Imperforate Anus and Cloacal Malformations
‘Imperforate anus’ has been a well-known condition since antiquity.1–3 For many centuries, physicians, as well as individuals who practiced medicine, have tried to help these children by creating an orifice in the perineum. Many patients survived, most likely because they suffered from a type of defect that is now recognized as ‘low.’ Those with a ‘high’ defect did not survive. In 1835, Amussat was the first to suture the rectal wall to the skin edges which was the first actual anoplasty.2 Stephens made a significant contribution by performing the first anatomic studies in human specimens. In 1953, he proposed an initial sacral approach followed by an abdominoperineal operation, if needed.4 The purpose of the sacral stage of this procedure was to preserve the puborectalis sling, considered a key factor in maintaining fecal incontinence. Over the next 25 years, different surgical techniques were described, with the common denominator being the protection and use of the puborectalis sling.5–8
The posterior sagittal approach for the treatment of imperforate anus was performed first in 1980, and its description was published in 1982.9 With this approach, the unique opportunity arose to correlate the external appearance of the perineum with the operative findings and, subsequently, with the clinical results.
Incidence, Types of Defects, and Terminology
An anorectal malformation occurs in one out of every 4000 to 5000 newborns and is slightly more common in males.10–12 The estimated risk for a couple having a second child with an anorectal malformation is approximately 1%.13–17 The most frequent defect in males is imperforate anus with a rectourethral fistula.18 In females, it is a rectovestibular fistula.18 Imperforate anus without a fistula is a rather unusual defect, occurring in about 5% of the entire group of malformations, and is associated with Down syndrome.18,19 Historically, a cloaca has been considered an unusual defect, whereas a high incidence of rectovaginal fistula has been reported in the literature.20 In retrospect, we now know that a cloaca is the third most common defect in female patients after vestibular and perineal fistulas, whereas a rectovaginal fistula is actually a rare defect, present in less than 1% of all cases.21,22 It is likely that most females with a persistent cloaca were erroneously thought to have a rectovaginal fistula. Many of these patients underwent repair of the rectal component but were left with a persistent urogenital sinus.21,23 Additionally, most rectovestibular fistulas were erroneously called ‘rectovaginal fistula’.21 A rectobladderneck fistula in males is the only true supralevator malformation and occurs in about 10%.18 As it is the only malformation in males in which the rectum is unreachable through a posterior sagittal incision, it requires an abdominal approach (via laparoscopy or a laparotomy) in addition to the perineal approach.
Anorectal malformations represent a wide spectrum of defects. The terms ‘low,’ ‘intermediate,’ and ‘high’ are arbitrary and not useful in current therapeutic or prognostic terminology. A therapeutic and prognostically oriented classification is depicted in Box 35-1.24
Male Anorectal Defects
Rectoperineal Fistulas
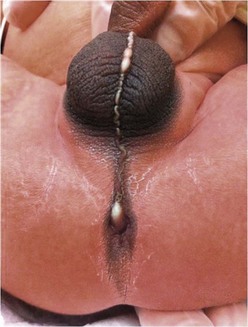
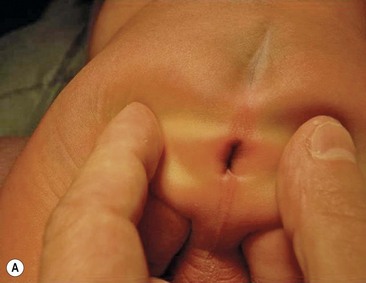
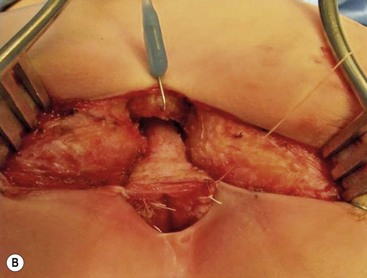
Rectoperineal fistula is the lowest defect. The rectum is located within most of the sphincter mechanism. Only the lowest part of the rectum is anteriorly mislocated (Fig. 35-1). Sometimes, the fistula does not open into the perineum but rather follows a subepithelial midline tract, opening somewhere along the midline perineal raphe, scrotum, or even at the base of the penis (Fig. 35-2). This diagnosis is established by perineal inspection. No further investigations are required. Usually, the anal fistula opening is stenotic. The terms covered anus, anal membrane, anteriorly mislocated anus, and bucket-handle malformations all refer to rectoperineal fistulas.
Rectourethral Fistulas
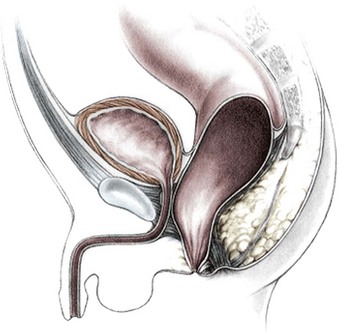
Imperforate anus with a rectourethral fistula is the most common defect in males.18 The fistula may be located at the lower (bulbar) (Fig. 35-3A) or the higher (prostatic) part of the urethra (Fig. 35-3B).
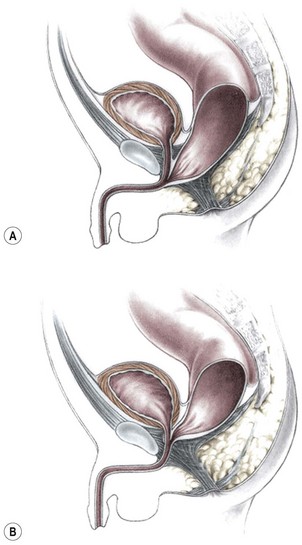
FIGURE 35-3 Anorectal atresia with rectourethral fistulas. (A) Rectourethrobulbar fistula; (B) rectourethroprostatic fistula.
Immediately above the fistula, the rectum and urethra share a common wall. The lower the fistula, the longer is the common wall. This is an important anatomic fact, which guides the operation. The rectum is usually distended and surrounded laterally and posteriorly by the levator muscle. Between the rectum and the perineal skin, a portion of striated voluntary muscle called the muscle complex is present. The contraction of these muscle fibers elevates the skin of the anal dimple. At the level of the skin, a group of voluntary muscle fibers, called parasagittal fibers, are located on both sides of the midline. Lower urethral fistulas are usually associated with good-quality muscles, a well-developed sacrum, a prominent midline groove, and a prominent anal dimple. Higher urethral fistulas are more frequently associated with poor-quality muscles, an abnormally developed sacrum, a flat perineum, a poor midline groove, and a barely visible anal dimple. Of course, exceptions to these rules exist. Occasionally, the infant passes meconium through the urethra, which is an unequivocal sign of a rectourinary fistula.
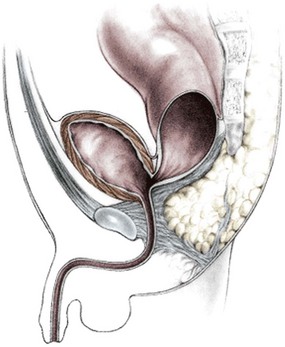
Rectobladderneck Fistulas
In this defect, the rectum opens into the bladder neck (Fig. 35-4). The patient usually has poor prognosis for bowel control because the levator muscles, the striated muscle complex, and the external sphincter frequently are poorly developed. The sacrum is often deformed and short. In fact, the entire pelvis seems to be underdeveloped. The perineum is often flat, which is evidence of poor muscle development. About 10% of males fall into this category.18
Imperforate Anus without Fistula
Interestingly, most patients with this unusual defect have a well-developed sacrum and good muscles, and have a good prognosis in terms of bowel function.18,19 The rectum usually terminates approximately 2 cm from the perineal skin. Although the rectum and urethra do not communicate, these two structures are separated only by a thin common wall. About half of the patients with no fistula also have Down syndrome, and more than 90% of patients with Down syndrome and imperforate anus have this specific defect, suggesting a chromosomal link.17–19 The fact that these patients have Down syndrome does not seem to interfere with their good prognosis for bowel control.19
Rectal Atresia/Rectal Stenosis
In this unusual defect in male patients (less than 1% of the entire group of malformations), the lumen of the rectum is totally (atresia) or partially (stenosis) interrupted.18 The upper pouch is represented by a dilated rectum, whereas the lower portion empties into a small anal canal that is in the normal location and is 1–2 cm long (Fig. 35-5A). These two rectal structures may be separated by a thin membrane or by dense fibrous tissue. The repair involves a primary anastomosis between the upper pouch and lower anal canal, and is ideally approached posterosagittally with splitting of the anal canal longitudinally (Fig.35-5B). Patients with this defect have all the necessary elements for continence and have an excellent functional prognosis because they have a well-developed anal canal, normal sensation in the anorectum, and normal voluntary sphincters. These patients must be screened for a presacral mass.25
Female Anorectal Defects
Rectoperineal Fistulas
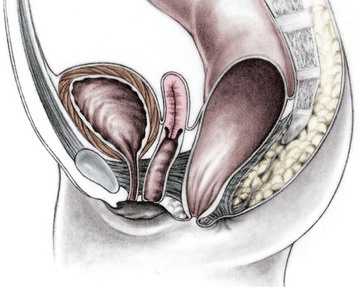
From the therapeutic and prognostic viewpoint, this common defect is equivalent to the perineal fistula described in the male patient.18 The rectum is well positioned within the sphincter mechanism, except for its lower portion, which is anteriorly located. The rectum and vagina are well separated (Fig. 35-6). The key anatomic issues are the anal opening in relation to the sphincter mechanism, and the length of the perineal body.
Rectovestibular Fistulas
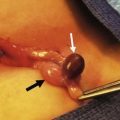
Rectovestibular fistula is the most common defect in females and has an excellent functional prognosis. The diagnosis is based on clinical examination. A meticulous inspection of the newborn’s genitalia allows the clinician to observe a normal urethral meatus and a normal vagina, with a third hole in the vestibule, which is the rectovestibular fistula (Fig. 35-7). About 5% of these patients will have two hemivaginas with a vaginal septum.26
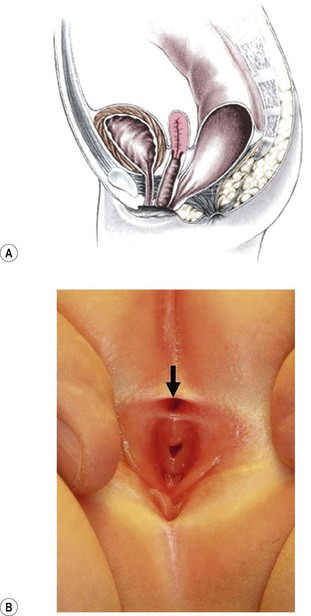
FIGURE 35-7 (A) Schematic drawing of a rectovestibular fistula. (B) Female neonate with a rectovestibular fistula is in the prone position. The rectal fistula (arrow) is located in the posterior aspect of the vestibule.
