient who has failed 2 to 4 weeks of empirical antitoxoplasma therapy (see Fig. 53A.3).
Progressive Multifocal Leukoencephalopathy
JC virus (JCV) is a DNA-containing human papillomavirus that is the causal agent for PML and an important opportunistic pathogen in patients with AIDS (Tan and Koralnik, 2010). Serological studies indicate that approximately 50% to 70% of the adult population harbors antibodies to JCV, typically acquired in childhood. PML is the most common complication of JCV infection. Rare JCV disorders include a granular cell cerebellar atrophy and encephalitis. PML is a late manifestation of AIDS and is seen in approximately 4% to 5% of patients with AIDS (Berger et al., 1987; Berger et al., 1998). Although its incidence seems to have decreased somewhat during the past several years, the decline has not been as marked as for other OIs.
PML is a demyelinating disease of the central nervous system that results from infection of oligodendrocytes with JCV. The lesions of PML begin as small foci of demyelination in subcortical white matter that gradually expand and may coalesce. The cerebellum, brainstem, and (exceptionally rarely) spinal cord can be involved in PML. Patients typically have a protracted course with focal neurological deficit, with or without changes in mental status. Visual field defects, hemiparesis, aphasia and other language disorders, sensory defects, and ataxia may occur. MRI typically reveals multiple nonenhancing white-matter lesions with a predilection for frontal, occipital, or parietal lobes (Fig. 53A.9); contrast enhancement on MRI is observed in as many as 15% of patients, even without IRIS (Berger et al., 1998). CSF is usually normal or shows nonspecific changes, but the viral DNA can be amplified from the CSF sample. CSF PCR for JCV DNA is specific for the diagnosis of PML and decreases the need for brain biopsy in the appropriate clinical context. In cases in which viral DNA is not detected in CSF, a brain biopsy is necessary to confirm the diagnosis. Brain biopsy reveals bizarre giant astrocytes with pleomorphic hyperchromatic nuclei, altered oligodendrocytes with enlarged nuclei that contain viral inclusions, and myelin loss.
There is no specific therapy for PML. Mean survival is 2 to 4 months; approximately 8% of patients experience spontaneous remission (Simpson and Berger, 1996). Despite anecdotal evidence of benefit from cytosine arabinoside and vidarabine, a controlled study showed no benefit. Other drugs including interferon alfa, topoisomerase inhibitors, and cidofovir have been used in AIDS-related PML, with generally unsatisfactory results. Improved understanding of the mechanism of JCV attachment and cellular entry has suggested that drugs that block the 5HT-2a receptor (mirtazapine, for example) necessary for the former or that block clathrin-dependent cell entry (e.g., chlorpromazine) may prove useful in treating PML but remain unproven (Verma et al., 2007). Regression of PML lesions and prolonged survival has been reported in patients with PML treated successfully with HAART for their HIV disease (Berger and Mucke, 1988; Clifford, 2002). Factors influencing a favorable outcome include higher CD4+ counts at baseline, the ability to maintain an HIV viral load of less than 500 copies per milliliter, undetectable JCV in CSF following HAART, contrast-enhancing lesions at time of diagnosis, and the presence of JCV-specific cytotoxic T lymphocytes.
Primary Central Nervous System Lymphoma
Primary CNS lymphomas (PCNSLs) of B-cell origin are considered opportunistic neoplasms that complicate the course of AIDS in up to 5% of patients (Gerstner and Batchelor, 2010). The incidence of PCNSL in HIV-infected individuals dropped substantially following the introduction of HAART in 1996, but now it appears to be cumulatively rising because of the increased longevity of HIV-infected persons following the efficacy of both prophylactic and therapeutic measures against OIs and HAART. Patients with PCNSL present with progressive focal or multifocal neurological deficits similar to those seen with toxoplasmosis and PML (DeAngelis, 2001). Autopsy studies have revealed that PCNSL is virtually always multifocal, even when solitary lesions are seen on neuroimaging studies. The tempo of symptom evolution in PCNSL is generally slower than in toxoplasmosis and faster than in PML, with patients with PCNSL presenting several days or a few weeks after the onset of symptoms, which may include headache, hemiparesis, aphasia, ataxia, behavioral changes, and altered mentation. Lymphomatous meningitis or ocular involvement occurs in about 15% of cases. Systemic dissemination of PCNSL occurs in about 5% of patients, mainly in the final stages of the disease. Fever and constitutional symptoms are absent except in patients with associated systemic infection.
MRI is more sensitive than CT scan for PCNSL and characteristically shows one or more lesions located deep in the brain, adjacent to the lateral ventricles and often in white rather than gray matter. MRI may show characteristic subependymal extension (Fig. 53A.10). In about 10% of patients, PCNSL is located in the posterior fossa. Mass effect may be present, but there is usually little contrast enhancement or surrounding edema. CSF cytology is frequently unhelpful, although the presence of monoclonal B lymphocytes by flow cytometry indicates PCNSL. PCR amplification of Epstein-Barr virus DNA in CSF corroborates the diagnosis of PCNSL. The definitive diagnosis of PCNSL generally requires brain biopsy. Most often, brain biopsy is undertaken after a therapeutic trial for cerebral toxoplasmosis. Uptake on thallium-201 SPECT supports the diagnosis of PCNSL and may prompt early biopsy. The use of stereotactic biopsy techniques has increased the access to these tumors and reduced the morbidity of biopsy.
