[level-membership-for-pediatrics-category]
121 Inborn Errors of Metabolism
Individually, IEM are rare, but they collectively make up a significant source of disease, particularly in infants and children. Most diseases are autosomal recessive, but autosomal dominant, X-linked, and mitochondrial inheritance patterns exist (see Chapter 115).
Clinical Presentation
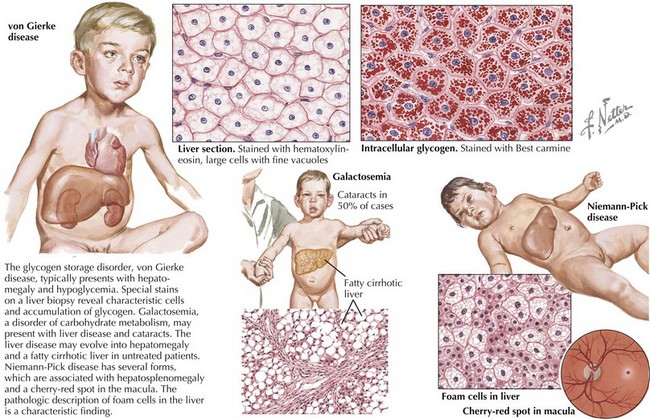
To accomplish this, a thorough history and physical examination will narrow the differential diagnosis before diagnostic testing. A complete dietary history may reveal symptoms instigated by certain food types or fasting. Food preferences or aversions may also be instructive. Other important information includes the frequency of and severity of response to illness, pattern of developmental delays, presence of consanguinity, and distinct body fluid odors. Although there are many physical examination and laboratory findings characteristic of specific disorders (Figure 121-1), several findings help to indicate general categories. For example, Kussmaul’s respirations may be found with metabolic acidosis or hyperventilation with hyperammonemia or cerebral edema. Organomegaly may also be evident as the result of a storage defect or organ dysfunction. Several disorders, such as the peroxisomal disorder Zellweger’s syndrome, have characteristic patterns of dysmorphia.
Evaluation And Management
Diagnostic Testing
When an IEM is suspected, it is often important to initiate treatment measures while an investigative workup is underway because a delay in therapy may affect the clinical outcome. Blood, urine, and cerebrospinal fluid (CSF) can be useful in the laboratory evaluation, and several laboratory studies are particularly helpful when an IEM is suspected in an ill child (Box 121-1). Serum electrolytes and blood gas analysis evaluate the acid-base status and anion gap. Ketone levels in urine, and sometimes blood, should be determined. Serum ammonia, lactate, and pyruvate, which can be tested in most hospitals, can also be informative, particularly in ill newborns. Metabolic laboratory studies such as plasma amino acids, plasma acylcarnitines, and urine organic acids, which provide important diagnostic information, must often be sent to specialized centers. Finally, one should consider other disease-specific testing, such as complete blood counts, to evaluate for bone marrow suppression in some organic acidurias. Molecular DNA testing and enzyme assays may require biopsy of specific tissues.
Common Presentations
Hypoglycemia
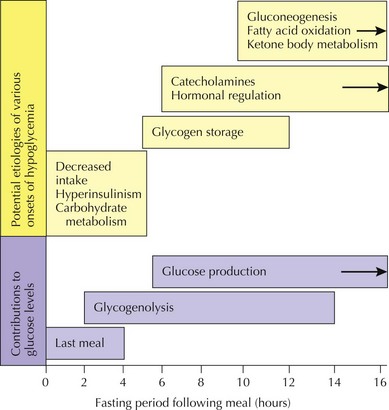
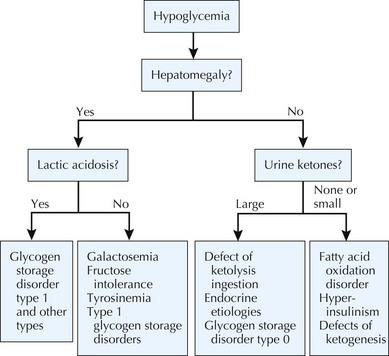
In general, hypoglycemia can be induced by lack of adequate glucose intake, poor homeostatic controls, iatrogenic causes, infection, and inborn errors of glucose metabolism. The brain is particularly dependent on glucose for energy but can alternatively use ketone bodies. Symptoms of hypoglycemia include lethargy, sweating, pallor, seizures, and mental status changes. Central to determining a metabolic cause of hypoglycemia is its onset after a carbohydrate load. For example, whereas early postprandial symptoms are more likely to be related to hyperinsulinism and glycogenoses, defects in fatty acid oxidation, ketone body synthesis, gluconeogenesis, and other IEM tend to take longer to present with hypoglycemia (Figure 121-2). Diagnostic pathways to approach the metabolic differential diagnosis for hypoglycemia are useful (Figure 121-3). A focused evaluation for an IEM may include electrolytes, glucose level, urine ketones, urine-reducing substances, ammonia, lactate, urine organic acids, and plasma acylcarnitines. One should also consider evaluation for non-IEM causes of hypoglycemia such as infection, drugs or toxins, and abnormal counterregulatory hormone mechanisms.
