Chapter 62 Inborn Errors of Metabolism of the Nervous System
Archibald Garrod introduced the concept of inborn errors of metabolism (IEM) in the 1908 Croonian Lectures and in his 1927 Huxley Lecture given at Charing Cross Hospital in London. The IEM are a heterogeneous group of disorders resulting from abnormalities of the synthesis, transport, and turnover of dietary and cellular components. Although individually uncommon, they collectively represent a significant cause of morbidity and mortality. In aggregate, approximately 1 in 1000 individuals is born with a metabolic disorder. IEM account for an estimated 20% of all deaths from genetic diseases; hereditary neurological or storage disorders account for 38%. The cost of care associated with the acute critical and chronic care of patients with IEM is substantial. Early diagnosis and intervention may influence patient quality of life, with potential accompanying healthcare cost savings.
Defects of intermediary metabolic pathways cause disease either by the accumulation of a toxic metabolite or depletion of a metabolic byproduct required to maintain proper cellular function. When an enzyme deficiency blocks normal catabolic routes, the diversion of metabolism to alternative pathways may disrupt cellular integrity. Deficient enzyme activity may arise from (1) mutations in the primary gene sequence for the protein, with loss of activity, (2) abnormal processing (i.e., defects of posttranslational modification), or (3) mistaken intracellular localization. Metabolic defects may also result from defects of a noncatalytic/cofactor, structural, or transport protein.
Most IEM are multiorgan disorders that usually involve the nervous system. The clinical course can be acute, subacute, or chronic. Disorders characterized by intoxication or energy depletion usually manifest acutely as altered mental status. Seizures and hypotonia may be associated. Vomiting, hepatic dysfunction, and renal dysfunction are other clinical features of acute intoxication. Some IEM follow an insidious course characterized by developmental delay or mental retardation, behavioral problems, sensory-motor impairment, or dementia. From a pathophysiological perspective, it helps to categorize the various IEM into one of three diagnostic groups: (1) disorders involving complex molecules (e.g., lysosomal storage disorders [LSDs]), peroxisomal diseases, congenital defects of glycosylation [CDG], and defects of cholesterol synthesis); (2) disorders involving “small molecules” (e.g., amino and organic acidurias, hyperammonemias, lactic acidemias); and (3) disorders associated with disruption of cellular energy metabolism (e.g., mitochondrial respiratory-chain defects, disorders of carbohydrate metabolism, disorders of fatty acid oxidation [FAO]). Metabolic defects involving complex molecules are usually progressive and not related to food intake, whereas those involving small molecules and cellular energy metabolism may be temporally related to food intake or metabolic states (e.g., fasting, postsurgical stress). The latter relationship accounts for the importance of dietary manipulation in the treatment of patients with certain IEM.
Most often, the inheritance of IEM is autosomal recessive, and the trait results in the deficiency of an enzyme or its cofactor. This may account for the absence of a family history when the sibship size is small. A few IEM are transmitted as autosomal dominant traits (e.g., acute intermittent porphyria, familial hypercholesterolemia), as X-linked traits (e.g., Fabry disease, Lesch-Nyhan syndrome, ornithine transcarbamylase deficiency, phosphorylase kinase deficiency, 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency), or segregate in a matrilineal fashion (e.g., mitochondrial DNA defects).
Early diagnosis is important for prognostication, genetic counseling, and providing treatment. A major goal of newborn screening is to reduce the burden of learning and functional impairment of affected children through early diagnosis and intervention. Most families may not be aware of their a priori risk, and early diagnosis of an affected child may enable consideration of prenatal diagnosis during future pregnancies. Therapeutic advances in recent years are considerable, and early diagnosis provides the best opportunity for a favorable outcome. Furthermore, treatment of secondary disabilities (e.g., seizures, sensory impairments, and behavioral, sleep/wake cycle, or communication problems) positively affects quality of life and helps address some of the sources of parental frustration.
This review covers the major IEM, except for the mitochondrial disorders. Most clinicians are neither metabolic specialists nor biochemical geneticists and not expected to be knowledgeable of the details of all biochemical pathways. This chapter deals with the IEM of the nervous system from the point of view of their pathogenesis and potential treatment. For fuller description of the clinical aspects of the individual diseases, the reader should consult the index.
General Considerations
Diagnostic Approach
When an IEM is possible, the clinical features help focus the approach to diagnosis. In the setting of acute illness, consider an IEM in parallel with more common disorders, even with a noninformative family history. Examine the blood and urine of patients with acute neurological deterioration for signs of acidosis, ketosis, hypoglycemia, and hyperammonemia (Tables 62.1 and 62.2). Also consider screening tests for abnormalities of amino or organic acids. Abnormal metabolites may not be present during stable periods or in samples obtained after the acute illness is over.
Table 62.1 Commonly Requested Tests for the Evaluation of a Patient Thought to Have an Inborn Error of Metabolism
| Tests | Clinical Utility |
|---|---|
| Ammonia | Urea cycle defects, organic acidemia |
| Carnitine, plasma or serum total and free (unesterified) urine levels | Deficiency may develop in carnitine transport defects, disorders of fatty acid oxidation and branched-chain amino acid metabolism, and valproic acid treatment |
| Acylcarnitine profile | Normal plasma acyl/free carnitine ratio: <0.25 |
| Ceruloplasmin | Decreased in Wilson and Menkes disease, aceruloplasminemia |
| Cholesterol | Low plasma levels in Smith-Lemli-Opitz syndrome, cerebrotendinous xanthomatosis; abnormal profile in the dyslipidemias |
| Free fatty acids (FFA); ketone bodies (KB): 3-hydroxybutyrate, acetoacetate | Disorders of fatty acid oxidation and ketolysis; supervised fasting and assessment of FFA/KB ratio and glucose and ketone levels enable distinction of hypo- and hyperketotic disorder |
| Lactate* | Defects of glycogen metabolism, gluconeogenesis and fatty acid oxidation (often seen with hypoglycemia); defects involving the electron transport chain, Krebs cycle, and pyruvate dehydrogenase (absence of hypoglycemic episodes) Lactate/pyruvate ratio (NI: <20 : 1) provides insight into oxidation-reduction status (increased in OXPHOS defects; normal in PDHC deficiency) Normal blood lactate <1.8 mmol/L Normal CSF lactate <2.2 mmol/L |
| Very long-chain fatty acid (VLCFA) | Disorders of peroxisomal metabolism |
| Phytanic acid | Elevated in Refsum disease and rhizomelic chondrodysplasia punctata |
| Uric acid | Elevated in Lesch-Nyhan syndrome† and other defects of purine metabolism and glycogen storage disorders; decreased in molybdenum cofactor deficiency and defects of pyrimidine metabolism CSF/plasma ratio |
| Glucose | <0.35 Glucose transport defect |
| Glycine | >0.6 Nonketotic hyperglycinemia; low in plasma and CSF glycine levels in 3-phosphoglycerate dehydrogenase deficiency; plasma glycine markedly elevated in patients with organic acidemias (MMA, PA, IVA) |
| Serine | <0.2 Serine deficiency syndromes (3-phosphoglycerate dehydrogenase and phosphoserine phosphatase deficiency) |
CSF, Cerebrospinal fluid; IVA, isovaleric acidemia; MMA, methylmalonic acidemia; OXPHOS, oxidative phosphorylation; PA, propionic acidemia; PDHC, pyruvate dehydrogenase complex.
* Presence or absence of hypoglycemia can be a useful aid to differential diagnosis of disorders that lead to lactic acidemia.
† Hyperuricemia may lead to formation of nephrolithiasis, obstructive nephropathy, and gout.
Certain situations require an analysis of cerebrospinal fluid (CSF) (Jones et al., 2006). For example, determination of CSF levels of biogenic monoamines and γ-aminobutyric acid (GABA) may be diagnostic in severe neonatal/infantile epileptic encephalopathy due to neurotransmitter defects (Table 62.3). Such defects are suggested in infants and children with (fluctuating) extrapyramidal disorders, in particular parkinsonism, dystonia or more general “athetoid cerebral palsy,” and vegetative disturbances. Other diagnoses for which CSF testing may be informative include nonketotic hyperglycinemia, GABA transaminase deficiency, and homocarnosinosis. Proper collection and handling of the CSF is important to avoid spurious results (Hyland, 2003).
Neurological deterioration is a characteristic feature of acute intoxication disorders (e.g., certain aminoacidopathies [maple syrup urine disease, MSUD], organic acidemias [methylmalonic acidemia, MMA; propionic acidemia, PA; isovaleric acidemia, IVA; multiple carboxylase deficiency, MCD], and the urea cycle defects). Abnormal urine odor is present in diseases associated with the excretion of volatile metabolites (maple syrup in MSUD; sweaty feet in IVA and glutaric acidemia type II). Isolated seizures are often the initial features of vitamin-responsive disorders (e.g., defects of pyridoxine and folinic acid metabolism, biotin-responsive MCD), and are a prominent feature in nonketotic hyperglycinemia, sulfite oxidase deficiency, and congenital malabsorption of magnesium. Congenital lactic acidosis and central hypotonia are features of deficiencies of pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) and disorders of the Krebs cycle and mitochondrial respiratory chain (Pithukpakorn, 2005). Recurrent hypoglycemia typically occurs in the glycogen storage disorders (GSD) affecting the liver and in defects of FAO (consequent to overutilization); these diseases are also often associated with signs of cardiac involvement (cardiomyopathy, arrhythmias). In the mitochondrial FAO defects, urinary ketone concentrations are low, and the ratio of 3-hydroxybutyrate/acetoacetate is usually less than 2.0. Analysis of plasma acylcarnitine and urinary acylglycine profile are particularly helpful in these cases. Renal tubular acidosis associated with HCO3− loss in urine can be encountered in PC deficiency, MMA, carbonic anhydrase deficiency, carnitine palmitoyltransferase I (CPT-I) deficiency, and cystinosis.
Ophthalmological examination often provides a clue to the diagnosis of IEM. Vertical supranuclear ophthalmoplegia occurs in Niemann-Pick disease type C, and saccadic initiation failure and defective optokinetic nystagmus occurs in Gaucher disease type III. Kayser-Fleisher rings (orange or greenish deposits around the limbus of the cornea due to copper deposition within the Descemet membrane) are a feature of Wilson disease. Table 62.4 shows additional ophthalmological findings characteristic of IEM.
Table 62.4 Ophthalmologic Findings Associated with Inborn Errors of Metabolism
| Ophthalmologic Abnormality | Associated Inborn Errors of Metabolism |
|---|---|
| Cataracts | Cerebrotendinous xanthomatosis |
| Cholesterol synthesis defects | |
| Galactokinase deficiency | |
| Galactosemia | |
| Lowe syndrome | |
| Menkes syndrome | |
| Mucopolysaccharidoses | |
| Peroxisomal disorders | |
| Serine deficiency disorders | |
| Tyrosinemia type II | |
| Cherry red spot | Galactosialidosis |
| GM1-gangliosidosis | |
| Niemann-Pick disease A | |
| Sandhoff disease | |
| Sialidosis type I | |
| Tay-Sachs disease | |
| Optic atrophy | Adrenoleukodystrophy |
| Canavan disease | |
| Hyperornithinemia with gyrate atrophy | |
| Lafora disease | |
| Lens dislocation | Homocystinuria |
| Molybdenum cofactor deficiency | |
| Sulfite oxidase deficiency | |
| Retinopathy | Carbohydrate-deficient glycoprotein syndrome |
| Neuronal ceroid lipofuscinosis | |
| Mitochondrial defects | |
| Peroxisomal disorders |
Hepatosplenomegaly and other signs of storage (e.g., coarse facies, nonimmune hydrops fetalis, dysostosis multiplex) occur with the lysosomal disorders. Liver dysfunction or hepatomegaly (or both) usually occur in defects of carbohydrate metabolism (galactosemia and hereditary fructose intolerance, GSDs, [particularly Pompe disease], and bile acid synthesis), and in tyrosinemia and CDG. Unconjugated hyperbilirubinemia associated with liver dysfunction or hemolysis in infancy may lead to permanent brain damage due to kernicterus.
Cardiomyopathy may develop in IEM associated with infiltrative (storage) disorders and defects of energy metabolism. The presence of hepatomegaly and other signs of systemic involvement (e.g., cataracts, coarse facies, dysostosis multiplex) may suggest storage disorders of glycosaminoglycans. Defects of energy metabolism may be associated with acute or chronic encephalopathy, hepatic dysfunction, and several biochemical abnormalities (e.g., hypoglycemia, lactic acidosis ± ketosis, and elevated liver transaminase levels).
Some disorders may have both a young and a later age of onset or follow an atypical course. Different allelic mutations with partial enzyme deficiencies or organ-specific expression may explain these differences. Examples include acid maltase deficiency (muscle weakness and respiratory problems in the absence of cardiomyopathy), FAO defects (myoglobinuria and rhabdomyolysis after extreme exercise), X-linked adrenomyeloneuropathy (spastic paraparesis secondary to demyelination of the spinal cord and peripheral nerves), glycogen brancher enzyme deficiency (adult polyglucosan body disease with progressive upper and lower motor neuron disease, sensory loss, neurogenic bladder, and dementia), and acute intermittent porphyria (abdominal pain, psychosis).
Tandem mass spectrometry (TMS) analysis of blood spots on filter paper is an effective means of screening for some defects of amino and organic acid metabolism and FAO defects (Pasquali et al., 2006) (Table 62.5). Methods for screening of lysosomal storage disorders by TMS have also been recently introduced (Matern, 2008). Metabolic profiling of infant urine through comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry for the diagnosis of organic acidurias and biomarker discovery is also an option (Kouremenos et al., 2009). The main advantages of TMS over previous methods of newborn screening are improved accuracy, sensitivity and specificity, and suitability for cost-effective multi-IEM screening. Furthermore, its use avoids the need for potentially harmful procedures (i.e., fasting, substrate loading).
Table 62.5 Metabolic Disorders Detected Through Tandem Mass Spectrometry
| Disorder | Enzyme Deficiency | Primary Metabolic Indicator |
|---|---|---|
| AMINO ACIDOPATHY | ||
| Phenylketonuria/hyperphenylalaninemia | Phenylalanine hydroxylase (and variants) | Phe |
| Maple syrup urine disease | Branched chain oxo- (or keto-) acid dehydrogenase | Leu/Ile, Val |
| Homocystinemia | Cystathionine β-synthase | Met |
| Hypermethioninemia | Methionine-S-adenosyltransferase | Met |
| Citrullinemia | Argininosuccinate synthetase | Cit |
| Argininosuccinic aciduria | Argininosuccinate lyase | Cit |
| Tyrosinemia type I | Fumarylacetoacetate hydrolase | Tyr |
| ORGANIC ACIDEMIA | ||
| Glutaric acidemia type 1 | Glutaryl-CoA dehydrogenase | C5DC |
| Propionic acidemia | Propionyl-CoA carboxylase | C3 |
| Methylmalonic acidemia | Methylmalonyl-CoA mutase | C3 |
| Isovaleric acidemia | Isovaleryl-CoA dehydrogenase | C5 |
| 3-Hydroxy-3-methylglutaryl CoA lyase deficiency | C5OH | |
| 3-Methylcrotonyl carboxylase deficiency | C5OH | |
| FATTY ACID OXIDATION DEFECTS | ||
| Medium-chain acyl-CoA dehydrogenase deficiency | C8, C10, C10:1, C6* | |
| Very long-chain acyl-CoA dehydrogenase deficiency | C14:1, C14, C16 | |
| Short-chain acyl-CoA dehydrogenase deficiency | C4 | |
| Multiple acyl-CoA dehydrogenase deficiency | C4, C5, C8, C12, C14, C16, C5DC | |
| Carnitine palmitoyltransferase deficiency | C16, C18:1, C18 | |
| Carnitine/acylcarnitine translocase deficiency | C160H, C18:1OH, C18OH | |
| Very long-chain hydroxyacyl-CoA dehydrogenase deficiency | C16OH, C18:1OH, C18OH | |
| Trifunctional protein deficiency | C16OH, C18:1OH, C18OH |
* The abbreviations for fatty acid oxidation products and organic acid intermediates (e.g., C5, C160H, C18:1, etc.) refer to carnitine esters of aliphatic monocarboxylic acids, with chain length indicated by the number adjacent to C, and the number of double bonds indicated by the number after the colon.
From Centers for Disease Control and Prevention, 2001. Using tandem mass spectrometry for metabolic disease screening among newborns. Recommendations and reports. MMWR Morb Mortal Wkly Rep 50, April 13, 2001.
Assessment of carnitine profile (total and esterified carnitine levels and urinary carnitine excretion patterns) may also prove useful when suspecting a primary or secondary carnitine deficiency. Carnitine plays an essential role in the transfer of long-chain fatty acids across the inner mitochondrial membrane, in the detoxification of acyl moieties, and in the maintenance of free coenzyme A levels. Dietary sources provide carnitine. Primary carnitine deficiency (due to defective transport protein encoded by the OCTN2 gene) leads to increased urinary loss and cardiac and skeletal muscle disease. Secondary carnitine deficiency occurs in several IEM and may be partially responsive to oral carnitine supplementation. Table 62.6 shows the differential diagnosis of disorders involving carnitine metabolism.