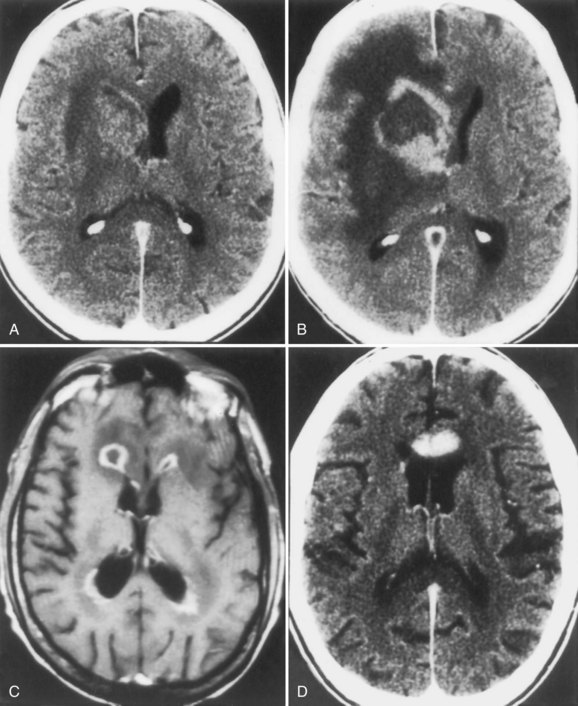
Before the HAART era, the outcome of AIDS-associated PCNSL had been dismal, with median survival of approximately 2 to 3 months. The most important factors related to survival are the clinical condition of the patient, the CD4+ count, and the absence of OIs. As with PML, the prognosis in PCNSL is improved by successful HAART and immune reconstitution. Vigorous attempts to suppress HIV replication are recommended in all patients (Cingolani et al., 2005). Mass effect is treated by high-dose corticosteroid therapy. Palliative whole-brain irradiation therapy is recommended. The use of chemotherapy remains controversial outside a clinical research trial setting.
Stroke
A wide spectrum of cerebrovascular disorders has been associated with HIV infection (Dobbs and Berger, 2009). Ischemic and hemorrhagic strokes have been reported in up to 4% of clinical series and up to 34% of autopsy series (Pinto, 1996). HIV infection itself appears to elevate stroke risk by a number of mechanisms. Thrombocytopenia, coagulopathy related to liver disease or disseminated intravascular coagulation, PCNSL, metastatic Kaposi sarcoma, and (rarely) toxoplasmosis may be associated with cerebral hemorrhage. Causes of ischemic stroke include HIV cerebral vasculitis, co-infection with syphilis with early meningovascular strokes (see previous discussion), bacterial endocarditis (particularly in IDUs), as well as nonbacterial thrombotic endocarditis and procoagulant states. Granulomatous angiitis of the nervous system has been reported in AIDS. VZV and tuberculous meningitis can cause infectious vasculitis, as can the angioinvasive fungi, Aspergillus and Mucor.
Other Focal Central Nervous System Disorders
Numerous other infections have been reported to cause focal cerebral dysfunction in HIV-infected patients. Bacteremia from indwelling catheters needed to manage other aspects of HIV infection or from parenteral drug use predisposes to bacterial brain abscess (Fig. 53A.11). Other bacterial causes of focal cerebral dysfunction include M. tuberculosis abscess, syphilitic gumma, B. henselae, and N. asteroides. Fungal causes of focal brain disease, in addition to the angioinvasive fungi discussed previously, include cryptococcoma, B. dermatitidis, and H. capsulatum. Among parasites, relevant diagnostic considerations in patients who have lived in or traveled through endemic areas are cysticercosis and intracerebral Chagas disease (T. cruzi). VZV can cause a demyelinating syndrome with lateralizing features, and CMV has been reported to cause mass lesions (Moulignier et al., 1996). Although HIV infection does not appear to significantly increase the risk for herpes simplex virus encephalitis, it may alter its clinical presentation. The long-term effect of the protease inhibitor–induced metabolic syndrome on atherosclerosis and stroke is currently the topic of clinical research.
HIV-Associated Vacuolar Myelopathy
VM is the most common cause of spinal cord dysfunction in untreated patients with AIDS, apparent pathologically in 25% to 55% of AIDS autopsy series (Di Rocco and Simpson, 1998; McArthur et al., 2005). VM complicates late HIV infection and frequently coexists with HAD and DSPN. Affected patients develop gait difficulty caused by spasticity, leg weakness, and impaired proprioception, often accompanied by sphincter dysfunction, evolving over several months. Back pain is not a prominent feature. Examination reveals spastic paraparesis with Babinski signs and hyperreflexia unless concomitant neuropathy is severe. Sensation in the legs, particularly proprioception and vibratory sense, is usually impaired, but a discrete sensory level should suggest another etiology of the myelopathy. The arms are typically spared. MRI may occasionally reveal cord atrophy but usually is unremarkable. Pathological findings are most striking in the dorsolateral thoracic cord and include vacuolar changes in myelin sheaths, with relative preservation of axons. Despite the clinical and pathological resemblance to combined systems degeneration, vitamin B12 levels are typically normal in affected patients. HIV-induced release of neurotoxic cytokines or abnormalities in vitamin B12 utilization may contribute to the development of VM.
HIV-Associated Neuromuscular Disorders
Neuropathies
Peripheral neuropathies are common in HIV infection (Cornblath and Hoke, 2006; Robinson-Papp and Simpson 2009; Sacktor, 2002; Verma, 2001). Peripheral neuropathies complicate all stages of HIV disease and cause considerable morbidity and disability in HIV-infected individuals and AIDS patients. Although symptomatic neuropathy occurs in approximately 10% to 25% of HIV-infected patients overall, pathological evidence of peripheral nerve involvement is present in virtually all end-stage AIDS patients. There are five major clinical types of HIV-associated neuropathies that are regularly seen in large HIV clinics: DSPN, acute and chronic inflammatory demyelination polyradiculoneuropathies (AIDP and CIDP), CMV-associated polyradiculomyelopathy, and nucleoside-associated toxic neuropathies. A vasculitic neuropathy is less common but often responds well to corticosteroid treatment (Bradley and Verma, 1996).