This defect can be repaired without a protective colostomy by experienced surgeons.27–29 The advantage of this approach is that it avoids the potential morbidity of a colostomy and reduces the number of operations to one from as many as three (colostomy, main repair, and colostomy closure). Many patients do very well with a primary neonatal operation without a protective colostomy. However, a perineal infection followed by dehiscence of the anal anastomosis or perineal body, or recurrence of the fistula provokes severe fibrosis that may interfere with the sphincter function. If these complications occur, the patient may have lost the best opportunity for an optimal functional result because secondary operations do not render the same prognosis as a successful primary operation.30 Thus, a protective colostomy is still the best way to avoid these complications for most surgeons. The decision to perform a colostomy or primary repair in these cases must be made individually by the surgeon based on experience and the clinical condition of the patient. At our institution, neonates without significant associated defects undergo repair without a colostomy.
Persistent Cloaca
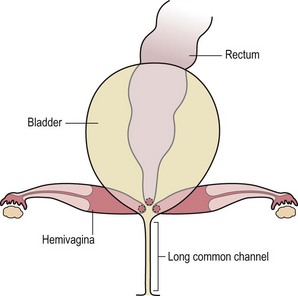
This group of defects represents the extreme in the spectrum of complexity of female malformations. A cloaca is a defect in which the distal portions of the rectum, vagina, and urinary tract fuse and create a single common perineal channel. The diagnosis of a cloaca is a clinical one. This defect should be suspected in a female born with imperforate anus and small-looking genitalia. Careful separation of the labia discloses a single perineal orifice. The length of the common channel varies from 1–7 cm, and is very important for operative and prognostic implications (Fig. 35-8). A common channel of less than 3 cm usually means that the defect can be repaired with a posterior sagittal operation without opening the abdomen. Common channels longer than 3 cm are more complex, mobilization of the vagina is often difficult, and some form of vaginal replacement may be needed during the definitive repair. When the rectum opens high into the dome of the vagina (Fig. 35-9), an abdominal approach must be utilized to mobilize the bowel.
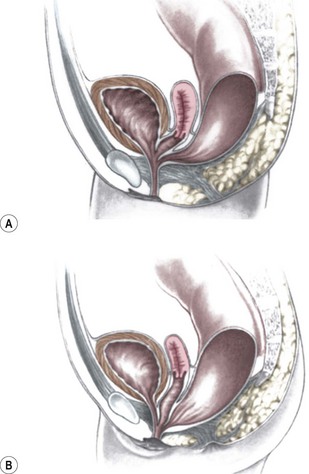
FIGURE 35-8 (A) Schematic diagram of a long common channel in a female with a cloacal anomaly. (B) The more commonly encountered short common channel cloaca is depicted.
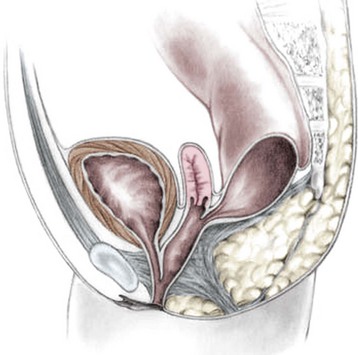
FIGURE 35-9 A schematic diagram of the rectum inserting high into the posterior vagina with a short common urethral and vaginal channel is shown.
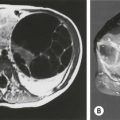
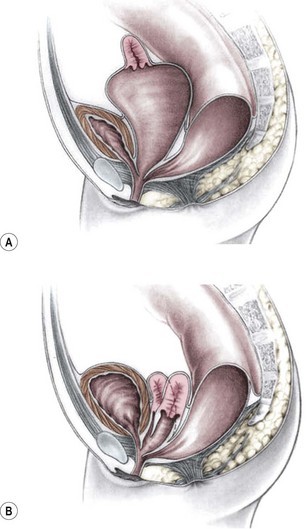
Frequently, the vagina is abnormally distended and full of secretions (hydrocolpos) (Fig. 35-10A). The distended vagina compresses the trigone and interferes with drainage of the ureters, and is frequently associated with hydronephrosis. This condition may be diagnosed prenatally.31,32 The dilated vagina can also become infected (pyocolpos) and may lead to perforation and peritonitis. Such a large vagina may represent a technical advantage for the repair as there is more vaginal tissue to facilitate the reconstruction. A frequent finding in cloacal malformations is the presence of different degrees of vaginal and uterine septation or duplication (Fig. 35-10B). In these cases, the rectum usually enters between the two hemivaginas. Rarely, patients have cervical atresia. During puberty, a variety of anatomic anomalies may mean that they are unable to drain menstrual blood through the vagina, can accumulate menstrual blood in the peritoneal cavity, and sometimes require emergency operations.26,33 An evaluation of the patient’s Müllerian anatomy, either at the time of the definitive repair or at colostomy closure, is very important and can prevent future problems.26 Short common channel cloacal malformations (<3 cm) are usually associated with a well-developed sacrum, a normal-appearing perineum, and adequate muscles and nerves. Therefore, a good functional prognosis can be expected.
Associated Defects
Sacrum and Spine
Sacral deformities are the most frequently associated defect.34 One or several sacral vertebrae may be missing. A single missing vertebrae does not seem to have important prognostic implications.18 However, more than two absent sacral vertebrae represent a poor prognostic sign in terms of bowel continence and, sometimes, urinary control, and is consistent with caudal regression. A hemisacrum is usually associated with a presacral mass and poor bowel control.25 Other sacral abnormalities, such as spinal hemivertebra, have negative implications for bowel control.
A sacral ratio is an objective evaluation of the sacrum (Fig. 35-11). The sacral ratio can range from 0.0 to 1.0. The normal sacral ratio in children is 0.77. Children with anorectal malformations suffer varying degrees of poor sacral development. We have never seen a patient have bowel control with a sacral ratio of less than 0.3. Greater than 0.7 is usually associated with good bowel control.
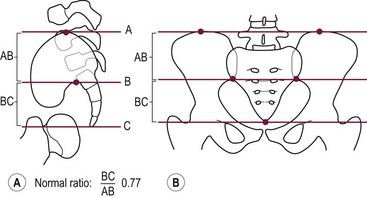
FIGURE 35-11 Drawings with landmarks necessary for the calculation of the sacral ratio. (A) Lateral view. (B) Anteroposterior view. The normal ratio is 0.77.
A tethered cord is frequently associated with anorectal malformations, and its presence has been assumed to be associated with a poor functional prognosis.35–44 Although it is true that most of these children have a poor prognosis, the presence of a tethered cord is usually found in patients with a very high defect, very abnormal sacrum, or spina bifida. Therefore, it is difficult to know whether the tethered cord itself is responsible for the poor prognosis. Although it is unclear whether the operation to release the tethered cord changes the functional bowel prognosis of the patient, it does seem to improve urinary function and certainly avoids motor and sensory deterioration of the lower extremeties.41,44
The reported frequency of associated genitourinary defects varies from 20–54%.45–57 The accuracy and thoroughness of the urologic evaluation may account for the reported variation.
It is clear that the higher the malformation, the more frequent are the associated urologic abnormalities. Patients with persistent cloacas or rectobladderneck fistulas have a 90% chance of having an associated genitourinary abnormality.51 Conversely, children with low defects such as perineal fistulas have less than a 10% chance of having an associated urologic defect. Hydronephrosis, urosepsis, and metabolic acidosis from poor renal function represent the main sources of morbidity. Thus, a thorough urologic investigation is particularly important in patients with high defects. The evaluation in every child with imperforate anus must include an ultrasound (US) of the kidneys and abdomen to evaluate for the presence of hydronephrosis or any other urologic obstructive process. In patients with a cloaca, this study is especially important to exclude the presence of hydrocolpos. Further urologic investigations after this initial screening may also be necessary.
Newborn Management
A decision-making algorithm for the initial management in male infants is seen in Figure 35-12.
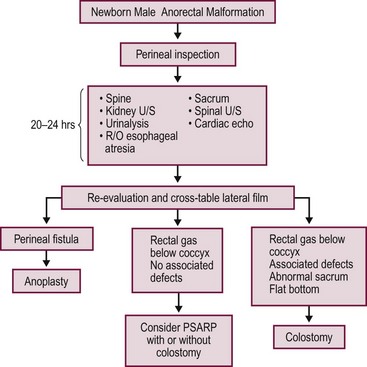
FIGURE 35-12 Algorithm for the management of male newborns with anorectal malformations based on the physical examination and radiographs. PSARP, posterior sagittal anorectoplasty; U/S, ultrasonography; R/O, rule out.
Radiographic evaluations may not show the correct anatomy before 24 hours because the rectum is collapsed. It takes a significant amount of intraluminal pressure to overcome the muscle tone of the sphincters that surround the lower part of the rectum. Therefore, imaging studies before 24 hours most likely will show a ‘very high rectum’ and may lead to an incorrect diagnosis. Historically, an invertogram was used to identify whether the anomaly is ‘high’ or ‘low’.58
During the first 24 hours, the neonate should receive intravenous fluids, antibiotics, and nasogastric decompression, and be evaluated for associated defects that may represent a threat to life. These include cardiac malformations, esophageal atresia, and urinary defects.34,59 A radiograph of the lumbar spine and the sacrum should be obtained as well as a spinal ultrasound to evaluate for a tethered cord. Renal/abdominal ultrasound should be done to evaluate for hydronephrosis.
If the neonate has signs of a rectoperineal fistula, an anoplasty can be performed in the newborn period without a protective colostomy. After 24 hours, if there is no meconium on the perineum, we recommend obtaining a cross-table lateral radiograph with the patient in the prone position (Fig. 35-13A). If air in the rectum is seen distal to the coccyx (Fig. 35-13B), and the patient is in good condition with no significant associated defects, one may consider performing a posterior sagittal operation without a protective colostomy. A more conservative alternative would be to perform the posterior sagittal repair and a protective colostomy at the same stage.
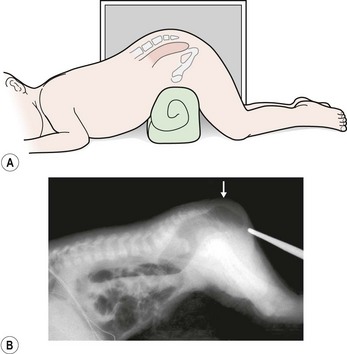
FIGURE 35-13 Technique for a cross-table lateral radiograph. (A) A roll has been placed beneath the hips of the infant to elevate the buttocks and allow air to migrate superiorly to the end of the rectum. (B) Actual cross-table lateral radiograph. Air is visualized distal to the coccyx (arrow).
