[level-membership-for-allergy-and-immunology-category]
Immunoproliferative Disorders
At the conclusion of this chapter, the reader should be able to:
• Compare the general characteristics of monoclonal and polyclonal gammopathies.
• Describe and compare the etiology, epidemiology, signs and symptoms, immunologic manifestations, diagnostic evaluation, and treatment of multiple myeloma and Waldenström’s primary macroglobulinemia.
• Explain and contrast the characteristics of other monoclonal disorders, such as monoclonal gammopathy of unknown significance.
• Analyze a case study related to immunoproliferation.
• Correctly answer case study related multiple choice questions.
• Be prepared to participate in a discussion of case study related critical thinking questions.
• Describe the principle and application of the Bence Jones Protein Screening Procedure.
Multiple Myeloma
Pathophysiology
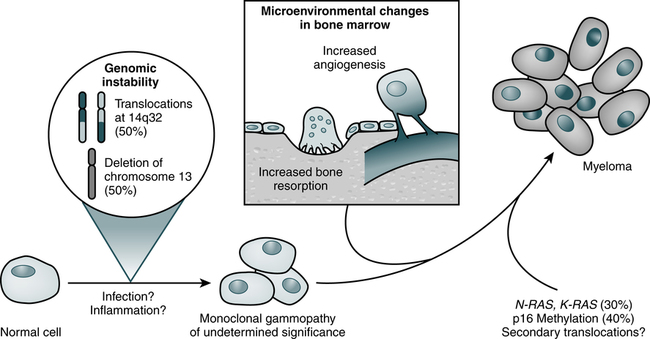
Myeloma cells proliferate slowly in the marrow (Fig. 27-1; Table 27-1). Less than 1% divide at any one time and myeloma cells do not differentiate. The absolute number of these cells correlates with disease activity and predicts the progression of disease in smoldering multiple myeloma. Circulating myeloma cells may disseminate the tumor within the bone marrow and elsewhere.
Table 27-1
Three Phases of Disease Progression in Multiple Myeloma
| Variable | Initial Phase | Medullary Relapse | Extramedullary Relapse |
| Site of myeloma-cell accumulation or proliferation | Bone marrow | Bone marrow | Blood, pleural effusion, skin, many other sites |
| Growth fraction∗ | <1% | ≥1% (1%-95%) | ≥1% (1%-95%) |
| Genetic or oncogenic events | Deregulation of c-myc Illegitimate switch recombinations |
N-ras and K-ras point mutations | p53 point mutations |
| Phenotypic changes | CD19 loss CD56 overexpression |
CD28 expression LFA-1 and VLA-5 loss |
CD28 expression CD56 loss |
| Cytologic changes | Detectable plasmablastic compartment in 15% of cases | Plasmablastic compartment growing | Major plasmablastic compartment |
| Circulating malignant plasma cells | <1% | Increasing | Increasing |
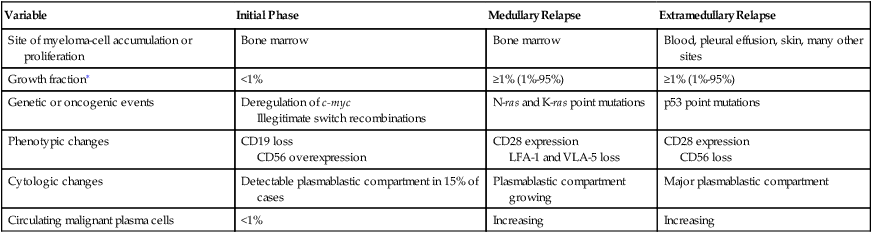
∗Growth fraction is the rate of atypical cells proliferating in the bone marrow.
From Bataille R, Harousseau JL: Medical progress: multiple myeloma, N Engl J Med 336:1657–1664, 1997.
Epidemiology
IgG myeloma is the most common form of MM (Table 27-2). Four subtypes of IgG heavy chains are known to exist among patients with IgG myeloma. Cases of IgG myeloma are distributed as follows: 65% are gamma G1, 23% gamma G2, 8% gamma G3, and 4% gamma G4 subclass. The only subclass-dependent difference is the greater propensity for patients with IgG3 myeloma to experience hyperviscosity syndrome, similar to the manifestation in WM.