Distal Sensory Polyneuropathy
Of the various peripheral nerve syndromes that complicate HIV infection, the most common is DSPN, also called HIV-associated neuropathy or AIDS neuropathy. This axonal, predominantly sensory, length-dependent polyneuropathy develops in approximately one-third of patients with AIDS, becoming more prevalent as the CD4+ count decreases. The 1-year incidence of DSPN showed a steep decline in the post-HAART compared with the pre-HAART era (Power et al., 2009). The overall prevalence of DSPN appears to be increasing, however, as HIV-infected persons live longer with the disease.
The rather typical clinical features usually obviate the need for electromyography and nerve conduction studies. Exposures to neurotoxins, including ethanol, should be reviewed. Neurotoxic drugs commonly used to manage HIV infection include the nucleoside analogs, didanosine, zalcitabine, and stavudine (see Table 53A.2), in addition to isoniazid, pyridoxine, dapsone, metronidazole, and vincristine. Screening for vitamin B12 deficiency and diabetes mellitus is important.
Nucleoside Analog–Associated Toxic Neuropathy
A painful polyneuropathy that closely resembles DSPN is the major dose-limiting toxicity of the nucleoside analog antiretroviral agents, didanosine, zalcitabine, and stavudine (Cornblath and Hoke, 2006; Robinson-Papp and Simpson 2009; Verma, 2001). Two clinical features can help distinguish nucleoside neuropathy from DSPN. First, nucleoside neuropathy typically evolves over weeks following initiation of therapy, in contrast to DSPN, which progresses over months or even years. Second, stopping the offending agent eventually leads to stabilization and regression of nucleoside neuropathy over several months, although “coasting,” in which symptoms worsen for several weeks before improvement, may complicate the evaluation of this strategy for diagnosis. Although preexisting DSPN increases the risk for nucleoside neuropathy, many patients with DSPN tolerate neurotoxic antiretrovirals, particularly if the dose is kept low. Similarly, many patients who develop nucleoside neuropathy can resume therapy at a lower dose or may tolerate a different neurotoxic drug. Other aspects of the evaluation and treatment are similar to those for DSPN.
Inflammatory Demyelinating Polyradiculoneuropathies
Less common than the painful neuropathies related to HIV or nucleoside antiretrovirals are the inflammatory demyelinating polyradiculoneuropathies, AIDP and CIDP (Cornblath and Hoke, 2006; Robinson-Papp and Simpson, 2009). These disorders resemble the syndromes seen in individuals without HIV infection with regard to pathogenesis and most clinical features. The precise prevalence is unknown, but case series from the United States and Africa suggest that they often develop during early HIV infection, sometimes around the time of seroconversion. AIDP, or the Guillain-Barré syndrome, typically presents as rapidly progressive ascending weakness with areflexia, variably accompanied by respiratory failure and dysautonomia. The Miller-Fisher variant, in which cranial nerve dysfunction, areflexia, and ataxia are more prominent than limb or respiratory weakness, has also been described. In CIDP, neuropathic weakness and sensory loss occur in a more indolent or episodic manner than in AIDP. In both AIDP and CIDP, electrophysiological studies reveal slowed conduction, temporal dispersion, multifocal block, and prolonged F waves, indicating demyelination. In HIV-infected patients with inflammatory demyelinating polyneuropathy, the CSF often shows a lymphocytic pleocytosis (10 to 50 cells/mL) in addition to increased protein. Clinical experience suggests that intravenous immunoglobulin, plasmapheresis, and corticosteroids (only in CIDP) are beneficial.
Lumbosacral Polyradiculomyelitis
Subacute lumbosacral polyradiculomyelitis with variable cord involvement is an uncommon HIV-related syndrome that results from a variety of infectious agents, most notably CMV. Clinical manifestations suggest a rapidly developing cauda equina syndrome, with leg weakness and later paralysis, sphincter dysfunction, sacral and leg paresthesias and sensory loss, and areflexia, typically evolving over several days. When such a syndrome develops in a patient with a CD4+ count less than 50/µL, and CSF reveals marked polymorphonuclear pleocytosis, elevated protein, and low to normal glucose levels, CMV infection of the nerve roots, with subsequent inflammation and necrosis, is the likely cause. CMV PCR or branched DNA assay in CSF is a helpful confirmatory test, but treatment should not be delayed pending these results. Intravenous ganciclovir (see Table 53A.4) can arrest and reverse the deficit in CMV polyradiculomyelitis, which is fatal without treatment. Polyradiculomyelitis due to ganciclovir-resistant CMV has been reported and may respond to foscarnet, either alone or with cidofovir. Other causes of polyradiculomyelopathy in AIDS include tuberculosis, neurosyphilis, and lymphomatous meningitis.
Other Neuropathies and Neuronopathies
Mononeuritis multiplex (MM) is a relatively rare peripheral nerve syndrome of HIV infection that manifests clinically as multifocal asymmetrical peripheral nerve lesions that may include cranial nerves. When the syndrome develops in early or midstage HIV infection, it often responds well to corticosteroid therapy, though it may be self-limited, not requiring immunosuppressive therapy (Bradley and Verma, 1996). MM complicating advanced HIV infection, with CD4+ count less than 50/mL, may be caused by CMV and responds to intravenous ganciclovir. Occurrence of classical amyotrophic lateral sclerosis (ALS) in HIV disease is probably fortuitous (Verma and Berger, 2006). An ALS-mimicking syndrome has also been reported in HIV disease (Moulignier et al., 2001).