Performing the definitive repair early in life has important advantages including less time with an abdominal stoma, less size discrepancy between the proximal and distal bowel at the time of colostomy closure, and easier anal dilation (because the infant is smaller). In addition, at least theoretically, placing the rectum in the right location early in life potentially may represent an advantage in terms of acquired local sensation.60
All of these potential advantages of an early operation must be weighed against the possible disadvantages of an inexperienced surgeon who is not familiar with the anatomic structures of an infant’s pelvis. Operating on patients with anorectal malformations primarily without a protective colostomy has been done successfully, but may have negative consequences if the surgeon does not have the necessary preoperative evaluation and experience.29,61,62
A temptation to repair these defects without a protective colostomy always exists.27,61,62 Repair without a colostomy does not allow a distal colostogram which may be very helpful to the surgeon. The worst complications involve infants who undergo repair without a colostomy or a properly performed distal colostogram.63 Proceeding with the posterior sagittal approach looking blindly for the rectum has resulted in a spectrum of serious complications, including damage to the urethra, complete division of the urethra, pull-through of the urethra, pull-through of the bladder neck, injury to the ureters, and division of the vas deferens or seminal vesicles.63
A decision-making algorithm for the initial management of newborn females is shown in Figure 35-14. Again, the perineal inspection is the most important step to guide diagnosis and decision making. The first 24 hours should also be used to evaluate for associated defects, as previously described. Perineal inspection may show the presence of a single perineal orifice, which establishes the diagnosis of a cloaca and carries a high risk of an associated urologic defect. Also, it should prompt a complete urologic evaluation, including abdominal and pelvic ultrasound, to look for hydronephrosis and hydrocolpos.
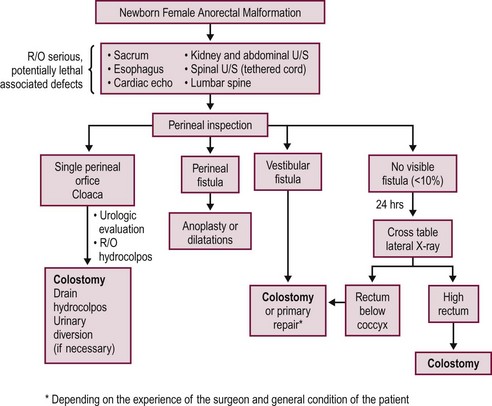
FIGURE 35-14 Decision-making algorithm for female newborns with anorectal malformations. U/S, ultrasonography; R/O, rule out.
Patients with a cloaca require a colostomy. It is important to perform the divided sigmoid colostomy in such a manner as to leave enough redundant, distal rectosigmoid colon to allow for the subsequent pull-through (Fig. 35-15). When performing the colostomy for a cloaca, it is important to drain a hydrocolpos when present. This can be achieved with a curled catheter. Because a significant number of these patients have two hemivaginas, the surgeon must be certain that both hemivaginas are drained. Occasionally, a vaginovaginostomy in the vaginal septum needs to be created to drain both hemivaginas with one catheter. At times, the hydrocolpos is so large that it may produce respiratory distress. Also, the hydrocolpos can compress the trigone and cause hydronephrosis, and drainage of the hydrocolpos allows for decompression of the urologic system. Rarely, if the common channel is very narrow and does not allow the bladder to drain, the neonate may require a vesicostomy or suprapubic cystostomy to decompress the bladder. However, in most cases, drainage of the hydrocolpos is all that is required. Endoscopic examination of the cloaca is recommended to delineate the anatomy. This evaluation is best done several months later during a separate anesthetic because the neonatal perineum is swollen and endoscopy is difficult in the newborn.
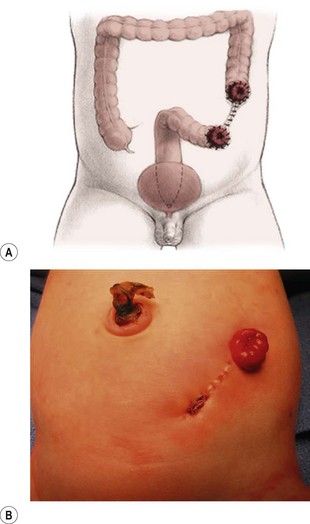
FIGURE 35-15 An ideal colostomy for infants with high anorectal malformations is seen in (A) the drawing and (B) infant. Note the colostomy and mucous fistula are separated and that adequate distal colonic length remains for the subsequent rectal pull-through.
The presence of a vestibular fistula represents the most common finding in female infants (see Fig. 35-7). When newborns with a vestibular fistula undergo primary repair at our institution, we hospitalize the patient for seven days with no oral intake, and use concentrated dextrose intravenous nutrition. When the patient undergoes a primary repair of a vestibular fistula or perineal fistula without colostomy later in life, we are particularly strict about preoperative bowel preparation to ensure that the intestine is completely clean.
If the perineal inspection shows a rectoperineal fistula, we recommend performing a primary anoplasty without a colostomy. In fewer than 5% of girls, there is no visible fistula and there is no evidence of meconium after 24 hours of observation. This small group of patients requires a cross-table lateral prone radiograph (see Fig. 35-13). If the radiograph shows gas in the rectum very close to the skin, it means that the baby likely has a very narrow perineal fistula or has no fistula defect. If the baby is in stable condition, one can perform a primary operation without a colostomy, depending on the surgeon’s experience. Most of these patients without a fistula also have Down syndrome.19 If associated conditions make the rectal repair not feasible in the newborn period, a colostomy should be performed, with definitive repair later.
Occasionally, if the infant with a rectoperineal or rectovestibular fistula has significant associated defects or is unstable, the surgeon may elect to dilate the fistula to facilitate emptying of the colon while these other issues are addressed. Definitive repair can be performed in a few months.
A divided descending colostomy is ideal for the management of anorectal malformations when diversion is needed (see Fig. 35-15). The completely diverting colostomy provides bowel decompression as well as protection for the final reconstruction. In addition, the colostomy is used for the distal colostogram, which is the most accurate diagnostic study to determine the detailed distal anatomy.64–66
The descending or upper sigmoid colostomy has definitive advantages over a right or transverse colostomy.65 It is important to have a relatively short segment of defunctionalized distal colon, but not too short as to interfere with the subsequent pull-through. The ideal location is just at the point where the proximal sigmoid comes off the left retroperitoneum. Mechanical cleansing of the distal colon at the time of colostomy is much less difficult when the colostomy is located in the descending portion of the colon. In the case of a large rectourethral fistula, the patient may pass urine into the colon. A more distal colostomy allows urine to escape through the distal stoma without significant absorption. With a more proximal colostomy, the urine remains in the colon and is absorbed, leading to metabolic acidosis. A loop colostomy permits the passage of stool from the proximal stoma into the distal bowel. This can lead to urinary tract infections, distal rectal pouch dilation, and fecal impaction (Fig. 35-16). Prolonged distention of the rectal pouch may produce a megarectosigmoid, leading to severe constipation later in life. Also, the problem of colostomy prolapse is more frequent with loop colostomies.65 The most common error is a colostomy established too distal in the rectosigmoid that will interfere with mobilization of the rectum during the pull-through (Fig. 35-17).
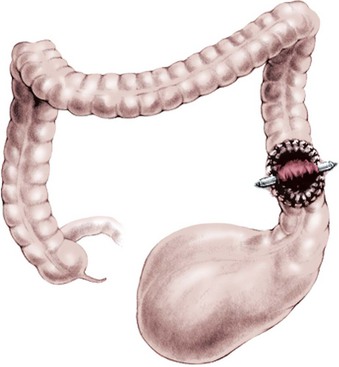
FIGURE 35-16 A markedly dilated distal rectal pouch secondary to fecal impaction. (Adapted from Prem Puri. In: Newborn Surgery. 3rd ed. Hodder Arnold; 2012. p. 580.)
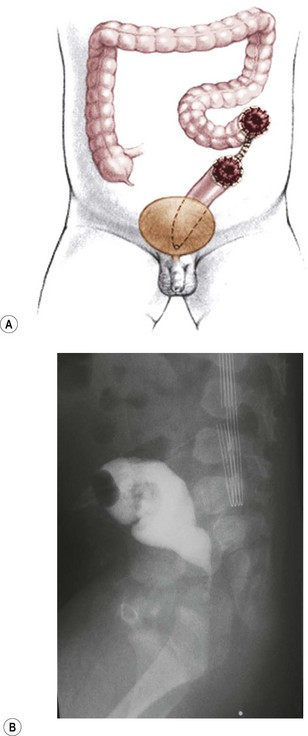
FIGURE 35-17 (A) It is important not to create the colostomy too distal because there will not be sufficient rectal length to allow for pull-through. (B) This problem is seen on the lateral view of the barium enema, where there is insufficient distal rectal length for the pull-through. This is because of an inappropriately placed colostomy and mucous fistula.
To perform the reconstruction, the patient is placed in prone position with the pelvis elevated. An electrical stimulator is used to elicit muscle contraction during the operation. The contraction serves to keep the incision in the midline, leaving an equal amount of muscle on both sides. The length of the incision varies with the type of defect and can be extended to achieve the exposure needed to result in a satisfactory repair. Thus, a perineal fistula requires a minimal posterior sagittal incision (2 cm) whereas higher defects may require a full posterior sagittal incision that runs from the lower portion of the sacrum toward the base of the scrotum in the male or to the single perineal orifice in females with a cloaca. The incision includes the skin and subcutaneous tissue and splits the parasagittal fibers, muscle complex, and levator muscles in the midline. With perineal and vestibular fistulas, the incision divides only the parasagittal fibers and the muscle complex in the midline. It is not usually necessary to open the levator muscle. Once the sphincter mechanism is divided, the next most important step of the operation is the separation of the rectum from the urogenital structures. This is the most delicate part of the procedure. Any blind maneuver at this point exposes the patient to the possibility of serious injury.63
If a colostomy has been created, the posterior sagittal approach should never be attempted without a technically adequate high-pressure distal colostogram to determine the exact position of the rectum and the fistula (Fig. 35-18).64 Attempting the repair without this critical information significantly increases the potential damage to the seminal vesicles, ureters, vas deferens, prostate, urethra, and innervation to the bladder.63
Repair of Specific Defects in Male Infants
Rectoperineal Fistula
The operation in these infants is performed in the prone position with the pelvis elevated. A urinary catheter should be inserted preoperatively. Multiple 6-0 silk stitches are placed in the fistula orifice. An incision is created dividing the sphincter mechanism located just posterior to the fistula. The incision usually measures about 2 cm in length. The sphincter is divided, and the posterior rectal wall is identified by its characteristic whitish appearance. Dissection of the rectum continues laterally following this specific plane. The last part of the dissection consists of separating the anterior rectal wall from its intimate relation to the urethra. The most common and serious complication in these relatively straightforward operations involves injury to the urethra.63 The best way to avoid urethral injury is to understand the fact that the common wall has no plane of dissection and that the surgeon must create two walls out of one.
