[level-membership-for-dermatology-category]
Infantile Hemangiomas and Other Vascular Tumors
Anita N. Haggstrom, Sarah L. Chamlin
Infantile hemangioma
Infantile hemangioma (IH) is the most common vascular tumor encountered during early infancy. It is a benign proliferation of endothelial cells that characteristically undergoes a phase of rapid growth followed by slow spontaneous involution. Although large population-based studies are lacking, infantile hemangiomas are estimated to occur in 2.6–5% of infants.1,2 The incidence is highest among Caucasian infants with lower rates in African-American, Hispanic, and Asian infants.3 Females are more commonly affected than males at ratios of 2–3 : 1, with the female-to-male ratio approximately 9 : 1 in patients with segmental hemangiomas associated with PHACE syndrome.4,5
Caucasian race, prematurity and low birthweight are well-established risk factors for the development of IH.3,6 The risk of IH increases by 40% for every 500 g decrease in birthweight.7 A prospective study of over 1000 children with infantile hemangiomas identified additional risk factors, including multiple gestation pregnancies, advanced maternal age, and pregnancies complicated with pre-eclampsia or placenta previa.5,8 Placental abnormalities that impair utero-placental circulation, including retroplacental hematoma, ischemic infarction, dilated vascular communications, vasculitis and chorioamnionitis have been observed to be more common in infants with IH.9 An earlier study reported a threefold increased incidence of hemangiomas in infants born to mothers who undergo chorionic villus sampling, compared with those whose mothers undergo amniocentesis, but subsequent studies have not been conclusive.10 In-vitro fertilization has been suggested as a possible risk factor in one study.2 The majority of hemangiomas arise sporadically; however, a family history can be elicited in some patients, and autosomal dominant transmission has been reported.11
Clinical subtypes
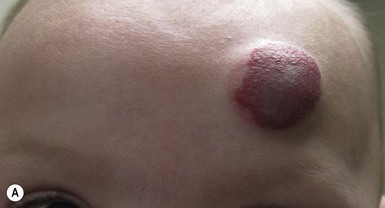
Infantile hemangiomas can be classified based on their location within the skin: superficial, deep, and mixed (superficial and deep). Superficial hemangiomas are the most common type, occurring in approximately 50–60% of cases. Mixed-type IH are estimated to occur in 25–35% of cases, and deep hemangiomas in 15%.12 Superficial hemangiomas are typically bright red with a lobulated surface. These characteristics have led to the use of the term ‘strawberry’ hemangioma. Deep hemangiomas are not commonly noted in the neonatal period, often appearing at 1–3 months of life, or later, as a warm, subcutaneous mass caused by proliferation of the tumor in the deeper portion of the dermis or subcutis. The overlying skin may appear normal or have relatively inconspicuous superficial changes, such as faint blue color, telangiectasias or dilated veins. Mixed-type hemangiomas frequently exhibit a configuration resembling a poached egg, with a well-circumscribed red superficial portion overlying a less well-defined bluish to violaceous deeper component.
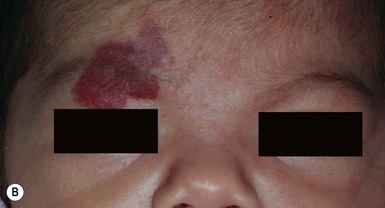
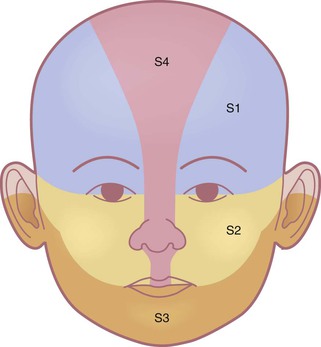
In addition to characterizing hemangiomas according to their depth and location within the skin, the pattern of involvement is a predictor of prognosis and risk for associated complications.5 Described patterns include ‘focal’, or localized, lesions that appear to arise from a central point, and ‘segmental’ lesions, which typically encompass a larger territory of skin, which in some anatomic sites clearly corresponds to a developmental subunit (Fig. 21.1).13,14 Some lesions are difficult to classify as either focal or segmental and are thus designated ‘indeterminate’. Segmental lesions more often require treatment and are more likely to be associated with complications, including underlying extracutaneous anomalies, including spinal dysraphism, genitourinary anomalies, and PHACE syndrome.14 The patterns of segmental hemangiomas correspond to known embryologic prominences, suggesting a neuroectodermal derivation for their distribution.15 The frontotemporal (S1), maxillary (S2), mandibular (S3), and frontonasal (S4) segments designate commonly observed patterns of facial hemangiomas (Fig. 21.2).15
Natural history
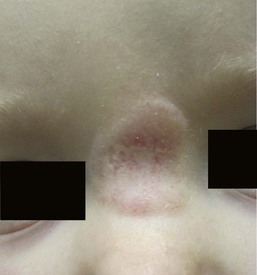
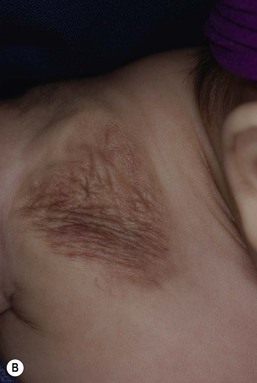
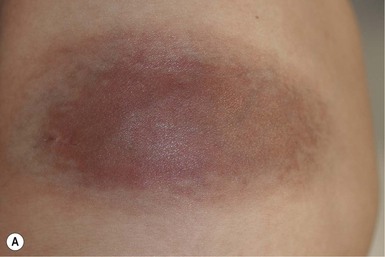
Infantile hemangiomas have a characteristic clinical course marked by rapid early proliferation, slower late proliferation, a plateau phase, and finally gradual involution. They are either present at birth or appear in the first few weeks of life. Precursor lesions (or so-called ‘premonitory marks’) are seen in as many as 65% of patients and may appear as discrete fine telangiectasias superimposed on a background of pallor, a faint erythematous patch, or a bruise-like area (Fig. 21.3).16 It may be difficult initially to differentiate an erythematous hemangioma precursor from a capillary malformation, but clues include a more ill-defined border, fine telangiectasias, and, if present, skin ulceration. Serial examinations may be necessary to establish the correct diagnosis. Rarely, a hemangioma on the perineum or lip may present in the neonate with ulceration, without an obvious hemangioma, mimicking bacterial or herpetic infection. In these cases, the hemangioma becomes evident over the subsequent days to weeks (see Chapters 10 and 17).
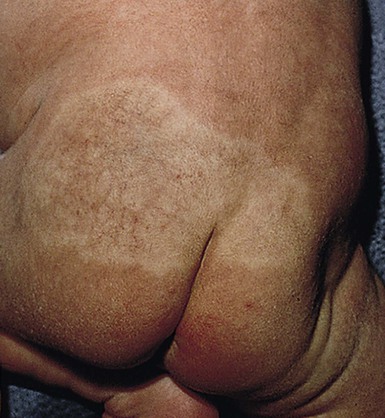
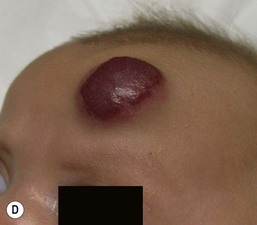
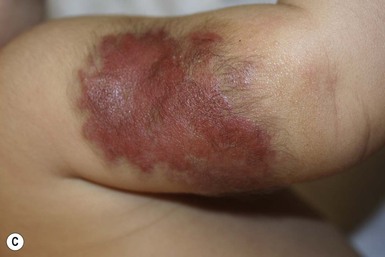
In the first few weeks of life, most infantile hemangiomas proliferate rapidly; 80% of superficial hemangioma growth occurs before 3 months of age, with a period of accelerated growth between 5 and 7 weeks of age.16,17 The late proliferative stage is marked by slower growth, but is more variable in its duration, with certain subsets of hemangiomas, including those that are large, segmental, or those with a significant deep component growing much longer, even beyond the age of 1 year.17,18 Most hemangiomas establish the boundaries of their anatomic territory relatively early, growing only in volume thereafter. Superficial hemangiomas initially have a bright red color early in proliferation. Both combined and deep hemangiomas may feel tense, and fluctuations in size and volume may be noted with crying or activity. The earliest signs of involution include a change in color from bright red to a violaceous gray, usually beginning in the center of the hemangioma. The deeper portion also regresses, but this is often not as apparent clinically. Parents may note less fluctuation in hemangioma size during crying and less tumor firmess.6 The warmth of the hemangioma diminishes with time. Some resolve leaving normal or nearly normal skin, whereas others resolve with residua of variable cosmetic significance (Fig. 21.4). Involuted hemangiomas often show residual telangiectasia, pallor or a yellowish color, fibrofatty tissue deposition, and atrophy (Fig. 21.5). Several studies have shown that completion of involution occurs at a rate of approximately 10% per year, with 30% completing involution at 3 years, 50% at 5 years, etc.4,19 Although not specifically mentioned in these studies, it seems that small hemangiomas generally involute sooner than very large, bulky ones.
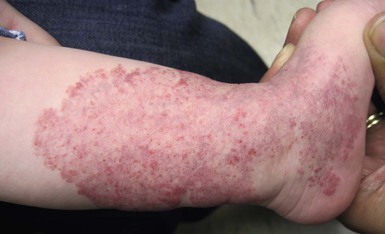
Less commonly, infantile hemangiomas display a more atypical growth pattern characterized by little, if any postnatal growth. These hemangiomas have been variously called infantile hemangiomas with minimal or arrested growth (IH-MAG), abortive hemangiomas, or reticular hemangiomas.20,21 IH-MAG present as telangiectatic patches that have minimal superficial proliferation, often manifested as 1–3 mm red papules at the periphery of the lesion and have a predilection for the lower extremities (Fig. 21.6).22 They may have violaceous to green ectatic vessels underlying the finer telangiectasias. The small proliferative vascular papules involute within the first few years of life but the ectatic vessels can be persistent. The vessels of IH-MAG stain positively for GLUT-1, similar to classic infantile hemangiomas.
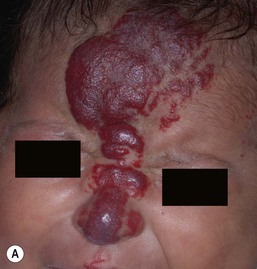
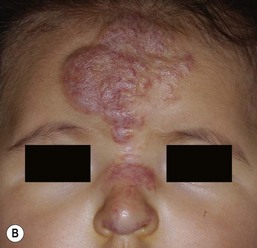
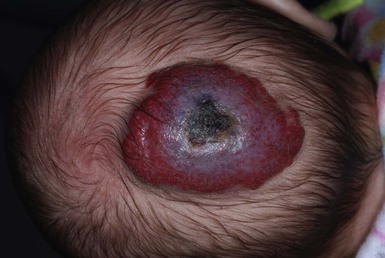
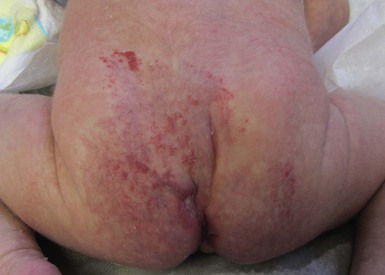
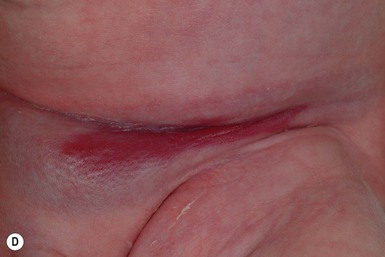
Ulceration is the most common complication of infantile hemangiomas, occurring in approximately 10–15%, most often during the rapid growth phase. The risk is much higher (approx. 50%) for hemangiomas involving the lip and perineum.23 When the ulceration heals, there is virtually always residual atrophy or scarring. Bleeding may occur, but profuse bleeding is surprisingly rare, occurring mainly with deeply ulcerated hemangiomas or those located on the scalp (Fig. 21.7). Superimposed bacterial infection most commonly with Staphylococcus aureus or Pseudomonas species may develop and may impede healing, but it is uncommon. Recurrent ulceration may complicate hemangiomas located in the perineum.
