Chapter 76 Inborn Errors of Metabolism
Metabolism can be defined as the sum of all biochemical processes that convert food to smaller molecules and energy for the purposes of structure and function. An inborn error of metabolism (IEM) is an inherited deficiency of any critical step in metabolism. Although genetic deficiency of catalytic enzymes in intermediary metabolic pathways is the classic paradigm for IEM, the pathophysiology of metabolic disorders may involve abnormalities of any number of cellular processes, including transmembrane transport, cell signaling, cell differentiation and development, energy production, and others. Many IEMs are individually rare, although a few, including phenylketonuria (PKU) and medium-chain acyl-coenzyme A (acyl-CoA) dehydrogenase deficiency (MCADD), a defect in fatty acid oxidation, exhibit a population incidence approaching 1:10,000 live births.1,2 Specific IEMs may be more common in certain ethnic groups with a history of relative reproductive isolation. Collectively, the population incidence of all IEMs may approach 1:1500 live births, depending upon how broadly IEM is defined. Many IEMs are associated with catastrophic illness necessitating advanced life support. Although IEMs may present very rarely within the professional lifetime of the average medical practitioner, critically ill children with IEMs will not be uncommon visitors to the pediatric intensive care unit (ICU), especially in a tertiary care center.
Other published textbooks on the diagnosis and treatment of IEM provide an exhaustive list of known disorders.3,4 Rather than recapitulate an encyclopedia of possible diseases, this chapter presents a diagnostic rationale based upon specific clinical symptom complexes that are likely to occur in the critically ill child. Algorithms for the differential diagnosis of specific clinical scenarios are given in support of this rationale. Symptoms often begin during early infancy in the biochemically most-severe IEMs; naturally, these IEMs with neonatal onset are the focus of our discussion in this chapter. However, “milder” or late-onset variants of virtually every IEM have been described, with onset of symptoms occurring at all ages, even during adulthood. Some IEMs uniformly present after the neonatal period; age of symptom onset (late infancy, childhood, or adulthood) often is an important clue to the specific diagnosis. The clinical presentation, diagnostic workup, and treatment of neonatal onset disorders provide a paradigm for the evaluation and management of possible IEM in a child of any age.
Pathophysiology of Inborn Errors of Metabolism
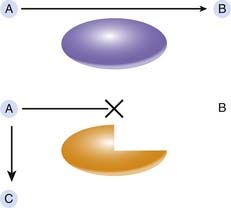
Under the classic paradigm, an IEM is associated with deficiency of a specific protein, often a catalytic enzyme, involved in a critical metabolic pathway (Figure 76-1). This deficiency leads to a block in the pathway and the accumulation of the enzyme substrate. In this model, three distinct pathogenic mechanisms are possible proximate causes of the symptoms associated with an IEM. The specific pathogenic mechanism involved in any given IEM dictates the appropriate treatment strategy. First, accumulation of the substrate may lead to toxic effects at very high levels; successful therapy requires effective elimination of the substrate or a method to block its toxic effects. An appropriate example for this mechanism is PKU, in which elevated phenylalanine levels adversely affect neuronal development, and the reduction of tissue phenylalanine content through dietary phenylalanine restriction largely prevents the major clinical features of PKU.5 Second, deficiency of the reaction product, should it be a critically important metabolite, may lead to disease. Supplementation with the essential metabolite, if possible, may cure the disease. Biotin is a required cofactor for four distinct carboxylase enzymes. Deficiency of free biotin develops in the face of genetic biotinidase deficiency and leads to symptoms of multiple carboxylase deficiency. Supplementation with oral biotin completely prevents the clinical manifestations of biotinidase deficiency.6 The final pathogenic mechanism involves the conversion of the enzyme substrate, through normally quiescent alternative pathways, to toxic secondary metabolites. Elimination or decreased production of these secondary metabolites may improve disease symptoms. For example, tyrosinemia type I (fumarylacetoacetate hydrolase [FAH] deficiency) is associated with recurrent attacks of abdominal pain and paresthesias reminiscent of acute intermittent porphyria. The accumulating substrate, fumarylacetoacetic acid, is converted through secondary pathways to succinylacetone, and succinylacetone in turn inhibits the heme synthetic pathway and causes porphyria-like symptoms. Pharmacologic inhibition of the tyrosine catabolic pathway proximal to the block at FAH decreases the production of fumarylacetoacetic acid and succinylacetone and alleviates the pathology associated with these toxic compounds.7
Signs and Symptoms of Inborn Errors of Metabolism
Clinical signs and symptoms frequently associated with IEMs are listed in Box 76-1. The symptom repertoire of the critically ill infant is limited, and the clinical presentation of metabolic disorders often is nonspecific. It is for this reason that the diagnosis of an IEM may be easily missed. To maintain maximum diagnostic sensitivity for IEMs, the clinician must maintain a high level of suspicion and be willing to initiate screening metabolic laboratory studies with little provocation. As was true for appendectomies in the era prior to the advent of ultrasound-based diagnosis of appendicitis, a certain number of nondiagnostic metabolic laboratory workups in sick children must be performed to ensure ascertainment of individuals with inherited metabolic disorders. In particular, IEM should be a strong diagnostic consideration in any neonate who has become catastrophically ill following a period of normalcy. This presentation may be clinically indistinguishable from bacterial or viral sepsis, and the nonspecific supportive therapy provided to potentially septic infants (fluid and glucose administration) may alleviate the symptoms and mask the presence of an IEM. Diagnostic metabolic laboratory studies are most likely to provide definitive information if performed on clinical samples obtained at initial presentation and before any therapy is initiated. Failure to obtain the necessary specimens at this time may miss an important diagnostic window of opportunity. Many children with IEM have been saved initially by intensive but nonspecific treatment but then suffered clinical relapse or even death in the absence of the correct diagnosis. Certainly, the possibility of an IEM should be considered in any child for whom the clinical picture suggests sepsis but the laboratory evaluation for sepsis is negative. Unfortunately, bacterial sepsis is often a complicating factor in critically ill children with IEM. For example, Escherichia coli infection (including pyelonephritis, bacteremia, or meningitis) is frequently detected at presentation in infants with galactosemia. The astute clinician remains ever vigilant for the signs and symptoms that may suggest an inherited metabolic disorder.
BOX 76–1 Signs And Symptoms of Inborn Errors of Metabolism
Acute illness after period of normal behavior and feeding (hours to weeks)
Recurrent decompensation with fasting, intercurrent illness, or specific food ingestion
Persistent or recurrent vomiting
Hepatomegaly or liver dysfunction
Unexplained hemorrhage or strokes
Developmental delay with unknown etiology
Seizures, especially if seizures are intractable
Chronic movement disorder (ataxia, dystonia, choreoathetosis)
Family history of unexplained death or recurrent illness in siblings
Recurrent episodes of vomiting and dehydration in response to fasting or intercurrent illness are an important clue to IEM in older infants and children. Feeding difficulties and failure to thrive are common chronic complications. Children with unexplained hypotonia, developmental delay, or movement disorder should be evaluated for possible IEM. Inherited neurodegenerative disorders, such as the lysosomal storage diseases, stereotypically cause developmental regression, specifically loss of previously attained developmental milestones. Several IEMs are associated with major physical anomalies (Table 76-1). When present, these anomalies are exceedingly valuable in suggesting a specific diagnosis and directing the diagnostic evaluation. More commonly, the child with IEM is morphologically normal, and the presenting symptoms are nonspecific. The clinician must then rely upon screening laboratory tests to evaluate the potential for IEM.