Rectourethral Fistulas
A posterior sagittal incision is made (Fig. 35-19). The parasagittal fibers, muscle complex, and levator muscle fibers are completely divided in the midline. Sometimes, the coccyx can be split in the midline or dissected on each side with cautery, particularly in those cases of a rectoprostatic fistula when the surgeon requires more exposure in the upper part of the incision. The higher the malformation, the deeper the levator muscle. With the entire sphincter mechanism divided, the surgeon should identify the rectum. It is at this point that the importance of a good high-pressure distal colostogram cannot be overstated. If the colostogram showed the presence of a bulbourethral fistula (see Fig. 35-18), the rectum is going to be found just below the levators, with little risk of inadvertent injury to the urinary tract. In this situation, the rectum actually bulges through the incision when the sphincter mechanism is divided (Fig. 35-20). Minimal mobilization of the rectum is needed because only a short gap exists between the rectum and perineum. The anterior rectal wall must be adequately dissected free of the urethra so as not to leave behind the rectourethral fistula.
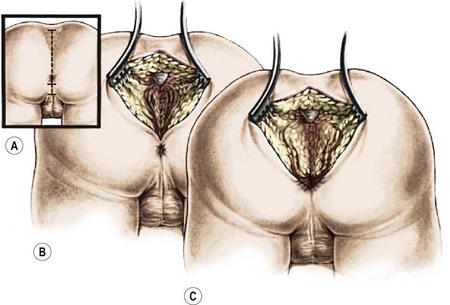
FIGURE 35-19 This drawing shows a posterior sagittal incision in a male patient with a rectourethral fistula. Separation of the parasagittal fibers and exposure of the muscle complex are shown.
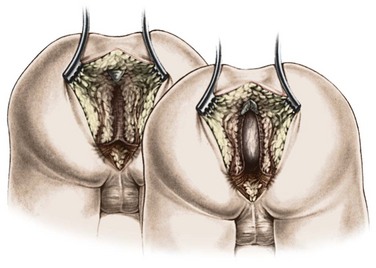
FIGURE 35-20 Schematic drawing of the posterior sagittal approach. The muscle complex and the levator muscles have been divided, and the rectum is visualized below the levator muscle complex.
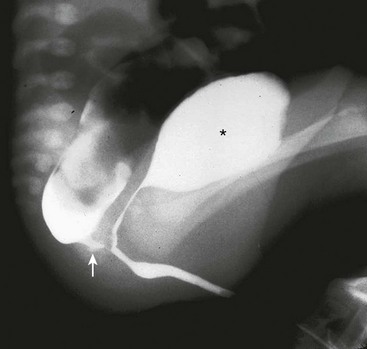
Conversely, if the preoperative distal colostogram shows a rectoprostatic fistula (Fig. 35-21), the surgeon must be particularly careful because the rectum joins the urinary tract much higher. The initial search for the rectum should be near the coccyx. Looking for the rectum lower than the coccyx risks injury to the urethra. If the colostogram shows a rectobladderneck fistula (Fig. 35-22), the posterior sagittal approach is not appropriate as a means of identifying the distal bowel. The rectum should be identified and separated from the urinary bladder through an abdominal approach (via laparoscopy or laparotomy). This is also true for a high rectoprostatic fistula.
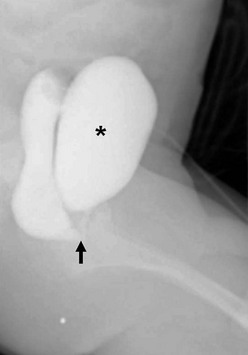
FIGURE 35-21 This distal colostogram shows the rectum entering the prostatic urethra (rectoprostatic urethral fistula) (arrow). Note filling of the bladder (asterisk) as well.
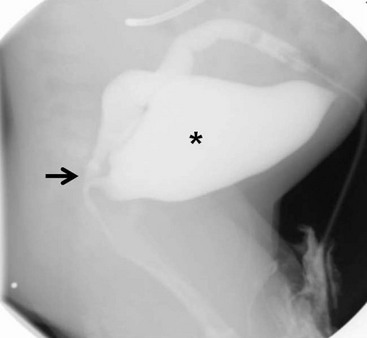
FIGURE 35-22 This distal colostogram shows a rectobladderneck fistula (arrow). The bladder is marked with an asterisk.
The anterior rectal wall above the fistula is part of a common wall, with no natural plane of separation between the urinary tract and the rectum. A plane of separation must be created in the common wall. For this, multiple 6-0 silk traction stitches are positioned in the rectal mucosa immediately above the fistula orifice. The rectal mucosa is then separated from the urethra for 5–10 mm above the fistula using a submucosal dissection (Fig. 35-23). It is this dissection that is the source of the most serious complications during this repair. We recommend creating a lateral plane of dissection on either side of the rectum to help delineate the rectal wall from the urethra and prostate. The rectum is covered by a thin fascia which contains fat, vessels, and nerves that must be preserved on the back side of the bladder. This tissue should be completely stripped from the rectum to be sure that one is working as close as possible to the rectal wall, which is the only way to prevent denervation of the bladder or injury to the vas deferens. Once the rectum is fully separated from the deep structures of the urinary tract, a circumferential perirectal dissection is performed to gain enough rectal length to reach the perineum.
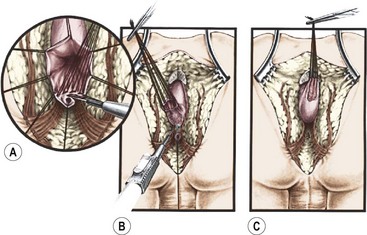
FIGURE 35-23 A schematic representation of the separation of the rectal fistula from the urethra is shown. (A) Separation of the rectum from the urethra using silk traction stitches in the rectum. (B) Proximal dissection of the rectum from the urethra. (C) Depiction of the rectum completely separated from the underlying urethra.
In babies with a rectoprostatic fistula, the perirectal dissection is considerably longer and more difficult, yet the separation from the posterior urethra is easier. During this dissection, uniform traction is applied on the multiple silk traction sutures that were placed on the rectal edges and also on the mucosa above the fistula. Uniform traction helps expose the rectal wall and allows identification of fibrous bands and vessels that hold the rectum in the pelvis. These bands must be carefully separated from the rectal wall using cautery because they contain vessels that tend to retract into the pelvis once divided. The dissection should be performed as close as possible to the rectal wall without injuring the wall. Laparoscopy can also be used here instead of a posterior sagittal dissection.62,67–69
At the completion of the dissection in prostatic fistula cases, many of the extrinsic vessels that supply the rectum have been sacrificed. The rectum should be viable, however, provided the intramural blood supply was preserved. One might think that this denervation would provoke a motility disorder, leading to severe constipation, but this has not been our experience. Patients with lower defects (who undergo less dissection) are bothered by more severe postoperative constipation than are patients with higher defects.70 There seems to be an inherent motility disorder that is more prevalent in lower defects.
The limits of the sphincter mechanism are electrically determined and marked with temporary silk stitches at the skin level. Sometimes, in patients with a good sphincter mechanism, these limits are easily visible, even without electrical stimulation. The limits of the sphincter are represented by the crossing of the muscle complex with the parasagittal fibers. One can use an electrical stimulator or a bipolar cautery to show the muscle contraction. These are the voluntary muscles that run from the levator all the way down to the skin parallel with the direction of the rectum. This muscle structure crosses the parasagittal fibers that run perpendicular and lateral to the muscle complex and parallel to the posterior sagittal incision. The perineal body is reconstructed, bringing together the anterior limits of the sphincter. The rectum must then be positioned in front of the levator and within the limits of the muscle complex (Fig. 35-24). Long-lasting 5-0 absorbable stitches are used to bring together the posterior edge of the levator muscle.
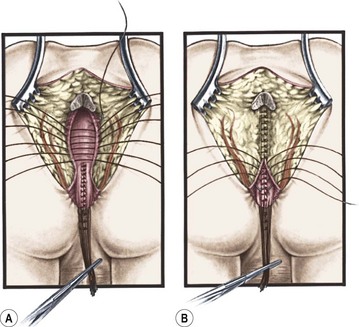
FIGURE 35-24 Position of the rectum after separation from the urethra is shown. (A) Technique of passing the rectum in front of the levator muscle complex. (B) Technique of anchoring the rectal wall to the levator complex to avoid rectal prolapse.
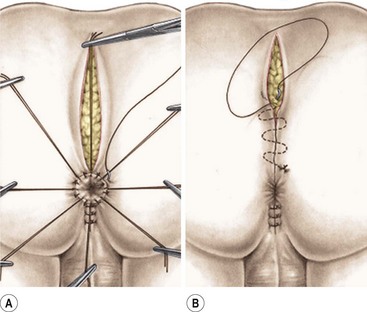
The posterior limit of the muscle complex is then reapproximated behind the rectum. These sutures should incorporate part of the rectal wall to anchor it and help avoid rectal prolapse.71 An anoplasty is performed with 16 interrupted long-lasting absorbable stitches. The ischiorectal fossa and the subcutaneous tissue are then reapproximated, and the wound is closed with subcuticular 5-0 absorbable monofilament suture (Fig. 35-25).
Rectobladderneck Fistulas
In these patients, the rectum enters the bladder neck approximately 2 cm below the peritoneal reflection. A very important anatomic feature is that the higher the malformation, the shorter the common wall between the rectum and the urinary tract. In such cases, this means that the rectum joins with the urinary tract at nearly a right angle, with little common wall. Thus, separation of the rectum from the bladder is much easier. The laparoscopic approach provides an excellent view of the peritoneal reflection, the ureters, and the vas deferens, which must be visualized to prevent injury.