The Kasabach–Merritt phenomenon (KMP) was originally believed to be a complication of large hemangiomas, but studies have proven that KMP is a complication of other vascular tumors, including kaposiform hemangioendothelioma and tufted angioma, rather than infantile hemangiomas (see below).
High-risk hemangiomas.
Although the vast majority of hemangiomas are not worrisome and do not necessitate treatment, it is important to recognize the subsets of hemangiomas that are at highest risk for causing permanent disfigurement, impairing vital functions including vision and feeding, and that may be associated with visceral hemangiomatosis or internal anomalies (Box 21.1).
Locations associated with high risk for disfigurement.
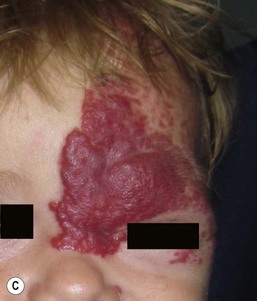
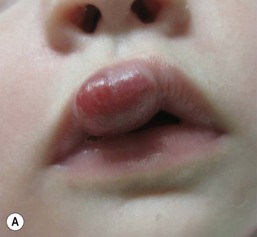
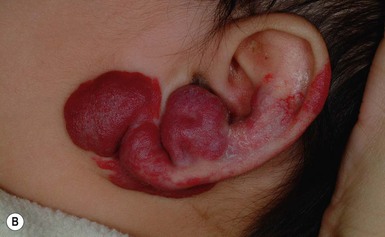
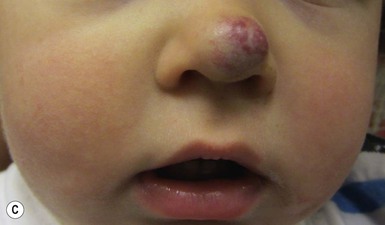
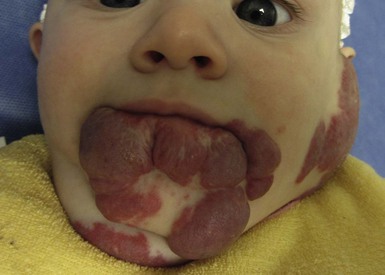
The most common reason infants with hemangiomas receive treatment is the potential risk for disfigurement (Fig. 21.8).5 Hemangiomas involving the central face may distort important anatomic landmarks and have a significant impact on future cosmesis. Although exophytic lesions on the medial cheek do not impair vital functions, they are often distressing because of their conspicuous locations and may benefit from more aggressive medical therapies. Distortion of the lip contour can occur even with small hemangiomas because of the disruption of the vermillion border or loss of the curvature of the philtrum. Mixed hemangiomas located on the nasal tip distort the underlying cartilage and leave residual bulk. Surgical repair is often necessary to correct the ‘Cyrano nose’ deformity that can result in splaying of the nasal cartilage. Ulceration involving the columella can lead to destruction of the nasal septum. Hemangiomas on the pinna may ulcerate and become secondarily infected, contributing to structural deformity of the involved ear.
Periorbital hemangiomas.
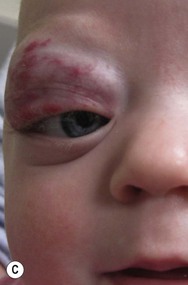
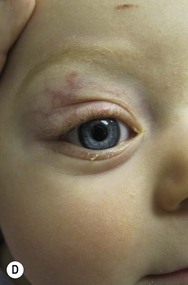
Growth of periorbital and lid hemangiomas may cause visual impairment principally by deformation of the cornea, creating refractive errors or less commonly by obstructing the visual axis. If severe or persistent, this can lead to amblyopia. Large and/or segmental hemangiomas in the periocular area have the greatest risk of these complications, but even small lesions may pose a threat to normal visual development, with astigmatism being the most common sight-threatening ocular complication.24,25 Proliferation of retrobulbar lesions may lead to proptosis, strabismus, and visual compromise.26
Large cervicofacial hemangiomas and PHACE syndrome.
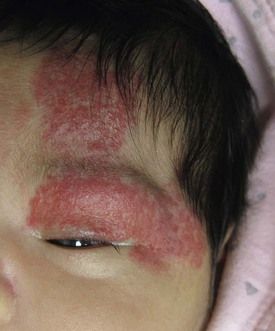
Large cervicofacial hemangiomas may impair vital functions, distort normal anatomy, or lead to congestive heart failure as they proliferate. These problematic hemangiomas are more common in females than males at a ratio approaching 9 : 1 in some series.27 The term PHACE syndrome refers to the association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities (Box 21.2). The term PHACE(S) is sometimes used when associated sternal anomalies or a supraumbilical raphe are present. Although the exact incidence of PHACE is unknown, it may be more common than Sturge–Weber syndrome.27 In 2009, a multidisciplinary panel of experts published diagnostic criteria for ‘definite’ and ‘possible’ PHACE using specific major and minor criteria.28 A multicenter prospective study of 108 infants with facial hemangiomas measuring 22 cm2 or larger, found that when applying published diagnostic criteria for PHACE, 31% had PHACE and 90% of affected infants had more than one extracutaneous finding. The most common extracutaneous manifestation in PHACE is anomalous cerebrovasculature, with the highest incidence seen in infants with upper face hemangiomas (S1, frontotemporal or S4, frontonasal) (Figs 21.4, 21.9).29
Central nervous system (CNS) anomalies include both structural malformations of the brain and anomalous vasculature of the head and neck, both of which are typically ipsilateral to the cutaneous hemangioma.30 Structural CNS malformations include the Dandy–Walker malformation and other posterior fossa anomalies, such as arachnoid cyst, enlarged fourth ventricle, enlarged cisterna magna, and cerebellar or vermian hypoplasia.31–33 Other CNS anomalies have been reported, including cerebral atrophy, gray matter heterotopia, pituitary abnormalities, and absent corpus callosum. Macrocephaly, ophthalmologic abnormalities, hypotonia, seizures, and psychomotor retardation may be a presenting sign of an underlying structural malformation. Cerebrovascular abnormalities often involve the ipsilateral internal carotid artery and its branches, though other vessels can be involved.30 Dysgenesis of the vessel, including looping, elongation, ectasia, kinking, and focal or fusiform aneurysmal enlargement of the vessel, is most frequently noted.30 Less commonly, vessels can have an anomalous course and/or origin, display narrowing or be completely absent.30 Rarely, patients with intracranial anomalous vessels may demonstrate progressive occlusive arterial changes and cerebral infarction.34 Stroke is a rare complication of PHACE and may be more common in patients with aplasia, hypoplasia or occlusion of a major cerebral artery.35 Other neurologic sequelae of PHACE may include seizures, migraine-like headaches, and developmental delay, including most commonly language and gross motor abilities.36
Cardiac abnormalities, seen in two-thirds of infants with PHACE, usually manifest as unusual forms of aortic arch coarctation, primarily involving long segment narrowing of the transverse aorta often associated with adjacent dilatation or aneurysms.29,37,38 Less frequently, structural anomalies of the heart may be seen, including atrial and ventricular septal defects.28
Less common manifestations include ocular, midline anomalies and hearing loss of the affected ear.39 Ocular findings may include persistent fetal vasculature, retinal vascular anomalies, optic nerve hypoplasia, morning glory deformity, peripapillary staphyloma and coloboma, with other less specific anomalies noted more rarely. Midline defects such as partial or complete sternal agenesis and supraumbilical raphe may also occur.28,40
Endocrine abnormalities, including pituitary dysfunction, thyroid dysfunction, and growth hormone deficiency have been reported in some patients with PHACE. A few infants with PHACE have been noted to have absent or lingual thyroid glands.41–43
Lumbosacral hemangiomas and regional anomalies.
Analogous to PHACE syndrome, segmental lumbosacral and perineal hemangiomas can be cutaneous signs of regional anomalies such as underlying structural malformations of the genitourinary, gastrointestinal, neurologic, and skeletal systems (Fig. 21.10).44 The acronyms for this association include LUMBAR, PELVIS, and SACRAL syndromes.44–46 A prospective study of 41 lumbosacral hemangiomas >2.5 cm reported 35% had evidence of spinal dysraphism.47 Tethered spinal cord, spinal lipomas and spinal hemangiomas are the most common underlying abnormalities.48 Imperforate anus, rectoscrotal fistula, renal anomalies, abnormal external genitalia, lipomyelomeningocele, and bony deformities of the sacrum have been reported.44–46 Ulceration is associated with a higher risk of underlying anomalies.47
Multifocal cutaneous and visceral hemangiomas.
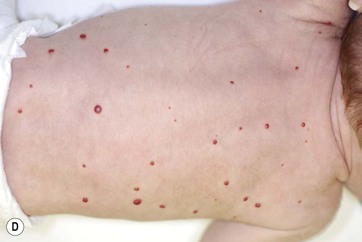
Multiple hemangiomas occur in approximately 10–25% of infants and are more common in premature infants than in term infants.49 Cutaneous lesions may range in size from a few millimeters to more than several centimeters in diameter. The presence of five or more cutaneous hemangiomas is a known risk factor for extracutaneous hemangiomas, mainly hepatic hemangiomas. Rarely, other organs such as the gastrointestinal tract, lungs, CNS, oral mucosa, and eyes may be affected. Screening ultrasound of the liver is recommended for young infants with five or more cutaneous hemangiomas.50 Serial radiologic evaluation of the liver, especially screening abdominal ultrasound, with further evaluation with CT or MRI, may be necessary to follow progression of visceral lesions. Hepatic hemangiomas (HH) may be solitary or multiple. A classification for hepatic hemangiomas has been proposed, including solitary, multifocal, and diffuse HH.51 Solitary HH in the absence of cutaneous hemangiomas – particularly if detected in utero – are usually GLUT1 negative, regress rapidly, and most likely represent a form of vascular tumor in the liver similar to rapidly involuting congenital hemangioma (RICH) rather than true infantile hemangioma (see below). Multifocal hepatic lesions usually occur in the setting of multiple small skin hemangiomas. They may be completely asymptomatic without the need for treatment, but if they have arteriovenous shunts, they may require liver embolization and/or medical therapy to manage congestive heart failure, as well as hemangioma-specific therapy (such as beta-blockers). Hepatic hemangiomas associated with an arteriovenous, arterioportal shunt or portovenous fistula are associated with greater morbidity.51 Diffuse liver hemangiomas cause massive hepatomegaly, abdominal compartment syndrome, and are associated with hypothyroidism (see below). They have a high rate of mortality and require aggressive medical therapy, and even consideration of liver transplantation.
Consumptive hypothyroidism may occur in association with hepatic hemangiomatosis due to the deactivation of thyroxine by a type 3 iodothyronine deiodinase produced by the hemangioma.52–54 This form of hypothyroidism can be detected by the presence of markedly elevated TSH and low T3. T4 levels may be normal or low. Neonatal screening for hypothyroidism is inadequate to assess for this complication, as the most active phase of hemangioma proliferation typically occurs after this period. In some cases, this consumptive form of hypothyroidism may require large doses of thyroid hormone replacement given intravenously. The hypothyroidism typically resolves as the hemangioma regresses, but may have already caused significant morbidity if not detected promptly. Type 3 iodothyronine deiodinase activity has also been found in association with large cutaneous hemangiomas, therefore thyroid function tests should also be considered in patients with large cutaneous hemangiomas.52
Cutaneous hemangiomas associated with airway hemangiomas.