Table 27-2
Distribution of Immunoglobulin Types in Patients With Multiple Myeloma
| Type of Protein | Multiple Myeloma (%) |
| IgM | 12 |
| IgG | 52 |
| IgA | 22 |
| IgD | 2 |
| IgE | Rare |
| Light chains (kappa or lambda) | 11 |
| Heavy chains | Rare |
| Monoclonal proteins | <1 |
| Nonsecretory myeloma | 1 |
Signs and Symptoms
Skeletal Abnormalities
About 90% of patients with MM have broadly disseminated destruction of the skeleton, which is responsible for the predominance of bone pain. These abnormalities consist of punched-out lytic areas (Fig. 27-2), osteoporosis, and fractures in about 80% of patients. The vertebrae, skull, thoracic cage, pelvis, and proximal humeri and femurs are the most frequent sites of involvement.
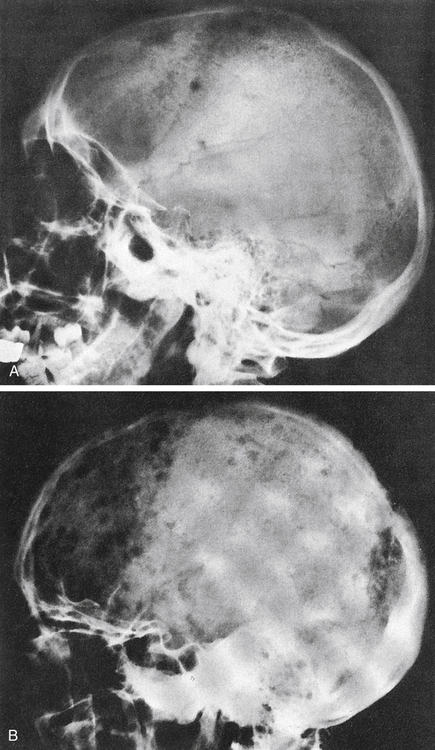
A, Several scattered, small, well-marginated lytic lesions appear in calvarium, located in normally mineralized bone. Multiple lytic lesions can also be seen in the mandible. B, Multiple circumscribed lytic lesions crowd bones throughout skull. Lesions are still discrete and margins of most are fairly sharp. (From Newton TH, Potts DG: Radiology of the skull and brain, St Louis, 1971, Mosby.)
Diagnostic Evaluation
Hematologic Assessment
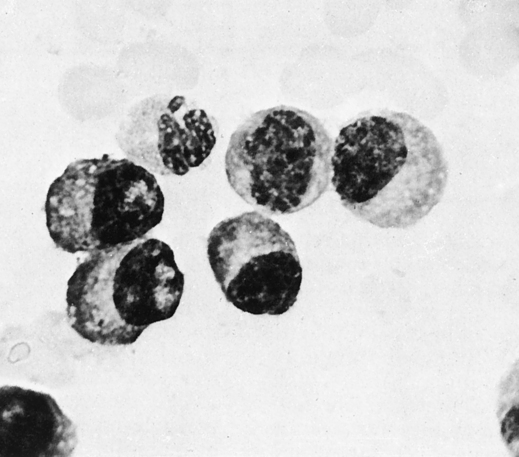
Diagnosis of MM, however, depends on the demonstration of an increased number (>10%) of plasma cells in a bone marrow aspirate (Fig. 27-3; see Color Plate 12) and/or biopsy and supporting laboratory results. Cytogenetic analysis or fluorescence in situ hybridization (FISH) of bone marrow aspirate is recommended.
Free Light Chains
Free light chains are incorporated into immunoglobulin molecules during B lymphocyte development and expressed initially on the surface of immature B cells. Production of FLCs occurs throughout the rest of B cell development and in plasma cells, in which secretion is highest. Tumors associated with the different stages of B cell maturation will secrete monoclonal FLCs into the serum, where they may be detected by FLC immunoassays (Box 27-1; Table 27-3).