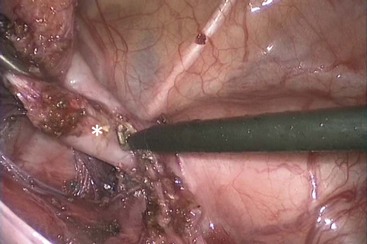
For this repair, a total body preparation is performed. The entire lower part of the patient’s body is included in the sterile field. The operation is begun laparoscopically by dividing the peritoneum around the distal rectum to create a plane of dissection to be followed distally. The dissection should occur on the rectal wall. The rectum rapidly narrows as it reaches its communication with the bladder neck (Fig. 35-26). We use as a reference the 3 mm Maryland, which should be able to completely cross the distal rectal fistula. At this point, the fistula is divided and the bladder side of the fistula is sutured or ligated with an endoloop. The vessels that supply the distal rectum are divided until there is enough length to pull the rectum comfortably down to the perineum. These vessels should be divided close to the rectal wall, preserving the inferior mesenteric artery trunk because the arcade may have been disrupted by the initial colostomy. This dissection preserves perfusion to the rectum because the rectum has an excellent intramural blood supply. In these cases, the distal rectum can be ectatic and may require tapering. If the colostomy was created too distal in the sigmoid, it may interfere with this mobilization. Once the rectum is mobile, the legs are lifted up, a small midsagittal incision is made, and a clamp is gently passed through the perineum, just anterior to the coccyx. This space is much easier to see with the opened perineum afforded by the posterior sagittal incision. Great care must be taken to avoid passing the clamp too anteriorly because injury to the bladder neck or ectopic ureters can occur. The distal rectum is grasped and pulled through to be situated in the center of the sphincter mechanism. Often, in these patients, the center of the sphincter is quite anterior, near the base of the scrotum. Traction on the rectum from below can help demonstrate further lines of tension that must be divided laparoscopically to allow the rectum to reach the perineum. The posterior edges of the muscle complex are visualized and tacked to the posterior rectal wall to help avoid prolapse.71
Rectal Atresia and Rectal Stenosis
The approach to these malformations is also posterior sagittal. The upper rectal pouch is opened and the distal anal canal is split in the posterior midline. An end-to-end anastomosis is performed (see Fig. 35-5B). If a presacral mass is identified, it is removed with presacral dissection at the same time.
Repair of Specific Defects in Female Infants
Rectovestibular Fistulas
The complexity of this defect is frequently underestimated. Multiple 5-0 silk sutures are placed at the mucocutaneous junction of the fistula. The incision is shorter than the one used to repair the male rectourethral fistula, and continues down to and around the fistula into the vestibule. Once the sphincter mechanism is divided, the surgeon identifies the posterior rectal wall by its characteristic whitish appearance. The fascia that surrounds the rectum must be removed to ensure that the dissection is as close as possible to the rectal wall. The dissection is aided by working along each side of the rectum, while applying tension on the silk traction sutures. A long common wall exists between the vagina and the rectum, and two walls must be created out of one using meticulous, delicate technique. The dissection continues cephalad until the rectal and vaginal walls are fully separated and an areolar plane between the two is encountered (Fig. 35-27). If the rectum and the vagina are not completely separated, a tense anal anastomosis predisposes the patient to dehiscence, retraction, and stricture.30,72
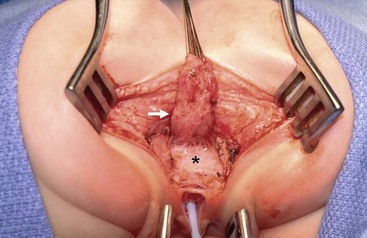
FIGURE 35-27 In this operative photograph of a female patient undergoing repair of a rectovestibular fistula, note the complete separation of the rectum (arrow) from the anteriorly positioned vagina (asterisk). A catheter has been inserted into the vagina.
Once the dissection is complete, the perineal body is repaired (Fig. 35-28). The anterior edge of the muscle complex is reapproximated as previously described. The sutures include the posterior edge of the muscle complex and the posterior rectal wall to avoid rectal prolapse.71 The anoplasty is performed as previously described for males.
Cloaca Repair
Before undertaking repair of a cloaca, the surgeon should perform an endoscopic study to determine the length of the common channel. Our review of 490 patients identified two well-characterized groups of patients with cloaca.73 These two groups have different technical challenges and must be recognized preoperatively. The first is represented by patients who are born with a common channel shorter than 3 cm. Fortunately, these patients comprise the majority and they can usually undergo repair with a posterior sagittal approach alone, avoiding a laparotomy. The operation to repair this particular variant is reproducible and can be accomplished by most pediatric surgeons. The second group is represented by patients with a long common channel (>3 cm). These patients usually need a laparotomy, and the intraoperative decision making requires a large experience, often a vaginal replacement, and special training in urology. Therefore, referral to a specialized center is recommended.
Cloacas with a Common Channel Shorter than 3 cm
The incision extends from the middle portion of the sacrum down to the single perineal orifice (Fig. 35-29). The sphincter mechanism is divided in the midline. The first structure that the surgeon finds after division of the sphincter mechanism is the rectum. However, because of the complexity of these malformations, bizarre anatomic arrangements of the rectum and vagina are sometimes encountered. The rectum is opened in the midline, and silk sutures are introduced along the edges of the posterior rectal wall. A mosquito clamp placed in the single perineal orifice facilitates extension of the incision through the posterior wall of the common channel. The entire common channel is exposed, which allows for measurement and visual confirmation of its length. The separation of the rectum from the vagina, which share a common wall, is performed as described for the repair of a rectovestibular fistula.
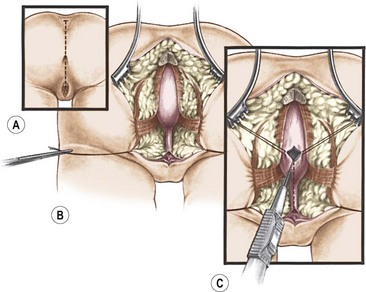
FIGURE 35-29 Drawings of the initial steps of the posterior sagittal approach for cloacal repair. (A) The incision extends from the sacral prominence to the common orifice. (B) The exposure of the rectum and common channel above the levator complex. (C) The technique of opening the rectum and placement of traction sutures in the rectal opening.
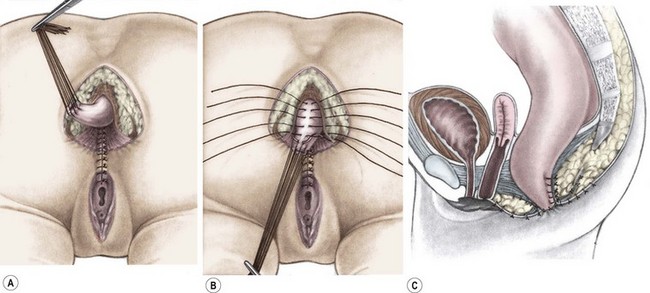
Once the rectum has been completely separated from the vagina, the total urogenital mobilization begins (Fig. 35-30), which consists of bringing both vagina and urethra to the perineum as a single unit.74 After the rectum has been mobilized from the urogenital complex, multiple silk traction sutures are placed at the edge of the vagina and the common channel in order to apply uniform traction on the urogenital sinus as it is mobilized. Another series of fine traction sutures is placed transversely approximately 5 mm proximal to the clitoris (Fig. 35-31). The urogenital sinus is transected between the last row of silk stitches and the clitoris. The anterior aspect is dissected full thickness from the pubic symphysis, taking advantage of the natural plane that exists between it and the pubis. This dissection is usually bloodless. At the upper edge of the pubis, there are fibrous, avascular suspensory ligaments that give support to the vagina and bladder. While applying traction to the multiple urogenital sinus sutures, the suspensory ligaments are divided, providing 2–3 cm of additional mobilization. Lateral and posterior dissection of the urogenital sinus will provide an additional 0.5–1.0 cm in length, allowing for complete urogenital mobilization (Fig. 35-32). Both the urethral meatus and the vaginal introitus can then be anastomosed to the perineum in the appropriate positions, which requires splitting of the common channel up to the urethra. Approximately 60% of all cloacas can be satisfactorily repaired with this technique, which has the additional advantage of preserving an excellent blood supply to both the urethra and vagina while placing the urethral opening and the smooth-walled urethra in a visible location to facilitate intermittent catheterization when necessary (Fig. 35-33). The common channel creates two lateral flaps that are sutured to the skin, creating the new labia. The vaginal edges are mobilized to reach the skin to create a surprisingly natural-looking introitus. The limits of the rectal sphincter are electrically determined, and the perineal body is reconstructed, bringing together the anterior limits of the sphincter. The rectum is placed within the margins of the sphincter (Fig. 35-34). As they have a colostomy, these neonates can eat the same day, and their pain is usually easily controlled. They are discharged 48 hours after surgery with their urinary catheter left for ten to 14 days.
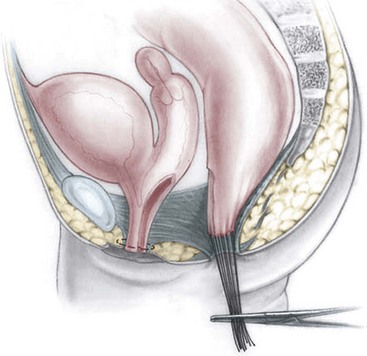
FIGURE 35-30 A schematic representation of total urogenital sinus mobilization with separation of the rectum from the vagina. (Adapted from Peña A. Total urogenital mobilization: An easier way to repair cloacas. J Pediatr Surg 1997;32:263–8.)
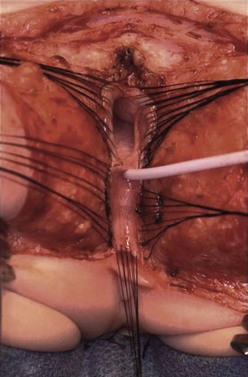
FIGURE 35-31 Total urogenital mobilization. The patient is in the prone position. Silk sutures have been placed around the vagina (top) and transversely across the dissection plane near the clitoris (bottom). A catheter has been inserted in the urethra. (From Peña A, Levitt, Treatment of Cloacas. In: Anorectal Malformations in Children. Ch 22 Holschneider AM, Hutson J, editors. Heidelberg: Springer; 2006. p. 307–14.)
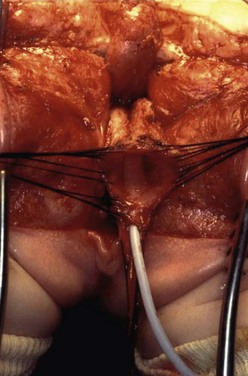
FIGURE 35-32 In this female infant with a cloaca who is in the prone position, the urogenital sinus has been fully mobilized and freed from all of its lateral, anterior, and posterior attachments. The catheter is in the urethra. (From Peña A, Levitt, Treatment of Cloacas. In: Anorectal Malformations in Children. Ch 22 Holschneider AM, Hutson J, editors. Heidelberg: Springer; 2006. p. 307–14.)
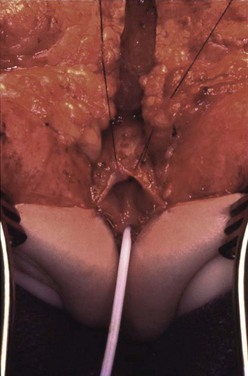
FIGURE 35-33 A nearly completed cloacal repair. The urethral meatus (with catheter) and vaginal introitus have been anastomosed to the perineum in their appropriate positions. The patient is in the prone position. (From Peña A, Levitt. In Imperforate Anus and Cloacal Malformations. In: Pediaric Surgery. 4th ed. Ashcraft, Whitfield and Murphy, editors. Philadelphia: Elsevier Saunders; 2005. p. 496–517.)