Table 62.6 Useful Measures to Aid in the Differential Diagnosis of Inborn Errors of Metabolism Involving Carnitine
Histological examination of appropriate tissue samples can provide clues to the nature of the storage materials found in certain IEM (e.g., lysosomal disorders) (Warren and Alroy, 2000). When performing a skin biopsy, it is advisable to obtain samples for microscopic examination and tissue culture. These are useful as source material for subsequent biochemical or molecular (genetic) testing. Disorders of amino and organic acid metabolism are not associated with deposition of storage material, and histological findings are nonspecific. For this group of disorders, use skin fibroblasts for confirmatory diagnosis by enzymatic assays. For some disorders, biochemical testing is not accurate for carrier detection, because residual enzyme activity in a significant proportion of carriers overlaps with values obtained from the general population. In certain IEM, molecular assays may be available for diagnostic purposes and carrier testing. Microscopic examination of scalp hair can provide important diagnostic information in some cases, such as in Menkes syndrome (pili torti) (Smith et al., 2005).
The GeneTests website (www.genetests.org) lists diagnostic laboratories performing specialized genetic tests. Careful attention to sample requirements and shipping and handling considerations is imperative. Clinical information is required with the specimen to receive expert advice in patient evaluation. In cases with an established diagnosis, detailed information may be obtained from several websites, including Online Mendelian Inheritance in Man (OMIM [www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM]), GeneClinics (www.geneclinics.org), and the National Organization for Rare Disorders (NORD [www.rarediseases.org]). Several support and patient advocacy groups provide information about community resources and ongoing clinical trials.
Mutation Analysis in the Diagnosis of Inborn Errors of Metabolism
Molecular genetic techniques offer an alternative means for the diagnostic confirmation of IEM. This is particularly true for diseases with a founder effect—common mutations in which one or a few alleles account for a significant proportion of cases (e.g., Finnish and Jewish “heritage” diseases). In diseases with a known causal mutation, testing of other family members permits accurate carrier identification. Deoxyribonucleic acid (DNA) analysis for prenatal diagnosis provides rapid diagnosis because the use of chorionic villi or amniocytes obviates the need for culture (although in practice, most laboratories insist on subsequent confirmatory testing of cultured cells and the need to exclude maternal cell contamination). Caution should be applied in assignment of causality, particularly for novel mutations or sequence alterations wherein functional impact has not been established.
Examples of disorders for which DNA testing has proven useful include medium-chain acyldehydrogenase [MCAD] deficiency, myophosphorylase deficiency (McArdle disease), and Gaucher disease (Gregersen et al., 2004; Madonna et al., 2002; Maire, 2001). Among patients with MCAD deficiency of northwestern European descent, 80% are homozygous for a single missense mutation (A985G), and 17% carry this mutation in combination with another less common defect. This finding has improved the reliability of MCAD carrier identification and diagnosis, particularly of siblings who may be affected but asymptomatic at the time of family screening. Certain metabolic disorders (e.g., Tay-Sachs disease, Gaucher disease, Niemann-Pick disease type A, and Canavan disease) have an increased prevalence among individuals of Ashkenazi Jewish ancestry (i.e., of Central and Eastern European descent). A limited number of “common” mutations in this population cause the disease. This has enabled targeted screening for appropriate counseling prior to marriage. In hereditary fructose intolerance and dihydropyrimidinase deficiency, screening for the disease mutation in blood obviates the need for liver biopsy. However, in mitochondrial disorders that cause myopathy, biopsy of a weak muscle is still required, because expression of the mutant gene may not occur in other tissues. Fresh muscle tissue is also the preferred sample for studies of respiratory chain function.
Special Considerations
Deficiency of succinate dehydrogenase or fumarase activity can lead to lactic acidosis. Interestingly, heterozygous mutations involving the succinate dehydrogenase (SDH) complex have been associated with hereditary paraganglioma and pheochromocytoma. Heterozygous mutations involving fumarase have been associated with hereditary leiomyomatosis and renal cell cancer, a genodermatosis, and familial renal cancer syndrome (Pollard, 2003). Thus, parents and at-risk family members of patients with SDH or fumarase deficiency require screening for these tumors.
Recent studies suggest an increased incidence of parkinsonism in families segregating for Gaucher disease (Neumann et al., 2009).
Inborn Errors of Metabolism Associated with Abnormal Brain Development and Encephaloclastic Lesions
Several metabolic disorders can cause a disruption of the normal sequence of brain development and lead to encephalocele, dysgenic corpus callosum, and neuronal migration defects (Hennekam, 2005; Jeng et al., 2001; Nissenkorn et al., 2001) (Tables 62.7 and 62.8). For instance, cystic necrosis of white matter, with or without basal ganglia involvement, occurs in PDH, PC, and molybdenum cofactor deficiencies. Nonsyndromic congenital microcephaly has been associated with maternal PKU, phosphoglycerate dehydrogenase deficiency, and 2-ketoglutaric aciduria (the cause of Amish lethal microcephaly). The proposed mechanisms to explain abnormal brain development and encephaloclastic lesions (such as porencephalic cysts) in IEM include the production of a toxic or energy-deficient intrauterine milieu, modification of the content and function of membranes, and disturbance of the normal expression of intrauterine genes responsible for neurulation and neuronal migration (Prasad et al., 2009).
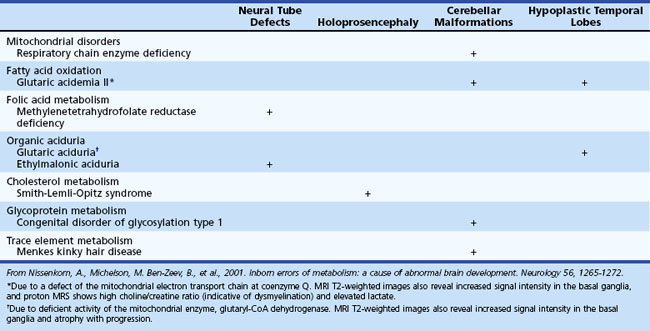
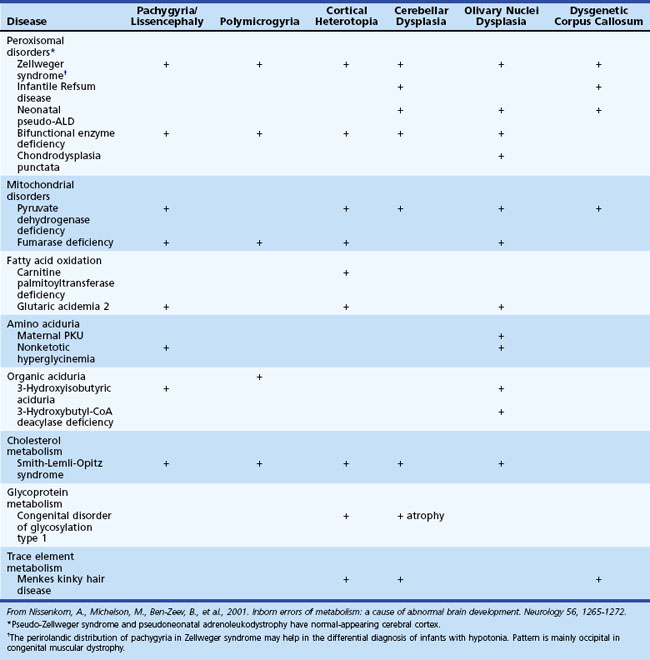
Neuroimaging in the Diagnosis and Management of Neurometabolic Brain Diseases
Magnetic resonance imaging (MRI) has been widely used to obtain brain anatomical data in patients with static and progressive encephalopathy. Advanced imaging techniques such as MR spectroscopy (MRS) and diffusion-weighted imaging (DWI) provide additional information relating to brain biochemistry and cell viability and point to the diagnosis of a neurometabolic disease (Cheon et al., 2002; Faerber and Poussaint, 2002; Kaye, 2001). MRS and DWI are quantitative techniques that may prove useful in assessing severity of the underlying pathological changes and potential response to treatment.
White-matter signal abnormalities consistent with leukodystrophy can be found in several metabolic disorders such as Krabbe disease, metachromatic leukodystrophy (MLD), X-linked adrenoleukodystrophy, and Canavan disease. In addition to signal hyperintensities noted on T2-weighted imaging, special features may provide additional clues to the diagnosis. Enlarged perivascular spaces or small cysts are characteristically noted in the mucopolysaccharidoses and Lowe syndrome. Gray-matter lesions may be found in Zellweger syndrome and other peroxisomal disorders. In glutaric aciduria type 1, imaging may show selective frontotemporal atrophy, especially involving subcortical white matter, with prominent extraaxial CSF collection, widening of the sylvian fissure with poor opercularization (“bat wing” appearance), and in some cases, subdural hemorrhage (Oguz et al., 2005). The presence of T2 hyperintense lesions in the subcortical cerebral white matter, basal ganglia, and dentate nuclei, together with cerebellar atrophy, is considered pathognomonic of l-2-hydroxyglutaric aciduria (Steenweg et al., 2009).
MRS has enabled definition of the creatine deficiency syndromes, a newly discovered group of disorders causing mental retardation and other neurological problems (i.e., extrapyramidal movement abnormalities, hypotonia). The common feature is severe depletion of brain creatine/phosphocreatine. With brain MRS, elevated brain levels of N-acetylaspartate (NAA) are seen in patients with Canavan disease, whereas patients with mitochondrial defects, defects of gluconeogenesis, and biotin-responsive multiple carboxylase deficiency may show elevated brain lactate levels. In MLD, MRS reveals decreased NAA and increased choline and myoinositol compatible with axonal loss, dysmyelination, and gliosis. A prominent signal at 8.3 ppm in gray-and white-matter brain regions has been described in patients with adenylosuccinate lyase deficiency (Henneke et al., 2010). In patients with MSUD and acute metabolic decompensation, MRS demonstrates decreased NAA, methyl resonance of branched chain amino acids at 0.9 ppm, and elevated lactate (Cakmakci et al., 2009). In patients with X-linked adrenoleukodystrophy, the NAA/choline ratio (at a cutoff point of 0.5) has been shown to predict disease progression in six cases with cerebral disease (Eichler et al., 2002). Multislice proton MRS imaging was useful in identifying impending or beginning degeneration in white matter that still appeared normal on conventional MRI.
Imminent Death Prior to Diagnosis in a Child with a Suspected Inborn Error of Metabolism
Obtain samples needed for diagnosis when a child develops acute fatal metabolic decompensation and in cases of sudden and unexpected death (Olpin, 2004). A correct diagnosis may help families as they cope with their loss and enables appropriate counseling and prenatal diagnosis for subsequent pregnancies. Freeze plasma (separated from whole blood) and urine (CDC, 2003). Blood and bile (obtained by direct puncture of the gall bladder) specimens can also be collected, spotted on filter paper (two circles for each, about 25 µL), and dried before being sent to the laboratory. Obtain a skin sample under sterile technique (use alcohol swabs and not iodine, which interferes with cell growth), and store it at room temperature in tissue culture medium. When suspecting a storage disorder, obtain a small snip of skin and place it in glutaraldehyde for subsequent electron microscopic studies.
Management Considerations
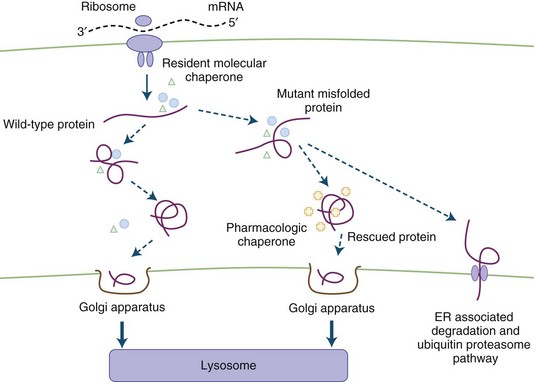
The appropriate management of IEM depends upon the particular metabolic and/or molecular derangement. Therapeutic strategies may include one or more of the following approaches: (1) substrate reduction by dietary manipulation or precursor synthesis inhibition, (2) removal (or enhanced clearance) of the toxic metabolites, (3) replenishment of depleted metabolites or cofactor supplementation (or both), (4) enzyme (replacement or enhancement) therapy, and (5) cellular replacement (e.g., bone marrow, liver, heart, or kidney transplantation) (Chakrapani and Wraith, 2002; Leonard, 2004; Wilcken, 2003). Previous active exploration of gene therapy ended with the unexpected death of a study subject with ornithine transcarbamylase (OTC) deficiency. Reexamination of this approach led to demands for greater caution in future clinical trials (Hsich et al., 2002; Sands and Davidson, 2006). Meanwhile, gene therapy experiments are ongoing in various animal (mouse) models of disease (Koeberl et al., 2009). Other approaches under consideration are liver repopulation (hepatocyte infusion), chaperone-mediated therapy for diseases associated with residual enzyme activity, and the transplantation of stem cells with directed differentiation along specific lines (Desnick, 2004; Grompe, 2002; Kawashita et al., 2005). The rationale for the use of pharmacological chaperones is the demonstration that certain small molecules may rescue misfolded proteins in the endoplasmic reticulum and restore functional conformation (Gregersen et al., 2001; Pastores and Sathe, 2006). Certain drugs have also been shown in vitro to have the potential of reading through stop codons and may induce the synthesis of a full-length protein. A short-term study of gentamicin in a patient with McArdle disease and the R50X mutation failed to show modification of muscle metabolism (Schroers et al., 2006); these observations may be influenced by the short duration of the study or underlying gene defect. Clearly, more studies are required to examine the potential of this approach. Bezafibrate has been shown to increase carnitine palmitoyl transferase II (CPT2) messenger ribonucleic acid (mRNA), with normalization of enzyme activity in CPT2-deficient cultured fibroblasts and myoblasts. In a pilot trial including six patients treated with bezafibrate, FAO concentrations normalized in all patients, palmitoyl-l-carnitine oxidation increased, episodes of rhabdomyolysis were reduced, and quality of life improved without adverse effects (Bonnefont et al., 2009). These advances have changed the attitude about treatment for patients with IEM away from nihilism and hopelessness toward cautious optimism.
Each patient requires an individualized approach; some disorders require more than one management option. The clinical response to most treatment plans may vary, and residual disturbances are common. Patients may remain at risk for metabolic decompensation when stressed by infection, trauma, or surgery. Reducing energy expenditure and promoting anabolism are immediate management goals. Emergency measures may prevent further deterioration, but most options are nutritionally incomplete, and extension beyond 48 hours without dietary review is wrong. In most situations, provision of symptomatic treatment requires specialized care units with expertise in the specific disease.
Special diets require attention to caloric needs and balanced nutrition; this is particularly true for minerals and supplements. Diseases managed with dietary restriction include the aminoacidopathies: phenylketonuria (PKU), MSUD, and homocystinuria. Recent clinical studies suggest that treatment of PKU patients with sapropterin (a cofactor of phenylalanine hydroxylase) provides better disease control and increases dietary phenylalanine tolerance, allowing significant relaxation, or even discontinuation, of dietary phenylalanine restriction (Blau et al., 2009). In classic Refsum disease, reduction in dietary phytanate results in normalization of the biochemical and clinical phenotype.
Some disorders require alternative dietary sources. Administer medium-chain triglycerides as a lipid source to patients with very long-chain acyldehydrogenase [VLCAD] and long-chain acyldehydrogenase [LCHAD] deficiency. In Smith-Lemli-Opitz syndrome (3β-hydroxysterol-Δ7-reductase deficiency), the use of a high-cholesterol diet (± bile acids) improves growth and neurodevelopmental status, although clinical response is variable. In children with GSDs, carbohydrate supplements prevent hypoglycemia and suppress secondary metabolic derangements (e.g., hyperlipidemia, hyperuricemia). In the urea cycle disorders, arginine or citrulline supplements make up for compounds that are not synthesized secondary to the metabolic block.
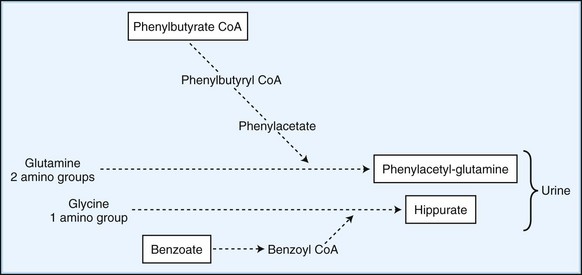
Employ methods to enhance the excretion or detoxification of toxic metabolites when dietary manipulation fails to correct their accumulation. In patients with hyperammonemia, give sodium benzoate and sodium phenylbutyrate, which conjugate with glycine and glutamine to facilitate nitrogen excretion (Fig. 62.1). In IVA, treatment with oral glycine (500 mg/kg) conjugates the highly toxic isovaleric acid to form a harmless byproduct excreted in the urine. Administration of cysteamine to patients with cystinosis promotes the formation of cysteine, which is subsequently excreted in the urine. Carnitine supplementation given to patients with organic acidemia prevents carnitine deficiency secondary to the formation and renal excretion of acylcarnitine compounds. During acute metabolic decompensation, dialysis and hemofiltration facilitates the rapid clearance of toxic metabolites. These techniques are already a part of the treatment of MSUD, carbamoylphosphate synthetase (CPS) deficiency, and hyperammonemic or leucine encephalopathy (Daschner and Schaefer, 2002). In patients with urea cycle defects, hemodialysis should be considered at ammonia levels above 600 mol/L (1000 g/L). Administration of glycerol trioleate and glycerol trierucate (Lorenzo oil) to asymptomatic patients with X-linked ALD may modify the disease course (Moser et al., 2005).