Myopathies
Myopathic symptoms in HIV-infected individuals can arise from toxic (zidovudine) or dysimmune (polymyositis) causes or from AIDS cachexia (muscle wasting syndrome) (Authier et al., 2005; Robinson-Papp and Simpson, 2009). In HIV-associated polymyositis, patients develop proximal weakness and, less commonly, myalgia—both of which are ascribed to a dysimmune response following HIV infection. On occasion it may occur with immune restoration following HAART (Sellier et al., 2000). Serum creatine kinase is elevated in most cases, and electrophysiological studies often reveal myopathic motor units and increased insertional activity and spontaneous activity typical of an inflammatory myopathy. Muscle biopsy reveals fiber-size variability, fiber degeneration, and endomysial infiltrates. Cytoplasmic bodies and nemaline rod bodies are other common histological features. HIV does not appear to directly infect muscle fibers, but rather induces them to express major histocompatibility complex I, triggering cell-mediated muscle fiber injury. Even so, inflammatory myopathy is among the few HIV-related neurological disorders that develop at any time during HIV infection. Despite the potential risks of corticosteroid therapy in the setting of HIV infection, such treatment is often well tolerated. Prednisone has helped in motor recovery and pain improvement in HIV-associated polymyositis. Starting with a dose of 1 mg/kg/day, the dose is titrated downward as strength improves. Inclusion body myositis also has been reported (Authier et al., 2005).
Zidovudine myopathy is a toxic mitochondrial disorder that presents with the insidious onset of proximal weakness and myalgia, making it difficult to distinguish clinically this condition from HIV-associated inflammatory myopathy (Robinson-Papp and Simpson, 2009). Though first described in patients taking zidovudine in doses of 1000 mg/day or more, zidovudine myopathy also develops on lower-dose regimens in current HAART regimens. Affected patients typically have taken zidovudine for at least 6 months. Serum creatine kinase may be normal or elevated, and muscle biopsy shows histological features suggesting mitochondrial dysfunction with no or scanty inflammation. Clinical response to a drug holiday or reduction in zidovudine dose often obviates the need for muscle biopsy.
Pyomyositis, a focal suppurative bacterial muscle infection, was more common in the tropics and rare in developed nations before the AIDS epidemic (Authier et al., 2005). Clinical features include fever accompanied by local muscle pain and swelling evolving over several weeks. A source of bacteremia may be evident by history or general examination. Typically the affected area is swollen, hot, and indurated, but not fluctuant. Peripheral white cell count and serum creatine kinase are usually normal, prompting consideration of cellulitis or deep venous thrombosis as the initial diagnosis. Ultrasound, CT, or MRI of the affected area helps establish the diagnosis. Blood cultures may reveal the causative organism, usually Staphylococcus aureus or, less commonly, Salmonella typhi or other gram-negative bacilli. Empirical intravenous antibiotic therapy should be given to cover these pathogens. Surgical drainage may be required. Intravenous therapy for 1 to 2 weeks is usually followed by oral antibiotic therapy for up to 8 weeks.
Ambroise-Thomas P. Parasitic diseases and immunodeficiencies. Parasitology. 2001;122(Suppl):S65-S71.
Apisarnthanarak A., Powderly W.G. Treatment of acute cryptococcal disease. Expert Opin Pharmacother. 2001;2:1259-1268.
Authier F.J., Chariot P., Gherardi R.K. Skeletal muscle involvement in human immunodeficiency virus (HIV)-infected patients in the era of highly active antiretroviral therapy (HAART). Muscle Nerve. 2005;32:247-260.
Berger J.R. Neurosyphilis in human immunodeficiency virus type 1-seropositive individuals. A prospective study. Arch Neurol. 1991;48:700-702.
Berger J.R., Kaszovitz B., Post M.J., et al. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann Intern Med. 1987;107:78-87.
Berger J.R., Levy R.M. AIDS and the Nervous Systems. Philadelphia: Lippincott-Raven; 1997.
Berger J.R., Moskowitz L., Fischl M., et al. Neurologic disease as the presenting manifestation of acquired immunodeficiency syndrome. South Med J. 1987;80:683-686.
Berger J.R., Mucke L. Prolonged survival and partial recovery in AIDS-associated progressive multifocal leukoencephalopathy. Neurology. 1988;38:1060-1065.
Berger J.R., Pall L., Lanska D., et al. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4:59-68.
Bicanic T., Harrison T.S. Cryptococcal meningitis. Br Med Bull. 2005;72:99-118.
Bradley W.G., Verma A. Painful vasculitic neuropathy in HIV-1 infection: relief of pain with prednisone therapy. Neurology. 1996;47:1446-1451.
Cardoso F. HIV-related movement disorders: epidemiology, pathogenesis and management. CNS Drugs. 2002;16:663-668.
Cingolani A., Fratino L., Scoppettuolo G., et al. Changing pattern of primary cerebral lymphoma in the highly active antiretroviral therapy era. J Neurovirol. 2005;11(Suppl 3):38-44.
Clifford D.B. AIDS dementia. Med Clin North Am. 2002;86:537-550.
Corder E.H., Robertson K., Lannfelt L., et al. HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med. 1998;4:1182-1184.
Cornblath D.R., Hoke A. Recent advances in HIV neuropathy. Curr Opin Neurol. 2006;19:446-450.
D’Aquila R.T., Schapiro J.M., Brun-Vezinet F., et al. Drug resistance mutations in HIV-1. Top HIV Med. 2003;11:92-96.
DeAngelis L.M. Primary central nervous system lymphomas. Curr Treat Options Oncol. 2001;2:309-318.
Di Rocco A., Simpson D.M. AIDS-associated vacuolar myelopathy. AIDS Patient Care STDS. 1998;12:457-461.
Dobbs M.R., Berger J.R. Stroke in HIV infection and AIDS. Expert Rev Cardiovasc Ther. 2009;7:1263-1271.
Ellis R., Langford D., Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33-44.
Fauci A.S., Lane H.C. HIV neurology. In: Hauser S.L., editor. Harrison’s Neurology in Clinical Medicine. New York: McGraw-Hill; 2006:467-480.
Gazzard B.G., and the BHVA Treatment Guidelines Writing Group. British HIV Association guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy. HIV Med. 2008;9:563-608.
Geraci A.P., Simpson D.M. Neurological manifestations of HIV-1 infection in the HAART era. Compr Ther. 2001;27:232-241.
Gray F., Bazille C., Adle-Biassette H., et al. Central nervous system immune reconstitution disease in acquired immunodeficiency syndrome patients receiving highly active antiretroviral treatment. J Neurovirol. 2005;11(Suppl 3):16-22.