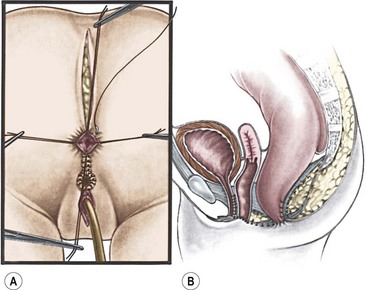
FIGURE 35-34 Drawings showing the final stages of cloacal repair. (A) The repaired urethra and vagina and the anoplasty are being completed. (B) Sagittal depiction of the finished cloacal repair. (Adapted from Peña A. Atlas of Surgical Management of Anorectal Malformations. New York: Springer-Verlag; 1990. p. 69.)
Cloacas with a Common Channel Longer than 3 cm
When endoscopy shows that the patient has a long common channel, the surgeon must be prepared to face a significant technical challenge. In the presence of a long common channel, the patient should be prepped so that the entire lower body is accessible because it is likely that the patient will require a laparotomy after initial exploration via the posterior sagittal approach. As described previously, the rectum is separated and the length of the common channel of the urogenital sinus is determined. If the vagina is low in the pelvis, a total urogenital mobilization is performed. However, if further dissection around the urogenital complex fails to gain adequate length, the urinary tract and genital tract must be separated. To accomplish this safely, a midline laparotomy is performed. The bladder is opened in the midline, and feeding tubes are placed into the ureters to protect them. The ureters run through the common wall between the vagina and bladder, and the ureteral stents allow for their identification during the difficult dissection of the vagina from the bladder neck. Sometimes, though, delivering the mobilized urogenital complex up into the abdomen allows for enough dissection to get it to reach. If further dissection does not allow it to reach then the vagina can be separated from the urinary tract and the urethra tubularized.
The patency of the Müllerian structures can be checked by injecting saline through a 3 French feeding tube through the fimbriae of the fallopian tubes. If one of the tubes is not patent, we recommend excising it, along with its hemiuterus if the system is bifid, and the ovary and its blood supply are preserved.26
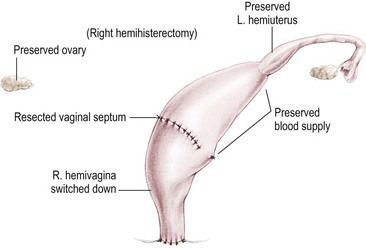
Vaginal Switch Maneuver
There is a specific group of patients who are born with hydrocolpos and two hemivaginas. The hemivaginas are very large, and the two hemiuteri are separated (Fig. 35-35). The distance between one hemiuterus and the other is longer than the vertical length of both hemivaginas. In these cases, it is ideal to perform a maneuver called a ‘vaginal switch’ (Fig. 35-36), whereby one of the uteri and its fallopian tube are resected, preserving the ovary and its blood supply. The blood supply of the ipsilateral hemivagina must be sacrificed, but collateral vessels from the opposite vagina should support both. The vaginal septum is resected, creating a single long vagina. The cut end of the ipsilateral vagina is turned down to the perineum. This is an excellent technique for constructing a viable and functional vagina.
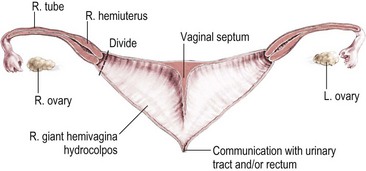
FIGURE 35-35 Diagram shows the constellation of persistent cloaca with hydrocolpos in the presence of hemivaginas and hemiuterus. Bilateral hydrocolpos is present with a very high vagina. This circumstance is the ideal anatomy for a subsequent repair via a vaginal switch maneuver. (Adapted from Kiely EM, Peña A. Anorectal malformations. In: O’Neil JA, Rowe MI, Grosfeld JL, et al, editors: Pediatric Surgery. St. Louis: Mosby–Year Book; 1998. p. 1442.)
Vaginal Augmentation and/or Replacement
1. Vaginal reconstruction with rectum. The vagina can be constructed with the rectum when the patient has a megarectum that can be divided longitudinally, preserving the mesenteric blood supply (Fig. 35-37). Occasionally, when there is adequate length, the rectum can be divided transversely, maintaining vascular pedicles for both the rectum and vagina. The blood supply of the rectum will be provided transmurally from branches off the inferior mesenteric vessels.
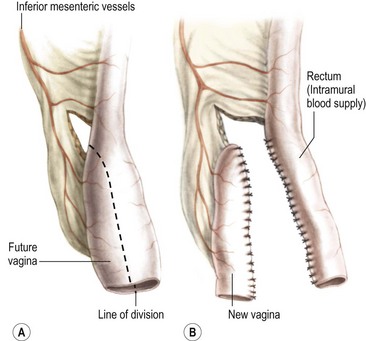
FIGURE 35-37 The technique of vaginal replacement using the rectum is depicted. The presence of a megarectum is necessary for successful vaginal reconstruction. (A) The portion of the existing rectum that will become the vagina. (B) Division of the rectum and creation of the vagina and remaining rectum.
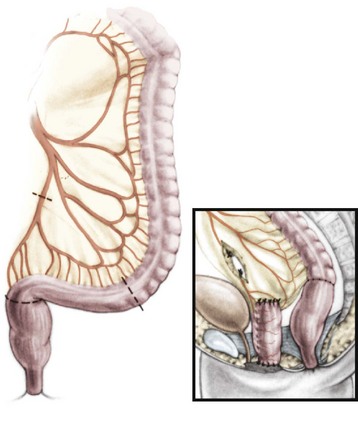
2. Vaginal replacement with colon. The colon is an ideal substitute for the vagina. When available, the left colon is preferable. This most mobile portion of the colon usually has a long mesentery. When the patient has internal genitalia or a little cuff of vagina or cervix, the upper part of the bowel used for replacement must be sutured to the vaginal cuff. When the patient has no vagina and no uterus, the neovagina is closed at its upper end and is used only for sexual purposes (Fig. 35-38).
3. Vaginal reconstruction with small bowel. If a colon segment is not available, the most mobile portion of the small bowel is utilized for vaginal reconstruction. Generally, the ileum located approximately 15 cm proximal to the ileocecal valve (Fig. 35-39) is isolated and pulled down, preserving its blood supply.
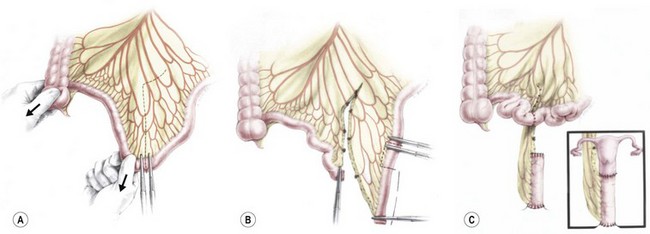
FIGURE 35-39 Technique of vaginal replacement using small bowel. (A) A segment of small bowel is chosen that has adequate mesenteric length for transposition to the pelvis. (B) The mesentery has been divided, the segment of small bowel chosen for vaginal reconstruction is identified, and the blood supply is evaluated to ensure adequate perfusion. (C) The completed anastomosis.
The most difficult type of cloacal malformation is one in which two little hemivaginas are attached to the bladder neck or even to the trigone. In these cases, the rectum also opens into the bladder neck/trigone (Fig. 35-40). Separation of these structures is performed in the abdomen, and the patient is frequently left with a nonfunctional bladder neck. The operative decision now lies between an attempt to reconstruct the bladder neck or to close it permanently. In the first situation, most of the patients will need intermittent catheterization to empty the bladder. If permanent closure of the bladder neck is elected, a vesicostomy is created, delaying a continent diversion until the patient is 3 to 4 years old. With this type of malformation, vaginal replacement should be performed using one of the substitutes described previously. Alternatively, the common channel can be left untouched, the vaginas separated from it, and the common channel then left as the catheterizable urethra.
Postoperative Management and Colostomy Closure
Postoperatively, these infants generally have a smooth course. Pain is rarely a complaint except for those who have undergone a laparotomy. After cloaca repair, the urinary catheter remains for ten to 14 days until the perineum is no longer swollen, and the patient can be recatheterized, if necessary. In very complex malformations, as with a bladder neck reconstruction, we prefer to leave a suprapubic tube or create a vesicostomy. Male infants with repaired rectourethral fistulas should have urinary catheter drainage for seven days. If the catheter becomes dislodged, the patients often can void without difficulty and do not require catheter replacement. Intravenous antibiotics are administered for 24 hours and antibiotic ointment is applied to the perineal sutures for 7 days. Most patients are discharged two days after posterior sagittal repair and 3 to 4 days if laparoscopy or laparotomy was needed.
Anal dilatations are started two weeks after repair with a dilator that fits gently into the anus. Dilation is performed twice daily by the parents at home, and the size of the dilator is increased weekly until the rectum reaches the desired size, which depends on the patient’s age (Table 35-1). Once this desired size is reached, the colostomy can be closed. The frequency of dilatation can be reduced once there is no resistance using the final dilator size. After that, the parents follow a tapering dilatation schedule that is once a day for one month, every third day for one month, twice a week for one month, once a week for one month, and once a month for three months. Anal strictures develop when the blood supply to the distal rectum is insufficient, or when the anoplasty was performed under tension.30,72
TABLE 35-1
Size of Dilator According to Age
| Age | Hegar Dilator (Number) |
| 1–4 months | 12 |
| 4–12 months | 13 |
| 8–12 months | 14 |
| 1–3 years | 15 |
| 3–12 years | 16 |
| >12 years | 17 |
At the time of colostomy closure in patients who have undergone cloacal repair, endoscopy should be performed to evaluate the repair. After the colostomy is closed, the patient may have multiple bowel movements and may develop perineal excoriation. A constipating diet may be helpful in treating this problem. After several weeks, the number of bowel movements decreases and most patients will develop constipation and need laxatives.70 After one to three months, the patient develops a more regular bowel movement pattern. A good prognosis can usually be predicted in a patient who has one to three bowel movements per day, remains clean between bowel movements, and shows evidence of feeling or pushing during bowel movements. This type of patient can be toilet trained. A patient with multiple bowel movements or one who passes stool constantly without showing any signs of sensation or pushing usually has a poor functional prognosis.