Table 76–1 Physical Anomalies Associated with Inborn Errors of Metabolism
| Dysmorphic facial features |
Laboratory Evaluation of Suspected Inborn Errors of Metabolism
Abnormal results of routine laboratory studies may provide clues to the presence and type of IEM (Table 76-2). Highly informative but sometimes subtle laboratory abnormalities are often overlooked, especially in a busy ICU or hospital ward. For instance, a clinically relevant newborn screening result may have been sent to the primary care provider or birth hospital but not efficiently communicated to the ICU, in a different hospital, to which the now critically ill infant has been admitted. It is imperative to verify the infant’s screening results with the primary care provider or newborn screening laboratory (Box 76-2). Calculation of the anion gap, another example of a routine and highly informative result, is key to the differential diagnosis of metabolic acidosis. The absence of urine ketones in hypoglycemic children older than 2 weeks strongly suggests impaired ketogenesis as a consequence of either hyperinsulinism or fatty acid oxidation disorder. On the other hand, fatty acid oxidation and ketogenesis are incompletely developed in neonates. The presence of ketones in the urine of infants younger than 2 weeks is very unusual even during fasting or hypoglycemia and suggests the presence of an unusual keto acid, such as those excreted in maple syrup disease or the organic acidemias. Keto acids, organic acids, and sugars such as galactose or fructose increase urine specific gravity. Urine specific gravity greater than 1.020 in any neonate or in a well-hydrated older child suggests the unexpected presence of an osmotically active substance. Routine urinalysis at many hospitals may not include use of the Clinitest to detect reducing substances. Urine Chemstrips utilize a colorimetric glucose oxidase-based method to specifically detect glucose. This test does not react with any other sugar (galactose or fructose). However, some bedside glucose monitoring systems do react with galactose or fructose; inappropriately elevated capillary blood “glucose” accompanied by a normal venous glucose as measured by chemistry analyzer suggests the presence of a sugar other than glucose in the blood. A comatose infant with a blood urea nitrogen (BUN) level below the limits of detection may have an inherited defect in the urea cycle. Blood ammonia measurement is crucial to confirming that suspicion. Failure to check the blood ammonia level has caused missed diagnoses, failure to appropriately treat hyperammonemia, and further morbidity and mortality in comatose infants with urea cycle disorders or organic acidemias. Finally, bacterial sepsis and meningitis are more common causes of severe lethargy and coma in infants than is IEM, but bacterial infection may also be a complicating feature in severely ill infants with IEM. Infants with galactosemia, for example, are particularly prone to pyelonephritis, bacteremia, sepsis, or meningitis, often with E. coli, as noted above. Antibiotic therapy without diagnosis and specific treatment of the underlying disorder may be useful in the short term but does not mitigate long-term IEM-specific effects.
Table 76–2 Initial Laboratory Evaluation of Suspected Inborn Errors of Metabolism
| Laboratory Test | Abnormality | Disorder |
|---|---|---|
| Complete blood count |
BUN, Blood urea nitrogen; FAO, fatty acid oxidation; MSUD, maple syrup urine disease.
Box 76–2 Screening Metabolic Laboratory Studies for Children with Suspected Inborn Errors of Metabolism
Suspicion of an IEM based upon clinical and routine laboratory findings should initiate specialized biochemical testing (Table 76–3). In the case of severely ill infants or when the clinical suspicion of IEM is very high, consultation with a biochemical geneticist, even if only by phone, is strongly advised to help direct the laboratory investigation and initial therapy. When the clinical presentation is nonspecific, that is, catastrophic illness in a previously well child without signs of any particular IEM, the “shotgun” diagnostic evaluation should minimally include plasma amino acid analysis, urine organic acid analysis by gas chromatography-mass spectrometry, and a so-called urine metabolic screen. The battery of qualitative assays included in a urine metabolic screen differs among laboratories, and the ordering clinician should be aware of which tests and disorders are included in the repertoire of the diagnostic laboratory chosen. Furthermore, although diagnostic laboratories in the United States must meet Clinical Laboratory Improvement Amendments requirements and often are accredited by the College of American Pathologists, the testing methodologies used, the quality of diagnostic testing for IEM, and more problematically, the availability of laboratory-associated consultants with experience in the diagnosis and treatment of IEM vary widely among laboratories. Although the ability of clinicians to direct clinical specimens toward specific diagnostic laboratories may be inhibited by contractual arrangements between the hospital and large referral laboratories, the critically ill patient is best served by diagnostic evaluation carried out in a timely manner by an experienced biochemical genetics laboratory, with laboratory staff available by phone for expert consultation on interpretation of test results.
Table 76–3 Biochemical Genetic Laboratory Studies
| Specimen | Test | Disorder |
|---|---|---|
| Blood | Plasma amino acid analysis | Aminoacidopathies |
| Plasma carnitine | ||
| Plasma acylcarnitine profile | ||
| Serum transferrin electrophoresis | Congenital disorders of glycosylation | |
| Urine | ||
| Organic acid analysis | ||
| Acylglycine profile | ||
| Quantitative mucopolysaccharide measurement and electrophoresis | Mucopolysaccharidoses | |
| Qualitative sulfites (Sulfitest) or quantitative sulfocysteine | ||
| Quantitative succinylacetone | Tyrosinemia type 1 | |
| Quantitative purines | Purine synthesis disorders |
PKU, Phenylketonuria; FAO, fatty acid oxygenation; MSUD, maple syrup urine disease.
The specific clinical presentation or specific screening laboratory findings may direct the intensivist or biochemical geneticist to order other more specialized metabolic tests (see Table 76-3). These analyses may provide diagnostic confirmation for specific disorders and supportive evidence alone for others. For several IEMs, confirmation of diagnosis may require enzyme activity analysis in tissue (red blood cells, lymphocytes, cultured skin fibroblasts, liver, or skeletal muscle depending upon the specific disorder in question) or molecular DNA testing for a specific gene defect. In general, these tertiary tests, which are often difficult, labor-intensive, and expensive, should be ordered following consultation with a biochemical geneticist. In some instances, confirmatory diagnostic biochemical or molecular tests are available only through specialized research laboratories.
Postmortem Evaluation of a Child with Suspected Inborn Errors of Metabolism
Some IEMs, particularly those exacerbated by fasting, may present as sudden infant death. For many IEMs, acute metabolic compensation may be rapid and lethal despite intensive medical intervention. The time after clinical presentation but prior to death may be insufficient to execute an adequate metabolic evaluation. Disease diagnosis is still possible postmortem and is important for fully understanding the cause of death and determining recurrence risk in the family. A protocol for postmortem evaluation of an infant or child with suspected IEM is given in Box 76-3. Many of the biochemical genetic analyses recommended for acutely ill children are still valid on postmortem specimens. Valuable information may be learned from amino acid, carnitine, and acylcarnitine analyses in blood and from metabolic screening and organic acid analysis in urine. However, collection of blood and urine may not be possible postmortem, especially if the autopsy is performed many hours after death. In these instances, metabolic testing may be obtained on alternative specimens such as vitreous humor or bile. In the event that screening biochemical studies suggest a specific diagnosis, disease confirmation by enzyme analysis in tissue is highly desirable. Many enzymes can be assayed in cultured fibroblasts; viable fibroblasts may be cultured from skin or Achilles tendon samples obtained as late as 24 hours after death. Biopsies of other organs may be necessary for analysis of certain other enzymes. Muscle, liver, and kidney specimens may be obtained postmortem for enzymatic analysis, but most enzymatic activities in solid organs deteriorate rapidly following death. Collection of specimens as soon as possible after death is critical for valid enzyme analyses.