Infantile hemangiomas of the airway are most commonly seen in the subglottis and can occur with or without cutaneous hemangiomas. Hemangiomas in the ‘beard distribution’ involving the skin overlying the mandible, chin, and neck, have a high risk of concomitant airway involvement (Fig. 21.11). There is a striking female predilection of 6–7 : 1 for ‘beard’ hemangiomas.55 Though hemangiomas in the mandibular region are most common, other hemangioma patterns that have been associated with airway hemangiomas include unilateral multisegment and hemifacial hemangiomas.56,57 Affected infants typically present within the first few weeks of life with an increasing degree of noisy breathing, stridor, hoarse cry, or other signs of airway obstruction. Having an airway hemangioma together with one or more large facial hemangiomas confers a greater than expected incidence of PHACE – possibly as high as 47%.57
Pathogenesis
Infantile hemangiomas are believed to represent localized areas of abnormal vascular growth, and several hypotheses have been proposed to explain their pathogenesis. These include endothelial cell defects, a placental origin, extrinsic abnormalities in neighboring cells and exposure to vascular growth stimulators. IH was long thought to be a disease of aberrant angiogenesis, however more recent research supports the idea that it may also represent a disorder of vasculogenesis, that is the formation of new blood vessels de novo, rather than sprouting from pre-existing cells. While some evidence supports many of the proposed hypotheses, no unifying paradigm has emerged: it is likely that the pathogenesis is multifactorial with many mechanisms contributing to their development, proliferation and involution.58–60
Some evidence suggests that hemangioma endothelial cells represent a clonal expansion of cells due to a somatic mutation in genes that play a significant role in vascular growth or vascular regulatory pathways. Mutations in several genes involved in the VEGF signaling pathway (VEGF receptors), Tie-2 and Dusp-5 have been noted supporting this hypothesis, but their exact role in hemangioma pathogenesis is not clear.61–63 Initial analysis of several pedigrees of familial hemangiomas revealed a linkage to chromosome 5q.62 Subsequent analysis of sporadic hemangiomas demonstrated loss of heterozygosity of 5q, further supporting the possibility that somatic mutations play a role in hemangioma formation.64
Infantile hemangiomas share unique immunohistochemical markers with human placenta microvasculature. North and coworkers reported that glucose transporter protein-1 (GLUT-1) is expressed by infantile hemangiomas during all phases of their development (proliferating, involuting, and involuted), distinguishing infantile hemangiomas from vascular malformations and other vascular tumors.65 Other placenta-associated vascular antigens, including merosin, FcγRII and Lewis Y antigen, are also present in hemangiomas and absent in microvessels of normal skin.66 In addition, studies using DNA microarray techniques have revealed that the gene expression profiles of hemangiomas and placenta vascular endothelium are remarkably similar.67 While it is speculated that hemangiomas may arise from embolized placental cells or angioblasts that abnormally differentiate toward a placental phenotype, this is challenged by the finding of negative placental trophoblastic markers in immunochemical analysis of hemangiomas.68
Multiple factors regulate the proliferation and involution of hemangiomas. During proliferation, hemangiomas demonstrate proliferating cell nuclear antigen (PCNA), as well as increased levels of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).69–71 The VEGF signaling pathway is likely one of the more important mechanisms contributing to hemangioma proliferation.72 Angiogenesis mediators, including monocyte chemoattractant protein-1 and the adhesion molecules E-selectin and ICAM-3, are also expressed at high levels.73–76 Proteins involved in extracellular matrix remodeling are expressed during the proliferating phase.59,69,77 Increased levels of urinary matrix metalloproteinase (MMP) proteins are noted in proliferating infantile hemangiomas, suggesting a role in extracellular matrix remodeling, and the expression of lymphatic endothelial hyaluronan receptor-1 (LYVE-1) in proliferating hemangiomas may explain the rapid growth.78,79 Local factors are also believed to play a significant role in hemangioma proliferation. Investigators have noted that hemangioma growth correlates with hyperplasia of the overlying epidermis, with these tissues elaborating angiogenic factors (e.g., VEGF, bFGF). Moreover, these tissues lacked expression of the angiogenesis inhibitor interferon (IFN)-β.80 Indoleamine 2,3-dioxygenase (IDO) enzyme activity may play a role in the transition from proliferation to involution.8,81
During involution, the levels of angiogenic factors found during proliferation decrease, whereas levels of the angiogenesis inhibitor TIMP-1 (tissue inhibitor of metalloproteinase-1) increase.69,75,76 Interferon-regulated genes are upregulated during involution.69,82 Factors that promote apoptosis increase during involution, whereas inhibitors of apoptosis are upregulated during proliferation.83–85 In addition, loss of Dusp-5 function increases apoptosis in human umbilical vein endothelial cells causing speculation for its role in hemangioma involution.72,86
Diagnosis
Most infantile hemangiomas can be diagnosed on the basis of history and clinical appearance. The differential diagnosis includes other vascular and nonvascular tumors that are discussed elsewhere. Where history and physical examination are not helpful, further studies such as imaging or even biopsy may be necessary to confirm the diagnosis. Doppler ultrasound and MRI are most helpful in confirming the diagnosis of hemangioma. Doppler studies demonstrate high-flow lesions. T1-weighted MRI sequences demonstrate flow voids as a result of high-flow vessels and enhancement on T2-weighted sequences.87–89
Histopathology
If there is significant diagnostic uncertainty, a biopsy and GLUT-1 staining may be necessary to exclude other soft tissue tumors or vascular malformations. Early in the proliferating stage this tumor consists predominantly of a mass of endothelial cells. Lumina, lined by normal-appearing endothelial cells, become evident somewhat later in the late proliferative phase. PAS stains reveal a thickened basement membrane. Mast cells are increased within proliferating hemangiomas compared to normal tissue. As hemangiomas mature, they show lobules of endothelial channels separated by fibrous septa. Actin-positive smooth muscle cells are deposited around the vessels. Fat cells may be prominent in some involuted hemangiomas. Immunohistochemical analysis with GLUT-1 can be used to confirm the diagnosis of infantile hemangioma.
Differential diagnosis
Deep hemangiomas may be particularly difficult to differentiate from other tumors and malformations (Box 21.3 ).90,91 Infantile myofibromatosis may mimic vascular tumors but is firmer to palpation and has distinct histopathologic features. Infantile fibrosarcoma, a rare tumor that is sometimes congenital, may resemble a deep hemangioma or lymphatic malformation. Rhabdomyosarcoma is the most common sarcoma of early childhood and may present in newborns as a rapidly enlarging red cutaneous mass, usually involving the head and neck, that may be difficult to differentiate from a deep hemangioma. Other benign and malignant tumors that may resemble hemangiomas include adrenal carcinoma, spindle and epithelioid nevi, hemangiopericytoma, dermatofibrosarcoma protuberans, lipoblastoma, neuroblastoma, and nasal glioma. Congenital Langerhans’ cell histiocytosis, multifocal lymphangioendotheliomatosis and disseminated pyogenic granulomas may mimic diffuse neonatal hemangiomatosis. Developmental anomalies such as encephaloceles, dermoid cysts, meningoceles, and teratomas may all be mistaken for deep hemangiomas.
).90,91 Infantile myofibromatosis may mimic vascular tumors but is firmer to palpation and has distinct histopathologic features. Infantile fibrosarcoma, a rare tumor that is sometimes congenital, may resemble a deep hemangioma or lymphatic malformation. Rhabdomyosarcoma is the most common sarcoma of early childhood and may present in newborns as a rapidly enlarging red cutaneous mass, usually involving the head and neck, that may be difficult to differentiate from a deep hemangioma. Other benign and malignant tumors that may resemble hemangiomas include adrenal carcinoma, spindle and epithelioid nevi, hemangiopericytoma, dermatofibrosarcoma protuberans, lipoblastoma, neuroblastoma, and nasal glioma. Congenital Langerhans’ cell histiocytosis, multifocal lymphangioendotheliomatosis and disseminated pyogenic granulomas may mimic diffuse neonatal hemangiomatosis. Developmental anomalies such as encephaloceles, dermoid cysts, meningoceles, and teratomas may all be mistaken for deep hemangiomas.
Management
Perhaps the greatest challenge in managing hemangiomas in infancy is the identification of those lesions that need treatment. Because of widely divergent sizes, location(s), and the rapid changes that can occur in early infancy, it may be difficult to predict prognosis at the time of initial evaluation. Frequent assessments during the first few weeks to months of life may be needed, particularly if high-risk features are present. The major goals of management are the prevention and treatment of life-threatening or function-threatening complications of the hemangioma as well as the prevention of permanent disfigurement that may have a long-term social and psychosocial impact on the patient and family (Table 21.1).92
TABLE 21.1
Hemangioma evaluation and management
| Hemangioma presentation | Association or complication | Evaluation and treatment |
| Facial segmental hemangiomas/facial hemangioma >5 cm | PHACE syndrome | MRI/MRA of brain and neck, echocardiogram, ophthalmology evaluation, and consider topical and/or systemic therapy |
| Periocular hemangioma | Ocular complications | Ophthalmology evaluation and consider topical and/or systemic therapy |
| Lumbosacral hemangioma | Spinal dysraphism, intraspinal hemangioma, lipoma | MRI of spine or ultrasound spine if neonate, neurosurgical intervention if indicated, consider topical and/or systemic therapy |
| Perineal segmental hemangioma | Spinal dysraphism and genitourinary anomalies (PELVIS syndrome) | MRI of lumbar spine and pelvis, urological and neurosurgical intervention if indicated, consider topical and/or systemic therapy |
| Ulceration | Pain, bleeding, scarring | See Box 21.6 |
| Central facial or other hemangiomas distorting normal anatomy | Disfigurement from skin and/or cartilage destruction and deformation | Consider topical and/or systemic therapy |
| Airway hemangiomas | Respiratory distress | Otolaryngology evaluation and/or MRI of neck, systemic therapy |
| Multifocal hemangiomas (>5) | Hepatic hemangiomas | Hepatic ultrasound and systemic treatment if symptomatic |
There are a number of treatments employed for infantile hemangiomas, although there is currently no treatment specifically FDA-approved for this indication. There are topical and systemic therapies that target hemangioma proliferation as well as adjunctive therapies that address hemangioma-related complications or the appearance of hemangioma residua after involution. The potential benefits of any form of treatment should be carefully weighed against the associated risks.
Several clinical features are important to consider when choosing treatment. It is important to consider not only anatomic location, but also the size, type, and pattern of the hemangioma and whether the hemangioma is actively proliferating. The following sections describe some of the treatment methods that have been employed.
Active non-intervention.
This term refers to the active observation and anticipatory guidance that can be given to parents, even if no specific therapy is instituted. Parents may feel significant distress as they await spontaneous involution and may react with disbelief, fear, and mourning. Some feel a degree of social stigmatization and many are accused of child abuse, either jokingly or in earnest. Parent–child interactions may be adversely affected.92 A careful discussion of the natural history of hemangiomas, with photographic examples to demonstrate natural involution, as well as a thorough discussion of therapeutic options, may help allay parental fears. Frequent visits, measurements, and photographs during the proliferating phase are recommended, and as the hemangioma begins to involute, visits can become less frequent. Acknowledgment of the intrusive and unsolicited questions and advice parents may receive can also be helpful.
Local and topical treatment.
For superficial hemangiomas that have not yet become large or exophytic, topical treatment may be beneficial, including topical steroids, beta-blockers, and imiquimod. Initially described for the treatment of superficial, periocular hemangiomas, ultrapotent topical steroids such as clobetasol may decrease hemangioma size as well as reduce the thickness and color intensity of the hemangioma.93–95 Steroid-induced cataracts and increased intraocular pressure are theoretical risks and therefore patients using potent topical steroids in the periocular area should have periodic ophthalmologic exams. Percutaneous absorption and hypothalamic-pituitary axis suppression may occur but the true risk for systemic absorption is unknown.