In our experience, about 20% of the patients with a cloacal malformation and a common channel shorter than 3 cm require intermittent catheterization to empty their bladder.73 Patients with common channels longer than 3 cm require intermittent catheterization 70% to 80% of the time. Upon removal of the urinary catheter, the patient is observed to see if the patient is capable of spontaneous bladder emptying. A kidney and bladder ultrasound can assess this emptying and should be performed two to three weeks after the catheter is removed and repeated every several months. If she cannot pass urine, or does not empty her bladder well, the parents need to learn how to perform intermittent catheterization.
Most patients with persistent cloaca have a flaccid, smooth, large bladder that does not empty completely.75 Fortunately, most patients with cloacas have a competent bladder neck. The combination of a competent bladder neck with a flaccid bladder makes these patients ideal candidates for intermittent catheterization, which keeps them completely dry. Two exceptions to this rule exist. One is the patient with a very long common channel and the hemivaginas attached to the bladder neck, leading to bladder neck damage during the reconstruction. The other is the rare baby born with separated pubic bones, a condition that can be described as a ‘covered exstrophy’.76,77 These patients have congenital absence of the bladder neck and will eventually require a continent urinary diversion.
Functional Disorders after Repair of Anorectal Malformations
Most patients who undergo repair of an anorectal malformation suffer from some degree of a functional defecating disorder.78,79
Fecal continence depends on three main factors:
1. Voluntary muscle structures. These structures are represented by the levator muscle, the striated muscle complex, and the external sphincter. Normally, they are used only for brief periods when the rectal fecal mass, pushed by the involuntary peristaltic contraction of the rectosigmoid, reaches the anorectal area. This contraction occurs only in the minutes prior to defecation. The voluntary muscle structures that close around the anus are used only occasionally during the rest of the day and night. Patients with anorectal malformations have abnormal voluntary striated muscles with varying degrees of hypodevelopment. Voluntary muscles can be used only when the patient feels that it is necessary to use them.
2. For that sensation, the patient needs information that can only be derived from an intact sensory mechanism, a mechanism lacking in many patients with anorectal malformations. Exquisite sensation in normal individuals resides in the anal canal. Except for patients with rectal atresia and stenosis, most patients with anorectal malformations are born without an anal canal. Therefore, sensation does not exist or is rudimentary. Distention of the rectum, however, can be felt in many of these patients, provided the rectum has been located accurately within the muscle structures. This proprioception seems to be a consequence of stretching of the voluntary muscle. The most important clinical implication is that liquid or soft fecal material may not be felt by the patient with anorectal malformations as the rectum is not distended. Thus, to achieve some degree of sensation and bowel control, the patient must be able to (or helped to) form solid stool.
3. Bowel motility. Perhaps the most important factor in fecal continence is bowel motility. In a normal individual, the rectosigmoid remains quiet for variable periods of time (one to several days), depending on an individual’s defecation habits. During that time, anorectal sensation and voluntary muscle structures are almost unnecessary because the stool remains in the rectosigmoid if it is solid.
The patient normally feels the rectosigmoid’s peristaltic contraction. Voluntarily, the normal individual can relax the striated muscles, which allows the rectal contents to migrate down into the highly sensitive area of the anal canal. There, accurate information is provided concerning the consistency and quality of the stool. The voluntary muscles are used to push the rectal contents back up into the rectosigmoid and hold them if desired until the appropriate time for evacuation. At the time of defecation, the voluntary muscle structures relax, allowing the fecal mass to pass into and through the anorectum.
The main factor that initiates emptying of the rectosigmoid is an involuntary peristaltic contraction that is helped sometimes by a Valsalva maneuver. Most patients with an anorectal malformation have a disturbance of this sophisticated bowel motility mechanism. Patients who have undergone a posterior sagittal anorectoplasty or any other type of sacroperineal approach, in which the most distal part of the bowel was preserved, often show evidence of an overefficient bowel reservoir (megarectum) (Fig. 35-41). The main clinical manifestation of this is constipation, which seems to be more severe in patients with lower defects.70 The enormously dilated rectosigmoid has normal ganglion cells, but behaves like it has a hypomotility disorder. The overflow fecal incontinence based on rectosigmoid constipation in patients with potential for bowel control can be managed with the appropriate dose of stimulant laxatives. Those with a poor sacrum, poor muscles, and thus with no potential for bowel control are treated with a daily enema.78
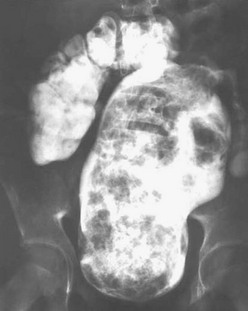
FIGURE 35-41 A contrast enema is shown in a patient with a megarectum. Note the markedly dilated rectum in relation to the more proximal normal-sized colon.
Those patients treated with older techniques in which the most distal part of the bowel was resected (abdominoperineal pull-through) behave clinically as individuals without a rectal reservoir (Fig. 35-42). This is a situation equivalent to a perineal colostomy. Depending on the amount of colon resected, the patient may have loose stools. In these cases, medical management consisting of a daily enema and a constipating diet, with medications to slow down the colonic motility, is indicated.78 See Chapter 36 for more information on bowel management.
Evaluation of Results
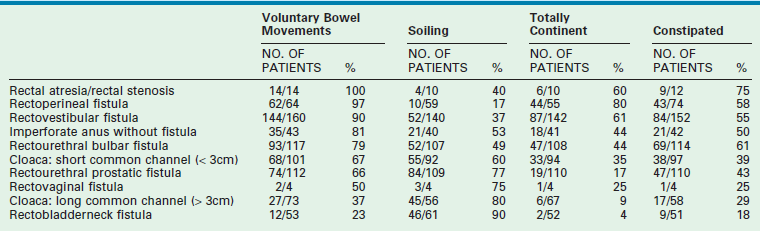
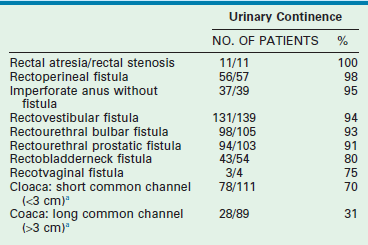
Tables 35-2 and 35-3 show our results. The patients with a sacral ratio of less than 0.3 and flat perineums have fecal incontinence regardless of the type of malformation or quality of the repair. Associated spinal anomalies negatively affect progress for bowel control.
Complications
There are several complications related to the operative repair of anorectal malformations. Wound infection in the immediate postoperative period can occur and, fortunately, usually affects only the skin and subcutaneous tissue. These usually heal secondarily without functional sequelae. Sometimes a wound separation can be resutured in the immediate postoperative period. Anal strictures may be the consequence of failure to follow the protocol of dilatations or leaving the anoplasty under tension and/or with inadequate blood supply. When trying to prevent discomfort for the patient, some surgeons only dilate the anus once a week, frequently under anesthesia. This protocol can eventually create a severe, intractable fibrous stricture.72 Constipation is the most common functional disorder observed in patients who undergo a posterior sagittal anorectoplasty.70 Patients with the best prognosis for bowel control have the highest incidence of constipation. Patients with a poor prognosis, such as those with bladder neck fistula, have a low incidence of constipation and a high rate of incontinence. Colostomies that do not allow cleaning and irrigation of the distal colon can lead to megarectum. Patients require proactive aggressive treatment of constipation after colostomy closure to avoid significant problems.70 Rectal prolapse occurs on occasion and is more prevalent in higher malformations with poor sphincters and is worsened by postoperative constipation.71 Finally, transient femoral nerve palsy can occur as a consequence of excessive pressure on the groin during the prone position. This problem can be avoided by adequate cushioning.
Specific to cloacal repairs, urethrovaginal fistulas had been the most common and feared complication prior to the introduction of the total urogenital mobilization technique.74 In cases in which vaginal mobilization and separation from the neourethra result in opposing suture lines, 90° rotation of the vagina can decrease the incidence of a postoperative fistula. Fibrosis of the vagina can develop secondary to an excessive dissection during the mobilization of a high vagina.72
In males, a review of over 500 patients showed significant urologic injuries.63 Failure to obtain a good distal colostogram to delineate the anatomy precisely was the most important reason for these complications. Neurogenic bladder in males, as a result of the anorectal malformation or the repair itself, must be extremely unusual because it only happens in patients with an abnormal sacrum or spine.75 Otherwise, it may reflect a poor operative technique with denervation of the bladder and bladder neck during the repair.63
Finally, fecal incontinence is a very common sequelae of any anorectal malformation repair and is discussed further in Chapter 36.
References
1. Aegineta, P, On the imperforate anus Adams F (trans). The Seven Books (book 6). Sydenham Society: London. 1844; 405–406.
2. Amussat, JZ. Gustiure d’une operation d’anus artifical practique avec success par un nouveau procede. Gaz Med Paris. 1835; 3:735–758.
3. Roux de Brignoles, JN. De l’imperforation de l’anus chez les nouveaux-nex: Rapport et discussion sur l’operation à tenter dans ces cas. Gaz Med Paris. 1834; 2:411–412.
4. Stephens, FD. Imperforate rectum: A new surgical technique. Med J Aust. 1953; 1:202–206.
5. Kiesewetter, WB. Imperforate anus: II. The rationale and technique of sacroabdominoperineal operation. J Pediatr Surg. 1967; 2:106–117.
6. Louw, JH, Cywes, S, Cremin, BJ. The management of anorectal agenesis. S Afr J Surg. 1971; 9:21–30.
7. Rehbein, F. Imperforate anus: Experiences with abdominoperineal and abdomino sacroperineal pull-through procedures. J Pediatr Surg. 1967; 2:99–105.
8. Soave, F. Surgery of the rectal anomalies with preservation of the relationship between the colonic muscular sleeve and puborectal muscle. J Pediatr Surg. 1969; 4:705–712.
9. Peña, A, deVries, P. Posterior sagittal anorectoplasty: Important technical considerations and new applications. J Pediatr Surg. 1982; 17:796–881.
10. Brenner, EC. Congenital defects of the anus and rectum. Surg Gynecol Obstet. 1915; 20:579–588.
11. Santulli, TV. Treatment of imperforate anus and associated fistulas. Surg Gynecol Obstet. 1952; 95:601–614.
12. Trusler, GA, Wilkinson, RH. Imperforate anus: A review of 147 cases. Can J Surg. 1962; 5:169–177.
13. Mundt, E, Bates, M. Genetics of Hirschsprung disease and anorectal malformations. Semin Pediatr Surg. 2010; 19:107–117.
14. Anderson, RC, Read, SC. The likelihood of recurrence of congenital malformations. Lancet. 1954; 74:175–176.
15. Cozzi, F, Wilinson, AW. Familial incidence of congenital malformations. Lancet. 1954; 74:175–176.
16. Murken, JD, Albert, A. Genetic counseling in cases of anal and rectal atresia. Progr Pediatr Surg. 1976; 9:115–118.
17. Falcone, RA, Levitt, MA, Peña, A, et al. Increased heritability of certain phenotypes. J Pediatr Surg. 2007; 42:124–128.