Table 27-3
| Assay | Advantages | Disadvantages |
| Total urine protein | Simple, inexpensive, widely used | Inadequate sensitivity for FLC detection |
| Urine dipstick | Simple, inexpensive, widely used | Inadequate sensitivity for FLC detection |
| Serum protein electrophoresis | Simple, manual or semiautomated method Well established, inexpensive Monoclonal bands observed Quantitative results with scanning |
Insensitive (<500-2000 mg/L) Cannot detect FLCs at low concentration Subjective interpretation of results |
| Urine protein electrophoresis | Simple, manual, or semiautomated method Well established, inexpensive Monoclonal bands observed Sensitive in concentrated urine (10 mg/L) Quantitative results with scanning |
Subjective interpretation of results Urine may require concentration, with possible protein loss False bands from concentrating urine Heavy proteinuria obscures results Cumbersome 24-hour urine collection |
| Immunofixation electrophoresis (IFE) on serum and urine | Well established Good sensitivity for serum, very sensitive for concentrated urine (5-30 mg/L) |
Nonquantitative Serum sensitivity (150-500 mg/L) inadequate for normal serum FLC levels Rather laborious to perform Visual interpretation may be difficult Expensive use of antisera Cannot be used to quantify monoclonal immunoglobulins because of precipitating antibody |
| Capillary zone electrophoresis | Automated technology Quantitative |
Less sensitive (400 mg/L) than IFE for serum FLCs Can fail to detect 5% of positive samples (false-negatives) |
| Total serum κ and λ assays | Automated immunoassay | Not sensitive enough for routine testing Specificity inadequate for detecting many patients with light-chain multiple myeloma |
Adapted from Bradwell AR: Serum free light chain analysis, Birmingham, UK, 2006, Binding Site, pp 23, 47-52.
Immunologic Testing
The identification of κ and λ molecules has been accomplished with the use of antibodies specific for each type of protein. Immunodiffusion was initially used, followed by immunoelectrophoresis (in 1953), radial immunodiffusion, and ultimately nephelometry and turbidimetry. An automated nephelometric assay, described in 2001, represented a major breakthrough. This methodology allows for the quantitation of both κ and λ free light chains and can be performed using automated chemistry analyzers (e.g., Dade Behring [now Siemens AG], Beckman Coulter, Roche Hitachi, Olympus) (see Chapter 13).
Each monoclonal protein (M protein or paraprotein) consists of two heavy-chain polypeptides of the same class and subclass and two light-chain polypeptides of the same type. The different monoclonal proteins are designated by capital letters corresponding to the class of their heavy chains, which are designated by Greek letters: gamma (γ) in IgG, alpha (α) in IgA, mu (µ) in IgM, delta (δ) in IgD, and epsilon (ε) in IgE. The subclasses are IgG1, IgG2, IgG, and IgG4, or IgA1 and IgA2, and their light-chain types are κ and λ. A monoclonal protein is characterized by a narrow peak or localized band on electrophoresis, by a thickened bowed arc on immunoelectrophoresis, and by a localized band on immunofixation. Many different entities are associated with M proteins (monoclonal gammopathies; Box 27-2).
Electrophoresis of the serum or urine reveals a tall sharp peak on the densitometer tracing or a dense localized band in most cases of multiple myeloma (Fig. 27-4). A monoclonal protein is demonstrable in the serum and urine in 90% of patients. In all, 60% of patients exhibit IgG, 20% IgA, 10% light chain only (BJ proteinemia), and 1% IgD. Electrophoresis of urine shows a globulin peak in 75% of cases, mainly albumin in 10% of patients, and a normal pattern in 15%. When an M spike is observed on serum protein electrophoresis, the suggested sequence of testing includes testing by immunoelectrophoresis and immunofixation (Table 27-4). Screening for cryoglobulins and viscosity may also be warranted.
