Chapter 19 Immunological Tolerance
• Immunological tolerance is the state of unresponsiveness to a particular antigen which is primarily established in T- and B-lymphocytes. The clonal receptors of lymphocytes are generated by random recombination of the many genes that code for the antigen binding regions. This creates the need to sort out dangerous receptors that could recognize and destroy self tissues. The breakdown of immunological tolerance to self-antigens is the cause of autoimmune diseases.
• Immunological tolerance is achieved by many different mechanisms operating on different cell types.
• Central tolerance refers to the selection processes which T cell precursors undergo in the thymus before they are released as mature naive T cells. Thymic epithelial cells and dendritic cells present self-antigens to the immature T cell precursors. Those T cell precursors that respond strongly to the self-antigens presented in the thymus undergo apoptosis. This is called negative selection. A specialized population of thymic epithelial cells is capable of expressing genes which are expressed in a strictly organ specific manner (e.g. insulin, which is expressed only in the pancreas and the thymus).
• Peripheral tolerance refers to the diverse mechanism that enforce and maintain T-cell tolerance outside the thymus. These include the prevention of contact between auto-reactive T-cells and their target antigens (immunological ignorance), the peripheral deletion of auto-reactive T-cells by activation induced cell death or cytokine withdrawal, the incapacity of T cells to mount effector responses upon recognizing their target antigen (anergy), and the suppression of immune responses by regulatory T cells.
• B cell tolerance is established by several mechanisms including clonal deletion of autoreactive B cells, mostly in the bone marrow; the rearrangement of autoreactive B cell receptors (receptor editing) or by B cell anergy. In addition B-cell tolerance is maintained by tolerant T cells. The production of high-affinity class-switched antibodies depends on T-cell help. Therefore, if tolerance to a particular antigen is firmly established in the T-cell compartment, B-cells that recognize this antigen will usually remain tolerant.
• To establish or re-establish tolerance is a major goal for innovative treatments for autoimmunity, allergy and transplantation. In contrast, to overcome immunological tolerance is one major goal for innovative treatments against cancer.
Generation of autoreactive antigen receptors during lymphocyte development
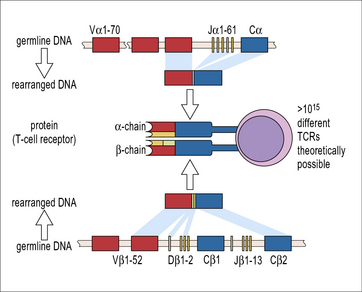
The specificity of the antigen receptors of T cells and B cells is the result of random shuffling of the many genes that encode the antigen-binding site of these receptors. Theoretically, this process could generate more than 1015 different T-cell receptors, including some that can bind to autoantigens (Fig. 19.1). Similar considerations apply to B-cell receptors. Cells expressing such receptors are often called self-reactive lymphocytes. The immune system has to fulfill two contradictory requirements: on the one hand the repertoire of different antigen receptors needs to be as large as possible to avoid ‘holes in the repertoire’ that could be exploited by pathogens to evade immune detection. On the other hand, the receptor repertoire must be shaped to prevent the immune system from attacking the organism that harbors it. Any disturbance in this delicately balanced system can have pathogenic or even lethal consequences, either from infections or from the unwanted reaction with autoantigens or harmless external antigens as in allergy. This paradox was recognized at the beginning of the last century by Paul Ehrlich who coined the term ‘horror autotoxicus’ for the necessity to avoid immunological reactions against self-antigens. Tolerance is the process that eliminates or neutralizes such autoreactive cells, and a breakdown of this system can cause autoimmunity. To avoid autoreactivity the randomly generated repertoire of T- and B-cell receptors is censored by several different mechanisms. CD4+ TH cells are pivotal for the multitude of immunological mechanisms that induce and maintain immunological tolerance. In this chapter we will discuss immunological tolerance in B cells and conventional αβ TCR expressing T cells.
T cell tolerance
Central T-cell tolerance develops in the thymus
The chief mechanism of T-cell tolerance is the deletion of self-reactive T cells in the thymus. Immature T cell precursors migrate from the bone marrow to the thymus. There, they proliferate, differentiate and undergo selection processes before a selected few re-enter the blood stream as mature naive T cells. These differentiation and selection processes depend on interactions with thymic epithelial cells and dendritic cells in specialized microenvironments within the thymus (Fig. 19.2).