Timolol is a nonselective beta-adrenergic receptor inhibitor originally formulated for ophthalmologic use (0.25–0.5% solution or gel-forming solution) and has increasingly been reported as an alternative to topical steroids for superficial hemangiomas and some small mixed-type hemangiomas.96 Although numerous cases have been reported, variability with regards to dose, frequency and vehicle exist and large clinical trials are still needed to elucidate its optimal use. Ophthalmologic doses (1–2 drops BID) have commonly been applied to avoid increasing the risk of systemic absorption. Timolol, when used intraocularly for glaucoma, is rarely associated with respiratory complications including bronchospasm.97 The gel-forming solution may have lower systemic absorption compared to solution formulations of timolol and thus may be preferable to the standard solution vehicle.98–100 Importantly, topical treatment is not appropriate for periocular lesions that cause visual axis compromise or that are at risk for inducing significant astigmatism and amblyopia, where systemic treatment is more appropriate.
There are a few case reports of treating infantile hemangioma with the topical immunomodulator 5% imiquimod cream.101–103 This cream is approved for the treatment of genital warts, actinic keratoses and superficial BCC in adults. Further studies are needed to determine the safety and efficacy of this therapy in infants. Crusting or ulceration has been reported as a side-effect of topical treatment in some cases.
Intralesional corticosteroids.
Intralesional steroid treatment may be useful in the treatment of small, localized hemangiomas in problematic locations such as the lips, tip of the nose, ear, or face. The frequency of injections is governed by clinical response: generally 1–3 treatments at 4–6-week intervals are needed. The dose of triamcinolone should not exceed 1–3 mg/kg per treatment session. Intralesional injection of hemangiomas on the eyelid with long-acting corticosteroid preparations may be complicated by occlusion of the central retinal or ophthalmic arteries, which may cause blindness.104 Other complications associated with intralesional treatment in this area include intraocular deposits, eyelid necrosis, and scleroderma-like linear atrophy of the skin. Therefore, caution is advised when using intralesional steroids in the periocular region.105 HPA axis suppression can occur in infants receiving large doses intralesionally.106
Pulsed dye laser.
The flashlamp-pumped pulsed dye laser (PDL) has been used to treat hemangiomas. Superficial lesions appear to respond best to this treatment.107–109 Hemangiomas with a deeper dermal component may show lightening of the superficial erythematous portion, but treatment does not affect the deeper portion. Early laser treatment and its impact on future hemangioma growth is controversial. Multiple treatment sessions over several months may be needed to treat superficial hemangiomas. Treatment appears to be relatively well tolerated, but the risk of scarring appears to be higher than when similarly aged infants with port-wine stains are treated, particularly for large hemangiomas in a rapidly proliferative state. A randomized controlled study of PDL for early hemangiomas failed to show substantial benefits at 1 year of age, but this study has been criticized by some because it used a type of PDL without more recent improvements, such as a cooling spray to spare epidermal injury.110 Side-effects include transient pigmentary alteration and atrophic scarring.110 Severe ulceration and scarring have been reported, especially when treating segmental hemangiomas in the proliferative phase, although these observations were made using older laser technology and with treatment fluencies higher than now recommended.111
The pulsed dye laser is an effective treatment for residual telangiectasia in involuted hemangiomas, and may also be effective for ulcerated hemangiomas. Some patients who fail to respond to topical treatment for ulceration respond with healing of the ulceration and subjective reduction of pain after laser treatment.112,113 Other laser light sources, including the argon and Nd : YAG lasers, have been used to treat hemangiomas in earlier series, but are associated with higher risks of ulceration and scarring.
Systemic therapy.
Although systemic corticosteroids had been the mainstay of systemic treatment, more recently, beta-blockers have become first-line treatment for complicated IH. Propranolol is a nonselective beta-adrenergic antagonist that inhibits both beta-1 and beta-2 adrenoreceptors. In 2008, Léauté-Labrèze and colleagues reported the serendipitous discovery that propranolol had efficacy for infantile hemangioma treatment.114 Since that initial report, numerous case series have supported propranolol use both in the proliferative phase (Fig. 21.12), and – unlike systemic steroids – for older children with hemangiomas that have plateaued or partially involuted.115–117 Airway hemangiomas and hepatic hemangiomas have similarly been successfully treated with systemic propranolol alone or in combination with systemic steroids.118
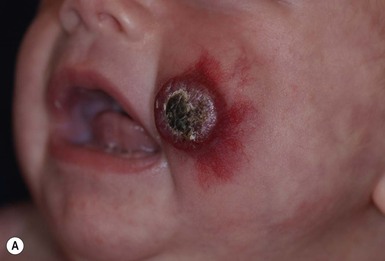
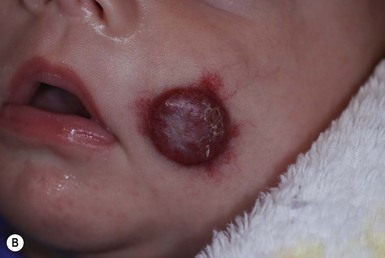
Although the exact mechanism by which propranolol works for infantile hemangiomas has not yet been elucidated, its vasoconstrictive effects on vascular smooth muscle are hypothesized to be responsible for the color changes and softening of hemangiomas often observed within in the first few days of treatment. In vitro, propranolol has been shown to decrease levels of vascular endothelial growth factor (VEGF), an important mediator in hemangioma proliferation.119 Propranolol may also induce apoptosis of endothelial cells and decrease the VEGF angiogenic effects.120
Because larger clinical trials are still in progress, use of propranolol is currently based on collective published experience and proposed consensus recommendations (Box 21.4).121 Propranolol is most commonly administered in initial oral doses of 0.5–1 mg/kg per day divided into twice or three times daily dosing with gradual titration over several weeks to achieve a target dose of 1–2 mg/kg per day in divided doses. A recent study has shown that doses of 3 mg/kg per day are also highly effective and well-tolerated, though some authors reserve its use for refractory cases. Before initiating treatment, evaluation of cardiovascular status includes exclusion of pre-existing heart block by a thorough cardiac exam and/or electrocardiogram, confirmation of normal vital signs, and formulation of a plan for monitoring vitals during the treatment period should be made.121 A history of bronchospasm or restrictive airway disease should be elicited and may be a relative contraindication to therapy. Recent guidelines for propranolol administration advocate caution with use in premature infants, infants under 8 weeks of age, and those with coexisting cerebrovascular or aortic anomalies.121 In these more vulnerable groups, consideration for inpatient observation during initiation is reasonable.121
Potential adverse effects of propranolol include hypotension, bradycardia, hypoglycemia, diarrhea, and sleep disturbance (Box 21.5). In patients treated for infantile hemangioma, only rarely has symptomatic hypotension or bradycardia occurred. Perhaps more concerning is hypoglycemia that has been reported in patients treated for infantile hemangioma who had recent illness or prolonged fasting.122 Of note, children as old as 18 months have been reported to have symptomatic hypoglycemia during propranolol therapy.122 Regular feeding is suggested with avoidance of prolonged fasting. Long-term steroid administration with the addition of propranolol therapy has been reported to induce hypoglycemia possibly due to associated adrenal insufficiency that may compound hypoglycemia risk.123 Propranolol should be stopped if a child is ill and not tolerated feeds, and can be restarted after illness resolves and routine feeding resumes.
The duration of propranolol therapy varies but is typically at least 6–12 months. Treatment should extend beyond the proliferative period, in order to try to avoid rebound growth with many patients requiring treatment until 12–15 months of age. Deep hemangiomas tend to grow longer and have a higher relapse rate compared with other hemangiomas.124 Although propranolol may be discontinued abruptly, tapering doses over several weeks may allow observation for potential early rebound in some patients.
In patients with large facial hemangiomas at risk for PHACE syndrome, imaging for significant cerebrovascular and aortic anomalies should be done prior to the initiation of propranolol, to assess the risk of potential complications if a hypotensive episode were to occur. Propranolol has been used successfully to treat patients with PHACE syndrome and the diagnosis of PHACE syndrome is not an absolute contraindication.125 However, its administration in these patients should proceed with caution and in collaboration with a neurologist or cardiologist as appropriate.
Corticosteroid therapy.
Systemic corticosteroids are also efficacious for hemangiomas, but have significant potential side-effects.126 Prednisone or prednisolone at a dosage of 2–3 mg/kg per day is used during the proliferating phase to slow or halt the growth of the hemangioma, and some authors advocate even higher doses.127 A systematic review of systemic corticosteroids used during the proliferating phase of infantile hemangioma128 found that most infants responded to treatment with cessation of growth. Therapy is slowly tapered over several months. The duration of treatment depends on response to therapy and the amount of time the hemangioma remains in the proliferating period.
A review of 62 children treated with prednisone or prednisolone at an initial dose of 2–3 mg/kg per day tapered over a mean of 7.9 months showed a low incidence of serious side-effects.129 Cushingoid facies, ‘personality changes,’ gastric irritation, and perineal and oral candidiasis are the most frequently reported short-term side-effects. Retardation of growth, including both height and weight, is also noted, with infants experiencing catch-up growth after cessation of therapy. Hypothalamopituitary–adrenal axis suppression is common, and the need to taper steroid doses prior to discontinuation is important. Hypertension is a potential complication of higher-dose therapy.130 In addition, patients should be cautioned about the risk of immunocompromise and susceptibility to infection, and live-virus vaccines should be avoided during corticosteroid treatment. Pneumocystis carinii pneumonia has been reported in an infant receiving corticosteroid therapy for infantile hemangioma.131 There are recent reports of neurotoxicity occurring in very premature infants receiving corticosteroid therapy within the first few weeks of life (in an attempt to prevent chronic lung disease).132 This type of neurotoxicity has not been reported to date in term infants receiving systemic corticosteroids; however, further studies would be helpful to determine optimal treatment protocols and to assess for short- and long-term side-effects.
Other therapies.
Recombinant interferon-α has been successful in the treatment of life-threatening hemangiomas that have failed to respond to corticosteroid therapy.133–135 Both interferon-α2a and -α2b have been used. The most common treatment regimen consists of a daily injection of 3 million U/m2 as needed.134 The response to therapy is variable, with some patients responding rapidly over the course of 3–6 months and others needing longer periods of treatment. Side-effects associated with treatment include fever, neutropenia, altered hepatic function chemistries, flu-like complaints, and agitation. Spastic diplegia, a side-effect reported in five of 26 infants receiving interferon-α2a for hemangiomas, is a particularly worrisome potential complication.136 Although this has led some to recommend the use of the α2b preparation, neurotoxicity has also been reported with this preparation. The risk of neurotoxicity after age one may be lower.136 Neurodevelopmental status should be assessed at baseline, and monthly during and after treatment with interferon-α.
Vincristine has been used to treat corticosteroid-resistant hemangiomas and ‘vascular tumors’ as well as Kasabach–Merritt syndrome.137 This chemotherapeutic agent is a vinca alkaloid that interferes with microtubule formation during mitosis by inhibiting tubulin. In vitro, it induces apoptosis of tumor and endothelial cells.138 Peripheral neuropathy, constipation, jaw pain, anemia, and leukopenia are potential toxicities. Administration through a central venous catheter may be necessary, and the participation of a pediatric oncologist/hematologist is advised. It is usually effective, but studies are needed to determine the indications for its use, the incidence of adverse reactions, and the optimal dosage regimen.139
Surgical excision.
Surgical excision is usually reserved for involuted hemangiomas to remove residual fibrofatty tissue and redundant skin. Early excision is indicated for a small subset of hemangiomas. These include periorbital hemangiomas that fail to respond to pharmacologic therapy, or patients in which medical therapy is believed to pose a greater risk, painful chronically ulcerated hemangiomas that have failed more conservative treatment, some large nasal tip hemangiomas, and some pedunculated hemangiomas that will inevitably result in prominent fibrofatty residual tissue even after involution is complete.
Treatment of ulceration.