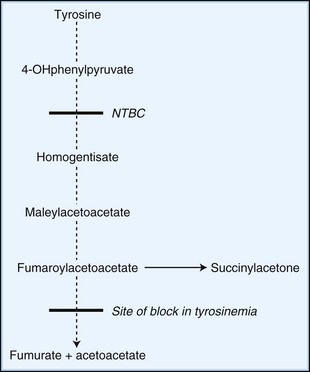
Novel therapeutic strategies for IEM use substrate synthesis inhibitors to block the production of toxic metabolites. In tyrosinemia type I (fumaroylacetoacetate hydrolase deficiency), NTBC (2-nitro-4-trifluoro-methylbenzoyl-1,3-cyclohexanedione) reduces the production of downstream metabolites of tyrosine degradation by inhibiting the enzyme, 4-hydroxyphenylpyruvate dioxygenase. This enzyme is involved in a reaction preceding the block (Fig. 62.2). In one study of NTBC in more than 300 patients with tyrosinemia type 1, 95% showed improvement of hepatic and kidney function (Grompe, 2001). In Gaucher disease type I, NB-DNJ (N-butyldeoxynojirimycin), miglustat administration leads to decreased liver and spleen volumes and a gradual but significant improvement in hematological parameters, with a decline in the levels of disease activity markers (Cox et al., 2000). NB-DNJ inhibits ceramide-specific glucosyltransferase, the first enzymatic step in glycosphingolipid (GSL) biosynthesis. Limiting substrate accumulation to a level that is sufficiently catabolizable by a mutant but partially active enzyme achieves metabolic homeostasis. Given this mechanism of action, miglustat may be potentially useful for other disorders of GSL metabolism (e.g., late-onset Tay-Sachs, Sandhoff disease, GM1-gangliosidosis, and Niemann-Pick disease type C [NPC]) (Pastores and Barnett, 2005).
Unfortunately, the miglustat trial in patients with either late-onset Tay-Sachs or Gaucher disease type 3 on enzyme replacement therapy was unable to prove any measurable benefit (Schiffmann et al., 2008; Shapiro et al., 2009). In patients with NPC, treatment with miglustat resulted in improvements in saccadic eye movements and swallowing, but impact on survival and eventual neurological prognosis appeared limited (Patterson et al., 2007).
Replenishing depleted substrates may partially correct the underlying defect in some IEM. In carnitine transport defects, the use of carnitine results in resolution of cardiomyopathy and prevention of further episodes of hypoketotic hypoglycemia. In other disorders, the production or binding affinity of a cofactor required for enzyme activity is impaired. Administration of pharmacological doses of the required supplement corrects the defect. Biotin, in some cases of PA and when given to children with biotinidase or holocarboxylase deficiency, leads to good clinical outcomes, except in the most severe forms with neonatal onset (Wolf, 2002). Vitamin B12 given for late-onset forms of MMA due to defects of adenosylcobalamin metabolism leads to a sustained decrease of toxic metabolites and a favorable developmental prognosis. Suppression of gut microbial propionate production (using metronidazole to inhibit anaerobic colonic flora) and dietary protein restriction are complementary approaches provided to children with MMA.
Additional examples of pharmacological correction include the use of tetrahydrobiopterin (BH4) to treat disorders of biopterin synthesis, rare variants of the hyperphenylalaninemia syndrome (Lambruschini et al., 2005). Although BH4 reduces elevated plasma phenylalanine levels by its action on liver phenylalanine hydroxylase, penetration of the blood-brain barrier is minimal. Only those with a peripheral-type defect respond. Tetrahydrofolate prevents demyelination in children with folate deficiency and dihydropteridine reductase deficiency. Assess the responsiveness to cofactor administration by controlled enzyme assays involving the patient’s cultured skin fibroblasts. Table 62.9 lists the cofactors used in various metabolic disorders and their recommended dosages.
Table 62.9 Cofactors Used in the Management of Various Inborn Errors of Metabolism
| Cofactor | Dose (mg/day) | Disorder |
|---|---|---|
| Betaine | 150* | Homocystinuria |
| MTHFR deficiency | ||
| Biotin | 10-20 | Propionic aciduria |
| Multiple carboxylase deficiency | ||
| Hyperlactacidemia due to pyruvate carboxylase deficiency | ||
| Carnitine | 50-100 PO | Branched-chain organic aciduria (MMA, PA, IVA) |
| 400 IV | Primary hyperammonemia | |
| Hyperlactacidemia | ||
| Fatty acid oxidation defects | ||
| Cobalamin (B12) | 1-2 | Methylmalonic aciduria |
| Folinic acid | 10-40 | Folinic-responsive seizures |
| Lorenzo oil | Asymptomatic X-linked (glycerol adrenoleukodystrophy trioleate and trierucate) | |
| Pyridoxine (B6) | 50-100 | Pyridoxine-responsive seizures, hyperoxaluria type 1, aromatic l-amino acid decarboxylase |
| Glutaric aciduria, homocystinemia | ||
| Riboflavin (B2) | 20-40 | Fatty acid oxidation defects† |
| Multiple acyl-CoA dehydrogenase deficiency | ||
| Thiamine (B1) | 10-50 | Maple syrup urine disease |
| Hyperlactacidemia due to pyruvate dehydrogenase deficiency |
IVA, Isovaleric acidemia; MMA, methylmalonic acidemia; MTHFR, methylene tetrahydrofolate reductase; PA, propionic acidemia.
* Dose is up to 150 mg/kg/day.
† Avoidance of fasting is critical; in emergency cases, where anorexia or vomiting precludes oral intake institute intravenous glucose administration.
Enzyme replacement therapy (ERT) reverses the hematological and visceral manifestations of Gaucher disease and may prevent or stabilize disease-related bone complications. This approach is also a consideration for the treatment of LSD due to single enzyme deficiencies. The relevant enzymes, produced from genetically manipulated mammalian cells in culture and subsequently modified, expose the appropriate sugar residues to facilitate targeted cell uptake. Once purified, management requires regular intravenous infusions of the recombinant enzyme. Beneficial effects have been noted in patients with Fabry disease, mucopolysaccharidosis (MPS) type I (Hurler-Scheie syndrome), MPS II (the mild variant of Hunter syndrome), MPS type VI (Maroteaux-Lamy syndrome), and GSD II (Pompe disease) (Pastores and Barnett, 2005). Enzyme therapy is also a consideration for other LSDs including Niemann-Pick disease.
Metabolic correction through cellular replacement by bone marrow transplantation (BMT)/hematopoietic stem cell transplantation (HSCT) has been performed in patients with LSD (e.g., MPS I, Gaucher disease type III) (Peters et al., 2003). In X-linked adrenoleukodystrophy, BMT has resulted in prolonged remission with reversal of MRI abnormalities and stabilization or improvement of motor function (Moser, 2006; Peters et al., 2004). Although BMT has altered the natural course of these diseases, donor limitation issues, procedural risks, and long-term care considerations (e.g., immunosuppression) exist. Advances in the methods of conditioning (e.g., nonmyeloablative procedures) prior to BMT and utilization of umbilical cord blood have addressed some of these concerns. Patients with IEM who may be suitable candidates for BMT should have serial follow-up visits incorporating neuropsychological and neuroradiological studies to time interventions prior to significant disease progression. This allows an outcome with minimal neurological sequelae. BMT is not appropriate for disorders characterized by rapid neurodegeneration, such as MPS II (severe Hunter syndrome) and MPS III (Sanfilippo syndrome).
Organ transplantation may be appropriate for disorders in which the metabolic defect is confined to the liver (e.g., Crigler-Najjar syndrome, hyperoxaluria type I) or leads to single organ failure (e.g., end-stage renal insufficiency in Fabry disease and hyperoxaluria type I, and liver failure in OTC deficiency, tyrosinemia, and GSD-IV) (Dhawan et al., 2005; Inderbitzin et al., 2005). In patients with tyrosinemia who do not respond to NTBC therapy or have evidence of hepatic malignancy, orthotopic liver transplantation is the treatment of choice. Microchimerism (the migration of donor-derived cells from the allograft) has occurred in a few patients following liver transplantation. However, this phenomenon is probably not sufficient to correct the systemic metabolic defect.
Symptomatic treatment remains a vital component of patient care. Indeed, several palliative measures improve quality of life and reduce the incidence and severity of disease-related complications. For instance, l-dopa improves motor function in patients with tyrosine hydroxylase deficiency. DDAVP (d-deamino-arginine-vasopressin) reduces the tendency for abnormal bleeding during surgery of patients with GSD type 1A (von Gierke disease). Granulocyte colony-stimulating factor (G-CSF) administered to patients with GSD IB and neutropenia minimizes the risk of recurrent bacterial infection and gastrointestinal tract ulceration. Corticosteroid and mineralocorticoid replacement are essential in patients with ALD and adrenal insufficiency.
Patients with certain metabolic disorders necessitate special considerations for anesthesia and surgery. For instance, instability of the atlantoaxial joint and upper airway obstructive disease in patients with mucopolysaccharidosis may lead to problems during induction and extubation. Hypoglycemia must be avoided in patients with fatty acid oxidation defects, glycogen storage disorders, and disorders of gluconeogenesis. This may require careful planning in terms of the time the procedure is undertaken and maintenance of euglycemia through intravenous administration of 10% glucose. Surgery and anesthesia may induce a metabolic crisis in patients with Refsum disease by mobilization of phytanic acid in fat stores.
In addition to dealing with the medical problems of an affected child, provide genetic counseling. The inheritance of approximately 90% of IEMs is autosomal recessive. Of the remaining 10%, approximately two-thirds are X-linked and one-third autosomal dominant traits. Prenatal diagnosis is available for most IEM. A study that looked at reproductive decisions made by parents of children with IEM noted that 56% were receptive to future prenatal diagnosis, and 41% would choose to take measures to prevent another affected pregnancy (Read, 2002). The study also found that parents of children with IEM had higher scores on a stress index, lower scores on an adaptive behavior scale, and fewer persons in their social support network. Furthermore, the parents expressed greater worry about their child’s future and perceived difficulty in meeting the child’s extra care needs. These observations underscore the importance of early intervention, supportive care, and appropriate genetic counseling.
Adolescent with an Inborn Error of Metabolism and Transition to Adulthood
Because of early diagnosis and intervention, many affected children have achieved longer survival. The care received as a child greatly influences the overall prognosis and quality of life for these individuals in adolescence and adulthood. An important goal is for societal integration and life fulfillment, which may be possible in certain cases.
In most instances, the provision of patient care has been by pediatricians and metabolic specialists. The majority of physicians caring for adults are not prepared to assume the care of these patients as they grow older. Familiarity with the natural history of the disease may lead to anticipatory guidance and appropriate monitoring, with early intervention at the first sign of trouble (Enns and Packman, 2002). Several examples illustrate this. Hepatic adenomas, which may become malignant, develop in the second and third decade of life in patients with GSD IA. Patients with tyrosinemia are also at risk for hepatocellular carcinoma; this requires monitoring by serial α-fetoprotein measurements and liver imaging.
Other late complications include acute and chronic recurrent pancreatitis which occurs in association with the hyperlipidemias, disorders of branched-chain amino acid degradation, homocystinuria, and acute intermittent porphyria (Simon et al., 2001). Cholelithiasis occurs in patients with metachromatic leukodystrophy and Gaucher disease, and in disorders of bile formation and biliary transport. Atherosclerosis and thromboembolism are potential causes of morbidity in homocystinuria and methylenetetrahydrofolate reductase (MTHFR) deficiency. Cardiomyopathy and retinopathy may complicate LCHAD. Renal insufficiency or failure may develop in patients with cystinosis, Fabry disease, GSD I, and MMA. Metabolic stroke with bilateral globus pallidus abnormalities and extrapyramidal signs may be a sequela of metabolic decompensation in MMA.
As most IEM affect multiple systems, the involvement of a multidisciplinary team with central coordination by a primary physician is essential. Patients and family members usually appreciate being included in the decision-making process. These moments of interaction provide an opportunity to assess the family’s understanding of the disease and its management as well as their coping mechanisms.
Direct your efforts to ensuring that the child reaches his or her maximum potential. Educational programs for the affected child must be adapted to developmental level and cognitive strengths to minimize frustration and associated behavioral problems. It is important to prepare for development of increasing handicap and take appropriate steps to facilitate individual performance during activities of daily living. Self-care, communications skills, and mobility issues require special attention.
All individuals become increasingly self-conscious of their body image during puberty. Enhance self-esteem in adolescents who feel stigmatized by their physical appearance, particularly when the IEM is associated with facial dysmorphic features and skeletal deformities. Dysarthria impedes communication, and disturbances of bowel and bladder continence undermine self-confidence and social interaction. Some disorders are associated with delayed puberty (GSD IA, galactosemia, and CDG-1a), or premature ovarian failure (hypergonadotropic hypogonadism). Osteoporosis is often underrecognized as an associated condition of IEM. This most often occurs in disorders causing poor mobility due to cognitive or neuromuscular impairment or those characterized by chronic acidosis and renal insufficiency. When dietary regimens are required for disease control, expect the adolescent to manage the diet without parental assistance. Peer pressure may lead to noncompliance, and the patient must understand the potential implications of this course of action.
Pregnancy is a critical time, requiring measures to ensure a good maternal-fetal outcome (Preece and Green, 2002). Women of childbearing age with an IEM require disease control before conception. Close follow-up is required during labor, delivery, and the postpartum period. Children born to women with poorly controlled PKU are at risk for microcephaly (70%), attention deficit disorder and mental retardation (>90%), intrauterine growth retardation (40%), and congenital heart disease (12%). Risk to the fetus correlates with maternal phenylalanine levels. The achievement of a maternal blood phenylalanine concentration of less than 10 mg/dL (600 mM) by 8 to 10 weeks’ gestation and maintaining that concentration throughout pregnancy provides the optimal outcome.
Women with homocystinuria (cystathionine β-synthase deficiency) may have an increased risk for spontaneous abortion and preeclampsia. Pregnancy may exacerbate the cutaneous lesions of porphyria cutanea tarda during the first trimester. Women who are carriers of OTC deficiency may develop a hyperammonemic encephalopathy during the postpartum period. Postpartum metabolic decompensation also occurs in MSUD.
Lysinuric protein intolerance (LPI) is associated with increased risk of anemia, toxemia, and intrauterine growth retardation during pregnancy, and bleeding complications during delivery. Children of mothers with LPI generally develop normally. Attention to maternal protein intake and control of hyperammonemia and other problems during pregnancy are essential (Tanner et al., 2006).
Psychiatric symptoms are primary features of some disorders or may develop secondary to metabolic decompensation (Gray et al., 2000). For instance, behavioral changes (e.g., agitation, delirium) occur in individuals with X-linked ALD, late-onset GM2 gangliosidosis, metachromatic leukodystrophy, urea cycle defects, and porphyria. Cognitive and behavioral problems also occur in children with PKU, especially in those who are not compliant with dietary restriction. Children subjected to BMT are at risk for neuropsychological complications secondary to chemotherapy and irradiation.
Animal Models of Human Inborn Errors of Metabolism
Several spontaneous animal models of human IEM exist, but successful breeding is uncommon. Over the last decade, recombinant genetic techniques have enabled the generation of animal (primarily mouse) models. In a genotype-driven approach, homologous recombination in embryonic stem cells incorporating a null allele accomplishes the generation of mouse (knockout) models. Alternatively, introducing single point mutations result in mice with partial rather than complete deficiencies. A recent example is that of a homozygous knockout mouse which lacked branched-chain ketoacid dehydrogenase (BCKAD) activity and metabolic derangements leading to neonatal lethality (Homanics et al., 2006). Transgenic expression of the human BCKAD cDNA in the liver of this knockout model of MSUD produced a mouse with an intermediate phenotype; BCKAD activity was 5% to 6% of normal, which was sufficient to allow survival but with elevated plasma branched-chain amino acid levels. The currently available animal models have proven useful in investigations of the natural history of the disease and in the preclinical testing of various drugs. In some cases, these investigations have provided the rationale for further clinical trials in humans. Insights into potential disease mechanisms have also come from several animal studies. For instance, transgenic mice overexpressing glycogen synthase on a background of α-glucosidase deficiency show structurally abnormal polysaccharides similar to those observed in patients with Lafora disease and glycogenoses IV and VII (Raben et al., 2001). Although similarities may exist between affected mice and men, species-specific differences in disease expression result from alternative pathways of substrate processing. These considerations highlight the need for caution in carrying over observations made in the animal models to humans.
Disorders Involving Complex Molecules
The metabolism of complex molecules in lysosomes and peroxisomes involves different biochemical pathways from those responsible for the processing of dietary constituents. This explains why dietary manipulation and vitamin or cofactor supplementation are not effective treatments. Tables 62.10 and 62.11 summarize an overview of the distinctive characteristics of these organelles and the associated general features. LSDs involve tissues and organs that develop normally but later malfunction. In contrast, the expression of early-onset peroxisomal disorders is often a severe developmental malformation.