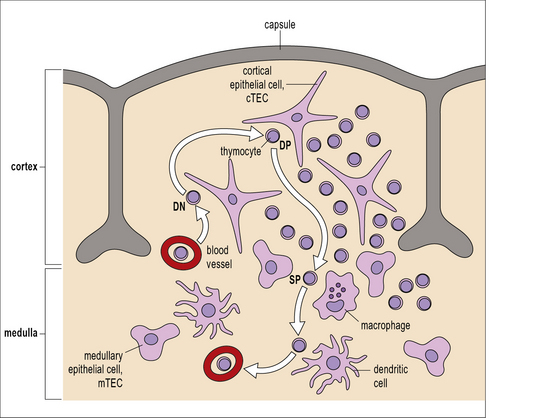
Fig. 19.2 T cell repertoire selection in the thymus
(Adapted from Kyewsky B, Klein L, Ann Rev Immunol 2006;24:571–606.)
Thymocytes are positively selected for their ability to interact with self MHC molecules
• in the DP T cells RAG is active and TCR α chains are continuously rearranged to maximize the chance of producing a TCR capable of interacting with self MHC;
• only upon recognition of a peptide/MHC complex via the TCR is RAG expression halted and the cell is committed to express a particular αβ TCR.
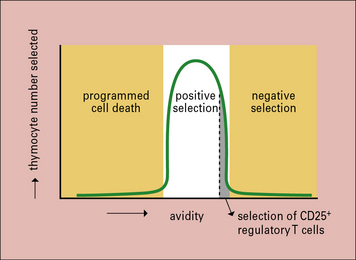
Moreover, recognition of a peptide/MHC complex via the TCR is necessary for the DP T cell to receive a survival signal. This is called positive selection (see Fig 19.2). Experiments have shown that T cells are positively selected by interaction with self-MHC to prepare them for subsequent activation by non-self-peptide/self-MHC complexes, indicating the biological benefit of positive selection.
Lack of survival signals leads to death by neglect
DP T cells whose receptors have a very low affinity for the peptide/MHC complexes they encounter during their approximately 3–4-day lifespan in the thymic cortex do not receive survival signals and undergo apoptosis in the thymus (see Fig. 19.w2) . This is called death by neglect. Accordingly, T cells do not progress beyond the DP stage in mice that have been manipulated to lack expression of MHC molecules. Analyses of the germ line repertoire of TCRs have revealed that this unselected repertoire contains more self-MHC reactive TCRs than expected by chance. In other words: co-evolution of TCR and MHC molecules has shaped the germline αβ TCR repertoire to favor the generation of receptors that can interact with self-MHC.
. This is called death by neglect. Accordingly, T cells do not progress beyond the DP stage in mice that have been manipulated to lack expression of MHC molecules. Analyses of the germ line repertoire of TCRs have revealed that this unselected repertoire contains more self-MHC reactive TCRs than expected by chance. In other words: co-evolution of TCR and MHC molecules has shaped the germline αβ TCR repertoire to favor the generation of receptors that can interact with self-MHC.
Thymocytes are negatively selected if they bind strongly to self-peptides on MHC molecules
After positive selection and commitment to the CD4 or CD8 lineage in the thymic epithelium, thymocytes express the chemokine receptor CCR7 and migrate towards the central region of the thymus, the thymic medulla, where the CCR7 ligands CCL19 and CCL20 are produced. In the thymic medulla the thymocytes are probed for another 4–5 days. T cells whose receptors have a high affinity for the peptide/MHC complexes encountered in the medulla are autoreactive and therefore, potentially dangerous. They receive death signals and undergo apoptosis. This is called negative selection (see Fig 19.2). Experimental evidence suggests that the majority of the DP T cells which were positively selected are later eliminated by negative selection.
A library of self antigens is presented to developing T cells in the thymus
The induction of central tolerance requires the presence of autoantigens in the thymus. This poses an obvious problem for thymic selection: Some autoantigens, e.g. insulin are expressed in a tissue-specific manner, are frequently called tissue-restricted antigens (TRAs). The question, then, is (how) do TRAs get into the thymus for presentation to developing T cells? Some might be brought into the thymus by immigrating antigen presenting cells but it is highly unlikely that this would yield a reliable representation of the organism’s TRAs. Moreover, developmentally regulated TRAs, e.g. antigens that are only expressed after puberty, would not gain access to the fetal thymus. The answer is that specialized cells in the thymus, the medullary thymic epithelial cells (mTECs) express proteins that are otherwise strictly tissue restricted. This has been called ectopic or promiscuous gene expression (Fig. 19.3).
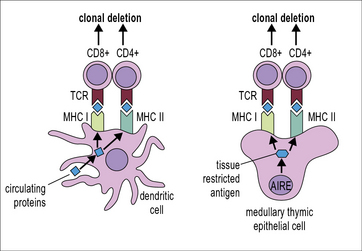
AIRE controls promiscuous expression of genes in the thymus
What enables mTECs to express a broad array of TRAs independently of spatial or developmental regulation? mTECs express the transcriptional regulator Aire (autoimmune regulator). Aire controls the expression of a large number of TRAs in mTECs (see Fig. 19.3).