Hyperammonemia
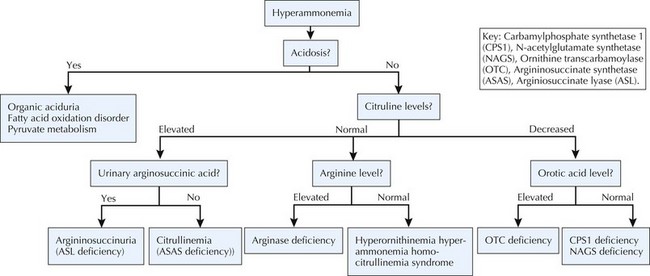
Ammonia is a product of protein metabolism that, in elevated levels, is toxic to the central nervous system (CNS), resulting in cerebral edema. Normally, ammonia is converted to urea in the liver via the urea cycle and is excreted in the urine. When the nitrogen load exceeds the clearance capacity of the liver, ammonia accumulates. The nitrogen load increases with dietary protein intake and endogenous protein breakdown, both of which can cause hyperammonemia in patients with primary or secondary urea cycle defects. Symptoms of hyperammonemia include vomiting, seizures, lethargy, coma, and hyperventilation associated with respiratory alkalosis. Nonspecific episodes of headache, vomiting, and mental status changes may be the only signs in later-onset cases. For extremely high ammonia levels, such as during neonatal presentations of urea cycle disorders (UCDs), dialysis to rapidly reduce the toxic levels may be necessary in addition to less invasive therapies. Nonmetabolic causes of hyperammonemia include sepsis; liver failure; use of medications such as valproate; and transient hyperammonemia of the newborn, a disorder associated with severe neonatal hyperammonemia that resolves perinatally, although residual neurologic sequelae may result. Classic metabolic causes of hyperammonemia include UCDs, organic acidurias, FAODs, hyperornithinemia-hyperammonemia-homocitrullinemia syndrome, pyruvate carboxylase deficiency, and hyperammonemia-hyperinsulinemia syndrome. Several of these disorders, including organic acidurias and FAODs, cause secondary urea cycle inhibition. Whereas hyperammonemia without metabolic acidosis and hypoglycemia is suggestive of UCD, an anion gap metabolic acidosis should arouse suspicion for an organic aciduria. Diagnostic pathways for biochemical causes of hyperammonemia are depicted in Figure 121-4.
Specific Inborn Errors Of Metabolism
This section covers some of the more common and well-described IEM encountered by metabolic physicians. A simplified diagram of some of the major biochemical pathways in the liver cell can be referred to as a visual aid (Figure 121-5).