Box 76–3 Postmortem Biochemical Genetic Evaluation
Modified from Steiner RD, Cederbaum SD: Laboratory evaluation of urea cycle disorders. J Pediatr 138 (Suppl 1):S21-29, 2001.
Analyses are most reliable if obtained within 6 hours after death.
Emergency Treatment of Children with Suspected Inborn Errors of Metabolism
Laboratory investigation of suspected IEM may require several days to complete, given that the biochemical genetics laboratory may be physically remote from the treating hospital and many of the tests involve complex specimen preparation and analysis. A general approach to the emergency treatment of children with suspected IEM, while awaiting diagnostic studies, is given in Table 76-4. For many IEMs associated with acute catastrophic illness, elimination of the offending metabolite is the key to therapy. Immediate cessation of oral feedings, to stop protein or fat intake, will begin to limit toxin production in disorders of amino acid or fatty acid metabolism. Adequate energy intake as carbohydrate must be supplied, usually parenterally, until a specific diagnosis and definitive treatment plan are available. Dextrose infusion at a high rate suppresses catabolism and reduces the consumption of endogenous protein or fatty acid stores. In extremely recalcitrant cases, insulin infusion drives anabolism and further decreases toxin production. Acute metabolic decompensation in some IEMs (e.g., maple syrup disease) is associated with mild peripheral insulin resistance. Insulin administration (often as little as 0.01 to 0.05 units/kg/hour given by continuous intravenous [IV] infusion or subcutaneous bolus injection) overcomes this resistance and has an immediate impact upon metabolic control. Some clinicians also use anabolic agents such as growth hormone or testosterone to acutely suppress protein and fat catabolism. In certain types of congenital lactic acidosis, particularly defects of pyruvate metabolism, carbohydrate infusion worsens lactic acidosis. Replacement of some carbohydrate with fat as an intralipid infusion may partly reduce blood lactate levels, but infants with this degree of sensitivity to glucose infusion often are difficult to treat and suffer high mortality. Severe hyperammonemia that does not respond to dietary protein restriction and dextrose infusion must be treated by hemodialysis. Ammonia clearance with exchange transfusion or peritoneal dialysis is insufficient to adequately decrease blood ammonia levels. If the results of specialized biochemical genetic diagnostic tests are expected within 2 to 3 days, then parenteral dextrose infusion alone should be adequate to maintain nutrition until a more definitive treatment plan is available. Beyond 3 days, developing essential amino acid and fatty acid deficiencies may induce catabolism of endogenous protein and fat. To prevent this occurrence, enteral or parenteral nutrition with minimal amounts of protein (0.5 g/kg body weight/day) and lipid (20% of total energy intake) should be considered. Empiric administration of cofactors such as the B vitamins is not harmful and may improve metabolite clearance, particularly in disorders caused by deficiency of enzymes that require specific cofactors. Carnitine is required for transport of long-chain fatty acids across the mitochondrial membrane and serves a secondary role in the disposal of excess and potentially toxic acyl-CoA species. Secondary carnitine deficiency is commonly associated with acute metabolic decompensation in organic acidemias and fatty acid oxidation defects. L-Carnitine administration prevents secondary carnitine deficiency and may improve clearance of toxic metabolites; it is lifesaving in specific inherited dilated cardiomyopathies.
Table 76–4 Emergency Treatment of Suspected Inborn Error of Metabolism
| Goal | Action |
|---|---|
| Suppress toxic metabolite production | Discontinue oral feedings |
| Correct fluid imbalance and electrolyte abnormalities | Appropriate IV fluid management |
| Correct hypoglycemia | IV dextrose-containing fluid infusion |
| Correct metabolic acidosis |
Classification of Inborn Errors of Metabolism by Clinical Presentation
As mentioned previously, the clinical presentation of IEM in neonates provides a paradigm for the suspicion and evaluation of potential IEM at all ages. The classification outlined here is adapted and expanded to include late-onset disorders from a neonatal IEM classification system first described by Jean-Marie Saudubray and colleagues.4,8
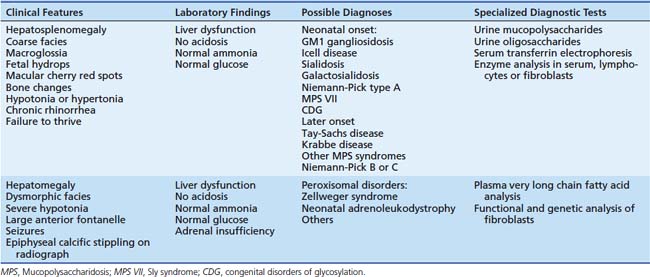
IEMs can be classified into one of three groups by pathogenic mechanism. In group 1 IEMs, the production or catabolism of complex molecules is disturbed. The lysosomal storage and peroxisomal disorders are included in this group. The symptoms of these disorders include permanent and progressive somatic and neurologic abnormalities that develop in utero, are often clinically apparent at birth, and are unaffected by food intake. This group is often distinguished by the presence of somatic abnormalities such as dysmorphic features or hepatosplenomegaly. Typical clinical features, potential neonatal and late-onset diagnoses, and confirmatory diagnostic tests are listed in Table 76-5.
Group 2 Inborn Errors of Metabolism
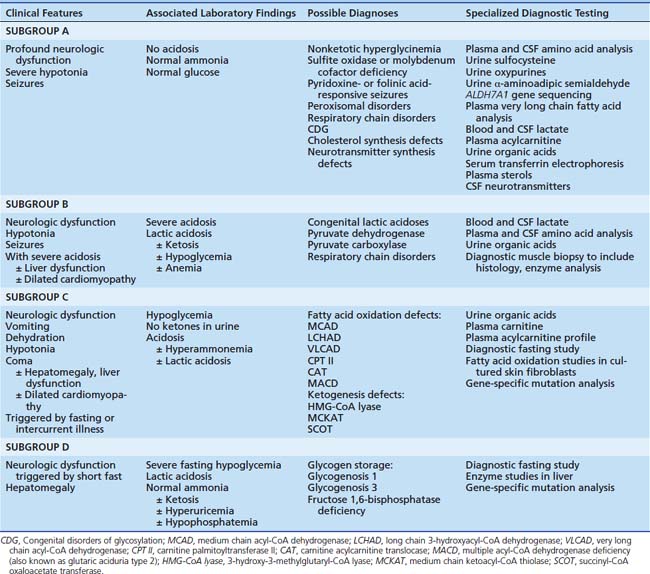
Systemic or tissue-specific impaired energy production from food substrates is the unifying feature of disorders classified in group 2. Generalized profound neurologic dysfunction, including severe central hypotonia, coma, and seizures, sometimes with peripheral spasticity or abnormal movements, typifies the clinical presentation. Children with these disorders present with similar clinical phenotypes but are easily separated into four subgroups (A through D) based upon associated results of routine laboratory studies (Table 76-6). Severe refractory generalized motor seizures, often beginning within the first hours after birth, sometimes even prenatally, are the hallmark of subgroup A. Routine laboratory studies (glucose, blood pH, electrolytes, ammonia) are generally normal unless the infant is near extremis, and secondary metabolic abnormalities are present. Several inherited disorders are associated with this phenotype; diagnostic differentiation depends upon clinical evaluation by an experienced pediatric neurologist or geneticist and the judicious use of specialized diagnostic laboratory tests.