Table 27-4
Suggested Sequence of Immunologic Testing for Monoclonal Proteins
| M Spike on Serum Protein Electrophoresis | |
| Serum | Urine |
| Immunoelectrophoresis | Screening of urine for increased protein, (e.g., sulfosalicylic acid) |
| Immunofixation | Total protein assay of a 24-hr urine specimen |
| Quantitation of immunoglobulins by radial immunodiffusion or nephelometry | Urinary protein electrophoresis |
| Screening for cryoglobulins | Urinary immunoelectrophoresis |
| Determination of serum viscosity if IgM, IgA, or IgG or signs and symptoms suggestive of hyperviscosity | Immunofixation |

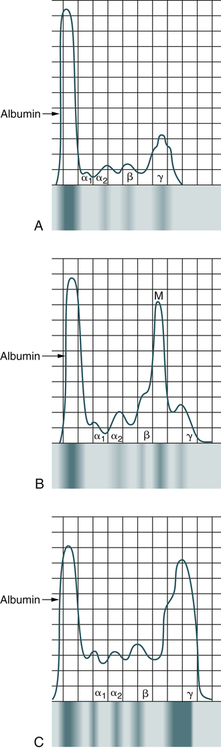
A, Normal patient. B, Patient with multiple myeloma. C, Patient with Waldenström’s macroglobulinemia.
Immunoelectrophoresis, also called gamma globulin electrophoresis or immunoglobulin electrophoresis, is a method of determining the blood levels of three major immunoglobulins—IgM, IgG, and IgA—based on their combined electrophoretic and immunologic properties (see Chapter 11). Immunoelectrophoresis is also used frequently to diagnose MM, which affects the bone marrow. Drugs that may cause increased immunoglobulin levels include therapeutic gamma globulin, hydralazine, isoniazid, phenytoin (Dilantin), procainamide, oral contraceptives, methadone, steroids, and tetanus toxoid and antitoxin. The laboratory should be notified if the patient has received any vaccinations or immunizations in the 6 months before the test. Prior immunizations lead to increased immunoglobulin levels, resulting in false-positive results. Because immunoelectrophoresis is not quantitative, it is being replaced by immunofixation, which is more sensitive and easier to interpret.
Treatment
Asymptomatic (smoldering) myeloma requires only clinical observation because early treatment with conventional chemotherapy has shown no benefit. Recently, the introduction of autologous stem cell transplantation (see Chapter 32) as a mainstay of myeloma therapy and the availability of agents such as thalidomide, lenalidomide, and bortezomib have changed the medical management of active (symptomatic) myeloma and extended overall survival. New proteasome inhibitors, immunomodulatory drugs (pomalidomide), targeted therapies, epigenetic agents, and humanized monoclonal antibodies are currently undergoing clinical trial investigations.
Waldenström’s Primary Macroglobulinemia
Etiology
Diagnostic Evaluation
Hematologic Assessment
• Faulty platelet adhesiveness
• Defective platelet aggregation
• Abnormal release of platelet factor 3
Immunologic Assessment
• Type I is composed of a single class. IgM and IgG classes are most common; IgA or light-chain, single cryoglobulins are seen less frequently. Type I constitutes about 25% of cryoglobulins and is generally associated with multiple myeloma, macroglobulinemia, and other, rarer neoplastic proliferations of plasma cells and lymphocytes.
• Type II cryoglobulins consist of two forms. The monoclonal form always has rheumatoid factor activity and usually is an IgM with κ light chains. The second form is polyclonal IgG, which reacts with the monoclonal IgM rheumatoid factor.
• Type III is a mixed cryoglobulin in which both constituent immunoglobulins are polyclonal. More than 90% of type III cryoglobulins contain IgM rheumatoid factor and IgG. Type III cryoglobulins are seen in a variety of autoimmune, systemic rheumatic diseases and persistent infections with immune complexes (e.g., bacterial endocarditis).
Other Monoclonal Disorders
Monoclonal Gammopathy of Undetermined Significance
The International Myeloma Working Group has established the differences between MGUS and plasma cell neoplasms (Table 27-5). Characteristics of MGUS include the following:
Table 27-5
| MGUS | Smoldering Multiple Myeloma | Multiple Myeloma | Waldenström’s Macroglobulinemia | |
| Bone marrow plasma cells | <10% and |
≥10% and/or |
≥10% and/or |
>10% and <10% lymphoplasmacytoid cells |
| Circulating monoclonal protein | <3 g/dL | ≥3 g/dL | ≥3 g/dL | >3 g/dL |
| Clinical signs and symptoms | Absent | Absent | Present | Present |

Adapted from International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders, Br J Haematol 121:749–757, 2003.