18. Levitt, MA, Peña, A. Anorectal Malformations. In: Orphanet, [serial online] http://www.OJRD.com/content/2/1/33.
19. Torres, P, Levitt, MA, Tovilla, JM, et al. Anorectal malformations and Down syndrome. J Pediatr Surg. 1998; 33:1–5.
20. Stephens, FD, Smith, ED. Incidence, frequency of types, etiology. In: Stephens FD, Smith ED, Paul NW, eds. Anorectal Malformations in Children. Chicago: Year Book Medical; 1971:160–171.
21. Rosen, NG, Hong, AR, Soffer, SZ, et al. Recto-vaginal fistula: A common diagnostic error with significant consequences in female patients with anorectal malformations. J Pediatr Surg. 2002; 37:961–965.
22. Bill, AH, Hall, DG, Johnson, RJ. Position of rectal fistula in relation to the hymen in 46 girls with imperforate anus. J Pediatr Surg. 1975; 10:361–365.
23. Levitt, MA, Peña, A. Pitfalls in the management of newborn cloacas. Pediatr Surg Int. 2005; 21:264–269.
24. Holschneider, A, Hutson, J, Peña, A, et al. Preliminary report on the international conference for the development of standards for the treatment of anorectal malformations. J Pediatr Surg. 2005; 40:1521–1526.
25. Lee, SC, Chen, YS, Jung, SE, et al. Currarino triad: Anorectal malformation, sacral bony abnormality, and presacral mass—A review of 11 cases. J Pediatr Surg. 1997; 32:58–61.
26. Breech, L. Gynecological concerns in patients with anorectal malformations. Semin Pediatr Surg. 2010; 19:139–145.
27. Moore, TC. Advantages of performing the sagittal anoplasty operation for imperforate anus at birth. J Pediatr Surg. 1990; 25:276–277.
28. Upadhyaya, VD, Gopal, SC, Gangopahyaya, AN, et al. Single stage repair of anovestibular fistula in neonate. Pediatr Surg Int. 2007; 23:737–740.
29. Menon, P, Rao, KL. Primary anorectoplasty in females with common anorectal malformations without colostomy. J Pediatr Surg. 2007; 42:1103–1106.
30. Peña, A, Grasshoff, S, Levitt, A. Reoperations for anorectal malformations. J Pediatr Surg. 2007; 42:318–325.
31. Bianchi, DW, Crombleholme, TM, D’Alton, ME, et al. Cloacal exstrophy. In Fetology: Diagnosis and Management of the Fetal Patient, 2nd ed, New York: McGraw-Hill; 2010:446–453.
32. Livingston, J, Eliçevik, M, Crombleholme, T, et al. Prenatal diagnosis of persistent cloaca: A 10-year review. Am J Obstet Gynecol. 2006; 195:S63.
33. Levitt, MA, Stein, DM, Peña, A. Gynecological concerns in the treatment of teenagers with cloaca. J Pediatr Surg. 1998; 33:188–193.
34. Stoll, C, Alembik, Y, Dott, B. Associated malformations in patients with anorectal anomalies. Eur J Med Genet. 2007; 50:281–290.
35. Karrer, FM, Flannery, AM, Nelson, MD, Jr., et al. Anorectal malformations: Evaluation of associated spinal dysraphic syndromes. J Pediatr Surg. 1988; 23:45–48.
36. Davidoff, AM, Thompson, CV, Grimm, JK, et al. Occult spinal dysraphism in patients with anal agenesis. J Pediatr Surg. 1991; 26:1001–1005.
37. Daskiewicz, P, Barszsc, S, Roskowski, M, et al. Tethered cord syndrome in children—impact of surgical treatment on functional neurological and urological outcome. Neurol Neurochir Pol. 2007; 41:427–435.
38. Bui, CJ, Tubbs, RS, Oakes, WJ. Tethered cord syndrome in children: A review. Neurosurg Focus. 2007; 23:1–9.
39. Tsuda, T, Iwai, N, Kimura, O, et al. Bowel function after surgery for anorectal malformations in patients with tethered spinal cord. Pediatr Surg Int. 2007; 23:1171–1174.
40. Kuo, MF, Tsai, Y, Hsu, WM, et al. Tethered spinal cord and VACTERL association. J Neurosurg. 2007; 106:201–204.
41. Steinbok, P, Garton, HJ, Gupta, N. Occult tethered cord syndrome: A survey of practice patterns. J Neurosurg. 2006; 104:309–313.
42. Drake, JM. Occult tethered cord syndrome: Not an indication for surgery. J Neurosurg. 2006; 104:305–308.
43. Seldon, NR. Occult tethered cord syndrome: The case for surgery. J Neurosurg. 2006; 104:302–304.
44. Levitt, MA, Patel, M, Rodriguez, G, et al. The tethered spinal cord in patients with anorectal malformations. J Pediatr Surg. 1997; 32:462–468.
45. Belman, BA, King, LR. Urinary tract abnormalities associated with imperforate anus. J Urol. 1972; 108:823–824.
46. Hoekstra, WJ, Scholtmeijer, RJ, Molenar, JC, et al. Urogenital tract abnormalities associated with congenital anorectal anomalies. J Urol. 1983; 130:962–963.
47. Munn, R, Schillinger, JF. Urologic abnormalities found with imperforate anus. Urology. 1983; 21:260–264.
48. Parrott, TS. Urologic implications of anorectal malformations. Urol Clin North Am. 1985; 12:13–21.
49. Wiener, ES, Kiesewetter, WB. Urologic abnormalities associated with imperforate anus. J Pediatr Surg. 1973; 8:151–157.
50. William, DI, Grant, J. Urological complications of imperforate anus. Br J Urol. 1969; 41:660–665.
51. Rich, MA, Brock, WA, Peña, A. Spectrum of genitourinary malformations in patients with imperforate anus. Pediatr Surg Int. 1988; 3:110–113.
52. Stephens, FD, Smith, ED. Incidence, frequency of types, etiology. In: Stephens FD, Smith ED, eds. Anorectal Malformations in Children. Chicago: Year Book Medical; 1971:289–292.
53. Spence, HM. Anomalies and complications of the urogenital tract associated with congenital imperforate anus. J Urol. 1954; 71:453–463.
54. Smith, ED. Urinary anomalies and complications in imperforate anus and rectum. J Pediatr Surg. 1968; 3:337–342.
55. Carcassonne, M, Monfort, G, Isman, H. Les problemes urologiques des malformations ano-rectales. Arch Fr Pediatr. 1971; 28:723–739.
56. Boemers, TM. Neurogenic bladder in infants born with anorectal malformations: Comparison with spinal and urologic status. J Pediatr Surg. 1999; 34:1889–1890.
57. Boemers, TM, de Jong, TP, van Gool, JD, et al. Urologic problems in anorectal malformations: II. Functional urologic sequelae. J Pediatr Surg. 1996; 31:534–537.
58. Wangensteen, OH, Rice, CO. Imperforate anus: A method of determining the surgical approach. Ann Surg. 1930; 92:77–81.
59. Shaul, DB, Harrison, EA. Classification of anorectal malformations—initial approach, diagnostic tests, and colostomy. Semin Pediatr Surg. 1997; 6:187–195.
60. Freeman, NV, Burge, DM, Soar, JS, et al. Anal evoked potentials. Z Kinderchir. 1980; 31:22–30.
61. Albanese, CT, Jennings, RW, Lopoo, JB, et al. One-stage correction of high imperforate anus in the male neonate. J Pediatr Surg. 1999; 34:834–836.
62. Vick, LR, Gosche, JR, Boulanger, SC, et al. Primary laparoscopic repair of high imperforate anus in neonatal males. J Pediatr Surg. 2007; 42:1877–1881.
63. Hong, AR, Rosen, N, Acuña, MF, et al. Urological injuries associated with the repair of anorectal malformations in male patients. J Pediatr Surg. 2002; 37:339–344.
64. Gross, GW, Wolfson, PJ, Peña, A. Augmented-pressure colostogram in imperforate anus with fistula. Pediatr Radiol. 1991; 21:560–563.
65. Peña, A, Migotto-Krieger, M, Levitt, MA. Colostomy in anorectal malformations: A procedure with serious but preventable complications. J Pediatr Surg. 2006; 41:748–756.
66. Wilkins, S, Peña, A. The role of colostomy in the management of anorectal malformations. Pediatr Surg Int. 1988; 3:105–109.
67. Sydorak, RM, Albanese, CT. Laparoscopic repair of high imperforate anus. Semin Pediatr Surg. 2002; 11:217–225.
68. Georgeson, K. Laparoscopic-assisted anorectal pull-through. Semin Pediatr Surg. 2007; 16:266–269.
69. Georgeson, KE, Inge, TH, Albanese, CT. Laparoscopically assisted anorectal pull-through for high imperforate anus: A new technique. J Pediatr Surg. 2000; 35:927–931.
70. Levitt, MA, Kant, A, Peña, A. The morbidity of constipation in patients with anorectal malformations. J Pediatr Surg. 2010; 45:1228–1233.
71. Belizon, A, Levitt, MA, Shoshany, G, et al. Rectal prolapse following posterior sagittal anorectoplasty for anorectal malformations. J Pediatr Surg. 2005; 40:192–196.
72. Levitt, MA, Peña, A. Reoperations in Anorectal Malformations. In: Teich S, Caniano D, eds. Reoperative Pediatric Surgery. Totowa: Humana Press; 2008:311–326.
73. Levitt, MA, Peña, A. Cloacal malformations: Lessons learned from 490 cases. Semin Pediatr Surg. 2010; 19:128–138.
74. Peña, A. Total urogenital mobilization—An easier way to repair cloacas. J Pediatr Surg. 1997; 32:263–268.
75. Peña, A, Levitt, MA. Neurogenic bladder and anorectal malformations. In: Esposito C, Guys JM, Gough D, et al, eds. Pediatric Neurogenic Bladder Dysfunction. Berlin: Springer; 2006:85–88.
76. Peña, A. New concepts in bowel reconstruction in cloacal exstrophy. Dialog Pediatr Urol. 2000; 23:3–4.
77. Levitt, MA, Mak, GA, Falcone, RA, et al. Cloacal exstrophy—pull through or permanent stoma? A review of 53 patients. J Pediatr Surg. 2008; 43:164–170.
78. Levitt, MA, Peña, A. Pediatric fecal incontinence: A surgeon’s perspective. Pediatr Review. 2010; 31:91–101.
79. Peña, A, Levitt, MA. Colonic inertia disorders. Curr Prob Surg. 2002; 39:661–730.