The amino acid glycine is an abundant neurotransmitter within the central nervous system (CNS). Inherited deficiency of the glycine cleavage system, which removes glycine from its receptor in the neuronal synapse, causes severe unrelenting generalized seizures and profound developmental arrest. The only ubiquitous laboratory finding is an elevated cerebrospinal fluid (CSF/plasma glycine ratio).9 Sulfite oxidase deficiency, either as a primary genetic defect or secondary to generalized deficiency of its molybdenum-containing cofactor, is another rare but important cause of neonatal-onset seizures. Recently, infantile-onset pyridoxine-dependent or folinic acid-dependent seizure disorders have both been found to be caused by recessively inherited deficiency of α-aminoadipic semialdehyde (α-AASA) dehydrogenase, an intermediate enzyme in the metabolism of the amino acid lysine.10 Consequently, all neonates with refractory seizures should be screened for these treatable disorders either through measurement of α-AASA in urine or sequencing of the ALDH7A1 (antiquitin) gene. Profound neurologic dysfunction with seizures is one of many possible clinical presentations of infants with peroxisomal or respiratory chain disorders. Some subtypes of a still expanding list of congenital disorders of glycosylation present with seizures,11 as do disorders of sterol production such as Smith-Lemli-Opitz syndrome,12 but these diagnoses are often associated with stereotypic dysmorphic features and anomalies. Finally, disorders of neurotransmitter synthesis should be considered in any infant with idiopathic seizures and neurologic dysfunction, especially if a movement disorder, most commonly dystonia, is also present. Abnormal CSF neurotransmitter levels (5-methyltetrahydrofolate, 5-hydroxyindoleacetic acid, homovanillic acid, 3-methyl-DOPA) are the only associated laboratory diagnostic clue in this latter category of disease.
Severe persistent lactic acidosis is the hallmark of the disorders in subgroup B of early-onset energy deficiency diseases. The presence of metabolic acidosis with an elevated anion gap suggests the possibility of lactic acidosis (subgroup B) or an organic acidemia (see group 3, intoxication types); these are differentiated by measurement of blood lactate and urine organic acid analysis. Blood lactate is most reliably measured on arterial blood or a free-flowing sample drawn from an indwelling central venous catheter. Artifactual elevation of lactate in peripheral venous blood samples is nearly ubiquitous and should be confirmed by lactate measurement in a more appropriate sample. Secondary lactic acidosis resulting from asphyxia, poor tissue perfusion, or tissue necrosis is much more common and may be difficult to differentiate from the congenital lactic acidoses. Occult cardiac disease, intracranial hemorrhage, or bowel necrosis must be considered and ruled out in infants with severe lactic acidosis. Congenital lactic acidosis generally persists despite adequate life support measures, including fluid resuscitation and ventilatory assistance. In certain enzyme deficiencies, the blood lactate level may further increase with IV dextrose infusion. Simultaneous measurements of blood and CSF lactate and amino acids are useful for differentiating primary from secondary lactic acidoses. In congenital lactic acidosis, the CSF lactate level often is higher than the blood lactate level, while the CNS is relatively protected from systemic acidosis in secondary lactic acidemias. The blood pyruvate level is elevated in some congenital lactic acidoses such as pyruvate dehydrogenase deficiency. However, accurate measurement of blood pyruvate is difficult and fraught with false-positive elevations. Elevated plasma alanine (which is measured as part of a plasma amino acid analysis) is a more stable and reliable indicator of pyruvic acidosis, as alanine and pyruvate are in equilibrium. Enzymatic analysis in cultured skin fibroblasts or mitochondria isolated from a fresh muscle biopsy often is necessary to confirm a specific enzyme deficiency.
Children with subgroup C defects present with hypoketotic hypoglycemia, triggered by fasting, metabolic stress, or intercurrent illness. In these disorders, utilization of fatty acids as fuel is impaired. The most common of the fatty acid oxidation defects is MCADD, which occurs in up to 1:10,000 white births. Although fatty acid oxidation and ketogenesis defects may present in the newborn period, particularly in the setting of delayed maternal milk production for exclusively breastfed infants, the first clinically significant episode may not occur for weeks to months or even years after birth. With extended fasting or intercurrent illness where metabolic demand exceeds available energy supply, severe lethargy acutely develops and then progresses to coma. Recurrent vomiting and consequent dehydration may be associated. Sudden infant death after an overnight fast is an all-too-frequent initial presentation in up to one third of infants with fatty acid oxidation defects. Infants who survive may suffer recurrent episodes of fasting or illness-induced coma, leading to progressive CNS damage and permanent disability. Metabolic acidosis (resulting from accumulation of partially oxidized fatty acids or secondary lactic acidosis), hyperammonemia, hepatomegaly and liver dysfunction, and hypertrophic cardiomyopathy may occur during acute metabolic decompensation episodes. Liver histology is typified by severe steatosis. Chronically affected children may exhibit recurrent vomiting, failure to thrive, developmental delay, and muscular hypotonia. Certain disorders that affect oxidation of long-chain fatty acids are frequently associated with recurrent rhabdomyolysis and myoglobinuria (long-chain 3-hydroxyacyl-CoA dehydrogenase [LCHAD] deficiency, trifunctional protein deficiency, very-long-chain acyl-CoA dehydrogenase [VLCAD] deficiency, or carnitine-palmitoyl transferase [CPT]-II deficiency) or pigmentary retinopathy and slowly progressive vision loss (LCHAD or trifunctional protein deficiency). Mothers of infants with fatty acid oxidation disorders (particularly LCHAD or trifunctional protein deficiency) may present with acute liver dysfunction during pregnancy with an affected fetus. This may manifest as acute fatty liver of pregnancy or maternal HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome. In the affected infant, hypoglycemia (serum glucose <40 mg/dL) with inappropriately low or absent ketone production during a symptomatic episode is the key laboratory finding that leads to suspicion of a disorder in this subgroup. Differentiation of the specific defects requires analysis of urine organic acids and plasma acylcarnitine species. Between episodes, when the child is clinically well, the urine organic acid profile may be completely normal. Acylcarnitine profiles are more consistently abnormal, but both tests, if normal initially, should be repeated on samples obtained during a symptomatic period to absolutely rule out the possibility of a fatty acid oxidation defect. Carnitine is required for normal fatty acid oxidation; long-chain fatty acids are activated to fatty acyl-CoA, then esterified to carnitine by CPT-I on the outer mitochondrial membrane. These acylcarnitine esters are then transported into mitochondria to complete the oxidation process. In fatty acid oxidation defects, the metabolic block leads to accumulation of the fatty acyl-CoA substrate specific to the deficient enzyme; these species appear in blood as acylcarnitine esters. Analysis of plasma acylcarnitine profiles by tandem mass spectrometry often suggests a specific enzyme deficiency in children with suspected fatty acid oxidation disorders.13 Diagnostic confirmation may require enzyme analysis in liver tissue or radiometric evaluation of fatty acid oxidation in cultured skin fibroblasts. For certain defects, molecular DNA analysis is clinically available. Two disorders, namely, MCADD14 and LCHAD deficiency,15 are associated with relatively common disease-causing mutations. Treatment of all disorders in this subgroup is based upon the provision of adequate nonfat calories and prevention of fasting. Generous IV glucose infusion is lifesaving and essential during acute episodes of metabolic decompensation. Chronic dietary therapy is tailored to the specific enzyme deficiency involved. Many practitioners prescribe carnitine supplementation, initially intravenously during an acute episode and later orally, but the efficacy of this intervention has not been formally investigated in any controlled clinical trial, and its use in disorders of long-chain fatty acid oxidation remains controversial.