• Serum monoclonal protein concentration less than 3 g/dL
• Fewer than 10% plasma cells in the bone marrow
• Absence of lytic bone lesions
• No clinical signs or symptoms related to the monoclonal gammopathy
Recommended laboratory testing includes the following:
• Bone marrow aspiration examination
• Total serum protein concentration and serum electrophoresis (serum monoclonal protein concentration)
• 24-hour urine protein excretion concentration and urine electrophoresis (urine monoclonal protein concentration)
• Serum and urine immunofixation (type of monoclonal protein)
• Determination of serum FLC ratio (κ and λ FLCs), particularly for the assessment of prognosis
Gammopathies With More Than One Band
CASE STUDY
1. To follow up with the diagnosis of this patient, a ______ would be of value.
2. A risk factor for an immunologic disease is significant in this patient because of:
See Appendix A for the answers to multiple choice questions.
Critical Thinking Group Discussion Questions
1. What follow-up laboratory tests might be ordered to assist in establishing a definitive diagnosis?
2. What is the nature of the protein found in the urine?
3. What is the most significant laboratory finding in this disorder?
4. What type of immunologic defect exists in this disease process?
See instructor site  website for the discussion of the answers to these questions.
website for the discussion of the answers to these questions.
 Bence Jones Protein Screening Procedure
Bence Jones Protein Screening Procedure website for the procedural protocol.
website for the procedural protocol.Chapter Highlights
• Hypergammaglobulinemias are monoclonal or polyclonal.
• A monoclonal gammopathy can be benign or malignant and results from a single clone of lymphoid plasma cells producing elevated levels of a single class and type of immunoglobulin, referred to as a monoclonal protein, M protein, or paraprotein. These disorders include multiple myeloma (MM) and Waldenström’s macroglobulinemia (WM). MM is the most common form of dysproteinemia.
• A polyclonal gammopathy is classified as a secondary disease and is characterized by the elevation of two or more immunoglobulins produced by several clones of plasma cells. Polyclonal protein consists of one or more heavy-chain classes; both light-chain types increase as secondary manifestations of infection or inflammation.
• The cause of MM is unknown, but radiation may be a factor; a viral cause has also been suggested. Other causes may include environmental stimuli or genetic factors.
• Signs and symptoms of MM include bone pain (back or chest), weakness, fatigue, and pallor associated with anemia or abnormal bleeding.
• Proteinuria is a common finding in more than 50% of patients excreting abnormal amounts of Bence Jones (BJ) protein (light chains). Patients have defects in humoral but not cellular immunity.
• Laboratory diagnosis of MM includes electrophoresis of the serum or urine. A monoclonal protein is seen in the serum and urine in 90% of patients. DNA hybridization or blotting technology can be used to detect abnormal genes in B cells. Although the gene product of monoclonal antibodies (MAbs) is the method of detection, DNA probes that detect the abnormal gene are available. Blotting techniques may replace the current laboratory evaluation of monoclonal gammopathies.
• WM is a malignant cell disorder that exhibits abnormally large amounts of 19S IgM. The cause is unknown, but a genetic predisposition may exist.
• WM has an indolent progression over many years. The basic abnormality is uncontrolled proliferation of B lymphocytes and plasma cells.
• Laboratory diagnosis of WM involves serum electrophoresis showing a homogeneous M component composed of monoclonal IgM. Blood samples characteristically display hyperviscosity. In addition, cryoglobulins can be detected.
• Other monoclonal disorders include light-chain disease (LCD), which represents about 10% to 15% of monoclonal gammopathies. In LCD, only κ or λ monoclonal light chains or BJ proteins are produced. A 24-hour urine specimen should be examined electrophoretically because almost all the protein may be BJ.