Ulceration is the most common complication of infantile hemangioma and should be treated promptly (Box 21.6). All ulcerations leave some degree of scarring. Ulceration can also cause pain, which can be severe, particularly in perioral and perineal locations,140 and medications to control pain, oral antibiotics, and other modalities such as pulsed dye laser may be necessary. There have also been reports of improvement after the application of 0.01% becaplermin (recombinant platelet-derived growth factor),140,141 but becaplermin is approved for the treatment of diabetic ulcers in adults and not approved for use in children. Its mechanism of action includes the promotion of angiogenesis, which raises questions about its potential for stimulating growth and worsening of hemangiomas. Topical beta-blockers have been helpful in some cases but the systemic absorption through the ulcer bed is not known. Systemic therapy may be indicated in some patients with ulcerated hemangiomas, and excisional surgery may need to be considered when medical therapy fails.
Other vascular tumors
The more rare vascular tumors discussed in the following sections are recognized by their clinical and histopathologic features and immunohistochemical markers. Recognition may require a skilled pathologist who is familiar with these vascular lesions.
Congenital hemangiomas
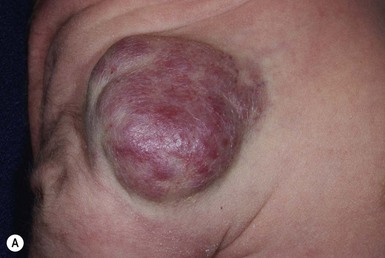
Hemangiomas that are fully formed tumors at birth and do not proliferate in postnatal life are distinct entities referred to as ‘congenital hemangiomas’.142,143 There are two subtypes of congenital hemangiomas which are differentiated based on their natural history: the rapidly involuting congenital hemangioma (RICH) and the non-involuting congenital hemangioma (NICH).
Cutaneous findings
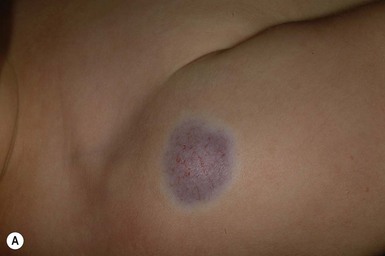
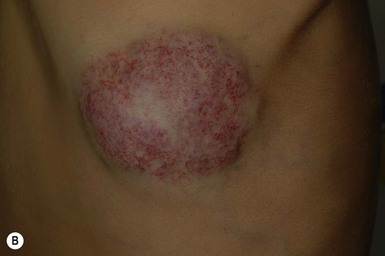
Both RICH and NICH are more common on the extremities or on the head and neck, vary in size from a few centimeters to 10–12 cm, and are often warm to palpation.142,144 They are usually solitary with rare cases of multifocal RICH reported.145 The proliferating phase of both RICH and NICH occurs in utero with no further growth occurring after birth. Three clinical morphologies of RICH have been described: (1) a raised violaceous tumor with prominent peripheral vasculature; (2) a raised tumor with overlying coarse telangiectasias with skin blanching, often a peripheral halo of pallor; and (3) a pink to violaceous tumor with deeper infiltrative papules or nodules (Fig. 21.13A–D ). The overlying skin may show hypertrichosis. Accelerated spontaneous involution starts in the first weeks of life,142,143 with regression by 14 months of age. After involution, atrophy, milia, telangiectasias and dilated veins may persist. Rarely, complete involution of RICH occurs in utero with atrophy at the site present at birth.146
). The overlying skin may show hypertrichosis. Accelerated spontaneous involution starts in the first weeks of life,142,143 with regression by 14 months of age. After involution, atrophy, milia, telangiectasias and dilated veins may persist. Rarely, complete involution of RICH occurs in utero with atrophy at the site present at birth.146
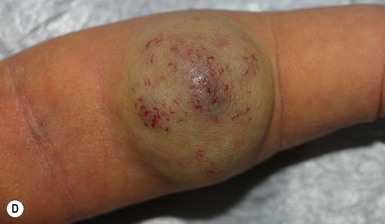
NICH may present as patches or large vascular tumors (Fig. 21.14). Enlarging draining veins are typical with both NICH morphologies. The lesions are usually less exophytic and less impressive in appearance than a RICH. They grow proportionately with the child and do not regress spontaneously. Some of these lesions clearly show arteriovenous microfistulas on Doppler examination, and these may develop increasing equatorial draining veins after years. They are often misdiagnosed in infancy as an infantile hemangioma, and the correct diagnosis is delayed until adolescence or adulthood, when they are excised, after having failed to involute. NICH may be painful in a minority of cases. Some cases of RICH involute only partially, and the residual tumor resembles NICH, supporting the concept that RICH and NICH may exist as a spectrum.147
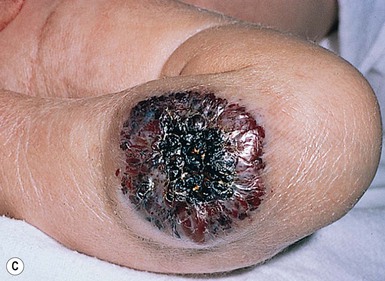
Ulceration is a rare complication in a congenital hemangioma and is primarily reported with a RICH soon after birth. Ulceration is usually noted in the center of a lesion and may be preceded by a necrotic crust. Severe bleeding episodes have been reported in association with ulceration.148,149 Necrosis and ulceration are rarely reported with a NICH.
Extracutaneous findings
A RICH may also present as a solitary hepatic lesion, often in an otherwise healthy infant. Some of these infants experience thrombocytopenia and anemia and rarely congestive heart failure due to arteriovenous shunting. In severe cases, treatment may be warranted with surgery, embolization and rarely liver transplantation.150,151 Transient coagulopathy with thrombocytopenia has been reported to occur with a cutaneous RICH and can be confused with mild Kasabach–Merritt phenomenon. Rarely, more severe coagulopathy and congestive heart failure are seen with cutaneous lesions alone.152,153
Etiology and pathogenesis
Congenital hemangiomas are reported equally in males and females with few case series reporting a male predominance. There are no known pre- or perinatal risk factors associated with having a congenital hemangioma and the pathogenesis is unknown.154 Traditionally, infantile hemangiomas and congenital hemangiomas were considered variants of the same tumor, but an increased understanding of clinical and immunohistochemical differences has clarified the classification and delineated them from one another.147,155 Both RICH and NICH demonstrate the proliferative elements seen in vascular tumors like infantile hemangiomas and dysplastic vessels more typically seen in vascular malformations. Moreover, they are Glut-1 negative, further distinguishing them from infantile hemangiomas.143,155
Differential diagnosis
Congenital hemangiomas are usually diagnosed based on clinical characteristics, but Doppler ultrasound and MRI can be helpful. MRI may be considered in the newborn whose tumor has a significant central crust or wound at birth, in order to evaluate the risk for hemorrhage: strikingly, some RICH are highly vascularized, with large tortuous flow voids on MRI, and others are relatively poorly vascularized, with MRI demonstrating a tumor of intermediate signal on T1-weighted signal-enhanced sequences. The main differential diagnosis of both RICH and NICH is common infantile hemangioma, but in atypical cases other soft tissue tumors, including malignancies, must be considered. Thrombocytopenia can lead to consideration of other vascular tumors associated with Kasabach–Merritt phenomenon (see below), but the drop in platelet count is usually brief, rather than progressive. A biopsy may occasionally be required.142
Occasionally, congenital hemangiomas are noted on routine prenatal ultrasound evaluation and may be mistaken for vascular malformations or other forms of neoplasia. Those that are detected usually reveal prominent vascularity and high flow.
Management
Indications for treatment of RICH include ulceration and bleeding, functional impairment (depending upon location), congestive heart failure, and prominent residual skin changes. Early excision may be particularly important in infants with ulceration or when a necrotic area is present, as there are often large, fast-flow vessels close to the surface, conferring a risk of severe hemorrhage.156 Indications for treatment of NICH (typically surgical excision) depend on whether their appearance is bothersome, or whether bleeding or pain are present.
Tufted angioma and kaposiform hemangioendothelioma
Tufted angioma (TA) and kaposiform hemangioendothelioma (KHE) are rare vascular tumors with many similar histologic and clinical features.157 Both are associated with Kasabach–Merritt phenomenon (described below).
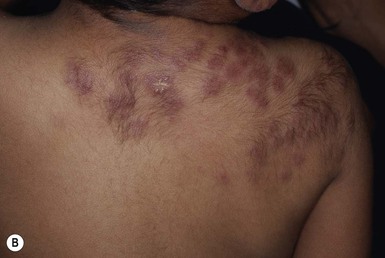
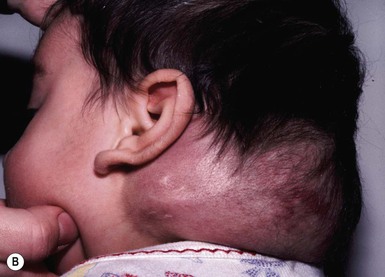
Tufted angioma, previously known in the Japanese literature as angioblastoma of Nakagawa, displays pathologic features of vascular tufts of tightly packed capillaries in a typical cannonball distribution, which are diagnostic for this benign vascular tumor.158–160 Most cases are acquired early in childhood and may have a protracted course with rare congenital forms reported.159 Common presenting clinical features are dusky erythematous to violaceous plaques or nodules with thickening over time. Rare multifocal TA are reported.161 Increased hair overlying this tumor is a relatively common finding with increased sweating or pain less commonly seen (Fig. 21.15A–D ). TAs may regress completely within a few years, shrink leaving a residuum, or persist unchanged.162,163
). TAs may regress completely within a few years, shrink leaving a residuum, or persist unchanged.162,163
Kaposiform hemangioendothelioma is a rare vascular tumor named for its histological resemblance to Kaposi’s sarcoma. KHE usually presents in infancy but may appear years later.164 It often presents as a violaceous to purpuric infiltrative, firm plaque or nodule with rare spontaneous involution. KHE may involve deep tissues and bones, and rare lymph node involvement has been reported without distant metastases.165 Most reported cases have had associated Kasabach–Merritt phenomenon, but KHE can occur in the absence of a coagulopathy.166 Both KHE and TA are more common on the extremities and trunk. In the absence of KMP, therapy for TA or KHE is individualized and may include observation, surgical excision, pulsed dye laser and other medical therapies.
Kasabach–merritt phenomenon
Since the first case report in 1940, the label Kasabach–Merritt phenomenon (KMP) has been used to describe infants with vascular anomalies and thrombocytopenia or other coagulopathy, and the syndrome was long considered to be a complication of a ‘hemangioma’.167 Several publications have now documented that this biologic phenomenon is not associated with a ‘true’ hemangioma of infancy, but rather with other vascular tumors, especially kaposiform hemangioendothelioma (KHE) or tufted angioma (TA), which may occur in association with a lymphatic malformation.164,168–171
Cutaneous findings
Affected infants have a congenital tumor or a lesion noted soon after birth, with subsequent development of an inflammatory, bruising, reddish or purple mass, and purpura. At times, the enlargement of the lesion and affected area can be massive (Fig. 21.16). Before the development of thrombocytopenia and platelet trapping, the clinical appearance may be quite variable as those described above for a TA or KHE. Rarely, an affected newborn may have a history of a congenital bulky tumor detected by prenatal ultrasound.172
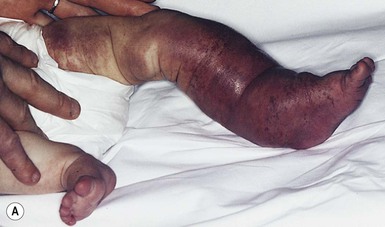
Extracutaneous findings
Severe thrombocytopenia due to platelet trapping is the hallmark of KMP. Consumption of fibrinogen, elevated d-dimers and anemia are common with KMP. The prothrombin time and the activated partial thromboplastin time are normal to elevated. The coagulopathy occurs to varying degrees, depending on the severity and phase of the disease. KMP has no anatomic site of predilection. Although more cases are within the skin and musculature, KMP can involve deeper visceral structures. Visceral KMP – cervicothoracic, abdominal, pelvic, or intracranial – is life-threatening.