Greene W.C., Peterlin B.M. Charting HIV’s remarkable voyage through the cell: basic science as a passport to future therapy. Nat Med. 2002;8:673-680.
Hansasuta P., Rowland-Jones S.L. HIV-1 transmission and acute HIV-1 infection. Br Med Bull. 2001;58:109-127.
Jarvis J.N., Harrison T.S. HIV-associated cryptoccal meningitis. AIDS. 2007;21:2119-2129.
Kinter A., Arthos J., Cicala C., et al. Chemokines, cytokines and HIV: a complex network of interactions that influence HIV pathogenesis. Immunol Rev. 2000;177:88-98.
Klein R.S., Williams K.C., Alvarez-Hernandez X., et al. Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J Immunol. 1999;163:1636-1646.
Letendtre S., Marquie-Beck J., Capparelli E., et al. Validation of the CNS Penetration-Effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008:65-70.
Liu P., Hudson L.C., Tompkins M.B., et al. Compartmentalization and evolution of feline immunodeficiency virus between the central nervous system and periphery following intracerebroventricular or systemic inoculation. J Neurovirol. 2006;12:307-321.
Marra C.M., Maxwell C.L., Collier A.C., et al. Interpreting cerebrospinal fluid pleocytosis in HIV in the era of potent artiretroviral therapy. BMC Infect Dis. 2007;7:37.
McArthur J.C., Brew B.J., Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543-555.
McArthur J.C., Steiner J., Sacktor N., et al. Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. 2010;67:699-714.
Moulignier A., Mikol J., Gonzalez-Canali G., et al. AIDS-associated cytomegalovirus infection mimicking central nervous system tumors: a diagnostic challenge. Clin Infect Dis. 1996;22:626-631.
Moulignier A., Moulonguet A., Pialoux G., et al. Reversible ALS-like disorder in HIV infection. Neurology. 2001;57:995-1001.
Pao D., Goh B.T., Bingham J.S. Management issues in syphilis. Drugs. 2002;62:1447-1461.
Park B.J., Wannemuehler K.A., Marston B.J., et al. Estimation of the current global burden of cryptoccal meningitis among persons living with HIV/AIDS. AIDS. 2009;23:525-530.
Pinto A.N. AIDS and cerebrovascular disease. Stroke. 1996;27:538-543.
Power C., Selnes O.A., Grim J.A., et al. HIV Dementia Scale: a rapid screening test. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;8:273-278.
Power C., Boissé L., Rourke S., et al. NeuroAIDS: an evolving epidemic. Can J Neurol Sci. 2009;36:285-295.
Price R.W., Spudich S. Antiretroviral therapy and central nervous system HIV type I infection. J Infect Dis. 2008;197(Suppl 3):S294-S308.
Riedel D.J., Pardo C.A., McArthur J., et al. Therapy insight: CNS manifestations of HIV-associated immune reconstitution inflammatory syndrome. Nat Clin Pract Neurol. 2006;2:557-565.
Robinson-Papp J., Simpson D.M. Neuromuscular diseases associated with HIV-1 infection. Muscle Nerve. 2009;40:1043-1053.
Sabri F., Titanji K., De Milito A., et al. Astrocyte activation and apoptosis: their roles in the neuropathology of HIV infection. Brain Pathol. 2003;13:84-94.
Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8(Suppl 2):115-121.
Sacktor N., Nakasujja N., Skolasky R., et al. Antiretroviral therapy improves cognitive impairment in HIV+ individuals in sub-Saharan Africa. Neurology. 2006;67:311-314.
Scaravilli F., Bazille C., Gray F. Neuropathologic contributions to understanding AIDS and the central nervous system. Brain Pathol. 2007;17:197-208.
Sellier P., Monsuez J.J., Evans J., et al. Human immunodeficiency virus-associated polymyositis during immune restoration with combination antiretroviral therapy. Am J Med. 2000;109:510-512.
Simpson D.M., Berger J.R. Neurological manifestations of HIV infection. Med Clin North Am. 1996;80:1363-1394.
Tan C.S., Koralnik I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9:425-437.
Verma A. Epidemiology and clinical features of HIV-1 associated neuropathies. J Peripher Nerv Syst. 2001;6:8-13.
Verma A., Berger J.R. ALS syndrome in patients with HIV-1 infection. J Neurol Sci. 2006;240:59-64.
Verma S., Kickurel K., Koranik I.J. Mirtazapine in progressive multifocal leukoencephalopathy associated with polycythemia vera. J Infect Dis. 2007;5:709-711.
Weiss J.M., Nath A., Major E.O., et al. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953-2959.
Williams I.G. Management of CMV disease in HIV infection. Int J STD AIDS. 1999;10:211-216.
Xiao X., Kinter A., Broder C.C., et al. Interactions of CCR5 and CXCR4 with CD4 and gp120 in human blood monocyte-derived dendritic cells. Exp Mol Pathol. 2000;68:133-138.
Yadav A., Collman R.G. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol. 2009;4:430-447.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 53A Infections of the Nervous System
Neurological Manifestations of Human Immunodeficiency Virus Infection
Epidemiology and Current Trends
HIV Genome, Replication, and Molecular Heterogeneity of HIV
Natural History of HIV Infection and Neuro-HIV Disease
Neuropathogenesis of HIV Disease
Antiretroviral Therapy and Its Effect on Neuro-HIV Disease
Clinical Spectrum of Neuro-HIV Disease
Epidemiology and Current Trends
Now, 3 decades after the first report of AIDS, this disease has become one of the greatest public health challenges globally. The HIV pandemic has resulted in an estimated 64 million infections worldwide, and it has claimed the lives of more than 30 million persons (www.unaids.org). In 2008, more than 1 million persons were living with HIV/AIDS in the United States, and based on new methods of determining incidence, approximately 56,000 new HIV infections and 25,000 deaths occurred (www.cdc.org). An estimated 44% of new HIV infections are in homosexual men, 17% are in IDUs, and in 35% the infection is transmitted by heterosexual contact. The demography of newly infected individuals has changed considerably since the 1990s in the United States; HIV infection is spreading rapidly in certain populations (racial and ethnic minority populations and women of color) and decreasing (IDUs) or fluctuating in others (homosexual men).