Peripheral T-cell tolerance
Immunological ignorance occurs if T cells do not encounter their cognate antigen
The importance of immunological ignorance was demonstrated in mice that express a transgene-encoded TCR which recognizes a peptide derived from the lymphocytic choriomeningitis virus (LCMV). These mice were bred with another transgenic strain that expressed the viral peptide on the surface of their pancreatic islet cells. Surprisingly, diabetes did not develop in the offspring even though in vitro their T cells could kill cells that displayed the viral peptide (Fig. 19.4). The T cells in these double-transgenic mice were therefore, not tolerant in vivo, instead they were ignorant of their target cells.
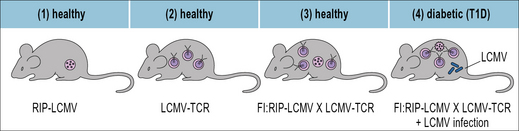
When the mice were infected with LCMV, the transgenic T cells became activated, invaded the pancreas and destroyed the islet cells. Consequently the mice succumbed to diabetes. Importantly, LCMV-infection did not cause diabetes in those mice that expressed the transgenic TCR but not the LCMV-peptide in the pancreas. Once activated by LCMV infection, the hitherto ignorant autoreactive T cells (autoreactive because the LCMV-derived peptide was transgenically expressed in the pancreas) acquired the capacity to migrate into their target tissue where they recognized and destroyed the LCMV-peptide expressing islet cells (see Fig. 19.4).
Self-reactive T cells and experimental autoimmunity
Q. Why might it be important to maintain these mice in germ-free conditions, to prevent them from getting EAE?
A. Activation of lymphocytes by microorganisms enhances the traffic of lymphocytes into tissues, including the brain (see Chapter 6). If an autoreactive cell then encounters its antigen (MBP) a cycle of inflammation can develop.
Some self antigens are sequestered in immunologically privileged tissues
One explanation is the sequestration of potentially harmful T cells from the tissues in which their target self-antigens are expressed. Sequestration can be achieved when antigens are physically separated from T cells (e.g. by the blood–brain barrier, see Chapter 12). The blood–brain barrier can be surmounted by activated lymphocytes, however, and many organs do not possess a physical barrier to prevent lymphocytes entering from the bloodstream. Instead, lymphocyte migration is controlled by chemokines, selectins and their receptors.

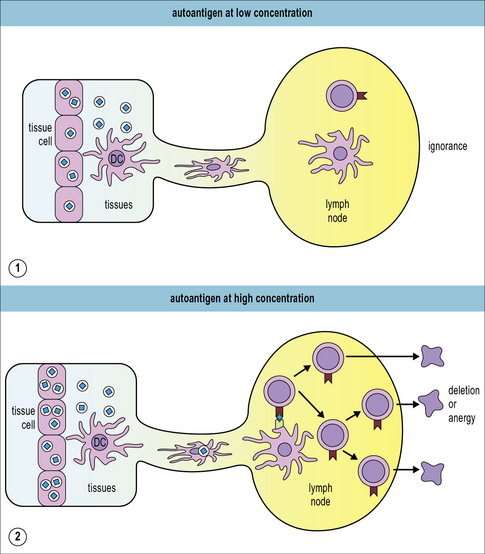
The amount of released self antigen critically affects sensitization
How does the immune system decide which autoreactive T cells may survive ignorantly and which need to be deleted? Antigen dose (Fig. 19.w1) and TCR avidity play a major role. One key experiment used two different strains of mice that expressed ovalbumin (OVA) specifically in the pancreas. One strain expressed OVA at low levels and the other expressed it at high levels. Only in the high-expressing strain was OVA presented to T cells in the draining lymph nodes, resulting in the deletion of adoptively transferred OVA-specific TC cells. In the low-expressing cells the OVA-specific T cells remained ignorant. Further in vitro assays revealed that the low-level OVA-expression was still sufficient to allow recognition and killing of the OVA-expressing pancreatic β cells by the OVA-specific CTL. Therefore, tissue restricted self-antigens need to be expressed at sufficiently high levels to be presented in the draining lymph nodes. These experiments also clearly showed that self-reactive T cells remain dangerous and poised to wreak havoc even if they are temporarily ignorant. In fact, destruction of pancreatic islet cells can induce the release of sufficient amounts of self-antigen to activate OVA-specific CTL in the OVA-low expressing mice.