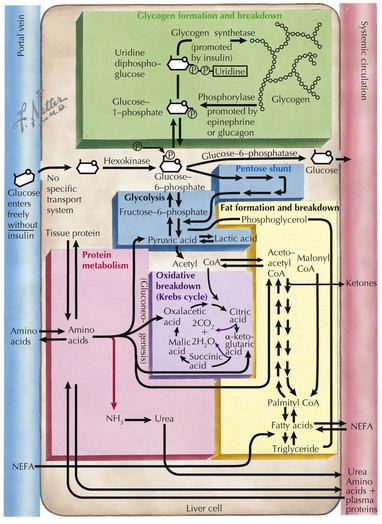
Disorders of Carbohydrate Metabolism
Disorders of Amino Acid Metabolism
Urea Cycle Disorders
The urea cycle converts ammonia, a product of protein breakdown, to water-soluble urea, which is excreted in urine. If deficient, ammonia elevations result in toxicity to the CNS. Several enzymes are involved in the hepatic mitochondrial urea cycle, each of which causes disease if deficient (Figure 121-6). Other than arginase deficiency, which has a chronic progressive neurologic presentation, the UCDs present during infancy with poor feeding, vomiting, and lethargy. Neonates classically manifest symptoms after 12 to 24 hours of life, presumably because of the toxic accumulation of ammonia. In addition, the effect of ammonia on brainstem respiratory control typically results in hyperventilation and respiratory alkalosis. However, patients with more residual enzyme activity may not be diagnosed until childhood or adulthood. All UCDs are autosomal recessive except for ornithine transcarbamoylase (OTC) deficiency, which is X-linked and the most common. Accordingly, a family history of male newborn deaths suggestive of X-linked disease is suggestive of OTC deficiency. The hair condition trichorrhexis nodosa is unique in argininosuccinate lyase deficiency.
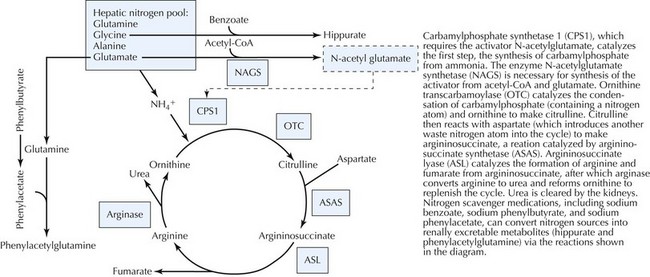
Classic patterns of biochemical findings assist with diagnosis. A low citrulline level (see Figure 121-6) is suggestive of enzymatic defects upstream in the urea cycle, of which only OTC deficiency has elevated urinary orotic acid, a product of accumulated carbamylphosphate. In contrast, citrulline levels are high in patients with downstream defects of the urea cycle. Molecular DNA analysis is available for many of the disorders.
Disorders of Organic Acid Metabolism
Disorders of Energy Metabolism
Primary Lactic Acidosis
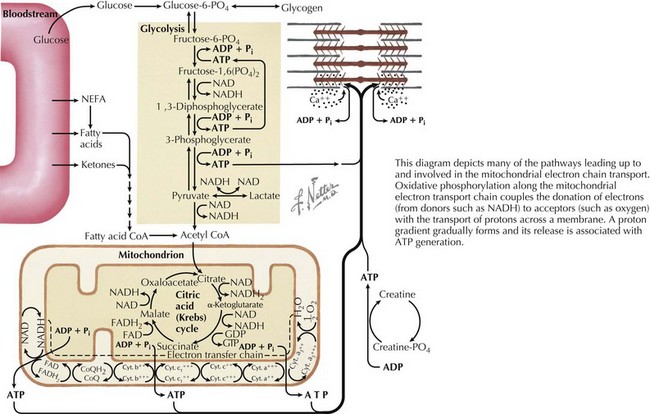
Lactate is formed from pyruvate during anaerobic glucose metabolism. Lactate and pyruvate exist in equilibrium and are affected by the redox state of cells and the generation of electron donors such as NADH (nicotinamide adenine dinucleotide), which contribute to oxidative phosphorylation. Oxidative phosphorylation occurs along the mitochondrial respiratory or electron transport chain (ETC). It couples the donation of electrons to acceptors (e.g., oxygen) with the transport of protons across a membrane, which then forms a proton gradient whose release is associated with adenosine triphosphate generation (Figure 121-7). Lactate accumulates because of blocks in pathways leading up to the ETC or defects along the ETC itself (which may result from circulatory collapse or hypoxia in addition to IEM). Primary lactic acidemias include defects in the mitochondrial respiratory chain, Krebs cycle, pyruvate metabolism, and several disorders of gluconeogenesis, and secondary lactic acidemias result from organic acidurias, FAODs, and UCDs that affect cellular redox potential or conversion of pyruvate to lactate. Several of these disorders can be inherited via maternal mitochondrial inheritance, as discussed in Chapter 115.