Group 3 Inborn Errors of Metabolism
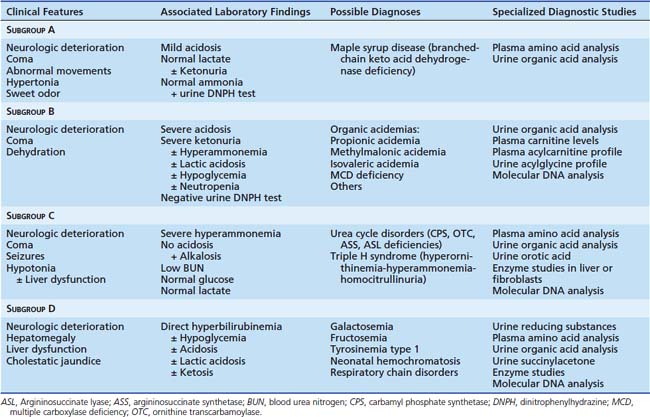
Infants with group 3 IEMs display symptoms and a progressive clinical course suggestive of intoxication. In these infants, who appear completely healthy at birth and for the first few days of life, neurologic dysfunction appears as toxic metabolites accumulate with increasing food intake. Initial symptoms may include vomiting and lethargy that progress, perhaps over only a few hours, to complete coma or shock. This specific clinical presentation in particular suggests the possibility of bacterial or viral sepsis; evaluation for infectious disease is entirely appropriate. However, the clinician must remain alert to the possibility of an underlying IEM in a previously healthy infant suffering catastrophic illness within the first days of life. Group 3 IEMs can be subdivided into four subgroups (A through D) based upon specific clinical and laboratory findings (Table 76-7).
Maple syrup urine disease (MSUD), or branched-chain keto acid dehydrogenase (BCKD) deficiency, affects the catabolism of the branched-chain amino acids leucine, isoleucine, and valine and is the only disorder in subgroup A. Affected infants present with coma; abnormal body movements including seizures; and, in contrast to many IEMs, hypertonia and opisthotonus. A severe burst-suppression pattern is the typical EEG abnormality. A sweet body odor, concentrated particularly in urine and cerumen, is often present. Mothers with previously affected children can often diagnose MSUD in a new infant by the presence of this odor. Routine laboratory studies may document mild metabolic acidosis and mild ketosis, but normal lactate and ammonia. The branched-chain keto acids that accumulate in MSUD react only slightly with the urine dipstick test for ketones but readily form a flocculent white precipitate with 2,4-dinitrophenylhydrazine (DNPH) in a urine metabolic screen. The presence and specific identities of branched-chain keto acids in urine are confirmed by urine organic acid analysis. Plasma amino acid analysis reveals tremendous elevation of leucine with lesser accumulations of valine and isoleucine. The neurologic symptoms associated with MSUD result entirely from leucine intoxication. Valine and isoleucine, which do not cross the blood-brain barrier as readily as leucine, seem to contribute little to the neurologic phenotype. Reduction of leucine levels in the body is the goal of MSUD treatment.16 Emergency therapy during the initial clinical episode includes dietary protein restriction and IV infusion of dextrose-containing fluids. Hyponatremia is a common associated feature; IV hydration with hypotonic fluids easily exacerbates this problem. Additionally, leucine accumulates in CSF and brain and is strongly osmotically active. Rapid IV infusion of hypotonic solutions in several instances has led to acute cerebral edema and death. Dextrose solutions containing a minimum of 0.45% saline (one-half normal saline) are essential, but 10% dextrose with normal saline is preferred if the serum sodium concentration is greater than 135 mEq/L. With administration of IV dextrose, mild hyperglycemia secondary to insulin resistance may occur; inclusion of regular insulin (often only 0.05 units/kg body weight/hour) by either IV infusion or subcutaneous injection promotes anabolism, suppresses endogenous protein catabolism, and accelerates leucine clearance. The vitamin thiamine is a cofactor for BCKD; some individuals with BCKD deficiency (usually with a late rather than neonatal presentation) may respond clinically to thiamine supplementation. Oral thiamine (100 mg/day) is often given empirically to determine whether there is any effect on leucine levels. Once the diagnosis of MSUD is confirmed by plasma amino acid analysis, enteral feedings with a medical food that is free of branched-chain amino acids should be initiated, even if the infant is comatose and nasogastric feedings are necessary. Parenteral hydration should continue until results of urine ketone and DNPH tests are negative and full enteral feeds are reestablished. On this regimen, plasma valine and isoleucine levels plummet rapidly, but several days may be required before plasma leucine normalizes. The valine and isoleucine deficiencies that frequently develop on this regimen stimulate endogenous protein catabolism, which impairs reduction of blood leucine, prolongs neurologic impairment, and chronically may be associated with symptoms of protein insufficiency (hair loss, skin breakdown, growth failure). Therefore valine and isoleucine supplementation (50 to 100 mg/kg/day) is required. Chronic lifelong therapy involves dietary protein restriction and provision of sufficient energy and amino acids in a leucine-free synthetic medical food. Despite this, infants who suffered prolonged severe leucinosis as neonates often exhibit significant developmental disability. Early diagnosis and appropriate therapy critically enhance neurodevelopmental outcome.
Hepatomegaly, liver dysfunction, and cholestatic jaundice in association with neurologic deterioration are the central presenting features of IEM in subgroup D. For all of these disorders, the accumulating toxin is particularly damaging to hepatocellular function. Hypoglycemia, acidosis, and mild ketosis may be present. Bacterial infection, particularly urinary tract infection, bacteremia, or meningitis, often caused by E. coli or other gram-negative enteral flora, is a frequent occurrence in infants with galactosemia. The specific diagnosis is suggested by the clinical scenario and by the results of screening laboratory studies. Infants with this clinical presentation who are breastfed or receiving cow’s milk-based infant formula are at risk for symptoms of galactosemia, given that lactose (milk sugar) is a disaccharide of galactose and glucose. Infants receiving exclusively soy milk-based formula ingest little galactose. The predominant dietary carbohydrates in soy formula are fructose and glucose, so infants fed soy formula who have this clinical presentation are likely to have fructosemia rather than galactosemia. More typically, infants with fructosemia present clinically after the introduction of fruit to their diet. In either galactosemia or fructosemia, reducing sugars are detected in urine following ingestion of the offending sugar by the urine reducing substance test (Clinitest). Plasma tyrosine level is elevated, urine organic acid analysis displays metabolites from the tyrosine pathway, and succinylacetone is detected in the urine of children with tyrosinemia type I (fumarylacetoacetate hydrolase deficiency). Neonatal hemochromatosis can be diagnosed only on liver biopsy by staining for iron. Diagnostic confirmation differs for each disorder but may include further metabolite analyses, enzymatic analysis in tissue, or molecular DNA testing. Initial therapy is nonspecific: cessation of enteral feeding and IV infusion of dextrose-containing fluid. Once the exact diagnosis is known, a specific therapy plan can be developed. For the carbohydrate disorders, the offending sugar must be reduced or eliminated from the diet. Galactosemic infants are fed soy-milk based formulas only. After weaning, ingestion of dairy products, including baked goods prepared with dairy products, is strictly avoided. Similarly, fructosemic individuals must strenuously avoid any fructose-containing foods. In prior eras, cirrhosis and liver failure were the inevitable outcomes in children with tyrosinemia type I unless they received a liver transplant. Effective therapy that prevents liver degeneration in tyrosinemia has now been developed. The oral drug 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC) blocks tyrosine metabolism upstream from FAH and prevents accumulation of the intermediate metabolites that are toxic to hepatocytes.17 This medication was highly successful in preventing cirrhosis in two separate clinical trials and has been approved by the United States Food and Drug Administration for general use. The long-term efficacy of NTBC therapy, particularly with regard to the incidence of hepatic adenoma, a common complication of tyrosinemia I, has yet to be proven.18
Metabolic Acidosis
The key to the differential diagnosis of metabolic acidosis is calculating the serum anion gap (Na+ − [Cl− + HCO3−]). This calculation, normally 10 to 15 mmol/L, represents the unmeasured negative ions, predominantly albumin, in blood. Normal anion gap acidosis (low serum HCO3 but normal anion gap) is caused by excess bicarbonate loss from either the gut (diarrhea) or kidney (renal tubular acidosis). An elevated or so-called positive anion gap suggests the presence of another unmeasured anion. Incidentally, a low serum anion gap may be seen in extreme hypoalbuminemia, as occurs in nephrotic syndrome (see Chapters 68 and 71).