HIV Genome, Replication, and Molecular Heterogeneity of HIV
HIV is a ribonucleic acid (RNA) virus belonging to the family of human retroviruses (Retroviridae) and the subfamily of lentiviruses. Electron microscopy shows that the HIV virion is an icosahedral structure. It contains numerous external spikes formed by the two major envelope proteins (gp120 and gp41) and one major core protein (gp24). Like other retroviruses, HIV has genes that encode the structural and enzyme proteins of the virus: env encodes the surface glycoproteins, gag encodes the core protein, and pol encodes for the enzyme responsible for reverse transcription and viral integration into the host genome. HIV also contains at least six other genes (tat, rev, nef, vif, vpr, and vpu) that encode for proteins involved in gene regulation. The replication cycle of HIV begins with high-affinity binding of the gp120 envelope protein to its receptor, the CD4 molecule, on the host cell surface (Greene and Peterlin, 2002). After gp120 binds to the CD4 molecule, it undergoes a conformational change that facilitates its binding to a co-receptor (CCR5, CXCR4) (Xiao et al., 2000). These co-receptors belong to the family of transmembrane G protein–coupled cellular receptors that determine the cellular tropism of the virus strain. Following surface attachment and fusion, the HIV genomic RNA is uncoated and internalized into the host cell. The subsequent steps in viral replication include activation of reverse transcriptase enzyme, reverse transcription of the genomic RNA into double-stranded deoxyribonucleic acid (DNA), and integration of DNA into the host cell chromosome through the action of another virally encoded enzyme—integrase. Molecular analyses of various HIV isolates from a single person over time or from cohorts of patients reveal sequence variations over many parts of the viral genome. The degree of difference in coding sequences of the viral envelope protein, for example, can vary from a few percent to 50%. The immune pressure and functional constraints on viral proteins (e.g., by antibodies) and the use of retroviral therapy influence the level of variation in viral genes and protein products.
Natural History of HIV Infection and Neuro-HIV Disease
The current Centers for Disease Control and Prevention (CDC) classification system of HIV-infected adolescents and adults categorizes persons on the basis of clinical conditions associated with HIV infection and CD4+ T-cell counts. The system is based on three ranges of such lymphocyte counts and three clinical categories, making a total of nine mutually exclusive categories (Table 53A.1). HIV disease follows a progressive course; once individuals have had a clinical condition in category B (or C), their disease cannot again be classified into a lower category (A or B). AIDS-defining conditions were originally designed for surveillance purposes, and clinicians should not focus on whether AIDS is present but instead should view HIV disease as a spectrum ranging from primary infection through asymptomatic stage to advanced disease.
The stage of systemic HIV infection influences both the risk and the nature of neurological disease; hence CD4+ T-cell count provides critical information that helps guide the evaluation (Fig. 53A.1). In early infection (corresponding to CD4+ T-cell counts >500/mL) and excess immune stimulation, autoimmune disorders such as demyelinating neuropathies may develop. During midstage infection (CD4+ T-cell counts of 200-500/mL), primary HIV-related disorders such as HIV-associated neurocognitive dysfunction (HAND) and certain opportunistic infections such as varicella-zoster virus (VZV) radiculitis (shingles) may appear. In advanced HIV infection (CD4+ T-cell count <200/mL), the risk of dementia, myelopathy, and painful neuropathy increases further, and patients become increasingly vulnerable to major opportunistic infections (OIs) such as cerebral toxoplasmosis, progressive multifocal leukoencephalopathy (PML), and cryptococcal meningitis, as well as to neoplasms—in particular, primary central nervous system lymphoma (PCNSL).
Primary Infection and Dissemination of Virus
HIV is transmitted by both homosexual and heterosexual contact, by sharing of contaminated needles in IDUs, by tainted blood and blood products, and from infected mothers to infants either intrapartum, perinatally, or via breast milk (Hansasuta and Rowland-Jones, 2001). Virus that enters directly into the bloodstream via infected blood or blood products is trapped rapidly by the spleen and lymphoid tissue, whereas entry of virus through a mucosal surface (sexual contact) requires mucosal dendritic cells to first carry the virus to the regional lymphoid tissue. In either case, virus replication in CD4+ T cells intensifies (more rapidly with bloodborne infection) prior to development of the HIV-specific immune response, leading to a burst of viremia and rapid dissemination of the virus to other lymphoid organs, the brain, and other tissues. The initial viremic burst generally results in “acute HIV syndrome.” Viral levels are measured in millions of HIV RNA virions per milliliter. Some 50% to 70% of individuals with acute HIV infection experience an “infectious mononucleosis”–type syndrome persisting 1 to 6 weeks and characterized by fever, erythematous or maculopapular rash, headache, nausea, anorexia, lethargy, arthralgia, sore throat, and lymphadenopathy.