Disorders of Complex Molecules
Lysosomal Disorders
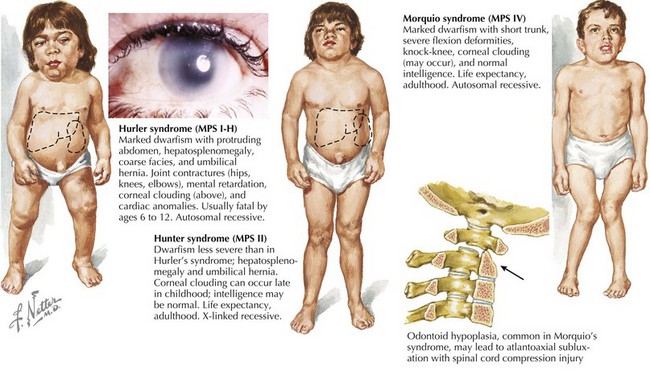
Lysosomes are cellular organelles that are important in the degradation of complex molecules. Various acid hydrolases within the lysosome may be deficient, leading to accumulation of enzyme substrates in various tissues. The particular storage material that accumulates defines the type of lysosomal storage disorder (LSDs), such as mucopolysaccharidoses, sphingolipidoses, and mucolipidoses. Lysosomal protein transport out of the lysosome may also be defective. Most but not all LSDs are autosomal recessive (i.e., Hunter and Fabry syndromes are X-linked). The clinical presentation and age of onset vary by disease. LSDs, particularly the mucopolysaccharidoses, are often associated with characteristic physical examination findings, including corneal clouding, coarse facies, organomegaly with liver disease, and skeletal abnormalities (Figure 121-8). Urine screening tests and enzyme assays are commonly used for diagnosis. Treatment is generally supportive in nature, but enzyme replacement and bone marrow transplantation have been effective in several disorders.
Berry GT. Inborn errors of carbohydrate, ammonia, amino acid, and organic acid metabolism. In: Taeusch HW, Ballard RA, Gleason CA, editors. Avery’s Diseases of the Newborn. ed 8. Philadelphia: Elsevier Saunders; 2005:227-257.
Berry GT. Introduction to the metabolic and biochemical genetic diseases. In: Taeusch HW, Ballard RA, Gleason CA, editors. Avery’s Diseases of the Newborn. ed 8. Philadelphia: Elsevier Saunders; 2005:271-326.
Blau N, Hoffmann GF, Leonard J, Clarke JTR, editors. Physician’s Guide to the Treatment and Follow-up of Metabolic Diseases, ed 1, Heidelberg, Germany: Springer Verlag, 2006.
Burton B. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics. 1998;102(6):E69.
Clarke JTR, editor. A Clinical Guide to Inherited Metabolic Diseases, ed 3, Cambridge, UK: Cambridge University Press, 2005.
Fernandes J, Saudubray JM, van den Berghe G, Walter JH. Inborn Metabolic Diseases: Diagnosis and Treatment, ed 4. Heidelberg, Germany: Springer Verlag; 2006.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
121 Inborn Errors of Metabolism
Individually, IEM are rare, but they collectively make up a significant source of disease, particularly in infants and children. Most diseases are autosomal recessive, but autosomal dominant, X-linked, and mitochondrial inheritance patterns exist (see Chapter 115).
Clinical Presentation
To accomplish this, a thorough history and physical examination will narrow the differential diagnosis before diagnostic testing. A complete dietary history may reveal symptoms instigated by certain food types or fasting. Food preferences or aversions may also be instructive. Other important information includes the frequency of and severity of response to illness, pattern of developmental delays, presence of consanguinity, and distinct body fluid odors. Although there are many physical examination and laboratory findings characteristic of specific disorders (Figure 121-1), several findings help to indicate general categories. For example, Kussmaul’s respirations may be found with metabolic acidosis or hyperventilation with hyperammonemia or cerebral edema. Organomegaly may also be evident as the result of a storage defect or organ dysfunction. Several disorders, such as the peroxisomal disorder Zellweger’s syndrome, have characteristic patterns of dysmorphia.