Hypoglycemia
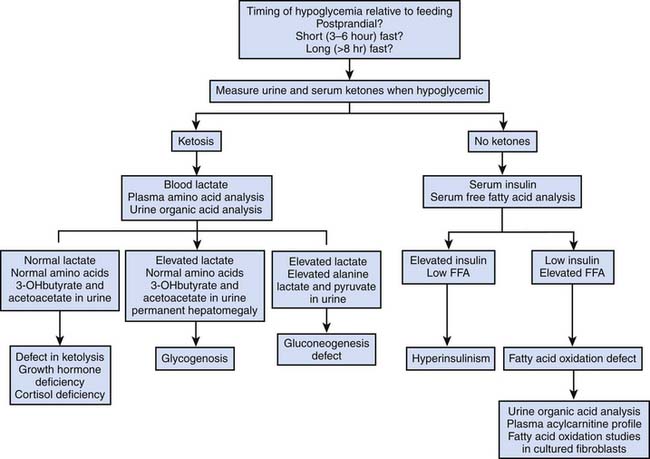
Hypoglycemia can be defined as a blood glucose concentration less than 40 mg/dL.19 Low blood glucose may be present within the first few hours after birth, especially in preterm or low-birth-weight infants, but the capacity for effective gluconeogenesis and fatty acid oxidation is induced within the first day after birth. Therefore blood glucose less than 40 mg/dL is distinctly unusual after the first 24 hours of life, particularly in infants who have started feeding, and should be thoroughly investigated (Fig. 76-2). A review of hypoglycemia in infants and children along with a useful diagnostic algorithm have been published.20 A detailed medical history and careful physical examination are essential to discovering the cause of hypoglycemia. The timing of hypoglycemia relative to feeding is a critical item of historical information. Persistent or postprandial hypoglycemia suggests hyperinsulinism. Hypoglycemia after a short fast (3 to 6 hours) along with permanent hepatomegaly suggests a glycogen storage disorder. Hypoglycemia following a longer fast (8 to 12 hours) suggests a defect in gluconeogenesis or a problem with utilization of fatty acids. The presence of ketones in urine (as measured qualitatively by urine dipstick) or in serum (quantitative measurement of 3-hydroxybutyrate or acetoacetate) is an important clue to the etiology of hypoglycemia. Ketosis during hypoglycemia demonstrates that insulin secretion is appropriately suppressed and that fatty acid mobilization and oxidation are intact. Glycogen storage disorders, gluconeogenic defects, and defects of ketone utilization all are associated with ketosis. The absence of ketogenesis during hypoglycemia suggests that either insulin levels are inappropriately elevated or fatty acid oxidation is blocked. An important caveat to this rule is that infants younger than approximately 1 week cannot normally produce enough ketones during fasting to trigger a positive urine dipstick test for ketones. The absence of urine ketones in an infant younger than 1 week does not contribute to the differential diagnosis of hypoglycemia. On the other hand, serum ketones increase with fasting even in neonates, and this test provides a valuable result in the investigation of hypoglycemia. In hypoketotic hypoglycemia, measurement of total serum free fatty acids provides further useful diagnostic information. During fasting, insulin secretion normally is suppressed, free fatty acids are mobilized into circulation from peripheral adipose tissues, and ketones are produced by oxidation of fatty acids in liver. A low serum total free fatty acid level during hypoketotic hypoglycemia strongly suggests inappropriate insulin secretion, even if insulin levels do not appear to be dramatically elevated. Hypoketotic hypoglycemia in association with elevated serum total free fatty acids suggests a defect in fatty acid oxidation.
The importance of treating hypoglycemia cannot be overemphasized as affected individuals are at high risk for seizures and permanent brain damage.21 After appropriate diagnostic studies are obtained, hypoglycemia should be treated with IV glucose administration at the rate of normal hepatic glucose production, approximately 10 mg glucose/kg body weight per minute or 150 mL/kg per day of a 10% solution until the underlying disorder is identified and more appropriate therapies can be initiated.
Hypoketotic hypoglycemia with low serum total free fatty acids suggests hyperinsulinism. Hyperinsulinism presenting in the newborn period may be caused by intrauterine exposure to elevated glucose levels (maternal diabetes mellitus), familial hyperinsulinemic hypoglycemia (defect in the sulfonylurea receptor), or hyperammonemia/hyperinsulinism syndrome (abnormality in regulation of insulin secretion secondary to mutation in glutamate dehydrogenase). Infants with hyperinsulinism often are obese and require glucose infusions greater than 10 mg/kg/min to maintain normoglycemia. Glucagon administration (0.03 mg/kg, up to 1 mg total dose) reverses hypoglycemia in hyperinsulinism. Oral diazoxide has not been shown to be efficacious in most neonatal cases; however, it can be effective in normalizing blood glucose levels in patients who have infantile forms of hyperinsulinism, including hyperammonemia/hyperinsulinism syndrome.22 This usually is given at doses of 5 to 10 mg/kg/day divided into three doses. When initially administered, it is given along with glucose and glucagon. The efficacy of diazoxide is defined by demonstrating normal preprandial and postprandial glucose concentrations after overnight fasting and after having stopped IV glucose and any other medications for 5 consecutive days.
Hypoketotic hypoglycemia with elevated serum total free fatty acids, usually occurring following an extended fast (8 to 12 hours) or in association with an intercurrent illness, suggests a defect in fatty acid oxidation. The clinical presentation of fatty acid oxidation disorders has been described. Although inherited deficiency of at least nine different enzymatic steps in the mitochondrial β-oxidation pathway has been described, the clinical presentation of infants and children with these diseases is stereotypically similar and can be differentiated only by appropriate metabolic testing. In all cases, vigorous hydration with dextrose-containing parenteral fluids is lifesaving. Fasting avoidance is key to long-term treatment and prevention of hypoglycemic episodes.