Table 62.10 Characteristic Biochemical Features of Defects of the Major Subcellular Organelles
| Lysosome | Peroxisome | Mitochondria |
|---|---|---|
| Acidic compartment actively maintained by proton ATPase | Metabolic functions: β-oxidation of fatty acids and derivatives, ether phospholipid synthesis | Site of coupling of oxidation and phosphorylation, generation of ATP |
| Terminal compartment in endocytic pathway | Increased VLCFA | Symptoms reflect tissue specificity for aerobic metabolism: brain > skeletal > cardiac muscle > kidney > eye |
| Rich in acid hydrolases (protease, glycosidase, sulfatase) | Disease often classified based on loss of single or multiple peroxisomal enzyme function | Has unique DNA that replicates independently of nuclear DNA |
| Enzymes use M6P targeting into prelysosome | Due to defects of biogenesis and targeting through PTS1 and 2 | Occurrence may be sporadic, matrilineal, or autosomal (dominant or recessive) inheritance |
| Autosomal recessive, except Fabry disease, Hunter syndrome (MPS-II), and Danon disease, which are X-linked traits | Autosomal recessive, except for X-linked adrenoleukodystrophy |
M6P, Mannose-6 phosphate; MPS, mucopolysaccharidosis; VLCFA, very-long-chain fatty acids.
Lysosomal Storage Disorders
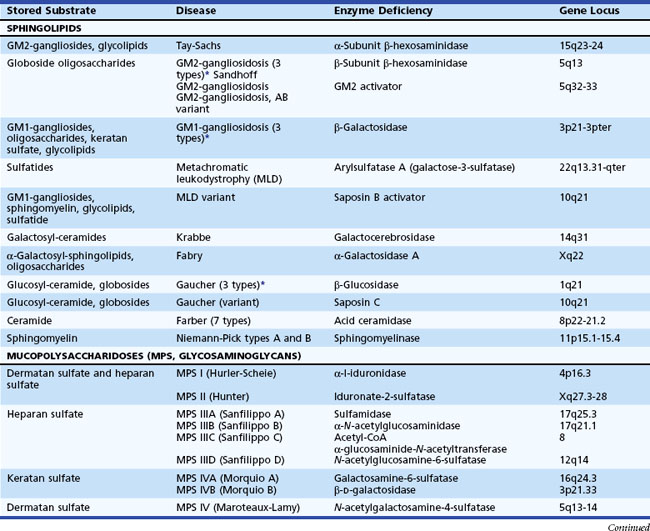
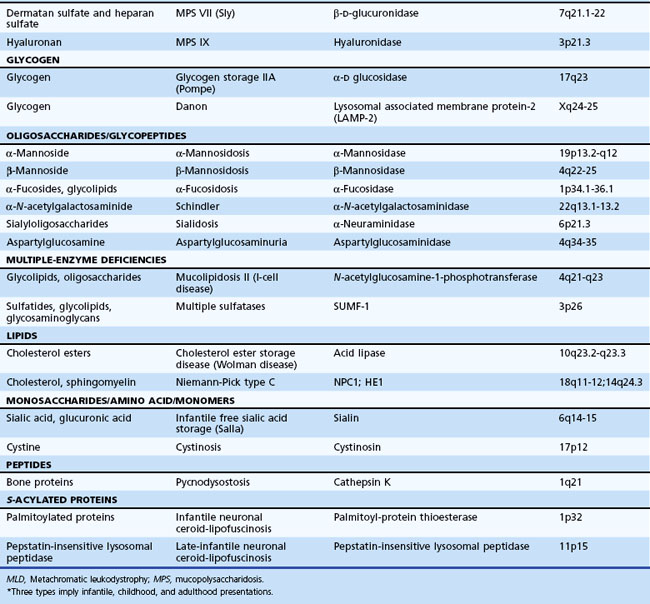
The lysosome is a membrane-bounded intracytoplasmic vacuole that contains enzymes required for the degradation of complex lipids, proteins, and nucleotides. Its acidic milieu (pH ≈ 5.4) is required for optimal activity of the contained hydrolytic enzymes and their cofactors and activators. Table 62.11 describes more than 40 different LSDs, with an estimated combined incidence of 1 in 5000 to 8000.
H.G. Hers first proposed the LSD concept in 1963, based on the detection of glycogen-filled vesicles of lysosomal origin in cells obtained from a patient with Pompe disease. Progressive lysosomal storage of incompletely metabolized substrates occurs either because of primary hydrolase deficiency (e.g., Fabry disease), deficiency of a protective protein that aids in the lysosomal targeting and prevention of premature degradation of enzymes (e.g., galactosialidosis), or the absence of an “activator protein” necessary for enzyme-substrate interaction and degradation (e.g., AB variant of GM2 gangliosidosis). Additional disease mechanisms include abnormal protein/enzyme processing in the endoplasmic reticulum, defects of posttranslational modification preventing functional enzyme maturation (e.g., multiple sulfatase deficiency), failure to attach the appropriate targeting signals (e.g., mannose-6-phosphate) in the Golgi apparatus (e.g., mucolipidosis II), and defective removal or transport of the substrate from lysosomes (e.g., Niemann-Pick disease type C and sialic acid storage disease) (Garver and Heidenreich, 2002; Gopaul and Crook, 2006). Certain enzymes are targeted to the lysosome via alternative routes; for example, glucocerebrosidase relies on LIMP-2 (Reczek et al., 2007). Recently, mutations in LIMP-2 have been identified in patients with focal glomerulosclerosis and progressive myoclonus epilepsy associated with storage material in the brain (Berkovic et al., 2008). In Danon disease, defects of lysosomal-associated membrane protein 2 (LAMP2) result in abnormalities in endocytosis, vesicle fusion, and the processing of autophagic elements (Sugie et al., 2002). LAMP2 is an integral membrane protein of endosomes and lysosomes. Other syndromes of intracellular vesicle damage that cause abnormal lysosomal formation and storage are Hermansky-Pudlak syndrome and Chédiak-Higashi syndrome.
Clinically the LSDs are a heterogeneous group of disorders involving multiple organ systems. The clinical features reflect the cellular sites of substrate storage and resultant organ dysfunction. There is incomplete understanding of disease pathogenesis, but there is increasing evidence for various mechanisms such as aberrant inflammation, induction of apoptosis, and defects of autophagy having a contributory role (Ballabio and Gieselmann, 2009).
In rapidly progressive forms, the onset of clinical features begins in the newborn or in early infancy. With later-onset forms, the initial features are delayed until adolescence or adult life, and the course can vary from acute to chronic. Acute and subacute courses are usually associated with primary central nervous system (CNS) involvement, developmental delay, and mental retardation.
Unlike the small-molecule diseases, the characteristic clinical features of LSDs are either a subacute or a chronic encephalopathy. Myoclonic seizures occur in the following disorders: fucosidosis, Gaucher disease types II and III, GM2 gangliosidosis, Schindler disease (α-N-acetylgalactosaminidase deficiency), and sialidosis type 1. Some LSDs do not have primary CNS involvement (e.g., Fabry disease, Gaucher disease type I, MPS I [Scheie syndrome], MPS IV [Morquio syndrome], mild MPS VI [Maroteaux-Lamy syndrome], and Niemann-Pick disease type B).
Defects in the enzyme cofactor/activator required for complete substrate hydrolysis, rather than a primary enzyme defect, cause rare variants of sphingolipid storage disorders. Two categories of sphingolipid activators exist. One represents the GM2 activator and the other a group of four molecules (saposin A, B, C, and D) derived by proteolytic cleavage of a common precursor, prosaposin. The gene localization for prosaposin is chromosome 10. Deficiency of the GM2 activator results in the AB variant of GM2 gangliosidosis. Saposin B activates arylsulfatase A. Deficiency of saposin B gives rise to a variant of metachromatic leukodystrophy (MLD variant). Saposin C activates glucocerebrosidase and β-galactocerebrosidase. The clinical picture of deficiency is an atypical form of Gaucher disease because of its clinical overlap with the type III variant (subacute neuropathic Gaucher disease). Characteristic of disorders resulting from cofactor deficiencies are normal enzymatic activities in vitro when using the synthetic (artificial) substrate. Therefore, routine biochemical testing misses the diagnosis. Molecular analysis may reveal the presence of mutations in the relevant encoding genes.
The transmission of all LSDs is autosomal recessive, except for Danon disease, Fabry disease, and Hunter syndrome (MPS-II), which are X-linked recessive traits. Biochemical assays are generally available for prenatal diagnosis. Take care in the interpretation of certain enzyme assay results (e.g., arylsulfatase activity, galactocerebrosidase), because low values may be obtained in the presence of pseudo-deficiencies. Diagnostic confirmation is also available by molecular (DNA) testing. In families with a known causal mutation, molecular testing enables accurate assignment of carrier or affected status. Prenatal diagnosis is possible for almost all LSDs. In certain cases, preimplantation genetic diagnosis may also be possible (Tomi et al., 2006).
Neufeld and colleagues showed that exchanging media from fibroblasts with different disease gene mutations (MPS I and MPS II) results in the clearance of intracellular storage material. The metabolic cross-correction was due to secretion of the functional enzyme from one cell line followed by intracellular uptake by the deficient cells. These studies provided the rationale for treatment of the LSD by ERT. Today, achieving cellular correction in certain clinical LSD subtypes is through BMT and ERT. Other therapeutic strategies under consideration include the use of substrate synthesis inhibitors, chaperone-mediated agents (Fig. 62.3), neuronal stem cell transplantation, and gene therapy (Sands and Davidson, 2006).
Peroxisomal Disorders
The peroxisome is an organelle involved in β-oxidation of very long-chain fatty acids (VLCFA), the synthesis of plasmalogen (an ether lipid) and bile acids, and oxidation of pipecolic, phytanic, and dicarboxylic acids (Fidaleo, 2009). The classification of peroxisomal disorders is generally by the presence of single or multiple enzyme deficiencies (Table 62.12). Estimates of their combined incidence are 1 in 25,000. The internalization of most peroxisomal matrix proteins is through one of two targeting sequences in a unique system allowing the importation of oligomerized proteins through a specific shuttle involving a receptor and its cargo. Defects of these cellular mechanisms (involving peroxins encoded by PEX genes) lead to disruption of peroxisomal metabolic functions (Oglesbee, 2005). Peroxisomal disorders are mostly autosomal recessive traits, except for X-linked ALD, which is also the most prevalent clinical type (1 : 17,000).
Table 62.12 Peroxisomal Disorders*
Cataracts, psychomotor retardation
Calcific stippling of the epiphysis (may disappear after age 2 years) and extraskeletal tissues
Intellectual regression, behavioral problems, spastic paraparesis, sphincter problems, adrenal insufficiency
ALD, Adrenoleukodystrophy; RD, Refsum disease.
* The clinical manifestations listed in the right-hand column are features suggestive of a particular subgroup of peroxisomal disorder and are not necessarily present in all of the individual conditions within each subgroup. There also is wide variability in disease expression within the different subgroups.
The spectrum of clinical findings in peroxisomal disorders includes craniofacial abnormalities, encephalopathy, neuronal migration and brain cortical defects, limb malformations, ocular abnormalities, and hepatic and intestinal dysfunction. In the late-onset types, the features are nonspecific and include behavioral changes and deterioration of intellectual function. Demyelination occurs in X-linked ALD, visual and hearing deficits in Refsum disease, and peripheral neuropathy and gait abnormality in Refsum disease and the atypical peroxisomal biogenesis defects.
In patients with ALD, MRI abnormalities become evident at least 12 months before onset of neurological symptoms. Thus, periodic neurological examination is not sufficient for monitoring. The best outcomes (92% 5-year survival) with BMT are in patients identified at an early stage of cerebral disease (performance intelligence quotient [IQ] above 80 and limited MRI abnormality). A recent report describes successful cord blood transplantation using a reduced-intensity conditioning regimen in a patients with advanced childhood-onset cerebral ALD (Awaya et al., 2009). The treatment was well tolerated, stopped disease progression, and contributed to a good neuropsychological outcome. Lentiviral-mediated gene therapy (encoding wild-type ABCD1) using autologous hematopoietic stem cells appears to be a promising approach for affected individuals (Cartier et al., 2009). Replacement therapy with adrenal corticosteroids is mandatory for all patients with ALD and impaired adrenal function.
The demonstration of elevated plasma VLCFA concentrations facilitates screening for peroxisomal disorders. Plasma VLCFA levels are normal in rhizomelic chondrodysplasia punctata [RCDP]), which is associated with impaired erythrocyte plasmalogen synthesis. Increased phytanic acid concentrations occur in Refsum disease and RCDP. Analysis of peroxisomal oxidation and plasmalogen synthesis in cultured chorionic villus sample (CVS) cells or amniocytes can be done for prenatal diagnosis in families at risk.
Disorders Involving Small Molecules
Disorders of intermediary metabolism often result in the accumulation of compounds that cause acute progressive neurological disorders. The term defects of small molecules is used because the compounds that build up proximal to the metabolic block are often elevated in blood and CSF and sometimes excreted in the urine or potentially cleared by dialysis. Detection of these compounds in blood, CSF, and urine enables diagnosis. Serial measurements monitor the effectiveness of disease control. Treatment often requires elimination of the accumulating toxic compounds by dietary restriction or the provision of vitamins or cofactors. During episodes of acute decompensation, metabolic homeostasis may be rapidly achieved by exchange transfusion or preferably by peritoneal or hemodialysis. In neonates with unstable hemodynamics, continuous venovenous hemodialysis may be an option for extracorporal ammonia detoxification (Arbeiter et al., 2010). Continuous control necessitates the use of compounds that bind with the toxic molecules and facilitate alternative pathways of clearance. Included in this group are the aminoacidopathies, organic acidemias, and the urea cycle defects. Table 62.13 provides examples for each category and their estimated incidence.
Table 62.13 Disorders of Small Molecules and Energy Metabolism
| Disorder | Incidence |
|---|---|
| AMINOACIDOPATHIES | |
| Phenylketonuria (PKU) | 1 : 10,000 (Northern European ancestry) |
| Tyrosinemia type I | 1 : 100,000 (in Quebec 1 : 16,700) |
| Maple syrup urine disease (MSUD) | 1 : 150,000 (in Old-Order Amish 1 : 176 births) |
| Homocystinuria | 1 : 300,000 |
| ORGANIC ACADEMIA | |
| Branched-chain (methylmalonic, propionic, isovaleric) MMA | 1 : 20,000 |
| UREA CYCLE | ~1 : 8,000 |
| Ornithine transcarbamylase deficiency | 1 : 15,000 |
| Arginase | 1 : 350,000 |
| CARBOHYDRATE (SUGAR) INTOLERANCE | |
| Galactosemia | 1 : 40,000 |
| Glycogen storage disease type la (von Gierke disease) | 1 : 100,000 |
| FATTY ACID OXIDATION DEFECTS | |
| Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency | ~1 : 20,000 |
MMA, Methylmalonic acidemia.
Disorders of Amino and Organic Acid Metabolism
Aminoacidopathies result from abnormalities in the breakdown of amino acids in the cytosol, whereas organic acidemias are distinguished by their involvement of coenzyme A (CoA)-activated metabolites and mitochondrial enzymes or function. The main clinical feature of these conditions is a symptom-free interval followed by an acute catastrophic event (e.g., vomiting, lethargy, coma). Characteristic of the acute episodes are metabolic acidosis, hypoglycemia, or hyperammonemia (Ogier de Baulny and Saudubray, 2002). These metabolic disorders can lead to progressive developmental regression and spasticity. Typifying MSUD is an acute encephalopathy without hyperammonemia or significant metabolic acidosis, failure to thrive, and mild to moderate psychomotor retardation. Although dysmorphic features suggest organelle pathologies, at least two disorders of organic acid metabolism (i.e., mevalonic aciduria, glutaric aciduria type II) often present with severe malformations. A specific diagnosis relies on the pattern of abnormalities displayed on the amino and organic acid screening profile and by detection of the relevant acylcarnitine compounds in plasma and urine. The usual treatment of these disorders is by dietary restriction, elimination of the toxic compounds, and adjunctive treatments using specific cofactor or vitamin supplements and carnitine as indicated.
Hyperammonemia
Blood ammonia is derived from protein catabolism and as a metabolic byproduct of bacterial reactions in the gastrointestinal tract. Ammonia is a neurotoxic metabolite that promotes excessive glutamine production in the cytosol of astrocytes by its action on glutamine synthetase. It can promote cellular swelling and brain edema by its osmotic effect. Blood ammonia concentrations are elevated by primary defects of the urea cycle (UCD) or secondarily in disorders of amino and organic acid metabolism. In the organic acidemias, intramitochondrial accumulation of acyl-CoA esters causes secondary inhibition of the urea cycle enzymes. Assessment of the plasma amino acid and ammonia levels and analysis of urine organic acid profile establishes the diagnosis. Elevated plasma glutamine levels are common to all the UCDs. Measurement of orotic acid levels in urine is useful in the differential diagnosis (Table 62.14). UCDs are all inherited as autosomal recessive traits except for OTC deficiency (the most common form of UCD), which is inherited as an X-linked trait. In addition to protein restriction and alternative pathway therapy, maintenance of arginine and ornithine levels is an important consideration.