Evaluation And Management
Diagnostic Testing
When an IEM is suspected, it is often important to initiate treatment measures while an investigative workup is underway because a delay in therapy may affect the clinical outcome. Blood, urine, and cerebrospinal fluid (CSF) can be useful in the laboratory evaluation, and several laboratory studies are particularly helpful when an IEM is suspected in an ill child (Box 121-1). Serum electrolytes and blood gas analysis evaluate the acid-base status and anion gap. Ketone levels in urine, and sometimes blood, should be determined. Serum ammonia, lactate, and pyruvate, which can be tested in most hospitals, can also be informative, particularly in ill newborns. Metabolic laboratory studies such as plasma amino acids, plasma acylcarnitines, and urine organic acids, which provide important diagnostic information, must often be sent to specialized centers. Finally, one should consider other disease-specific testing, such as complete blood counts, to evaluate for bone marrow suppression in some organic acidurias. Molecular DNA testing and enzyme assays may require biopsy of specific tissues.
Common Presentations
Hypoglycemia
In general, hypoglycemia can be induced by lack of adequate glucose intake, poor homeostatic controls, iatrogenic causes, infection, and inborn errors of glucose metabolism. The brain is particularly dependent on glucose for energy but can alternatively use ketone bodies. Symptoms of hypoglycemia include lethargy, sweating, pallor, seizures, and mental status changes. Central to determining a metabolic cause of hypoglycemia is its onset after a carbohydrate load. For example, whereas early postprandial symptoms are more likely to be related to hyperinsulinism and glycogenoses, defects in fatty acid oxidation, ketone body synthesis, gluconeogenesis, and other IEM tend to take longer to present with hypoglycemia (Figure 121-2). Diagnostic pathways to approach the metabolic differential diagnosis for hypoglycemia are useful (Figure 121-3). A focused evaluation for an IEM may include electrolytes, glucose level, urine ketones, urine-reducing substances, ammonia, lactate, urine organic acids, and plasma acylcarnitines. One should also consider evaluation for non-IEM causes of hypoglycemia such as infection, drugs or toxins, and abnormal counterregulatory hormone mechanisms.
Hyperammonemia
Ammonia is a product of protein metabolism that, in elevated levels, is toxic to the central nervous system (CNS), resulting in cerebral edema. Normally, ammonia is converted to urea in the liver via the urea cycle and is excreted in the urine. When the nitrogen load exceeds the clearance capacity of the liver, ammonia accumulates. The nitrogen load increases with dietary protein intake and endogenous protein breakdown, both of which can cause hyperammonemia in patients with primary or secondary urea cycle defects. Symptoms of hyperammonemia include vomiting, seizures, lethargy, coma, and hyperventilation associated with respiratory alkalosis. Nonspecific episodes of headache, vomiting, and mental status changes may be the only signs in later-onset cases. For extremely high ammonia levels, such as during neonatal presentations of urea cycle disorders (UCDs), dialysis to rapidly reduce the toxic levels may be necessary in addition to less invasive therapies. Nonmetabolic causes of hyperammonemia include sepsis; liver failure; use of medications such as valproate; and transient hyperammonemia of the newborn, a disorder associated with severe neonatal hyperammonemia that resolves perinatally, although residual neurologic sequelae may result. Classic metabolic causes of hyperammonemia include UCDs, organic acidurias, FAODs, hyperornithinemia-hyperammonemia-homocitrullinemia syndrome, pyruvate carboxylase deficiency, and hyperammonemia-hyperinsulinemia syndrome. Several of these disorders, including organic acidurias and FAODs, cause secondary urea cycle inhibition. Whereas hyperammonemia without metabolic acidosis and hypoglycemia is suggestive of UCD, an anion gap metabolic acidosis should arouse suspicion for an organic aciduria. Diagnostic pathways for biochemical causes of hyperammonemia are depicted in Figure 121-4.