Chronic Persistent HIV Infection
In HIV disease, a chronic infection develops and persists with varying degrees of virus replication and progressive immunological impairment for a median of 10 years before the patient becomes clinically ill. Chronic persistent infection is the biological hallmark of HIV disease. Establishment of chronic HIV infection requires that the virus must evade control and elimination by the immune system. The virus accomplishes this via a number of mechanisms, including high rates of genomic mutation and molecular heterogeneity (see earlier section on the HIV genome), sequestration of infected cells in immunologically privileged sites (e.g., brain), down-regulation of human leukocyte antigen (HLA) class I molecules on the surface of HIV-infected cells by viral proteins (e.g., nef), conformational masking of receptor-binding sites that fails to be neutralized by antibodies, and perhaps deletion of the initially expanded CD8+ T-cell clones by the overwhelming initial burst of viremia and antigenemia. Despite a vigorous immune response and the marked down-regulation of virus replication following primary HIV infection, HIV establishes a state of chronic low-grade infection. The half-life of a productively infected cell is approximately 1 day, and that of a circulating virion is 30 to 60 minutes. Given the relatively steady level of plasma viremia and of infected cells in an individual, it is estimated that extremely large amounts of virus (over a billion or so copies) are produced and cleared from the circulation each day. Thus, clinical latency should not be confused with microbial latency. Vigorous virus replication, though with low-level viremia, is present during the period of clinical latency. Even the term clinical latency is misleading because immunological progression of the HIV disease is generally relentless during this period. The CNS continues to harbor and mount a host reaction to the HIV throughout the asymptomatic or latent stage, yet without apparent immediate clinical sequelae (McArthur et al., 2010). The CSF in patients with latent HIV infection generally shows abnormalities including abnormal cell count, protein and immunoglobulin elevation, unique oligoclonal bands, and local synthesis of anti-HIV antibodies within the CNS compartment; the intact virus can be recovered from the CSF (Price and Spudich, 2008). Pathological studies have shown evidence of inflammatory reactions in the CNS, with perivascular mononuclear cell infiltrations, although the HIV RNA burden, as measured by polymerase chain reaction (PCR) from CSF and brain samples, appears to be low at this stage. Neither overt nor subclinical cognitive or motor dysfunction appears to be common in the early latent stage. From a practical standpoint, the risk of cognitive decline in asymptomatic individuals is sufficiently small as to provide no basis for disability or disqualification from work based simply on HIV-positive status.
The length of the asymptomatic stage is determined by viral and host factors (Kinter et al., 2000; McArthur et al., 2010; Price and Spudich, 2008; Xiao et al., 2000). Some patients who are termed long-term nonprogressors show little if any decline in CD4+ T-cell counts over many years. These patients generally have extremely low levels of HIV RNA. Other patients remain entirely asymptomatic despite progressive declines in the CD4+ count to extremely low levels. In these patients, the appearance of a systemic or CNS OI may be the first manifestation of HIV disease.
Neuropathogenesis of HIV Disease
Neurons lack the conventional surface receptor for HIV binding and fusion and therefore are not directly infected by the virus. The main cell types infected in the brain in vivo are those of the monocyte-macrophage lineage. These include monocytes that have migrated into the brain from the peripheral blood, perivascular macrophages, and resident microglial cells (Sabri et al., 2003; Yadav and Collman, 2009). Although there have been reports of infrequent HIV infection of astrocytes, there is no convincing evidence that brain cells other than those of monocyte-macrophage lineage can be productively infected in vivo (Price and Spudich, 2008). Nevertheless, in vitro infection of a neural cell line has been reported (Klein et al., 1999), and it appears that galactosylceramide on the neuronal surface is an essential component of the HIV gp120 receptor; antibodies to galactosylceramide inhibit HIV entry into the neural cell lines. Some studies have demonstrated that viral entry is due in part to the ability of virus-infected and immune-activated macrophages to induce adhesion molecules such as vascular adhesion molecule 1 (VCAM-1) on brain endothelium. Other studies have demonstrated that HIV gp120 enhances the expression of intercellular adhesion molecule 1 (ICAM-1) in glial cells; this effect may facilitate entry of HIV-infected cells into the CNS and may promote syncytia formation (Fig. 53A.2).
The R5 (CCR5 co-receptor) virus strains that are macrophage-tropic preferentially gain access into the brain rather than R4 (CXCR4 co-receptor) strains. HIV-infected individuals who are heterozygous for CCR5-del.32 (co-receptor gene with 32-base deletion) appear to be relatively protected against the development of HIV encephalopathy, compared to persons with homozygous wild-type CCR5 (Weiss et al., 1999). Distinct HIV envelope sequences are also linked with the clinical manifestation of HAND (McArthur et al., 2010).
Both white-matter changes and neuronal loss are observed in HIV infection. It is unlikely that direct infection of neurons or glial cells accounts for this cell loss. Rather, the brain pathology is thought to be due to a combination of direct effects, either toxic or function-inhibitory, of virus or viral antigens on neuronal cells and effects of a variety of neurotoxins released from the infiltrating monocytes, resident microglial cells, and astrocytes (McArthur et al., 2010; Price and Spudich, 2008; Scaravilli et al., 2007; Yadav and Collman, 2009). Neurotoxins can be released from monocytes as a consequence of other HIV-associated infections or immune activation. Activated monocyte-derived neurotoxic factors have been reported to injure neurons via the N-methyl-d-aspartate (NMDA) receptor. Additionally, HIV gp120 and tat shed by virus-infected monocytes and a variety of cytokines including tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, interferon alpha (IFN-α), and endothelin can contribute directly or indirectly to the neurotoxic effects in HIV infection. Activation of microglial cells, leading to increased production of eicosanoids, nitric oxide, and quinolinic acid, may also be neurotoxic.
Astrocytes may play diverse roles in HIV neuropathogenesis. Astrocytosis and reactive gliosis occur in brains of HIV-infected individuals; TNF-α and IL-6 have been shown to induce astrocyte proliferation. In addition, astrocyte-derived IL-6 can induce HIV expression in infected cells in vitro. Older HIV-infected individuals and those with the E4 allele for apolipoprotein E (APOE) appear to have an increased risk of HIV encephalitis and polyneuropathy (Corder et al., 1998). Neuropsychiatric abnormalities may rapidly improve following the initiation of antiretroviral therapy, indicating that HIV or products are involved in the neuropathogenesis of these disorders and suggesting that the dominant changes are either structural or functional at the synapses and dendritic spines (Yadav and Collman, 2009).