Table 62.14 Defects of the Urea Cycle
| Enzyme Deficiency | Plasma Amino Acid Profile | Urine Orotic Acid |
|---|---|---|
| Carbamoylphosphate synthase | Absent citrulline, decreased arginine Increased glutamine, alanine |
Normal or low |
| Ornithine transcarbamylase | Absent citrulline, decreased arginine Increased glutamine, alanine, uracil |
Increased |
| Argininosuccinic synthase (citrullinemia) | Markedly elevated citrulline; decreased arginine | Increased |
| Argininosuccinic lyase | Moderately elevated citrulline, argininosuccinic acid; decreased arginine | Increased |
| Arginase | Increased glutamine, alanine | Increased |
The clinical features are variable; newborns may exhibit rapidly progressive neurological deterioration, with irritability or lethargy, seizures, coma, and respiratory arrest (Singh et al., 2005; Steiner and Cederbaum, 2001). Transitory hyperammonemia of the newborn (THAN) must also be considered. Compared with neonates affected with UCD, those with THAN have a significantly lower birth weight for gestational age, and chest radiographic findings are usually abnormal. Treatment of affected newborns is by exchange transfusion and preferably by peritoneal dialysis or continuous arteriovenous hemofiltration if necessary. Most THAN survivors have normal neurological and developmental examinations later on and do not experience recurrent episodes of hyperammonemia.
Later-onset clinical UCD manifestations include developmental delay, behavioral problems, hepatomegaly, and gastrointestinal symptoms. The mortality rate is 28% (Bachmann, 2005; Nassogne et al., 2005). Affected children and adults may show behavioral problems, confusion, irritability, and cyclic vomiting, with deterioration in mental status during metabolic stress. Among the UCDs, the unique clinical characteristics of arginase deficiency are spastic diplegia, dystonia, ataxia, and seizures.
Two disorders of amino acid metabolism, lysinuric protein intolerance (LPI) and hyperammonemia-hyperornithinemia-homocitrullinemia (HHH syndrome), are associated with hyperammonemic encephalopathy. The initial features of LPI are growth retardation, hepatic and renal dysfunction, and hematological and pulmonary abnormalities. The cause is a defect in dibasic amino acid transport that leads to an increased urinary excretion of arginine, ornithine, and lysine. LPI is secondary to a functional disorder of the urea cycle. HHH syndrome, caused by mutations in the SLC25A15 (ORNT1) gene encoding the mitochondrial ornithine transporter, is associated with an elevation of plasma ornithine and increased urinary excretion of homocitrulline (a derivative of lysine and carbamoylphosphate conjugation) (Korman et al., 2004). The clinical features include intolerance to protein feeding, vomiting, seizures, and developmental delay. Ornithine administration improves urea cycle function in the HHH syndrome by providing the required precursor for uninterrupted completion of the sequential metabolic steps (Summar, 2001). Progressive spastic paraparesis is a late complication.
Hyperinsulinism-Hyperammonemia Syndrome
Hyperinsulinism-hyperammonemia syndrome is a form of congenital hyperinsulinism caused by a missense mutations in GLUD1 (located on 10q23.3), which encodes glutamate dehydrogenase (GDH). Mutations can occur de novo or be inherited in dominant mode. The mutations are “activating” and lead to a gain in enzyme function by reducing the sensitivity of GDH to allosteric inhibition by guanosine triphosphate (GTP) and adenosine triphosphate (ATP). The main clinical feature is recurrent episodes of symptomatic hypoglycemia, which may occur during fasting or be provoked by high protein intake (Kelly and Stanley, 2008). The hypoglycemia is usually not as severe as in infants with congenital hyperinsulinism due to defective ATP-sensitive potassium channels (mutations in SUR1 or Kir6.2). Children with HHS respond well to medical treatment with diazoxide and to protein restriction. The hyperammonemia is typically mild to moderate and resistant to detoxifying drugs and protein restriction. In contrast to patients with hyperammonemia due to urea cycle disorders, patients with HHS do not suffer from lethargy, headaches, or acute hyperammonemic crises, but they may have neurological complications such as epilepsy and learning disability.
Citrin Deficiency
Citrin deficiency encompasses both adult-onset type II citrullinemia (CTLN2) and neonatal intrahepatic cholestasis (NICCD), which are caused by mutations in the SLC25A13 gene that encodes citrin. Adult patients with CTLN2 suffer from recurring neuropsychiatric symptoms associated with hyperammonemia, including disorientation, delirium, seizures, and coma. Patients with NICCD show multiple metabolic abnormalities: aminoacidemias with an increased threonine/serine ratio, galactosemia, hypoproteinemia, cholestasis, and fatty liver (Kimura et al., 2010). Conventional therapeutic procedures for hyperammonemia, such as low-protein and high-carbohydrate diets, are harmful to patients with citrin deficiency, as carbohydrates cause generation of cytosolic NADH, which inhibits glycolysis and energy production and leads to suppression of ureagenesis (Fukushima et al., 2010). Instead, low-carbohydrate and high-protein/-fat diets are recommended. Liver transplantation has been shown to be beneficial.
Disorders of Energy Metabolism
The energy requirements of cellular metabolism derive from carbohydrates in the nourished state and glycogen and fatty acids stores during fasting. Cellular energy is stored in the form of ATP and creatine phosphate, generated in the cytoplasmic and mitochondrial compartments from glucose and FAO. Hormones mainly mediate the relevant metabolic pathways. Tissues with high aerobic metabolic rates, such as the brain, skeletal muscle, and cardiac muscle, are most vulnerable to defects of energy metabolism.
Several clinical presentations suggest an underlying defect of energy metabolism (Sim et al., 2002). In the amino and organic acidemias, deficiency in substrates for gluconeogenesis and the limited availability of free coenzyme A (CoA) for mitochondrial FAO may compromise energy metabolism, leading to an acute metabolic crisis. Acute or recurrent exercise intolerance and myoglobinuria, with or without cramps, are features of the glycogen and FAO disorders. An inability to perform sudden intense exercise suggests a problem with glycogenolysis or glycolysis, while inability to perform at a sustained level suggests an FAO defect. Progressive neuromuscular weakness and hypotonia are features of the glycogenoses (acid maltase, debrancher enzyme, and brancher enzyme deficiencies), FAO defects (involving carnitine uptake and carnitine acylcarnitine translocase defects), and mitochondrial disorders (cytochrome oxidase deficiency) (Vockley and Whiteman, 2002). Acute or chronic weakness occurs in VLCAD or LCAD, short-chain l-3-hydroxyacyl-CoA dehydrogenase, and trifunctional protein deficiencies.
Avoidance of fasting is an important consideration in the management of disorders of carbohydrate metabolism (glycogenolysis), FAO, and ketogenesis. In some cases, nasogastric or gastrostomy tube feedings are required to maintain the calories for energy metabolism.
Glycogen Storage Diseases
Enzyme defects of glycogen degradation/substrate utilization cause the GSDs. The usual method to designate a GSD is by a type number reflecting the historical sequence of their clinical characterization. The designations of several subtypes recognize the individual first calling attention to the condition. For instance, Pompe disease is the eponymous designation for GSD type II, a lysosomal storage disorder. Hepatomegaly and hypoglycemia characterize the GSD resulting from liver enzyme defects. Cramps on exertion and progressive weakness characterize the GSD resulting from muscle enzyme defects (e.g., McArdle disease caused by muscle glycogen phosphorylase deficiency) (Dimauro and Lamperti, 2001); rhabdomyolysis and myoglobinuria are known complications (Scarlato and Comi, 2002). The inheritance of disorders of carbohydrate metabolism is as autosomal recessive traits, except for the X-linked deficiencies of phosphorylase b kinase (GSD VIII) and phosphoglycerate kinase (GSD type IX). GSD VIII is associated with hepatomegaly, with no evidence of hypoglycemia or skeletal muscle disease, and normal mental development. GSD IX manifests with hemolytic anemia, seizures, mental retardation, and exercise intolerance, with myoglobinuria.
Disorders of Glycolysis
Deficiency of glycolytic enzyme in muscle is a rare cause of myopathy; hemolytic anemia can occur in all subtypes (Berardo et al., 2010). This category includes deficiency of phosphoglycerate kinase (PGK), phosphoglycerate mutase, and α-enolase. These disorders are inherited as autosomal recessive traits, except for PGK deficiency, which is an X-linked disease (Naini et al., 2009). Patients with PGK deficiency may have neurological problems including mental retardation, behavioral abnormalities, seizures, and stroke.
Disorders of Gluconeogenesis
Defects of gluconeogenesis result in recurrent hypoglycemia with lactic acidosis, with or without ketosis. Neurodegenerative features occur in deficiencies of pyruvate carboxylase (PC) and phosphoenolpyruvate carboxykinase (PEPCK). In the severe neonatal form of PC deficiency, there is congenital lactic acidosis along with citrullinemia and hyperammonemia. The glucagon stimulation test can be informative in the evaluation of the integrity of glycogenolysis and gluconeogenesis. In patients with glycogenosis type I (von Gierke disease), the glucose curve following glucagon administration (0.5 g intramuscularly) is usually flat, or there may be a decline associated with an increase in lactate and alanine levels.
Fatty Acid Oxidation Defects
The oxidation of fatty acids involves four components: the carnitine cycle, mitochondrial β-oxidation, electron transfer, and the synthesis of ketone bodies (KB). Some tissues use fatty acids and KB as alternative energy sources to spare the consumption of glucose. Before undergoing β-oxidation, free fatty acids activate to their corresponding acyl-CoA thioesters. In contrast to muscle, the brain cannot fully oxidize fatty acids but can utilize KB synthesized by the liver. The different acyl-CoA dehydrogenases utilize electron-transferring flavoprotein (ETF) as a final electron acceptor. Characteristic of the clinical course in early-onset cases are episodes of hypoketotic, hypoglycemic coma, and periods of metabolic decompensation during prolonged fasting, operations, or infections (Olpin, 2005). Deficiency of mitochondrial trifunctional protein (MTP) and carnitine palmitoyltransferase (CPT) II are disorders that may present in the neonatal period as an acute fulminant illness. Congenital anomalies occur with deficiency states for ETF, ETF-CoQ oxidoreductase, and multiple acyl-CoA dehydrogenase deficiency (glutaric aciduria II [GA-II]). In GA-II (also known as multiple acyl-CoA dehydrogenase deficiency), affected neonates can present with dysmorphic facial features, muscular defects of the abdominal wall, hypospadias (in males), and cystic disease of the kidneys. Rhabdomyolysis, cardiomyopathy, and skeletal muscle weakness are features of chronic late-onset cases of FAO defects. Carrier women heterozygous for a mutation associated with LCHAD deficiency and possibly other disorders of FAO, when pregnant with a homozygous (affected) fetus, may be at risk for acute fatty liver.
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common FAO defect. The initial crisis is fatal in up to 25% of cases. The basis for diagnosis of MCAD deficiency is an organic acid profile showing elevated levels of 5-hydroxyhexanoate, 7-hydroxyoctanoate, hexanoylglycine, suberylglycine, and phenylpropionylglycine.
Disorders of Ketogenesis and Ketolysis
Excess acetyl CoA, a byproduct of FAO, converts in the liver to ketones (3-hydroxybutyrate and acetoacetate) and subsequently is transported to and oxidized in the peripheral tissues. Ketone utilization by the brain spares glucose for use by other tissues, such as erythrocytes, that cannot meet their energy requirements from nonglucose substrates. Ketoacidosis is a prominent secondary feature of several defects of intermediary metabolism.
Primary disorders of ketogenesis and ketolysis are rare. Deficiencies of hydroxymethylglutarate (HMG)-CoA synthase and HMG-CoA lyase (active in leucine metabolism) cause recurrent episodes of hypoketotic hypoglycemia. Mitochondrial β-ketothiolase deficiency (a disorder of isoleucine catabolism) leads to severe episodes of ketoacidosis and encephalopathy, which shows a rapid response to intravenous glucose administration. Reports of dystonia and bilateral striatal necrosis are limited to a few patients. Urine organic acid analysis reveals the presence of 2-methyl-3-hydroxybutyrate, 2-methylacetoacetate, 2-butanone, and tiglylglycine (derived from the metabolism of isoleucine). Cytosolic acetoacetyl-CoA thiolase deficiency is a rare cause of psychomotor retardation and hypotonia, features that reflect the importance of the involved enzyme in sterol and isoprenoid synthesis. Conjugation of acyl-CoAs to glycine and carnitine are important mechanisms of CoA scavenging and detoxification, respectively.
Miscellaneous Metabolic Disorders
Dyslipidemias
The transport of lipids in plasma is on lipoproteins (consisting of a hydrophobic core of triglycerides and cholesterol esters, wrapped in an amphiphilic coating of apolipoproteins, phospholipids, and unesterified cholesterol). Diseases associated with abnormal lipid absorption and metabolism can lead to low blood levels of the fat-soluble vitamins (A, D, E, and K). This is correctable by oral or parenteral supplementation.
Abetalipoproteinemia and Hypobetalipoproteinemia
Abetalipoproteinemia and hypobetalipoproteinemia are disorders of reduced low-density lipoprotein (LDL) cholesterol metabolism. In abetalipoproteinemia (Bassen-Kornzweig syndrome), a rare autosomal recessive disorder, plasma apolipoprotein B (apoB) levels are undetectable, and total cholesterol levels are low (usually < 50 mg/dL). The absence of MTP, a heterodimeric protein involved in the transfer of lipid to apoB, causes the condition. Patients with abetalipoproteinemia have fat malabsorption and neurological disturbances that include symptoms like Friedreich ataxia with dysmetria, cerebellar ataxia, and a spastic gait. Ocular disturbances are common and include retinitis pigmentosa, impaired night vision, nystagmus, and ophthalmoplegia. Failure to thrive and steatorrhea occurs in infancy, whereas the neurological complications appear during adolescence. Other features include anemia (acanthocytosis), a consequence of reduced erythrocyte membrane fluidity. Acanthocytosis also occurs in vitamin E deficiency and neuroacanthocytosis (Rampoldi et al., 2002).
Hypobetalipoproteinemia, due to mutations in the apoB gene, causes a clinical syndrome similar to Friedreich ataxia (ataxia and peripheral neuropathy). Transmission is as an autosomal dominant trait. Most patients have low LDL cholesterol concentrations (usually <60 mg/dL). Rare patients homozygous for the defect have clinical features indistinguishable from those of abetalipoproteinemia. Vitamin supplements (A and E) have a beneficial influence on the neurological and ocular symptoms. Serial monitoring of vitamin levels avoids vitamin toxicity.
Tangier Disease
Tangier disease is a rare autosomal recessive disorder caused by mutations in a cell membrane protein called ABCA1, which normally mediates the secretion of excess cholesterol from cells into the high-density lipoprotein (HDL) metabolic pathway. It belongs to a family of ATP-binding cassette (ABC) transporters involved in the recognition of substrates and their transport into and out of cell membranes. Cystic fibrosis, age-related macular degeneration, and X-linked ALD are other disorders associated with defects of genes that encode this class of proteins. Characteristic of Tangier disease is severe deficiency of HDL and tissue storage of cholesterol esters. Clinical features include enlarged, orange-yellow tonsils (filled with foam cells representing deposits of β-carotene cholesterol esters), splenomegaly, and a relapsing sensorimotor neuropathy. Patients may develop distal weakness, hyporeflexia, decreased pain and temperature sensation, with relative preservation of position and vibration sense. There can be wide intrafamilial variability in clinical expression (Pichit et al., 2010).
Neutral Lipid Disease: Chanarin-Dorfman Syndrome
Chanarin-Dorfman syndrome is a rare autosomal recessive disorder of triglyceride metabolism resulting from mutations of CGI-58 gene, which encodes a protein of the esterase/lipase/thioesterase subfamily (Lass et al., 2006). Characteristic findings include neutral lipid storage in several tissues, nonbullous ichthyosiform erythroderma, and neurological manifestations such as psychomotor delay, ataxia, neurosensory defects, and proximal myopathy (Selimoglu et al., 2009). A second neutral lipid disease has been described, attributed to mutations in adipose triglyceride lipase (ATGL/PNPLA2). Compared to patients with CGI-58 gene mutations, those with defective ATGL function suffer from more severe myopathy but do not develop ichthyosis (Schweiger et al., 2009).
Sterol Synthesis Defects
A defect in sterol synthesis is the hallmark of Smith-Lemli-Opitz syndrome (SLOS), a disorder characterized by multiple malformations, growth and psychomotor retardation, and behavioral disturbances (Yu and Patel, 2005). It is a disorder of post-squalene cholesterol biosynthesis. Mutations of the 7-dehydrocholestrol (DHC)-D7 reductase (DHCR7) gene cause the disease. Inheritance is as an autosomal recessive trait. Biochemical tests reveal low total cholesterol and elevated serum levels of 7DHC.