Etiology and pathogenesis
The pathogenesis of TA and KHE is not well understood. They are thought to be derived from lymphatic endothelium, and lymphatic markers. D2-40 is positive in KHE supporting this hypothesis.173 These tumors are GLUT-1 negative distinguishing them from infantile hemangiomas.65,165 The pathogenesis of KMP is related to platelet trapping within the tumor with later hypofibrinogenemia and fibrinolysis. Surgical procedures or other trauma and infection seem to worsen the coagulopathy in many cases.
Course
Thrombocytopenia may persist for years, but more commonly, it resolves with treatment by 12–18 months of age or sooner. Hemorrhage, sepsis, organ failure or invasion of vital structures may cause death in up to 10–30% of cases. When the hematologic phenomenon is cured, the tumor shrinks but the patient usually has residual cutaneous changes. The tumor appearance varies from that of a pseudo-port-wine stain, with infiltrated areas and papules, stain-like areas with haloes, and nodular residua, to a poorly-delineated, fibrotic-feeling plaque. In some cases, muscle and joint fibrosis remains with an associated morbidity.174
Diagnosis
The diagnosis should be suspected in a newborn or young infant with an atypical vascular lesion with the clinical findings described for a TA or KHE. Characteristic thrombocytopenia and hypofibrinogenemia support the diagnosis of TA or KHE. MR imaging may be useful in determining the extent of disease and response to treatment. Skin biopsy may be necessary in atypical cases with the consideration for excess bleeding during the procedure due to the coagulopathy.
Differential diagnosis
Newborns or young infants with malignant soft tissue tumors will occasionally present with a large soft tissue mass and thrombocytopenia. Rarely, mild thrombocytopenia can be present in congenital hemangiomas due to platelet trapping, but this does not become as severe or as persistent as in KMP. In addition, large venous or mixed malformations can have an associated coagulopathy, but elevated d-dimers and consumption of clotting factors, rather than platelet trapping, is noted. Multifocal vascular lesions with an associated thrombocytopenia are more likely to be due to ‘multifocal lymphangioendotheliomatosis with thrombocytopenia’ than to KMP. Infantile hemangiomas are not associated with a coagulopathy.
Management
Infants with KMP exhibit an inconsistent and variable response to therapeutic regimens and are most often treated with multiple medical agents. Medical therapies include systemic corticosteroids, vincristine, interferon, anti-fibrinolytic agents (tranexamic acid and aminocaproic acid), antiplatelet agents (ticlopidine and aspirin), multi-agent chemotherapy, propranolol and rapamycin.175–177 Recent expert consensus opinion suggests first-line therapy for KMP with the combination therapy of intravenous vincristine and oral or intravenous corticosteroids.178 Surgical excision, arterial embolization, and radiation therapy may also benefit some patients. Of note, complete surgical excision is curative but rarely feasible. While platelet transfusions may be required for active bleeding or surgical intervention, they should be otherwise avoided due to the risk of further platelet trapping and tumor enlargement. Care of KMP patients should include an experienced multidisciplinary team that includes a hematologist/oncologist when available.179
Multifocal lymphangioendotheliomatosis with thrombocytopenia
This rare condition, also called cutaneo-visceral angiomatosis with thrombocytopenia, is characterized by multifocal congenital and progressive cutaneous and extracutaneous vascular papules and plaques. The vascular skin lesions are variable in number and range in morphology from telangiectatic macules to red-brown plaques to exophytic nodules. Affected infants typically have chronic fluctuating thrombocytopenia and bleeding due to vascular lesions within the GI tract.180–182 GI bleeding can be significant with mortality reported. Management is multimodal using both medical and surgical approaches.183–185 Other sites of involvement include bones, synovium, lungs, liver, muscle, kidney, spleen, and brain. Skin biopsy specimens demonstrate thin-walled vessels, hobnailed endothelial cells, and intraluminal papillary projections. Vessels stain positive for lymphatic vessel endothelial receptor-1 (LYVE-1).180,186
Infantile hemangiopericytoma
Childhood hemangiopericytoma (HPC) is a rare vascularized tumor which is no longer thought of as a vascular tumor, but rather of fibroblastic or myofibroblastic origin.187 The infantile type, presenting at <1 year of age, is differentiated from the adult type because of its more favorable prognosis with a less aggressive clinical course and good response to chemotherapy.134,188 Onset after a year of age holds a similar poorer prognosis to the adult type. The infantile type is more common in boys and usually presents as a firm skin-colored, blue or violaceous dermal or subcutaneous nodule that may become necrotic. Most often cutaneous lesions are located on the head, neck or extremities and are less commonly multicentric with intracranial, mediastinal, abdominal, and lung lesions reported.189 The better prognosis of the infantile type of HPC and its pathologic features suggests a continuum with infantile myofibromatosis, but this concept and the classification of HPC remains controversial.190
Spindle-cell hemangioendothelioma
Spindle-cell hemangioendothelioma (SCHE) occurs at any age and site but is most commonly reported on the distal extremities. Lesions are solitary or multiple and described as red-brown or blue nodules. Pathology is consistent with a nodular, dense, spindle cell proliferation associated with ‘cavernous’ vessels of attenuated, irregularly thickened walls. Lesions are treated with excision with local recurrence being common. Metastases have not been reported. Few reported patients with SCHE also had Maffucci syndrome.191
Congenital eccrine angiomatous hamartoma
Congenital forms of this tumor present as a solitary ill-defined, red-to-blue plaque or nodule, sometimes with lanugo, hyperhidrosis or pain at the site.192,193 Lesions are usually located on the extremities or trunk and may spontaneously involute. Rarely are they multifocal. It is not clear whether the reported congenital cases are identical to the slowly growing, persistent acquired form that is characterized by a bluish, brown or erythematous firm nodule or plaque with excess hair, hyperhidrosis, and pain overlying a slow-flow vascular malformation.192,194 Diagnosis is established on the basis of characteristic histologic findings which include hyperplasia of normal or dilated eccrine glands in association with dilated capillaries, a variable presence of pilar, lipomatous, mucinous, and lymphatic structure.
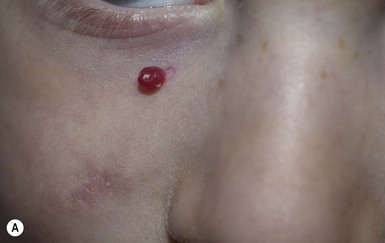
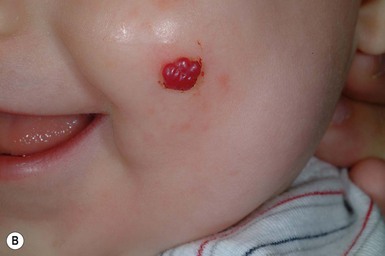
Pyogenic granuloma (lobular capillary hemangioma)
Pyogenic granuloma (PG) – also known by its correct histopathologic description ‘lobular capillary hemangioma’, rather than its historic misnomer – is a common vascular tumor of infants and children, albeit relatively rare in the newborn period. PGs usually present as a solitary, red, rapidly growing papule or nodule, often located on the head and neck (Fig. 21.17A,B ). They can be pedunculated and often develop an eroded surface and bleed easily and profusely. PGs can occur within an existing port-wine stain. When a PG occurs in a very young infant, it may be mistaken for an infantile hemangioma, and close examination for the collarette of scale seen in many PGs may help differentiate the two.195 The differential diagnosis in infants and older children also includes a Spitz nevus. Rare multifocal forms have been observed in newborns, but before specific immunohistochemical identification of infantile hemangiomas was possible they were often mistaken for infantile hemangiomas.196–199 PGs do not involute spontaneously, but simple curettage or shave excision followed by electrocautery is usually curative.200,201 This quick procedure can often be done under local anesthesia even in young children. Other treatment options include pulsed dye laser for small thin lesions or imiquimod but repeated or prolonged treatment, respectively, may be needed with these modalities.202
). They can be pedunculated and often develop an eroded surface and bleed easily and profusely. PGs can occur within an existing port-wine stain. When a PG occurs in a very young infant, it may be mistaken for an infantile hemangioma, and close examination for the collarette of scale seen in many PGs may help differentiate the two.195 The differential diagnosis in infants and older children also includes a Spitz nevus. Rare multifocal forms have been observed in newborns, but before specific immunohistochemical identification of infantile hemangiomas was possible they were often mistaken for infantile hemangiomas.196–199 PGs do not involute spontaneously, but simple curettage or shave excision followed by electrocautery is usually curative.200,201 This quick procedure can often be done under local anesthesia even in young children. Other treatment options include pulsed dye laser for small thin lesions or imiquimod but repeated or prolonged treatment, respectively, may be needed with these modalities.202
Access the full reference list at ExpertConsult.com 
Figures 13C, D, 15C, D and 17B are available online at ExpertConsult.com 
Box 3 is available online at ExpertConsult.com 
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Infantile Hemangiomas and Other Vascular Tumors
Anita N. Haggstrom, Sarah L. Chamlin
Infantile hemangioma
Infantile hemangioma (IH) is the most common vascular tumor encountered during early infancy. It is a benign proliferation of endothelial cells that characteristically undergoes a phase of rapid growth followed by slow spontaneous involution. Although large population-based studies are lacking, infantile hemangiomas are estimated to occur in 2.6–5% of infants.1,2 The incidence is highest among Caucasian infants with lower rates in African-American, Hispanic, and Asian infants.3 Females are more commonly affected than males at ratios of 2–3 : 1, with the female-to-male ratio approximately 9 : 1 in patients with segmental hemangiomas associated with PHACE syndrome.4,5
Caucasian race, prematurity and low birthweight are well-established risk factors for the development of IH.3,6 The risk of IH increases by 40% for every 500 g decrease in birthweight.7 A prospective study of over 1000 children with infantile hemangiomas identified additional risk factors, including multiple gestation pregnancies, advanced maternal age, and pregnancies complicated with pre-eclampsia or placenta previa.5,8 Placental abnormalities that impair utero-placental circulation, including retroplacental hematoma, ischemic infarction, dilated vascular communications, vasculitis and chorioamnionitis have been observed to be more common in infants with IH.9 An earlier study reported a threefold increased incidence of hemangiomas in infants born to mothers who undergo chorionic villus sampling, compared with those whose mothers undergo amniocentesis, but subsequent studies have not been conclusive.10 In-vitro fertilization has been suggested as a possible risk factor in one study.2 The majority of hemangiomas arise sporadically; however, a family history can be elicited in some patients, and autosomal dominant transmission has been reported.11
Clinical subtypes
Infantile hemangiomas can be classified based on their location within the skin: superficial, deep, and mixed (superficial and deep). Superficial hemangiomas are the most common type, occurring in approximately 50–60% of cases. Mixed-type IH are estimated to occur in 25–35% of cases, and deep hemangiomas in 15%.12 Superficial hemangiomas are typically bright red with a lobulated surface. These characteristics have led to the use of the term ‘strawberry’ hemangioma. Deep hemangiomas are not commonly noted in the neonatal period, often appearing at 1–3 months of life, or later, as a warm, subcutaneous mass caused by proliferation of the tumor in the deeper portion of the dermis or subcutis. The overlying skin may appear normal or have relatively inconspicuous superficial changes, such as faint blue color, telangiectasias or dilated veins. Mixed-type hemangiomas frequently exhibit a configuration resembling a poached egg, with a well-circumscribed red superficial portion overlying a less well-defined bluish to violaceous deeper component.