• Heavy-chain disease is characterized by monoclonal proteins composed of the heavy-chain portion of the immunoglobulin molecule. Alpha heavy-chain disease is most common.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Immunoproliferative Disorders
At the conclusion of this chapter, the reader should be able to:
• Compare the general characteristics of monoclonal and polyclonal gammopathies.
• Describe and compare the etiology, epidemiology, signs and symptoms, immunologic manifestations, diagnostic evaluation, and treatment of multiple myeloma and Waldenström’s primary macroglobulinemia.
• Explain and contrast the characteristics of other monoclonal disorders, such as monoclonal gammopathy of unknown significance.
• Analyze a case study related to immunoproliferation.
• Correctly answer case study related multiple choice questions.
• Be prepared to participate in a discussion of case study related critical thinking questions.
• Describe the principle and application of the Bence Jones Protein Screening Procedure.
Multiple Myeloma
Pathophysiology
Myeloma cells proliferate slowly in the marrow (Fig. 27-1; Table 27-1). Less than 1% divide at any one time and myeloma cells do not differentiate. The absolute number of these cells correlates with disease activity and predicts the progression of disease in smoldering multiple myeloma. Circulating myeloma cells may disseminate the tumor within the bone marrow and elsewhere.
Table 27-1
Three Phases of Disease Progression in Multiple Myeloma
| Variable | Initial Phase | Medullary Relapse | Extramedullary Relapse |
| Site of myeloma-cell accumulation or proliferation | Bone marrow | Bone marrow | Blood, pleural effusion, skin, many other sites |
| Growth fraction∗ | <1% | ≥1% (1%-95%) | ≥1% (1%-95%) |
| Genetic or oncogenic events | Deregulation of c-myc Illegitimate switch recombinations |
N-ras and K-ras point mutations | p53 point mutations |
| Phenotypic changes | CD19 loss CD56 overexpression |
CD28 expression LFA-1 and VLA-5 loss |
CD28 expression CD56 loss |
| Cytologic changes | Detectable plasmablastic compartment in 15% of cases | Plasmablastic compartment growing | Major plasmablastic compartment |
| Circulating malignant plasma cells | <1% | Increasing | Increasing |
∗Growth fraction is the rate of atypical cells proliferating in the bone marrow.
From Bataille R, Harousseau JL: Medical progress: multiple myeloma, N Engl J Med 336:1657–1664, 1997.
Epidemiology
IgG myeloma is the most common form of MM (Table 27-2). Four subtypes of IgG heavy chains are known to exist among patients with IgG myeloma. Cases of IgG myeloma are distributed as follows: 65% are gamma G1, 23% gamma G2, 8% gamma G3, and 4% gamma G4 subclass. The only subclass-dependent difference is the greater propensity for patients with IgG3 myeloma to experience hyperviscosity syndrome, similar to the manifestation in WM.
Table 27-2
Distribution of Immunoglobulin Types in Patients With Multiple Myeloma
| Type of Protein | Multiple Myeloma (%) |
| IgM | 12 |
| IgG | 52 |
| IgA | 22 |
| IgD | 2 |
| IgE | Rare |
| Light chains (kappa or lambda) | 11 |
| Heavy chains | Rare |
| Monoclonal proteins | <1 |
| Nonsecretory myeloma | 1 |
Signs and Symptoms
Skeletal Abnormalities
About 90% of patients with MM have broadly disseminated destruction of the skeleton, which is responsible for the predominance of bone pain. These abnormalities consist of punched-out lytic areas (Fig. 27-2), osteoporosis, and fractures in about 80% of patients. The vertebrae, skull, thoracic cage, pelvis, and proximal humeri and femurs are the most frequent sites of involvement.
A, Several scattered, small, well-marginated lytic lesions appear in calvarium, located in normally mineralized bone. Multiple lytic lesions can also be seen in the mandible. B, Multiple circumscribed lytic lesions crowd bones throughout skull. Lesions are still discrete and margins of most are fairly sharp. (From Newton TH, Potts DG: Radiology of the skull and brain, St Louis, 1971, Mosby.)
[/not-level-membership-for-allergy-and-immunology-category]