Sterol abnormalities that result from deficiencies of cholesterol biosynthesis also explain other recently recognized disorders of morphogenesis (mevalonic aciduria [MVA], desmosterolosis, X-linked chondrodysplasia punctata, and CHILD [congenital hemidysplasia with ichthyosiform erythroderma and limb defects] syndrome) (Haas et al., 2001). Sterol synthesis (SS) takes place partly in the peroxisome and explains why SS defects show some overlap in clinical features with the peroxisomal biogenesis disorders. Clinical features of MVA include progressive ataxia, psychomotor retardation, and retinitis pigmentosa. Affected patients manifest delayed closure of the cranial sutures and fontanelles, associated with atrophy of the cerebellar hemispheres and vermis. The diagnosis of MVA is based on demonstration of decreased mevalonate kinase activity in fibroblasts. The diagnosis of other disorders secondary to distally located defects of cholesterol biosynthesis requires sterol analysis in blood or tissues by gas chromatography–mass spectrometry (GC-MS). Perturbed hedgehog signaling likely underlies some of the malformations found in SLOS (Porter et al., 1996). Treatment is primarily symptomatic. Cholesterol supplementation may mitigate some symptoms, and simvastatin (an HMG-CoA reductase inhibitor) may avoid the formation of potentially toxic metabolites (7 and 8DHC) in patients with residual DHCR7 activity.
Cerebrotendinous Xanthomatosis (Cholestanolosis)
The transmission of cerebrotendinous xanthomatosis, or cholestanolosis (CTX), which is caused by mutations in the sterol 27-hydroxylase gene, is as an autosomal recessive trait. The primary feature is the formation of xanthomatous lesions in the brain and tendons. Neurological impairments include mental retardation, progressive spasticity, pseudobulbar palsy, and cerebellar dysfunction (Moghadasian, 2004). Brain MRI shows hyperintense signals that involve the corticospinal tracts in the brainstem, the white matter of both internal capsules, and the peritrigonal white matter. Bilateral cataracts and chronic diarrhea may precede the neurological deterioration. Biochemical findings include elevated plasma and bile cholestanol levels and increased urinary excretion of bile alcohol glucuronides associated with diminished biliary chenodeoxycholic acid (CDCA). Therapy with CDCA reverses the neurological features. In one study of CDCA therapy in 17 patients, dementia cleared in 10, pyramidal and cerebellar signs resolved in 5 and improved in another 8. Further, cerebral CT scans improved in 7 (Samenuk and Koffman, 2001).
Lowe Oculocerebrorenal Syndrome
Characteristic of Lowe syndrome, an X-linked trait, are bilateral congenital cataracts, mental retardation, and a renal ion transport defect (Fanconi syndrome) (Schneider et al., 2001). It results from mutations in the OCRL gene, which normally encodes a Golgi-associated protein (inositol polyphosphate-5-phosphatase) that regulates the cellular levels of a metabolite (phosphatidylinositol 4,5-biphosphate) involved in vesicular transport. Female carrier identification is by detection through slit-lamp examination of lens opacities. Symptomatic treatment with phosphate and vitamin D prevents the development of severe rickets. Carnitine supplementation may be required, as patients with generalized Fanconi syndrome can experience severe carnitine depletion. Patients are at risk for glaucoma and should have serial monitoring of intraocular pressure.
Molybdenum Cofactor and Sulfite Oxidase Deficiency
These two distinct autosomal recessive traits have overlapping presentations consisting of acute or subacute, severe, neonatal-onset epileptic encephalopathy with diffuse cavitary leukomalacia. Consider these disorders in the differential diagnosis of ischemic/anoxic perinatal leukomalacia.
Molybdenum cofactor is essential for xanthine dehydrogenase, sulfite oxidase, and aldehyde oxidase activity. Deficiency of the cofactor causes refractory seizures, axial hypotonia, and limb rigidity (Macaya et al., 2005). Urinary excretion of xanthine and sulfite is increased. Brain imaging shows multiple cystic cavities in the white matter. The clinical findings are believed to be caused by the accumulation of toxic sulfites leading to oxidation of SH groups and formation of disulfide groups, or indirectly via excessive NMDA receptor activation or inhibition of glutamate dehydrogenase activity (Sass et al., 2009).
The cause of sulfite oxidase deficiency is defects involving SUOX, the gene encoding the enzyme that catalyzes the terminal reaction in the sulfur amino acid degradation pathway (Johnson et al., 2002). Dislocated ocular lens is characteristic.
Disorders of Copper Metabolism
Copper is an essential element for the activity of several enzymes (cytochrome c oxidase, Cu/Zn-superoxide dismutase, dopamine-β-hydroxylase). Menkes disease and Wilson disease result from mutations in two distinct but highly homologous copper transporters (P-type ATPases).
Menkes Syndrome
Transmission of Menkes syndrome (also known as Menkes kinky hair syndrome) is as an X-linked trait. The membrane copper transporter, ATP7A, is defective, causing a functional copper deficiency and low levels of serum copper and ceruloplasmin (Daniel et al., 2004). Progressive neurodegeneration and marked connective tissue abnormalities are characteristic. The disease is usually lethal in infancy or childhood. Neuropathological findings include neovascularization and extreme reduplication of the cerebral arteries, in conjunction with cystic medial degeneration, bilateral cerebellar hypoplasia, focal cortical dysplasia, and cerebellar heterotopias. Daily intravenous copper histidine administration restores serum copper and ceruloplasmin levels and leads to favorable clinical results when started prior to neurodegeneration. Occipital horn syndrome, formerly known as Ehlers-Danlos syndrome type IX or X-linked cutis laxa, is an allelic variant of Menkes syndrome.
Wilson Disease
Wilson disease is an autosomal recessive disorder caused by mutations in the copper transporter (ATP7B) gene. Over 350 mutations in ATP7B have been reported (Davies et al., 2008). Excessive copper accumulates in the liver and brain. Neurological features include tremors, loss of fine motor control, poor coordination, rigid dystonia, dysarthria, and swallowing difficulties (Kitzberger et al., 2005). Diagnostic findings include the presence of Kayser-Fleischer rings, increased urine copper excretion, with low serum ceruloplasmin and increased liver tissue copper content. Basal ganglia degeneration is evident on MRI as increased T2 intensity in the caudate and putamen. The results of therapy by liver transplantation are mixed.
Disorders of Purine and Pyrimidine Metabolism
Purine and pyrimidine nucleotides are essential cellular components involved in energy transfer and the regulation and synthesis of DNA and RNA. Defects of purine and pyrimidine metabolism can result from disruption of biosynthetic, catabolic, and salvage pathways (Table 62.15, A and B). Consider these disorders in patients presenting with psychomotor delay and behavioral problems, abnormalities of muscle tone, extrapyramidal features, and seizures (Jurecka, 2009).
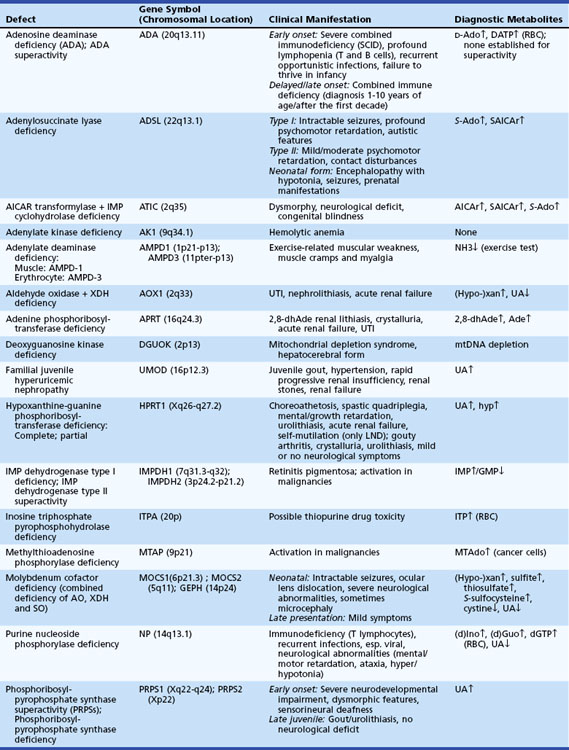
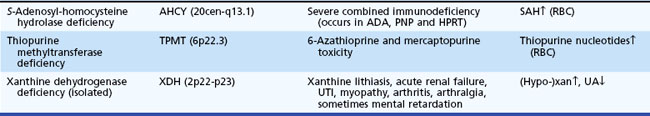
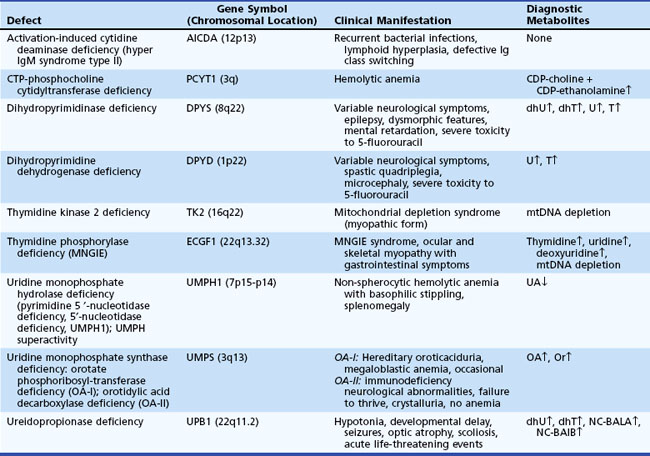
Transmission of Lesch-Nyhan syndrome is X-linked recessive. Hypoxanthine guanine phosphoribosyltransferase (HGPRT), required in the purine salvage pathway, is deficient, causing hyperuricemia. Clinical features include chorea and athetosis, dysarthria, hyperreflexia and hypertonia, cognitive impairment, and behavioral disturbances (including impulsive and self-injuring activity) (Schretlen et al., 2001). Reduced dopamine concentrations in the basal ganglia and CSF may account for some of the neurological features. Patients with partial HGPRT deficiency always have hyperuricemia and often have neurological abnormalities, but they do not show self-injuring behavior and usually have normal intelligence.
Transmission of adenylosuccinatelyase deficiency is as an autosomal recessive trait. De novo purine and adenosine monophosphate (AMP) synthesis are defective, and succinyl-aminoimidazole carboxamide (SAICA) riboside and adenylosuccinate accumulate in cells and are detectable in CSF and urine. Severe developmental delay, seizures, and growth retardation are characteristic. Psychomotor retardation with autistic-like behavior occurs in about half of the cases (Salerno and Crifo, 2004).
Dihydropyrimidine dehydrogenase deficiency is an inborn error of pyrimidine metabolism characterized by developmental delay, autistic-like behavior, and seizures. It is associated with increased urinary excretion of thymine and uracil, detectable by high-performance liquid chromatography/electrospray tandem mass spectrometry (van Kuilenburg et al., 2004). Deficiencies of two other enzymes involved in pyrimidine catabolism, dihydropyrimidinase and β-ureidopropionase, exist (Nyhan, 2005). These disorders have been associated with severe toxicity in patients with cancer, following the administration of the chemotherapeutic agent, 5-fluorouracil. Confirmatory diagnosis requires demonstration of deficient enzyme activity, which necessitates a liver biopsy in cases due to deficiency of dihydropyrimidinase or β-ureidopropionase.
Porphyrias
Porphyrins play an important role in the formation of metalloporphyrin complexes including hemoglobin, myoglobin, cytochromes, peroxidases, oxidases, and catalases. The cause of porphyria is deficiency of a number of specific enzymes involved in the heme biosynthetic pathway, and these disorders are characterized by the accumulation and excess excretion of byproducts of intermediary heme metabolism and their oxidized products (Table 62.16). The heme synthetic pathway normally occurs in bone marrow elements (85%) or in the liver. Consider a diagnosis of porphyria in patients with unexplained neuropsychiatric signs, visceral (gastrointestinal and hepatic) symptoms, or cutaneous photosensitivity (Dombeck and Satonik, 2005). Cutaneous manifestations are prominent in hereditary coproporphyria and variegate porphyria, but the neurovisceral disturbances may be indistinguishable from those of acute intermittent porphyria (AIP). Acute peripheral neuropathy and encephalopathy may develop in patients with AIP (Pischik and Kauppinen, 2009). Acute porphyric neuropathy is predominantly motor and associated with a history of abdominal pain and dysautonomia, CNS involvement, and mild hepatopathy. Acute encephalopathy manifests as a combination of mental symptoms, seizures, and SIADH, but rarely focal CNS deficits. Porphyrias with neurological features have either a constant or an intermittent excretion of aminolevulinic acid (ALA) and porphobilinogen. The pathogenesis of porphyric neuropathy is complex; overproduction of ALA can lead to direct neurotoxicity, oxidative damage, and modification of glutamatergic release. The elimination of precipitating factors is important to reduce the frequency and intensity of acute exacerbations.
Table 62.16 Clinical and Biochemical Features of Acute Porphyrias
| Disease\Prevalence | Clinical Features | Biochemical Features |
|---|---|---|
| Acute intermittent porphyria\1-2/10,000 | Autosomal dominant; low penetrance; no skin lesions | Normal fecal porphyrins (COPRO and PROTO); increased urinary levels of ALA and PBG (PBG > ALA) (higher during acute attacks); increased urinary porphyrins (URO and COPRO) (in overt AIP) |
| Hereditary coproporphyria\0.1/10,000 | Autosomal dominant; low penetrance; blister skin lesions during acute attacks and skin fragility (about 30% patients) | Increased fecal porphyrins (COPRO > > PROTO); increased urinary levels of ALA and PBG (PBG > ALA) (higher during acute attack); increased urinary porphyrins (URO and COPRO) |
| Variegate porphyria | Autosomal dominant; low penetrance; acute attacks (50% with blisters skin lesions) in 20%-30% patients; skin lesions only (blisters after sun exposure and skin fragility) in 70%-80% of patients | Increased fecal porphyrins (PROTO > or COPRO); increased urinary levels of ALA and PBG (PBG > ALA) mostly only during acute attacks; increased urinary porphyrins (COPRO prevalence); plasma fluorescence (specific emission at wavelength of 626-628 nm) |
| ALA dehydrase deficiency porphyria (Doss porphyria) |
Very rare autosomal recessive; acute neurovisceral attacks, neuropathy, or both; no skin lesions only | Normal fecal porphyrins; increased urinary levels of ALA (ALA > PBG); increased urinary porphyrins (COPRO) |
| Plumboporphyria (lead poisoning) | Lead exposition (incidental, professional); features resembling ALA dehydrase deficiency porphyria (see above) | Anemia; high level of lead in serum and urine |
ALA, δ-Aminolevulinic acid; COPRO, coproporphyrins; PBG, porphobilinogen; PROTO, protoporphyrins; URO, uroporphyrins.
Transmission of AIP is as an autosomal dominant trait. Mutations occur in the porphobilinogen deaminase (PBD) gene. The demonstration of deficient activity of erythrocyte PBD activity confirms the diagnosis. Bouts of abdominal pain, paresthesias, seizures, and peripheral neuropathy are prominent clinical features. Neurotic or psychotic behavior also occurs. Certain drugs precipitate acute attacks; provide patients with a list of medications to avoid (Table 62.17). Carbohydrate loading and administration of heme analogs manage the acute crises (Ventura et al., 2009).
Table 62.17 Medications Patients with Porphyria Should Avoid and Permissible Alternatives
| Contraindicated | Alternative |
|---|---|
| Sedatives, tranquilizers | Chloral hydrate |
| Barbiturates | Chlorpromazine |
| Anticonvulsants (ethosuximide, clonazepam, sodium valproate, phenytoin, primidone) | |
| Anticoagulants | Aspirin |
| Centrally acting agents (imipramine, nikethamide, metoclopramide, methyldopa) | Droperidol, methadone (Physeptone), chlorpromazine, promethazine |
| Analgesics/antiinflammatory agents (diclofenac, pentazocine, phenylbutazone, phenazone) | Morphine and derivatives |
Congenital Defects of Glycosylation
The carbohydrate-deficient glycoprotein syndromes (otherwise known as congenital defects of glycosylation [CDG]) are a heterogenous group of autosomal recessive disorders resulting from defects of the N-linked glycosylation pathway (Table 62.18). These disorders are subclassified based on underlying defects that can involve either the assembly of the oligosaccharide precursor, which leads to under-occupancy of N-linked glycosylation sites (group I), or glycan remodeling (group II). Many proteins undergo glycosylation (a posttranslational modification step) to render them functional. The initial features of CDG syndromes may include ataxia, strabismus, unusual fat distribution, severe liver dysfunction, seizures, and stroke-like episodes. All of these disorders, except for CDG-Ib (phosphomannose isomerase deficiency), show some degree of developmental and psychomotor retardation, and many have gastrointestinal dysfunction (Eklund and Freeze, 2006). Severe brain involvement occurs in phosphomannomutase deficiency (CDG-Ia), which accounts for about 80% of cases, and N-acetylglucosaminyl transferase-II deficiency, a disorder with craniofacial abnormalities (Pearl and Krasnewich, 2001). Testing for abnormal protein glycosylation requires isoelectric focusing analysis of serum transferrin. The Sebia Capillarys CDT system, a new capillary zone electrophoresis method, has been shown to be a simple and reliable method to screen for CDG (Parente et al., 2010). Mass spectrometry techniques have also been applied in the diagnosis of CDG (Barone et al., 2009). Treatment is not available. Oral mannose given to two children with CDG-Ib provided clinical improvement and normalization of blood glucose, aminotransferases, and coagulation factor levels in one child and resolution of gastrointestinal bleeding in another (Rush et al., 2000).