In addition to characterizing hemangiomas according to their depth and location within the skin, the pattern of involvement is a predictor of prognosis and risk for associated complications.5 Described patterns include ‘focal’, or localized, lesions that appear to arise from a central point, and ‘segmental’ lesions, which typically encompass a larger territory of skin, which in some anatomic sites clearly corresponds to a developmental subunit (Fig. 21.1).13,14 Some lesions are difficult to classify as either focal or segmental and are thus designated ‘indeterminate’. Segmental lesions more often require treatment and are more likely to be associated with complications, including underlying extracutaneous anomalies, including spinal dysraphism, genitourinary anomalies, and PHACE syndrome.14 The patterns of segmental hemangiomas correspond to known embryologic prominences, suggesting a neuroectodermal derivation for their distribution.15 The frontotemporal (S1), maxillary (S2), mandibular (S3), and frontonasal (S4) segments designate commonly observed patterns of facial hemangiomas (Fig. 21.2).15
Natural history
Infantile hemangiomas have a characteristic clinical course marked by rapid early proliferation, slower late proliferation, a plateau phase, and finally gradual involution. They are either present at birth or appear in the first few weeks of life. Precursor lesions (or so-called ‘premonitory marks’) are seen in as many as 65% of patients and may appear as discrete fine telangiectasias superimposed on a background of pallor, a faint erythematous patch, or a bruise-like area (Fig. 21.3).16 It may be difficult initially to differentiate an erythematous hemangioma precursor from a capillary malformation, but clues include a more ill-defined border, fine telangiectasias, and, if present, skin ulceration. Serial examinations may be necessary to establish the correct diagnosis. Rarely, a hemangioma on the perineum or lip may present in the neonate with ulceration, without an obvious hemangioma, mimicking bacterial or herpetic infection. In these cases, the hemangioma becomes evident over the subsequent days to weeks (see Chapters 10 and 17).
In the first few weeks of life, most infantile hemangiomas proliferate rapidly; 80% of superficial hemangioma growth occurs before 3 months of age, with a period of accelerated growth between 5 and 7 weeks of age.16,17 The late proliferative stage is marked by slower growth, but is more variable in its duration, with certain subsets of hemangiomas, including those that are large, segmental, or those with a significant deep component growing much longer, even beyond the age of 1 year.17,18 Most hemangiomas establish the boundaries of their anatomic territory relatively early, growing only in volume thereafter. Superficial hemangiomas initially have a bright red color early in proliferation. Both combined and deep hemangiomas may feel tense, and fluctuations in size and volume may be noted with crying or activity. The earliest signs of involution include a change in color from bright red to a violaceous gray, usually beginning in the center of the hemangioma. The deeper portion also regresses, but this is often not as apparent clinically. Parents may note less fluctuation in hemangioma size during crying and less tumor firmess.6 The warmth of the hemangioma diminishes with time. Some resolve leaving normal or nearly normal skin, whereas others resolve with residua of variable cosmetic significance (Fig. 21.4). Involuted hemangiomas often show residual telangiectasia, pallor or a yellowish color, fibrofatty tissue deposition, and atrophy (Fig. 21.5). Several studies have shown that completion of involution occurs at a rate of approximately 10% per year, with 30% completing involution at 3 years, 50% at 5 years, etc.4,19 Although not specifically mentioned in these studies, it seems that small hemangiomas generally involute sooner than very large, bulky ones.
Less commonly, infantile hemangiomas display a more atypical growth pattern characterized by little, if any postnatal growth. These hemangiomas have been variously called infantile hemangiomas with minimal or arrested growth (IH-MAG), abortive hemangiomas, or reticular hemangiomas.20,21 IH-MAG present as telangiectatic patches that have minimal superficial proliferation, often manifested as 1–3 mm red papules at the periphery of the lesion and have a predilection for the lower extremities (Fig. 21.6).22 They may have violaceous to green ectatic vessels underlying the finer telangiectasias. The small proliferative vascular papules involute within the first few years of life but the ectatic vessels can be persistent. The vessels of IH-MAG stain positively for GLUT-1, similar to classic infantile hemangiomas.
Ulceration is the most common complication of infantile hemangiomas, occurring in approximately 10–15%, most often during the rapid growth phase. The risk is much higher (approx. 50%) for hemangiomas involving the lip and perineum.23 When the ulceration heals, there is virtually always residual atrophy or scarring. Bleeding may occur, but profuse bleeding is surprisingly rare, occurring mainly with deeply ulcerated hemangiomas or those located on the scalp (Fig. 21.7). Superimposed bacterial infection most commonly with Staphylococcus aureus or Pseudomonas species may develop and may impede healing, but it is uncommon. Recurrent ulceration may complicate hemangiomas located in the perineum.
The Kasabach–Merritt phenomenon (KMP) was originally believed to be a complication of large hemangiomas, but studies have proven that KMP is a complication of other vascular tumors, including kaposiform hemangioendothelioma and tufted angioma, rather than infantile hemangiomas (see below).
High-risk hemangiomas.
Although the vast majority of hemangiomas are not worrisome and do not necessitate treatment, it is important to recognize the subsets of hemangiomas that are at highest risk for causing permanent disfigurement, impairing vital functions including vision and feeding, and that may be associated with visceral hemangiomatosis or internal anomalies (Box 21.1).
Locations associated with high risk for disfigurement.
The most common reason infants with hemangiomas receive treatment is the potential risk for disfigurement (Fig. 21.8).5 Hemangiomas involving the central face may distort important anatomic landmarks and have a significant impact on future cosmesis. Although exophytic lesions on the medial cheek do not impair vital functions, they are often distressing because of their conspicuous locations and may benefit from more aggressive medical therapies. Distortion of the lip contour can occur even with small hemangiomas because of the disruption of the vermillion border or loss of the curvature of the philtrum. Mixed hemangiomas located on the nasal tip distort the underlying cartilage and leave residual bulk. Surgical repair is often necessary to correct the ‘Cyrano nose’ deformity that can result in splaying of the nasal cartilage. Ulceration involving the columella can lead to destruction of the nasal septum. Hemangiomas on the pinna may ulcerate and become secondarily infected, contributing to structural deformity of the involved ear.
Periorbital hemangiomas.
Growth of periorbital and lid hemangiomas may cause visual impairment principally by deformation of the cornea, creating refractive errors or less commonly by obstructing the visual axis. If severe or persistent, this can lead to amblyopia. Large and/or segmental hemangiomas in the periocular area have the greatest risk of these complications, but even small lesions may pose a threat to normal visual development, with astigmatism being the most common sight-threatening ocular complication.24,25 Proliferation of retrobulbar lesions may lead to proptosis, strabismus, and visual compromise.26
Large cervicofacial hemangiomas and PHACE syndrome.
Large cervicofacial hemangiomas may impair vital functions, distort normal anatomy, or lead to congestive heart failure as they proliferate. These problematic hemangiomas are more common in females than males at a ratio approaching 9 : 1 in some series.27 The term PHACE syndrome refers to the association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities (Box 21.2). The term PHACE(S) is sometimes used when associated sternal anomalies or a supraumbilical raphe are present. Although the exact incidence of PHACE is unknown, it may be more common than Sturge–Weber syndrome.27 In 2009, a multidisciplinary panel of experts published diagnostic criteria for ‘definite’ and ‘possible’ PHACE using specific major and minor criteria.28 A multicenter prospective study of 108 infants with facial hemangiomas measuring 22 cm2 or larger, found that when applying published diagnostic criteria for PHACE, 31% had PHACE and 90% of affected infants had more than one extracutaneous finding. The most common extracutaneous manifestation in PHACE is anomalous cerebrovasculature, with the highest incidence seen in infants with upper face hemangiomas (S1, frontotemporal or S4, frontonasal) (Figs 21.4, 21.9).29
Central nervous system (CNS) anomalies include both structural malformations of the brain and anomalous vasculature of the head and neck, both of which are typically ipsilateral to the cutaneous hemangioma.30 Structural CNS malformations include the Dandy–Walker malformation and other posterior fossa anomalies, such as arachnoid cyst, enlarged fourth ventricle, enlarged cisterna magna, and cerebellar or vermian hypoplasia.31–33 Other CNS anomalies have been reported, including cerebral atrophy, gray matter heterotopia, pituitary abnormalities, and absent corpus callosum. Macrocephaly, ophthalmologic abnormalities, hypotonia, seizures, and psychomotor retardation may be a presenting sign of an underlying structural malformation. Cerebrovascular abnormalities often involve the ipsilateral internal carotid artery and its branches, though other vessels can be involved.30 Dysgenesis of the vessel, including looping, elongation, ectasia, kinking, and focal or fusiform aneurysmal enlargement of the vessel, is most frequently noted.30 Less commonly, vessels can have an anomalous course and/or origin, display narrowing or be completely absent.30 Rarely, patients with intracranial anomalous vessels may demonstrate progressive occlusive arterial changes and cerebral infarction.34 Stroke is a rare complication of PHACE and may be more common in patients with aplasia, hypoplasia or occlusion of a major cerebral artery.35 Other neurologic sequelae of PHACE may include seizures, migraine-like headaches, and developmental delay, including most commonly language and gross motor abilities.36
Cardiac abnormalities, seen in two-thirds of infants with PHACE, usually manifest as unusual forms of aortic arch coarctation, primarily involving long segment narrowing of the transverse aorta often associated with adjacent dilatation or aneurysms.29,37,38 Less frequently, structural anomalies of the heart may be seen, including atrial and ventricular septal defects.28
Less common manifestations include ocular, midline anomalies and hearing loss of the affected ear.39 Ocular findings may include persistent fetal vasculature, retinal vascular anomalies, optic nerve hypoplasia, morning glory deformity, peripapillary staphyloma and coloboma, with other less specific anomalies noted more rarely. Midline defects such as partial or complete sternal agenesis and supraumbilical raphe may also occur.28,40
Endocrine abnormalities, including pituitary dysfunction, thyroid dysfunction, and growth hormone deficiency have been reported in some patients with PHACE. A few infants with PHACE have been noted to have absent or lingual thyroid glands.41–43
Lumbosacral hemangiomas and regional anomalies.
Analogous to PHACE syndrome, segmental lumbosacral and perineal hemangiomas can be cutaneous signs of regional anomalies such as underlying structural malformations of the genitourinary, gastrointestinal, neurologic, and skeletal systems (Fig. 21.10).44 The acronyms for this association include LUMBAR, PELVIS, and SACRAL syndromes.44–46 A prospective study of 41 lumbosacral hemangiomas >2.5 cm reported 35% had evidence of spinal dysraphism.47 Tethered spinal cord, spinal lipomas and spinal hemangiomas are the most common underlying abnormalities.48 Imperforate anus, rectoscrotal fistula, renal anomalies, abnormal external genitalia, lipomyelomeningocele, and bony deformities of the sacrum have been reported.44–46 Ulceration is associated with a higher risk of underlying anomalies.47
Multifocal cutaneous and visceral hemangiomas.
Multiple hemangiomas occur in approximately 10–25% of infants and are more common in premature infants than in term infants.49 Cutaneous lesions may range in size from a few millimeters to more than several centimeters in diameter. The presence of five or more cutaneous hemangiomas is a known risk factor for extracutaneous hemangiomas, mainly hepatic hemangiomas. Rarely, other organs such as the gastrointestinal tract, lungs, CNS, oral mucosa, and eyes may be affected. Screening ultrasound of the liver is recommended for young infants with five or more cutaneous hemangiomas.50 Serial radiologic evaluation of the liver, especially screening abdominal ultrasound, with further evaluation with CT or MRI, may be necessary to follow progression of visceral lesions. Hepatic hemangiomas (HH) may be solitary or multiple. A classification for hepatic hemangiomas has been proposed, including solitary, multifocal, and diffuse HH.51 Solitary HH in the absence of cutaneous hemangiomas – particularly if detected in utero – are usually GLUT1 negative, regress rapidly, and most likely represent a form of vascular tumor in the liver similar to rapidly involuting congenital hemangioma (RICH) rather than true infantile hemangioma (see below). Multifocal hepatic lesions usually occur in the setting of multiple small skin hemangiomas. They may be completely asymptomatic without the need for treatment, but if they have arteriovenous shunts, they may require liver embolization and/or medical therapy to manage congestive heart failure, as well as hemangioma-specific therapy (such as beta-blockers). Hepatic hemangiomas associated with an arteriovenous, arterioportal shunt or portovenous fistula are associated with greater morbidity.51 Diffuse liver hemangiomas cause massive hepatomegaly, abdominal compartment syndrome, and are associated with hypothyroidism (see below). They have a high rate of mortality and require aggressive medical therapy, and even consideration of liver transplantation.