Antiretroviral Therapy and Its Effect on Neuro-HIV Disease
The dynamics of in vivo HIV production and turnover have been quantified using mathematical modeling in the setting of HAART. Treatment with effective HAART typically results in precipitous decline in the level of plasma viremia, often by well over 95% within a few weeks. The number of CD4+ T cells increases concurrently. Combination antiretroviral therapy is the cornerstone of management of HIV infection (Fauci and Lane, 2006). Suppression of HIV replication prolongs and improves the quality of life in patients with HIV infection.
Following the widespread use of HAART in the United States since 1996, dramatic declines have been noted in the incidence of most AIDS-defining conditions, including neurological diseases. Reconstitution of immune defenses with HAART has enabled some patients to discontinue secondary prophylaxis against CNS opportunistic pathogens. Primary neurological diseases can be prevented or delayed by successful HAART (Geraci and Simpson, 2001; Sacktor et al., 2006), although the evolution of drug-resistant viral strains may limit the sustained benefits of antiretroviral therapy (D’Aquila et al., 2003).
Once the decision has been made to initiate therapy, the physician must decide which drugs to use as the first therapeutic regimen. Initial choice of drugs will determine the immediate response to therapy, and it will have implications regarding options for future therapeutic regimens. The goal of treatment is to achieve a viral load of fewer than 50 copies/mL within 4 to 6 months of initiation (Gazzard et al., 2008). The two options for initial therapy most commonly in use today involve a three-drug regimen from two different antiretroviral classes (Table 53A.2). Recommendations regarding specific regimens are evolving. Combination therapy must be employed.
For HIV-associated brain disease, drugs that have good blood-brain barrier penetration should be preferred (see Table 53A.2). Following the initiation of therapy, one should monitor virological (HIV RNA levels) and immunological (CD4+ counts) responses periodically. The HIV RNA levels in serum generally reflect viral levels in CSF, at least until late stages of the HIV disease. In terminal stages, the CNS compartment may harbor slightly different and divergent HIV strains, with different degrees of drug susceptibility (Ellis et al., 2007; Liu et al., 2006; McArthur et al., 2010; Price and Spudich, 2008). In an attempt to determine an optimal therapeutic regimen, antiretroviral drug susceptibility through genotyping or phenotyping of HIV quasispecies has been attempted. Maximal suppression of viral replication is the goal of therapy, not just to prevent the disease progression but also to prevent the appearance of drug-resistant HIV quasispecies. The principles of current therapy for HIV infection are well articulated in publications of the U.S. Department of Health and Human Services (see updates at www.aidsinfo.nih.gov/guidelines/). As HAART has become widely available and has prolonged the life of patients with AIDS, an increasingly commonly observed manifestation was noted, characterized by a paradoxical clinical deterioration following therapy referred to as the immune reconstitution inflammatory syndrome (IRIS) and attributed to immunological recovery. Although typically observed with opportunistic infection, it may also occur as a response to noninfectious antigens. In the CNS, IRIS has been well described with Cryptococcus neoformans cytomegalovirus and PML. Patients with IRIS either present with clinical deterioration of a recognized infection following initiation of HAART, or the neurological disease may first manifest upon immunological recovery following the initiation of effective antiretroviral therapy. Neuroimaging generally shows worsening of previously observed lesions, frequently with contrast enhancement. Neuropathological findings at autopsy or from brain biopsy specimens reveal severe inflammatory reaction, with intraparenchymal and perivascular infiltration by lymphocytes (predominantly CD8+ T lymphocytes) and macrophages (Gray et al., 2005). The symptoms mostly regress with continued HAART, although a short course of high-dose parenteral corticosteroids (Riedel et al., 2006) has been suggested as effective in abrogating the response. Death may complicate IRIS (Gray et al., 2005).
Clinical Spectrum of Neuro-HIV Disease
The high prevalence and striking diversity of neurological disorders complicating AIDS were recognized early in the epidemic (Berger et al., 1987a). Clinical description of neurological OIs and malignancies predominated in early reports, but it also became clear that AIDS was associated with distinct neurological syndromes such as dementia and painful neuropathy that appeared to result solely from HIV. It also became recognized that the risk of neurological complications increased with the progression of HIV infection and decline of CD4+ counts. Clinically apparent neurological disease develops in approximately half of HIV-infected patients. Neuropathological abnormalities are nearly universal in patients dying with AIDS, suggesting subclinical disease, underdiagnosis, or both in many cases. Neurological disorders cause significant morbidity and mortality, and they may be the AIDS-defining illnesses in previously asymptomatic HIV disease or, occasionally, herald unrecognized HIV infection. Nervous system complications may directly threaten life as well as impair a patient’s ability to work or comply with complex HAART regimens necessary to manage HIV disease optimally. These disorders affect every level of the neuraxis, and a given patient may suffer more than one HIV-associated neurological disease.
The neurological complications of HIV infection occur at all stages of the disease (Simpson and Berger, 1996). Disorders of both the CNS and peripheral nervous system (PNS) can complicate HIV infection from the period after initial infection through the end stages of the severe immunosuppression. These neurological complications can be classified in a number of ways. A classification based on the underlying pathophysiology and HIV disease stages is summarized in Fig. 53A.1 and Table 53A.3. Clinicians must be aware of the possibility that more than one nervous system site can be involved at the same time, and deficits from one site may be masked by other lesions elsewhere in the neuraxis. The clinician must also be vigilant for other common conditions that are not necessarily associated with HIV or AIDS. Experience in large HIV clinics indicates that the diagnosis of neurological complications of HIV infection and AIDS is far from an academic exercise. Rather, precise diagnosis is critical, and it frequently leads to specific therapy with resultant reduction in morbidity and mortality and in preservation of meaningful function and quality of life. Fig. 53A.3 summarizes a diagnostic algorithm for CNS manifestations in HIV-infected patients.
Table 53A.3 Major HIV-Associated CNS Disorders Classified by Neuroanatomical Localization
| MENINGES |
[/not-level-membership-for-neurology-category]