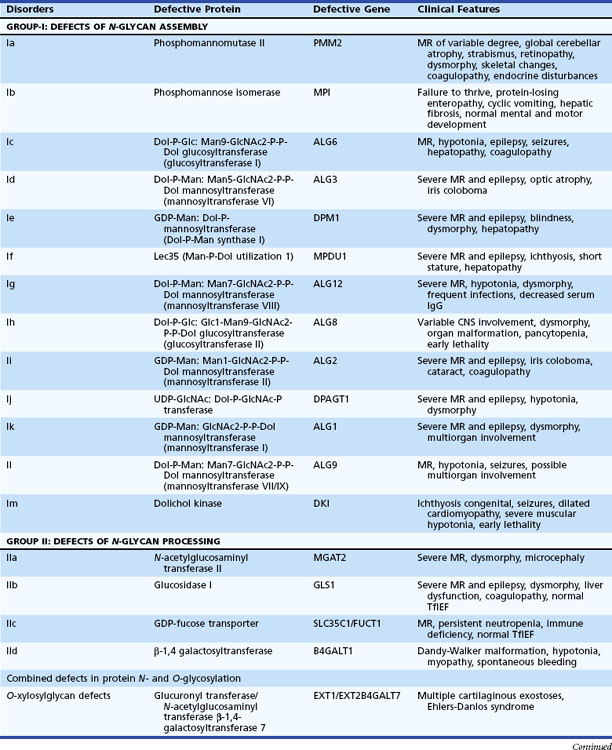
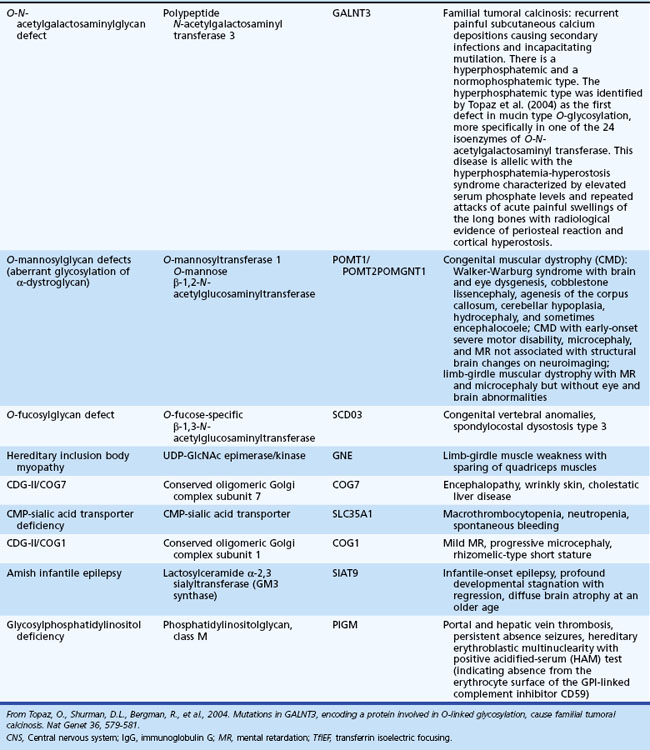
Canavan Disease
Canavan disease (CD) transmission is as an autosomal recessive trait. The cause is deficiency of aspartoacylase. N-acetyl aspartic acid (NAA), which is a normal intraneuronal compound, acts as a neurotoxic compound at high concentrations. Patients with CD show NAA accumulation in the brain and excessive amount of NAA excretion in the urine. CD mainly occurs in Ashkenazi Jews (carrier frequency 1 : 37 to 58). The pathological buildup of NAA in limited white-matter extracellular fluid (ECF) is hypothesized to lead to increased osmotic-hydrostatic pressure and initiation of the demyelination process (Baslow et al., 2009). The NAA in white matter ECF is believed to be a product of the catabolism of axon-released NAAG at nodes of Ranvier by astrocyte NAAG peptidase after it has docked with the astrocyte surface metabotropic glutamate receptor 3.
Clinical features include hypotonia, delayed development, optic atrophy, and seizures (Matalon and Matalon, 2002). CD shares several features (e.g., progressive macrocephaly, demyelination) with Alexander disease, a rare progressive leukoencephalopathy characterized by the widespread accumulation of Rosenthal fibers. However, Alexander disease has a later onset and slower course. Missense mutations in the coding region of the glial fibrillary acidic protein (GFAP) gene occur in patients with Alexander disease. Specific treatment is not available for either CD or Alexander disease. Gene therapy is being studied in children with CD.
Neurotransmitter and Small Peptide Defects
Neurotransmitters are divisible into the following groups: inhibitory (GABA, glycine), excitatory (aspartate, glutamate), cholinergic (acetylcholine), monoaminergic (epinephrine, norepinephrine, dopamine, and serotonin), and purinergic (adenosine, AMP, ADP, ATP). The causes of inherited defects of neurotransmission include deficient synthesis, release, breakdown, or reuptake of neurotransmitters. In addition, defects of receptors, failure of signaling pathways of glial cells, and the inability to maintain an appropriate milieu may cause disease. Clinical features vary from mild to severe and include psychomotor retardation, developmental delay, oculogyric crises, hypotonia, and ataxia (Pearl et al., 2004). Analysis of neurotransmitters and related metabolites in CSF can be informative (see Table 62.3).
Succinic semialdehyde dehydrogenase deficiency disrupts the normal metabolism of GABA, resulting in the accumulation of β-hydroxybutyrate, a compound with neuromodulatory properties in plasma, CSF, and urine. Neuroimaging reveals increased T2-weighted MRI signal usually affecting the globus pallidus, cerebellar dentate nucleus, and subthalamic nucleus, and often cerebral and cerebellar atrophy. Studies in the mouse model has revealed use-dependent down-regulation of GABAA and GABAB receptors; translational human studies similarly reveal down-regulation of GABAergic activity in patients, utilizing flumazenil-PET (positron emission tomography) and transcranial magnetic stimulation for GABAA and GABAB activity, respectively (Pearl et al., 2009). Vigabatrin inhibits GABA transaminase and causes variable improvement of the ataxia and behavioral disturbances. Additional defects of GABA metabolism include glutamic acid decarboxylase deficiency with nonsyndromic cleft lip/palate and GABA-transaminase deficiency.
Aromatic l-amino acid decarboxylase (AADC) deficiency causes a deficiency of serotonin and catecholamines, leading to a subacute epileptic encephalopathy. Autonomic dysfunction leads to ptosis, hypotension, gastric and intestinal dysmotility, and poor temperature regulation. Pterin and phenylalanine metabolisms are normal. The basis for diagnosis is the demonstration of low CSF homovanillic (HMV) acid, 5-hydroxy-3-indole acetic acid (5HIAA), and 3-methoxy-4-hydroxy-phenylethylene glycol, associated with increased levels of l-dopa. Urine organic acid profile often reveals elevated levels of vanillactic acid (a byproduct of alternative degradative pathway for l-dopa). AADC is a pyridoxal-5-phosphate-dependent enzyme, and patients with pyridoxal phosphate-dependent seizures show a similar neurotransmitter profile. Combined treatment with pyridoxine (vitamin B6), tranylcypromine (a monoamine oxidase inhibitor), and bromocriptine promotes clinical improvement. Additional defects of dopamine and serotonin synthesis include tyrosine hydroxylase deficiency and disorders of BH4 synthesis. AADC, tyrosine hydroxylase, sepiapterin reductase, and guanosine triphosphate cyclohydrolase (Segawa disease) deficiencies do not feature elevated serum phenylalanine and require CSF analysis for diagnosis (Pearl et al., 2007).
Nonketotic Hyperglycinemia
The cause of glycine encephalopathy (the preferred term) is a defect in the tetrameric protein (glycine decarboxylase) gene that encodes components of the mitochondrial glycine cleavage system (Toone et al., 2002). Glycine is a simple amino acid that functions as a neurotransmitter with dual excitatory (cortical) and inhibitory (spinal cord and brainstem) effects. Additional properties include a role in gluconeogenesis via pyruvate and with detoxification via glycine conjugation. Secondary causes of hyperglycinemia include valproic acid and d-glyceric acidemia. The clinical characteristic of nonketotic hyperglycinemia (NKH) is a neonatal encephalopathy associated with mutations in the gene encoding P protein. Newborns develop lethargy, hypotonia, myoclonic seizures, and apnea. The electroencephalogram shows a burst suppression pattern. CSF glycine is elevated, and the ratio of CSF to plasma glycine concentration (normally < 0.4) is above 0.6. Brain MRI reveals a hypoplastic or absent corpus callosum and gyral malformations; cerebellar hypoplasia is an associated feature. Neuropathological examination has revealed spongiform white-matter degeneration. Brain MRS may reveal a peak corresponding to glycine (Novotny et al., 2003). The excitatory effects of glycine on the N-methyl-d-aspartate (NMDA) receptor may cause the neurological symptoms. Treatment is with dextromethorphan (as an NMDA receptor blocker) and ketamine to inhibit receptor excitation. Prognosis is poor with early onset and in the presence of cerebral dysgenesis, and most affected children are mentally retarded.
Additional clinical variants have been described, including an infantile presentation after age 6 months with partial seizures or hypsarrhythmia; a childhood variant with mild mental retardation, delirium, chorea, and vertical gaze palsies; and a late-onset pattern in adults with progressive spastic diplegia and optic atrophy (Dinopoulos et al., 2005). Although H- and T-protein defects are associated with later-onset forms, there are also milder phenotypes of the neonatal onset form showing mutations in the glycine decarboxylase gene. Diagnosis is based on measurements of glycine cleavage activity in liver or cultured lymphoblasts, or identification of the relevant gene defect.
Serine Deficiency Syndromes
l-Serine is a precursor of several metabolites such as nucleotides, phospholipids, and the neurotransmitters, glycine and d-serine. Deficiency of 3-phosphoglycerate dehydrogenase (3PHGDH), involved with l-serine biosynthesis, leads to congenital microcephaly, intractable seizures, and severe psychomotor retardation (de Koning, 2006). Studies involving the mouse model of 3PHGDH deficiency have shown that serine deficiency impairs normal cell cycle progression and the subsequent neurogenesis of radial glia cells (Kawakami et al., 2009). Oral supplementation of the deficient amino acids (l-serine [500 mg/kg] and glycine [200 mg/kg] daily) reduces seizure frequency and prevents psychomotor delay if given at an early age.
Creatine Deficiency Syndromes
Creatine and phosphocreatine play essential roles in energy storage and transmission; several metabolic disorders of creatine synthesis or transport exist (Stromberger et al., 2003). Primary creatine deficiency can result from defects in one of two synthetic enzymes: arginine:glycine amidinotransferase (AGAT) and guanidinoacetate methyltransferase (GAMT). Transport problems result from an X-linked defect in the creatine transporter (CRTR). Affected children usually present with mental retardation and epilepsy. Patients with GAMT deficiency also exhibit dystonic hyperkinetic movements and epilepsy that in some cases is unresponsive to pharmacological treatment. Demonstration of cerebral creatine deficiency requires proton MRS (Dezortova et al., 2008). Measurement of guanidinoacetate in body fluids enables discrimination of GAMT (high concentration) and AGAT (low concentration) deficiencies from CRTR (normal concentration). GAMT and AGAT deficiency are treatable by oral creatine supplementation, but patients with CRTR deficiency do not respond to this type of treatment (Stockler et al., 2007).
Glucose Transporter Protein Deficiency (DeVivo Disease)
A defect of glucose transporter protein (GLUT1) leads to impaired glucose transport into the brain and a disease that usually starts in infancy with severe seizures, typically resistant to conventional anticonvulsant medications (Wang et al., 2005). This condition is caused by a spontaneous dominant mutation in the gene encoding GLUT1 on chromosome 1. At a later age, the child exhibits signs of mental retardation, motor delay, and impaired language development. Spasticity, ataxia, and dystonia have also been reported. In at least three cases, affected patients presented with intermittent ataxia without epilepsy (Joshi et al., 2008). The ketogenic diet effectively controls the seizures and other paroxysmal activities in patients with GLUT1 deficiency, but it has less effect on the cognitive symptoms. There is reduced CSF glucose and lactate, and CSF/blood glucose ratio is low (0.33 ± 0.07). Other defects of glucose transport across cell membranes include glucose-galactose malabsorption and Fanconi-Bickel syndrome (Pascual et al., 2004).
Defects in Leukotriene Synthesis
Leukotrienes (LTE) are active lipid mediators derived from the 5-lipoxygenase pathway. Cysteinyl leukotrienes have neuromodulatory functions apart from their role in the mediation of inflammation and host defense. Leukotriene synthesis defects due to γ-glutamyl transpeptidase deficiency result in severe muscular hypotonia, psychomotor retardation, failure to thrive, microcephaly, and glutathionuria, as well as in increased levels of γ-glutamyl cysteine and cysteine (Mayatepek et al., 2004). Another defect involving a deficiency of membrane-bound dipeptidase was described in a 15-year-old male patient with mental retardation, mild motor impairment, and partial deafness (Mayatepek et al., 2005). Biochemical investigations showed highly increased concentrations of LTED4, which is usually not detectable, whereas LTE4, the major urinary metabolite in humans, was completely absent.
Arbeiter A.K., Kranz B., Wingen A.M., et al. Continuous venovenous haemodialysis (CVVHD) and continuous peritoneal dialysis (CPD) in the acute management of 21 children with inborn errors of metabolism. Nephrol Dial Transplant. 2010;25:1257-1265.
Awaya T., Kato T., Niwa A., et al. Successful cord blood transplantation using a reduced-intensity conditioning regimen for advanced childhood-onset cerebral adrenoleukodystrophy. Pediatr Transplant. 2009. doi: 10.1111/j.1399-3046.2009.01188.x. [Epub ahead of print]
Bachmann C. Long-term outcome of urea cycle disorders. Acta Gastroenterol Belg. 2005;68:466-468.
Ballabio A., Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793(4):684-696.
Barone R., Sturiale L., Garozzo D. Mass spectrometry in the characterization of human genetic N-glycosylation defects. Mass Spectrom Rev. 2009;28(3):517-542.
Baslow M.H., Guilfoyle D.N. Are astrocytes the missing link between lack of brain aspartoacylase activity and the spongiform leukodystrophy in Canavan disease? Neurochem Res. 2009;34:1523-1534.
Berardo A., DiMauro S., Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10(2):118-126.
Berkovic S.F., Dibbens L.M., Oshlack A., et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82(3):673-684.
Blau N., Bélanger-Quintana A., Demirkol M., et al. Optimizing the use of sapropterin (BH(4)) in the management of phenylketonuria. Mol Genet Metab. 2009;96(4):158-163.
Bonnefont J.P., Bastin J., Behin A., et al. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360:838-840.
Cakmakci H., Pekcevik Y., Yis U., et al. Diagnostic value of proton MR spectroscopy and diffusion-weighted MR imaging in childhood inherited neurometabolic brain diseases and review of the literature. Eur J Radiol. 2009;74:e161-e171.
Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818-823.
Centers for Disease Control and Prevention (CDC). Contribution of selected metabolic diseases to early childhood deaths—Virginia, 1996-2001. MMWR Morb Mortal Wkly Rep. 2003;52:677-679.
Chakrapani A., Wraith J.E. Principles of management of the more common metabolic disorders. Curr Paediatr. 2002;12:117-124.
Cheon J.E., Kim I.O., Hwang Y.S., et al. Leukodystrophy in children: a pictorial review of MR imaging features. Radiographics. 2002;22:461-476.
Cox T., Lachmann R., Hollak C., et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481-1485.
Daniel K.G., Harbach R.H., Guida W.C., et al. Copper storage diseases: Menkes, Wilsons, and cancer. Front Biosci. 2004;1:2652-2662.
Daschner M., Schaefer F. Emergency dialysis in neonatal metabolic crises. Adv Ren Replace Ther. 2002;9:63-69.
Davies L.P., Macintyre G., Cox D.W. New mutations in the Wilson disease gene, ATP7B: implications for molecular testing. Genet Test. 2008;12(1):139-145.
de Koning T.J. Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis. 2006;29:347-351.
Desnick R.J. Enzyme replacement and enhancement therapies for lysosomal diseases. J Inherit Metab Dis. 2004;27:385-410.
Dezortova M., Jiru F., Petrasek J., et al. 1H MR spectroscopy as a diagnostic tool for cerebral creatine deficiency. MAGMA. 2008;21(5):327-332.
Dhawan A., Mitry R.R., Hughes R.D. Hepatocyte transplantation for metabolic disorders, experience at King’s College hospital and review of literature. Acta Gastroenterol Belg. 2005;68:457-460.
DiMauro S., Lamperti C. Muscle glycogenoses. Muscle Nerve. 2001;24:984-999.
Dinopoulos A., Matsubara Y., Kure S. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab. 2005;86(1-2):61-69.
Dombeck T.A., Satonik R.C. The porphyrias. Emerg Med Clin North Am. 2005;23:885-899.
Eichler F., Barker P.B., Cox C., et al. Proton MR spectroscopic imaging predicts lesion progression on MRI in X-linked adrenoleukodystrophy. Neurology. 2002;58:901-907.
Eklund E.A., Freeze H.H. The congenital disorders of glycosylation: a multifaceted group of syndromes. Neuro Rx. 2006;3:254-263.
Enns G.M., Packman W. The adolescent with an inborn error of metabolism: medical issues and transition to adulthood. Adolesc Med. 2002;13:315-330.