Consumptive hypothyroidism may occur in association with hepatic hemangiomatosis due to the deactivation of thyroxine by a type 3 iodothyronine deiodinase produced by the hemangioma.52–54 This form of hypothyroidism can be detected by the presence of markedly elevated TSH and low T3. T4 levels may be normal or low. Neonatal screening for hypothyroidism is inadequate to assess for this complication, as the most active phase of hemangioma proliferation typically occurs after this period. In some cases, this consumptive form of hypothyroidism may require large doses of thyroid hormone replacement given intravenously. The hypothyroidism typically resolves as the hemangioma regresses, but may have already caused significant morbidity if not detected promptly. Type 3 iodothyronine deiodinase activity has also been found in association with large cutaneous hemangiomas, therefore thyroid function tests should also be considered in patients with large cutaneous hemangiomas.52
Cutaneous hemangiomas associated with airway hemangiomas.
Infantile hemangiomas of the airway are most commonly seen in the subglottis and can occur with or without cutaneous hemangiomas. Hemangiomas in the ‘beard distribution’ involving the skin overlying the mandible, chin, and neck, have a high risk of concomitant airway involvement (Fig. 21.11). There is a striking female predilection of 6–7 : 1 for ‘beard’ hemangiomas.55 Though hemangiomas in the mandibular region are most common, other hemangioma patterns that have been associated with airway hemangiomas include unilateral multisegment and hemifacial hemangiomas.56,57 Affected infants typically present within the first few weeks of life with an increasing degree of noisy breathing, stridor, hoarse cry, or other signs of airway obstruction. Having an airway hemangioma together with one or more large facial hemangiomas confers a greater than expected incidence of PHACE – possibly as high as 47%.57
Pathogenesis
Infantile hemangiomas are believed to represent localized areas of abnormal vascular growth, and several hypotheses have been proposed to explain their pathogenesis. These include endothelial cell defects, a placental origin, extrinsic abnormalities in neighboring cells and exposure to vascular growth stimulators. IH was long thought to be a disease of aberrant angiogenesis, however more recent research supports the idea that it may also represent a disorder of vasculogenesis, that is the formation of new blood vessels de novo, rather than sprouting from pre-existing cells. While some evidence supports many of the proposed hypotheses, no unifying paradigm has emerged: it is likely that the pathogenesis is multifactorial with many mechanisms contributing to their development, proliferation and involution.58–60
Some evidence suggests that hemangioma endothelial cells represent a clonal expansion of cells due to a somatic mutation in genes that play a significant role in vascular growth or vascular regulatory pathways. Mutations in several genes involved in the VEGF signaling pathway (VEGF receptors), Tie-2 and Dusp-5 have been noted supporting this hypothesis, but their exact role in hemangioma pathogenesis is not clear.61–63 Initial analysis of several pedigrees of familial hemangiomas revealed a linkage to chromosome 5q.62 Subsequent analysis of sporadic hemangiomas demonstrated loss of heterozygosity of 5q, further supporting the possibility that somatic mutations play a role in hemangioma formation.64
Infantile hemangiomas share unique immunohistochemical markers with human placenta microvasculature. North and coworkers reported that glucose transporter protein-1 (GLUT-1) is expressed by infantile hemangiomas during all phases of their development (proliferating, involuting, and involuted), distinguishing infantile hemangiomas from vascular malformations and other vascular tumors.65 Other placenta-associated vascular antigens, including merosin, FcγRII and Lewis Y antigen, are also present in hemangiomas and absent in microvessels of normal skin.66 In addition, studies using DNA microarray techniques have revealed that the gene expression profiles of hemangiomas and placenta vascular endothelium are remarkably similar.67 While it is speculated that hemangiomas may arise from embolized placental cells or angioblasts that abnormally differentiate toward a placental phenotype, this is challenged by the finding of negative placental trophoblastic markers in immunochemical analysis of hemangiomas.68
Multiple factors regulate the proliferation and involution of hemangiomas. During proliferation, hemangiomas demonstrate proliferating cell nuclear antigen (PCNA), as well as increased levels of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF).69–71 The VEGF signaling pathway is likely one of the more important mechanisms contributing to hemangioma proliferation.72 Angiogenesis mediators, including monocyte chemoattractant protein-1 and the adhesion molecules E-selectin and ICAM-3, are also expressed at high levels.73–76 Proteins involved in extracellular matrix remodeling are expressed during the proliferating phase.59,69,77 Increased levels of urinary matrix metalloproteinase (MMP) proteins are noted in proliferating infantile hemangiomas, suggesting a role in extracellular matrix remodeling, and the expression of lymphatic endothelial hyaluronan receptor-1 (LYVE-1) in proliferating hemangiomas may explain the rapid growth.78,79 Local factors are also believed to play a significant role in hemangioma proliferation. Investigators have noted that hemangioma growth correlates with hyperplasia of the overlying epidermis, with these tissues elaborating angiogenic factors (e.g., VEGF, bFGF). Moreover, these tissues lacked expression of the angiogenesis inhibitor interferon (IFN)-β.80 Indoleamine 2,3-dioxygenase (IDO) enzyme activity may play a role in the transition from proliferation to involution.8,81
During involution, the levels of angiogenic factors found during proliferation decrease, whereas levels of the angiogenesis inhibitor TIMP-1 (tissue inhibitor of metalloproteinase-1) increase.69,75,76 Interferon-regulated genes are upregulated during involution.69,82 Factors that promote apoptosis increase during involution, whereas inhibitors of apoptosis are upregulated during proliferation.83–85 In addition, loss of Dusp-5 function increases apoptosis in human umbilical vein endothelial cells causing speculation for its role in hemangioma involution.72,86
Diagnosis
Most infantile hemangiomas can be diagnosed on the basis of history and clinical appearance. The differential diagnosis includes other vascular and nonvascular tumors that are discussed elsewhere. Where history and physical examination are not helpful, further studies such as imaging or even biopsy may be necessary to confirm the diagnosis. Doppler ultrasound and MRI are most helpful in confirming the diagnosis of hemangioma. Doppler studies demonstrate high-flow lesions. T1-weighted MRI sequences demonstrate flow voids as a result of high-flow vessels and enhancement on T2-weighted sequences.87–89
Histopathology
If there is significant diagnostic uncertainty, a biopsy and GLUT-1 staining may be necessary to exclude other soft tissue tumors or vascular malformations. Early in the proliferating stage this tumor consists predominantly of a mass of endothelial cells. Lumina, lined by normal-appearing endothelial cells, become evident somewhat later in the late proliferative phase. PAS stains reveal a thickened basement membrane. Mast cells are increased within proliferating hemangiomas compared to normal tissue. As hemangiomas mature, they show lobules of endothelial channels separated by fibrous septa. Actin-positive smooth muscle cells are deposited around the vessels. Fat cells may be prominent in some involuted hemangiomas. Immunohistochemical analysis with GLUT-1 can be used to confirm the diagnosis of infantile hemangioma.
Differential diagnosis
Deep hemangiomas may be particularly difficult to differentiate from other tumors and malformations (Box 21.3).90,91 Infantile myofibromatosis may mimic vascular tumors but is firmer to palpation and has distinct histopathologic features. Infantile fibrosarcoma, a rare tumor that is sometimes congenital, may resemble a deep hemangioma or lymphatic malformation. Rhabdomyosarcoma is the most common sarcoma of early childhood and may present in newborns as a rapidly enlarging red cutaneous mass, usually involving the head and neck, that may be difficult to differentiate from a deep hemangioma. Other benign and malignant tumors that may resemble hemangiomas include adrenal carcinoma, spindle and epithelioid nevi, hemangiopericytoma, dermatofibrosarcoma protuberans, lipoblastoma, neuroblastoma, and nasal glioma. Congenital Langerhans’ cell histiocytosis, multifocal lymphangioendotheliomatosis and disseminated pyogenic granulomas may mimic diffuse neonatal hemangiomatosis. Developmental anomalies such as encephaloceles, dermoid cysts, meningoceles, and teratomas may all be mistaken for deep hemangiomas.
Management
Perhaps the greatest challenge in managing hemangiomas in infancy is the identification of those lesions that need treatment. Because of widely divergent sizes, location(s), and the rapid changes that can occur in early infancy, it may be difficult to predict prognosis at the time of initial evaluation. Frequent assessments during the first few weeks to months of life may be needed, particularly if high-risk features are present. The major goals of management are the prevention and treatment of life-threatening or function-threatening complications of the hemangioma as well as the prevention of permanent disfigurement that may have a long-term social and psychosocial impact on the patient and family (Table 21.1).92
TABLE 21.1
Hemangioma evaluation and management
| Hemangioma presentation | Association or complication | Evaluation and treatment |
| Facial segmental hemangiomas/facial hemangioma >5 cm | PHACE syndrome | MRI/MRA of brain and neck, echocardiogram, ophthalmology evaluation, and consider topical and/or systemic therapy |
| Periocular hemangioma | Ocular complications | Ophthalmology evaluation and consider topical and/or systemic therapy |
| Lumbosacral hemangioma | Spinal dysraphism, intraspinal hemangioma, lipoma | MRI of spine or ultrasound spine if neonate, neurosurgical intervention if indicated, consider topical and/or systemic therapy |
| Perineal segmental hemangioma | Spinal dysraphism and genitourinary anomalies (PELVIS syndrome) | MRI of lumbar spine and pelvis, urological and neurosurgical intervention if indicated, consider topical and/or systemic therapy |
| Ulceration | Pain, bleeding, scarring | See Box 21.6 |
| Central facial or other hemangiomas distorting normal anatomy | Disfigurement from skin and/or cartilage destruction and deformation | Consider topical and/or systemic therapy |
| Airway hemangiomas | Respiratory distress | Otolaryngology evaluation and/or MRI of neck, systemic therapy |
| Multifocal hemangiomas (>5) | Hepatic hemangiomas | Hepatic ultrasound and systemic treatment if symptomatic |
There are a number of treatments employed for infantile hemangiomas, although there is currently no treatment specifically FDA-approved for this indication. There are topical and systemic therapies that target hemangioma proliferation as well as adjunctive therapies that address hemangioma-related complications or the appearance of hemangioma residua after involution. The potential benefits of any form of treatment should be carefully weighed against the associated risks.
Several clinical features are important to consider when choosing treatment. It is important to consider not only anatomic location, but also the size, type, and pattern of the hemangioma and whether the hemangioma is actively proliferating. The following sections describe some of the treatment methods that have been employed.
Active non-intervention.
This term refers to the active observation and anticipatory guidance that can be given to parents, even if no specific therapy is instituted. Parents may feel significant distress as they await spontaneous involution and may react with disbelief, fear, and mourning. Some feel a degree of social stigmatization and many are accused of child abuse, either jokingly or in earnest. Parent–child interactions may be adversely affected.92 A careful discussion of the natural history of hemangiomas, with photographic examples to demonstrate natural involution, as well as a thorough discussion of therapeutic options, may help allay parental fears. Frequent visits, measurements, and photographs during the proliferating phase are recommended, and as the hemangioma begins to involute, visits can become less frequent. Acknowledgment of the intrusive and unsolicited questions and advice parents may receive can also be helpful.
Local and topical treatment.
[/not-level-membership-for-dermatology-category]