Cardiomyopathy and Inborn Errors of Metabolism
Cardiomyopathies, as a rule, are rare. Studies undertaken by the Pediatric Cardiomyopathy Registry have determined that the overall annual incidence is 11.8 per 1 million patient-years and that the incidence was higher in children younger than 1 year than in those between 1 and 18 years old.23 In this regional study, 40% of cases were hypertrophic cardiomyopathies, 49% of cases were dilated cardiomyopathies, 3% of cases were restrictive or other types, and 8% were unspecified. Further study revealed that of cases of hypertrophic cardiomyopathy, 16% had an identifiable IEM as the underlying cause. These causes included disorders of glycogen metabolism (5%), mucopolysaccharide metabolism (4%), oxidative phosphorylation (5%), and fatty acid metabolism (2%). In the cases of dilated cardiomyopathy, 5% were found to be of a metabolic etiology with disorders of glycogen metabolism (1%), mucopolysaccharide metabolism (2%), and oxidative phosphorylation (2%) as the recognizable underlying cause. Thus it is important to consider IEM in the differential diagnosis of any child with dilated or hypertrophic cardiomyopathy. Because the prevalence of underlying metabolic disorders is so high, some authors have recommended that all children with cardiomyopathy undergo metabolic screening, including blood lactate, plasma amino acid analysis, urine organic acid analysis, urine metabolic screening (particularly for the detection of excessive urinary mucopolysaccharides), plasma carnitine levels, and plasma acylcarnitine profile (Box 76-4).24 Additionally, serum creatine kinase (CK) should be measured to exclude muscular dystrophy.
Autosomal dominant hypertrophic cardiomyopathy has an incidence of 1:500 but demonstrates extremely variable penetrance. Mutations in genes encoding structural sarcomeric proteins are frequent causes of dominant hypertrophic cardiomyopathy. More than 140 mutations in 15 different genes have been identified.25
Cardiomyopathy may be a complicating feature of several IEMs (Table 76-8), but with a few exceptions, other associated symptoms or physical examination findings at the time of presentation point toward the appropriate diagnosis. Very broadly, the pathogenesis of cardiomyopathy in IEMs is either myocardial energy deficiency, as occurs in the dilated cardiomyopathy associated with several organic acidemias, or excessive storage of complex molecules in the heart, as occurs in the hypertrophic cardiomyopathy of mucopolysaccharidoses such as Hurler syndrome. Cardiomyopathy occurs as the sole initial clinical manifestation in a relatively restricted list of metabolic diseases, including autosomal recessively inherited deficiency of the cellular carnitine transporter, fatty acid oxidation disorders, glycogenosis types II and IX, and disorders of oxidative phosphorylation. The carnitine transporter defect is caused by deficiency of the sodium-dependent transporter OCTN2, which is responsible for transporting carnitine from the circulation into tissues including cardiac and skeletal muscle.26 Dilated cardiomyopathy with symptoms of heart failure generally presents within the first years of life and is associated with severely low plasma total carnitine levels. Cardiac function improves dramatically after carnitine supplementation, and cardiomyopathy rarely recurs if carnitine is continued.
| Cardiomyopathy as the sole or key presenting feature | Carnitine transport defect |
VLCAD, Very long chain acyl-CoA dehydrogenase.
Hypertrophic cardiomyopathy resulting from myocardial steatosis may be an isolated presenting feature in several disorders of fatty acid oxidation, particularly those affecting long-chain fatty acid metabolism such as VLCAD or mitochondrial trifunctional protein deficiencies. Saudubray et al.27 examined a series of 109 patients with fatty acid oxidation defects and found that cardiac involvement, including hypertrophic cardiomyopathy or arrhythmia, was apparent at presentation in 51% of cases. Fatty acid oxidation disorders are most reliably detected by analysis of plasma acylcarnitine profiles by tandem mass spectrometry. Long-chain fatty acids are activated to CoA derivatives and then esterified to carnitine prior to transport into mitochondria for β-oxidation. In fatty acid oxidation disorders, especially during acute metabolic decompensation, acylcarnitine species accumulate in plasma and provide a diagnostic profile that is specific to a given enzyme deficiency. Confirmation of the diagnosis may require enzymatic analysis in cultured fibroblasts or mutation analysis. Once the diagnosis of a long-chain fatty acid oxidation disorder has been established, restriction of dietary long-chain fat intake and provision of medium-chain triglyceride oil as an alternative fuel source for the myocardium often reverses cardiomyopathy. Cardiac support measures, including extracorporeal membrane oxygenation, may be necessary for as long as 1 to 2 weeks after presentation before heart function improves.
Glycogen storage disease type II (acid α-glucosidase deficiency; Pompe disease) is a disorder of lysosomal glycogen accumulation that frequently presents as hypertrophic cardiomyopathy, yielding the classic “boot-shaped” radiographic appearance of the cardiac silhouette. Skeletal myopathy manifesting as severe hypotonia may complicate the presentation. Confirmation of the diagnosis requires measurement of enzyme activity in skeletal or cardiac muscle or cultured fibroblasts. In the past, treatment has only been supportive, but enzyme replacement therapy is now available. IV infusion of recombinant acid a-glucosidase every other week has led to improved cardiac function, neuromuscular development, and survival in infants with Pompe disease.28 However, antibody formation against the drug in some infants, with subsequently decreased treatment effectiveness, remains a clinical problem.
Myocardial function is highly dependent upon mitochondrial oxidative phosphorylation; up to 30% of the total myocardial volume is composed of mitochondria.29 Dilated or hypertrophic cardiomyopathy is a frequent presenting feature in infants with severe defects of mitochondrial oxidative phosphorylation. Skeletal muscle myopathy, liver dysfunction, renal tubulopathy, bone marrow failure, or CNS abnormalities may occur. Chronic lactic acidosis, if present, is an important indicator of mitochondrial dysfunction. Screening metabolic laboratory studies demonstrate nonspecific abnormalities associated with chronic lactic acidosis. Definitive diagnosis requires histologic evaluation of skeletal muscle and measurement of respiratory chain enzyme activities. Isolated deficiency of cytochrome c oxidase (COX or complex IV) and reduced nicotinamide adenine dinucleotide (NADH)-ubiquinone oxidoreductase (complex I) of the mitochondrial respiratory chain are the most common oxidative phosphorylation defects presenting with cardiomyopathy. Although some protein subunits of complexes I and IV are encoded by mtDNA, most infant-onset isolated complex deficiencies probably are the result of autosomal-recessively inherited deficiency of nuclear-encoded respiratory chain subunits or of chaperone proteins that ensure proper assembly of functional complexes. For instance, hypertrophic cardiomyopathy caused by functional COX deficiency has been associated with mutations in nuclear COX subunit genes30,31 or nuclear genes for COX associated-proteins SCO1 and SCO2.32
Metabolic Myopathies and Rhabdomyolysis
Rhabdomyolysis is a clinical syndrome resulting from skeletal muscle injury and release of potentially toxic substances into the circulatory system. Acute onset of severe muscle pain associated with increased serum CK levels is the hallmark of the disorder. In extreme cases, massive myoglobinuria may cause acute renal insufficiency. Although trauma and direct muscle injury are by far the most common causes of rhabdomyolysis, inborn errors of muscle metabolism should be considered in the differential diagnosis of rhabdomyolysis occurring at any age. In the absence of a history of trauma, the differential diagnosis of acute rhabdomyolysis should include drug or toxin exposure, muscle hypoxia (often associated with seizures), temperature alterations, inflammatory diseases, and IEMs. Because muscle contraction depends upon adenosine triphosphate (ATP) generated by the mitochondrial electron transport chain, it follows that any process that impairs muscle ATP synthesis or which results in energy expenditure that surpasses ATP production could lead to rhabdomyolysis. The clinical history should lead toward the appropriate diagnosis. A family history that includes rhabdomyolysis or a history in which more than one episode of exercise-induced rhabdomyolysis has been observed should induce suspicion of a metabolic disorder. Along with muscular dystrophy and endocrine etiologies (hypothyroidism, hyperthyroidism, diabetic ketoacidosis, pheochromocytoma), glycolytic defects, fatty acid oxidation disorders, purine biosynthetic disorders, and disorders of mitochondrial oxidative phosphorylation should be considered if historical elements do not direct toward the more common etiologies. As described previously, the fatty acid oxidation disorders can be detected by urine organic acid analysis and plasma acylcarnitine profile. Chronic lactic acidosis may be a clue to a disorder of oxidative phosphorylation. Measurement of blood lactate level before and after an exercise treadmill protocol may help detect a respiratory chain defect if the postexercise lactate level is severely elevated. Definitive diagnosis of a mitochondrial disorder requires histologic and enzymatic analysis of a fresh muscle biopsy. The glycolytic defects of phosphofructokinase and phosphoglycerate mutase deficiencies along with myophosphorylase deficiency (glycogen storage disease type V or McArdle disease) cause severe recurrent rhabdomyolysis; their detection requires enzymatic analysis of muscle tissue. Likewise, myoadenylate deaminase deficiency, a defect in purine catabolism, and CPT-II deficiency are also diagnosed by measurement of the enzyme activities in muscle.