Faerber E.N., Poussaint T.Y. Magnetic resonance of metabolic and degenerative diseases in children. Top Magn Reson Imaging. 2002;13:3-21.
Fidaleo M. Peroxisomes and peroxisomal disorders: The main facts. Exp Toxicol Pathol. 2009;62:615-625.
Fukushima K., Yazaki M., Nakamura M., et al. Conventional diet therapy for hyperammonemia is risky in the treatment of hepatic encephalopathy associated with citrin deficiency. Intern Med. 2010;49(3):243-247.
Garver W.S., Heidenreich R.A. The Niemann-Pick C proteins and trafficking of cholesterol through the late endosomal/lysosomal system. Curr Mol Med. 2002;2:485-505.
Gopaul K.P., Crook M.A. The inborn errors of sialic acid metabolism and their laboratory investigation. Clin Lab. 2006;52:155-169.
Gray R.G., Preece M.A., Green S.H., et al. Inborn errors of metabolism as a cause of neurological disease in adults: an approach to investigation. J Neurol Neurosurg Psychiatry. 2000;69:5-12.
Gregersen N., Bross P., Andresen B.S. Genetic defects in fatty acid beta-oxidation and acyl-CoA dehydrogenases. Molecular pathogenesis and genotype-phenotype relationships. Eur J Biochem. 2004;271:470-482.
Gregersen N., Bross P., Andresen B.S., et al. The role of chaperone-assisted folding and quality control in inborn errors of metabolism: protein folding disorders. J Inherit Metab Dis. 2001;24:189-212.
Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21:563-571.
Grompe M. Transition of stem cells to therapeutically functional tissue-specific cells. Ann N Y Acad Sci. 2002;961:305-306.
Haas D., Kelley R.I., Hoffmann G.F. Inherited disorders of cholesterol biosynthesis. Neuropediatrics. 2001;32:113-122.
Hennekam R.C. Congenital brain anomalies in distal cholesterol biosynthesis defects. J Inherit Metab Dis. 2005;28:385-392.
Henneke M., Dreha-Kulaczewski S., Brockmann K., et al. In vivo proton MR spectroscopy findings specific for adenylosuccinate lyase deficiency. NMR Biomed. 2010;23:441-445.
Homanics G.E., Skvorak K., Ferguson C., et al. Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med Genet. 2006;31:33-37.
Hsich G., Sena-Esteves M., Breakefield X.O. Critical issues in gene therapy for neurological disease. Hum Gene Ther. 2002;13:579-604.
Hyland K. The lumbar puncture for diagnosis of pediatric neurotransmitter diseases. Ann Neurol. 2003;54(Suppl. 6):S13-S17.
Inderbitzin D., Avital I., Largiader F., et al. Kidney transplantation improves survival and is indicated in Fabry’s disease. Transplant Proc. 2005;37:4211-4214.
Jeng L.B., Tarvin R., Robin N.H. Genetic advances in central nervous system malformations in the fetus and neonate. Semin Pediatr Neurol. 2001;8:89-99.
Johnson J.L., Coyne K.E., Garrett R.M., et al. Isolated sulfite oxidase deficiency: identification of 12 novel SUOX mutations in 10 patients. Hum Mutat. 2002;20:74.
Jones C.M., Smith M., Henderson M.J. Reference data for cerebrospinal fluid and the utility of amino acid measurement for the diagnosis of inborn errors of metabolism. Ann Clin Biochem. 2006;43:63-66.
Joshi C., Greenberg C.R., De Vivo D., et al. GLUT1 deficiency without epilepsy: yet another case. J Child Neurol. 2008;23(7):832-834.
Jurecka A. Inborn errors of purine and pyrimidine metabolism. J Inherit Metab Dis. 2009;32(2):247-263.
Kawakami Y., Yoshida K., Yang J.H., et al. Impaired neurogenesis in embryonic spinal cord of Phgdh knockout mice, a serine deficiency disorder model. Neurosci Res. 2009;63(3):184-193.
Kawashita Y., Guha C., Yamanouchi K., et al. Liver repopulation: a new concept of hepatocyte transplantation. Surg Today. 2005;35:705-710.
Kaye E.M. Update on genetic disorders affecting white matter. Pediatr Neurol. 2001;24:11-24.
Kelly A., Stanley C.A. Neurological aspects in hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol. 2008;50(12):888.
Kimura A., Kage M., Nagata I., et al. Histological findings in the livers of patients with neonatal intrahepatic cholestasis caused by citrin deficiency. Hepatol Res. 2010;40:295-303.
Kitzberger R., Madl C., Ferenci P. Wilson disease. Metab Brain Dis. 2005;20:295-302.
Koeberl D.D., Pinto C., Brown T., et al. Gene therapy for inherited metabolic disorders in companion animals. ILAR J. 2009;50(2):122-127.
Korman S.H., Kanazawa N., Abu-Libdeh B., et al. Hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome with evidence of mitochondrial dysfunction due to a novel SLC25A15 (ORNT1) gene mutation in a Palestinian family. J Neurol Sci. 2004;218:53-58.
Kouremenos K.A., Pitt J., Marriott P.J. Metabolic profiling of infant urine using comprehensive two-dimensional gas chromatography: Application to the diagnosis of organic acidurias and biomarker discovery. J Chromatogr A. 2009;1217:104-111.
Lambruschini N., Perez-Duenas B., Vilaseca M.A., et al. Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab. 2005;86:S54-S60.
Lass A., Zimmermann R., Haemmerle G., et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309-319.
Leonard J.V., McKiernan P.J. The role of liver transplantation in urea cycle disorders. Mol Genet Metab. 2004;81:S74-S78.
Macaya A., Brunso L., Fernandez-Castillo N., et al. Molybdenum cofactor deficiency presenting as neonatal hyperekplexia: a clinical, biochemical and genetic study. Neuropediatrics. 2005;36:389-394.
Madonna P., de Stefano V., Coppola A., et al. Hyperhomocysteinemia and other inherited prothrombotic conditions in young adults with a history of ischemic stroke. Stroke. 2002;33:51-56.
Maire I. Is genotype determination useful in predicting the clinical phenotype in lysosomal storage diseases? J Inherit Metab Dis. 2001;24(Suppl 2):57-61.
Matalon R., Matalon K.M. Canavan disease prenatal diagnosis and genetic counseling. Obstet Gynecol Clin North Am. 2002;29:297-304.
Matern D. Newborn screening for lysosomal storage disorders. Acta Paediatr. 2008;457(Suppl 97):33-37.
Mayatepek E., Okun J.G., Meissner T., et al. Synthesis and metabolism of leukotrienes in gamma-glutamyl transpeptidase deficiency. J Lipid Res. 2004;45:900-904.
Mayatepek E., Badiou S., Bellet H., et al. A patient with neurological symptoms and abnormal leukotriene metabolism: a new defect in leukotriene biosynthesis. Ann Neurol. 2005;58(6):s968-s970.
Moghadasian M.H. Cerebrotendinous xanthomatosis: clinical course, genotypes and metabolic backgrounds. Clin Invest Med. 2004;27:42-50.
Moser H.W. Therapy of x-linked adrenoleukodystrophy. NeuroRx. 2006;3:246-253.
Moser H.W., Raymond G.V., Lu S.E., et al. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch Neurol. 2005;62:1073-1080.
Naini A., Toscano A., Musumeci O., et al. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol. 2009;66(3):394-398.
Nassogne M.C., Heron B., Touati G., et al. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28:407-414.
Neumann J., Bras J., Deas E., et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132:1783-1794.
Nissenkorn A., Michelson M., Ben-Zeev B., et al. Inborn errors of metabolism: a cause of abnormal brain development. Neurology. 2001;56:1265-1272.
Novotny E.J.Jr, Fulbright R.K., Pearl P.L., et al. Magnetic resonance spectroscopy of neurotransmitters in human brain. Ann Neurol. 2003;54(Suppl. 6):S25-S31.
Nyhan W.L. Disorders of purine and pyrimidine metabolism. Mol Genet Metab. 2005;86:25-33.
Ogier de Baulny H., Saudubray J.M. Branched-chain organic acidurias. Semin Neonatol. 2002;7:65-74.
Oglesbee D. An overview of peroxisomal biogenesis disorders. Mol Genet Metab. 2005;84:299-301.
Oguz K.K., Ozturk A., Cila A. Diffusion-weighted MR imaging and MR spectroscopy in glutaric aciduria type 1. Neuroradiology. 2005;47(3):229-234.
Olpin S.E. The metabolic investigation of sudden infant death. Ann Clin Biochem. 2004;41:282-293.
Olpin S.E. Fatty acid oxidation defects as a cause of neuromyopathic disease in infants and adults. Clin Lab. 2005;51:289-306.
Parente F., Mew N.A., Jaeken J., et al. A new capillary zone electrophoresis method for the screening of congenital disorders of glycosylation (CDG). Clin Chim Acta. 2010;411:64-66.
Pascual J.M., Wang D., Lecumberri B., et al. GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol. 2004;150:627-633.
Pasquali M., Monsen G., Richardson L., et al. Biochemical findings in common inborn errors of metabolism. Am J Med Genet C Semin Med Genet. 2006;142:64-76.
Pastores G.M., Barnett N.L. Current and emerging therapies for the lysosomal storage disorders. Expert Opin Emerg Drugs. 2005;10:891-902.
Pastores G.M., Sathe S. A chaperone-mediated approach to enzyme enhancement as a therapeutic option for the lysosomal storage disorders. Drugs R D. 2006;7(6):339-348.
Patterson M.C., Vecchio D., Prady H., et al. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6(9):765-772.
Pearl P.L., Gibson K.M., Cortez M.A., et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32(3):343-352.
Pearl P.L., Krasnewich D. Neurological course of congenital disorders of glycosylation. J Child Neurol. 2001;16:409-413.
Pearl P.L., Taylor J.L., Trzcinski S., et al. The pediatric neurotransmitter disorders. J Child Neurol. 2007;22(5):606-616.
Pearl P.L., Wallis D.D., Gibson K.M. Pediatric neurotransmitter diseases. Curr Neurol Neurosci Rep. 2004;4:147-152.
Peters C., Charnas L.R., Tan Y., et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881-888.
Peters C., Steward C.G., et al. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines, National Marrow Donor Program. Bone Marrow Transplant. 2003;31:229-239.
Pichit P., Quillard M., Couvert P., et al. Tangier disease phenotype diversity in dizygous twin sisters. Rev Neurol (Paris). 2010.
Pischik E., Kauppinen R. Neurological manifestations of acute intermittent porphyria. Cell Mol Biol. 2009;16(55):72-83.
Pithukpakorn M. Disorders of pyruvate metabolism and the tricarboxylic acid cycle. Mol Genet Metab. 2005;85:243-246.
Pollard P.J., Wortham N.C., Tomlinson I.P. The TCA cycle and tumorigenesis: the examples of fumarate hydratase and succinate dehydrogenase. Ann Med. 2003;35(8):632-639.
Porter J.A., Young K.E., Beachy P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255-259.
Prasad A.N., Malinger G., Lerman-Sagie T. Primary disorders of metabolism and disturbed fetal brain development. Clin Perinatol. 2009;36(3):621-638.
Preece M.A., Green A. Pregnancy and inherited metabolic disorders: maternal and fetal complications. Ann Clin Biochem. 2002;39:444-455.
Raben N., Danon M., Lu N., et al. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739-1745.
Rampoldi L., Danek A., Monaco A.P. Clinical features and molecular bases of neuroacanthocytosis. J Mol Med. 2002;80:475-491.
Read C.Y. Reproductive decisions of parents of children with metabolic disorders. Clin Genet. 2002;61:268-276.
Reczek D., Schwake M., Schröder J., et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131(4):770-783.
Rush J.S., Panneerselvam K., Waechter C.J., et al. Mannose supplementation corrects GDP-mannose deficiency in cultured fibroblasts from some patients with congenital disorders of glycosylation (CDG). Glycobiology. 2000;10:829-835.
Salerno C., Crifo C. Biochemical and molecular genetic correlation in adenylosuccinate lyase deficiency. Nucleosides Nucleotides Nucleic Acids. 2004;23:1253-1255.
Samenuk P., Koffman B.M. Chenodeoxycholic treatment of cerebrotendinous xanthomatosis. Neurology. 2001;56:695-696.
Sands M.S., Davidson B.L. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839-849.
Sass J.O., Gunduz A., Araujo Rodrigues Funayama C., et al. Functional deficiencies of sulfite oxidase: Differential diagnoses in neonates presenting with intractable seizures and cystic encephalomalacia. Brain Dev. 2009;32:544-549.
Scarlato G., Comi G.P. Metabolic and drug-induced muscle disorders. Curr Opin Neurol. 2002;15:533-538.
Schiffmann R., Fitzgibbon E.J., Harris C., et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol. 2008;64(5):514-522.
Schneider J.F., Boltshauser E., Neuhaus T.J., et al. MRI and proton spectroscopy in Lowe syndrome. Neuropediatrics. 2001;32:45-48.
Schretlen D.J., Harris J.C., Park K.S., et al. Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc. 2001;7:805-812.
Schroers A., Kley R.A., Stachon A., et al. Gentamicin treatment in McArdle disease: failure to correct myophosphorylase deficiency. Neurology. 2006;66:285-286.
Schweiger M., Lass A., Zimmermann R., et al. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am J Physiol Endocrinol Metab. 2009;297:E289-E296.
Selimoglu M.A., Esrefoglu M., Gul M., et al. Chanarin-Dorfman syndrome: clinical features of a rare lipid metabolism disorder. Pediatr Dermatol. 2009;26:40-43.
Shapiro B.E., Pastores G.M., Gianutsos J., et al. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med. 2009;11(6):425-433.
Sim K.G., Hammond J., Wilcken B. Strategies for the diagnosis of mitochondrial fatty acid beta-oxidation disorders. Clin Chim Acta. 2002;323:37-58.
Simon P., Weiss F.U., Zimmer K.P., et al. Acute and chronic pancreatitis in patients with inborn errors of metabolism. Pancreatology. 2001;1:448-456.
Singh R.H., Rhead W.J., Smith W., et al. Nutritional management of urea cycle disorders. Crit Care Clin. 2005;21(Suppl 4):S27-S35.
Smith V.V., Anderson G., Malone M., et al. Light microscopic examination of scalp hair samples as an aid in the diagnosis of paediatric disorders: retrospective review of more than 300 cases from a single centre. J Clin Pathol. 2005;58:1294-1298.
Steenweg M.E., Salomons G.S., Yapici Z., et al. L-2-Hydroxyglutaric aciduria: pattern of MR imaging abnormalities in 56 patients. Radiology. 2009;251(3):856-865.
Steiner R.D., Cederbaum S.D. Laboratory evaluation of urea cycle disorders. J Pediatr. 2001;138(Suppl 1):S21-S29.
Stockler S., Schutz P.W., Salomons G.S. Cerebral creatine deficiency syndromes: clinical aspects, treatment and pathophysiology. Subcell Biochem. 2007;46:149-166.
Stromberger C., Bodamer O.A., Stockler-Ipsiroglu S. Clinical characteristics and diagnostic clues in inborn errors of creatine metabolism. J Inherit Metab Dis. 2003;26:299-308.
Sugie K., Yamamoto A., Murayama K., et al. Clinicopathological features of genetically confirmed Danon disease. Neurology. 2002;58:1773-1778.
Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138(Suppl 1):S30-S39.
Tanner L., Nanto-Salonen K., Niinikoski H., et al. Hazards associated with pregnancies and deliveries in lysinuric protein intolerance. Metabolism. 2006;55:224-231.
Tomi D., Schultze-Mosgau A., Eckhold J., et al. First pregnancy and life after preimplantation genetic diagnosis by polar body analysis for mucopolysaccharidosis type I. Reprod Biomed Online. 2006;12(2):215-220.
Toone J.R., Applegarth D.A., Kure S., et al. Novel mutations in the P-protein (glycine decarboxylase) gene in patients with glycine encephalopathy (non-ketotic hyperglycinemia). Mol Genet Metab. 2002;76:243-249.
Topaz O., Shurman D.L., Bergman R., et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, caused familial tumoral calcinosis. Nat Genet. 2004;36:579-581.
van Kuilenburg A.B., van Lenthe H., Loffler M., et al. Analysis of pyrimidine synthesis “de novo” intermediates in urine and dried urine filter- paper strips with HPLC-electrospray tandem mass spectrometry. Clin Chem. 2004;50:2117-2124.
Ventura P., Cappellini M.D., Rocchi E. The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med. 2009;4(4):297-308.
Vockley J., Whiteman D.A. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12:235-246.
Wang D., Pascual J.M., Yang H., et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005;57(1):111-118.
Warren C.D., Alroy J. Morphological, biochemical and molecular biology approaches for the diagnosis of lysosomal storage diseases. J Vet Diagn Invest. 2000;12:483-496.
Wilcken B. An introduction to nutritional treatment in inborn errors of metabolism—different disorders, different approaches. Southeast Asian J Trop Med Public Health. 2003;34(Suppl 3):198-201.
Wolf B. Children with profound biotinidase deficiency should be treated with biotin regardless of their residual enzyme activity or genotype. Eur J Pediatr. 2002;161:167-168.
Yu H., Patel S.B. Recent insights into the Smith-Lemli-Opitz syndrome. Clin Genet. 2005;68:383-391.