Neonatal Screening for Inborn Errors of Metabolism
Newborn screening for IEMs was first introduced in the 1960s, with screening for phenylketonuria. Technological advances, most significantly the introduction of tandem mass spectrometry to mass screening, have greatly increased the number of disorders that can be identified by analysis of a dried blood spot on a filter paper card.33 An expert review conducted by the American College of Medical Genetics led to the recommendation that 29 core conditions, including several aminoacidopathies, fatty acid oxidation defects, and organic acidurias detectable by tandem mass spectrometry, should be included in the panel of disorders screened.34 As of 2009, all states in the United States and most of Europe have now adopted this recommendation. The cost versus benefits of expanded screening, whether to include specific very rare or poorly treatable disorders in the screening panel, and the availability of adequate follow-up resources continue to be debated, but a general consensus has emerged that expanded newborn screening is an effective tool for identifying IEMs early in life, allowing for the initiation of therapy, often before the infant becomes symptomatic, and for preventing morbidity and mortality associated with IEMs.35 It must be remembered, however, that newborn screening is just that—a screen, and both false-positive and false-negative results are possible. Thus the astute clinician must remain cognizant of the fact that in an ill infant, a normal newborn screen is reassuring but should not be taken as absolute proof-positive that an IEM identifiable on newborn screen is not present. Appropriate screening laboratory evaluation and emergency treatment should be instituted if clinical signs and symptoms of an IEM are present in a sick child.
1. Eisensmith R.C., Woo S.L. Population genetics of phenylketonuria. Acta Paediatr Suppl. 1994;407:19-26.
2. Hoffmann G.F., von Kries R., Klose D., et al. Frequencies of inherited organic acidurias and disorders of mitochondrial fatty acid transport and oxidation in Germany. Eur J Pediatr. 2004;163(2):76-80.
3. Nyhan W.L., Ozand P.T. Atlas of metabolic diseases. London: Chapman & Hall; 1998.
4. Fernandes J., Saudubray J.M., van den Berghe G., et al. Inborn metabolic diseases, ed 4. Heidelberg, Germany: Springer; 2004.
5. Phenylketonuria. screening and management. NIH Consensus Statement Online. 2000;17(3):1-27.
6. Baumgartner E.R., Suormala T. Multiple carboxylase deficiency: inherited and acquired disorders of biotin metabolism. Int J Vitam Nutr Res. 1997;67(5):377-384.
7. Russo P.A., Mitchell G.A., Tanguay R.M. Tyrosinemia: a review. Pediatr Dev Pathol. 2001;4(3):212-221.
8. Saudubray J.M., Narcy C., Lyonnet L., et al. Clinical approach to inherited metabolic disorders in neonates. Biol Neonate. 1990;58(Suppl 1):44-53.
9. Hayasaka K., Tada K., Fueki N., et al. Nonketotic hyperglycinemia: analyses of glycine cleavage system in typical and atypical cases. J Pediatr. 1987;110(6):873-877.
10. Gallagher R.C., Van Hove J.L., Scharer G., et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65(5):550-556.
11. Marquardt T., Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr. 2003;162(6):359-379.
12. Opitz J.M., Gilbert-Barness E., Ackerman J., et al. Cholesterol and development: the RSH (“Smith-Lemli-Opitz”) syndrome and related conditions. Pediatr Pathol Mol Med. 2002;21(2):153-181.
13. Millington D.S., Kodo N., Norwood D.L., et al. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321-324.
14. Yokota I., Indo Y., Coates P.M., et al. Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J Clin Invest. 1990;86(3):1000-1003.
15. Sims H.F., Brackett J.C., Powell C.K., et al. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc Natl Acad Sci U S A. 1995;92(3):841-845.
16. Morton D.H., Strauss K.A., Robinson D.L., et al. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999-1008.
17. Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21(4):563-571.
18. Luijerink M.C., Jacobs S.M., van Beurden E.A., et al. Extensive changes in liver gene expression induced by hereditary tyrosinemia type I are not normalized by treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC). J Hepatol. 2003;39(6):901-909.
19. LaFranchi S. Hypoglycemia of infancy and childhood. Pediatr Clin North Am. 1987;34(4):961-982.
20. Lteif A.N., Schwenk W.F. Hypoglycemia in infants and children. Endocrinol Metab Clin North Am. 1999;28(3):619-646.
21. de Lonlay P., Giurgea I., Touati G., et al. Neonatal hypoglycaemia: aetiologies. Semin Neonatol. 2004;9(1):49-58.
22. Stanley C.A. Hyperinsulinism in infants and children. Pediatr Clin North Am. 1997;44(2):363-374.
23. Lipshultz S.E., Sleeper L.A., Towbin J.A., et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348(17):1647-1655.
24. Bonnet D., de Lonlay P., Gautier I., et al. Efficiency of metabolic screening in childhood cardiomyopathies. Eur Heart J. 1998;19(5):790-793.
25. Ackerman M.J., VanDriest S.L., Ommen S.R., et al. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39(12):2042-2048.
26. Nezu J., Tamai I., Oku A., et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21(1):91-94.
27. Saudubray J.M., Martin D., de Lonlay P., et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22(4):488-502.
28. Nicolino M., Byrne B., Wraith J.E., et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11(3):210-219.
29. Page E., Polimeni P.I., Zak R., et al. Myofibrillar mass in rat and rabbit heart muscle. Correlation of microchemical and stereological measurements in normal and hypertrophic hearts. Circ Res. 1972;30(4):430-439.
30. Antonicka H., Leary S.C., Guercin G.H., et al. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12(20):2693-2702.
31. Antonicka H., Mattman A., Carlson C.G., et al. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72(1):101-114.
32. Leary S.C., Kaufman B.A., Pellecchia G., et al. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum Mol Genet. 2004;13(17):1839-1848.
33. Schulze A., Lindner M., Kohlmuller D., et al. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399-1406.
34. Newborn screening toward a uniform screening panel and system. Genet Med. 2006;8(Suppl 1):1S-252S.
35. Fearing M.K., Marsden D. Expanded newborn screening. Pediatr Ann. 2003;32(8):509-515.