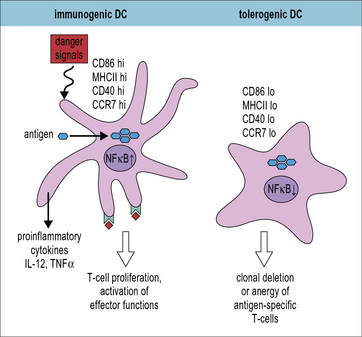
Antigen presenting cells reinforce self tolerance
Dendritic cells can present antigen in a tolerogenic manner
Functional maturation of DCs, characterized by strong expression of MHC and co-stimulatory molecules is induced by microbial or self-derived stimuli, which are sometimes called danger signals (Fig. 19.5). In the absence of such stimuli immature DCs express MHC and costimulatory molecules at low levels and antigen presentation induces T cell anergy or deletion depending upon the expression of high or low levels of self-antigen respectively (see Fig. 19.w1). The signals that induce tolerogenic DC maturation as well as the tolerogenic interactions between DC and T cells are only incompletely understood. Still there are some clear candidates. Several molecules have already been identified that are necessary for tolerogenic DC:T cell interactions. These include surface molecules such as E-cadherin, PD-1 L, CD103, CD152 (CTLA-4) and ICOS-L (CD275) and cytokines, including IL-10 and TGF-β.
The signals that induce tolerogenic DC maturation as well as the tolerogenic interactions between DC and T cells are only incompletely understood. Still there are some clear candidates. Several molecules have already been identified that are necessary for tolerogenic DC:T cell interactions. These include surface molecules such as E-cadherin, PD-1 L, CD103, CD152 (CTLA-4) and ICOS-L (CD275) and cytokines, including IL-10 and TGF-β.

Regulatory T cells
Regulatory T cells suppress immune responses
• normal mice harbor self-reactive cells, which can cause autoimmune damage;
• normal mice produce CD4+ T cells in the thymus, which can suppress the autoreactive cells;
• depletion of these suppressive cells can cause autoimmune disease.
The transcription factor FoxP3 controls Treg development
Natural Treg cells differentiate in the thymus
During thymic development FoxP3 expression starts in CD4+CD8+ double positive thymocytes. Approximately 5% of the more mature CD4+CD8− single positive thymocytes express FoxP3. These Treg cells which acquire their phenotype and functional capacities in the thymus and are released into the periphery as CD4+CD25+FoxP3+ T cells, are called natural Treg cells or nTregs (Fig. 19.6).
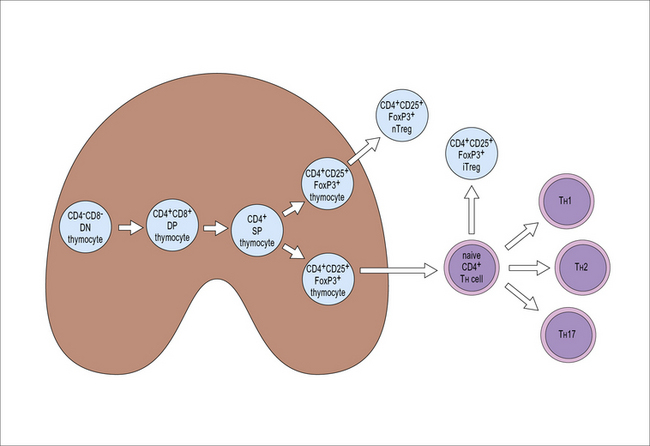

Selection of nTregs is partly related to the affinity for antigen/MHC
• the antigen is expressed at high levels in the thymus; or
• the TCR possesses a high affinity for the transgenic antigen.
In contrast, if the TCR’s affinity for the transgenic peptide is low or if the transgenic peptide is present in low concentration in the thymus, few T cells bearing the transgene-encoded TCR will develop into nTregs (Fig. 19.w2). It is currently a matter of debate if these findings reflect positive selection of T cells with strongly self reactive TCRs to the Treg lineage or the resistance of FoxP3+ thymocytes to negative selection.
iTreg cells differentiate in the periphery
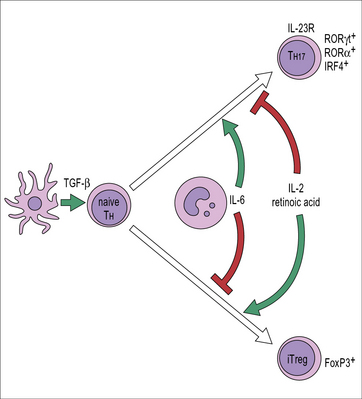
In addition to the natural Tregs which differentiate in the thymus, mature T cells outside the thymus can also acquire Treg phenotype and function. These are called induced Treg cells (iTregs) (see Fig. 19.6). FoxP3 expression can be induced in naive CD4+ cells in vitro by antigen recognition in the presence of TGF-β. There is a close developmental relationship between iTregs and TH17 cells. Antigen recognition in the presence of TGF-β induces FoxP3 expression if IL-6 is not present. In contrast, antigen recognition in the presence of TGF-β and IL-6 prevents FoxP3 expression, induces expression of the retinoic acid receptor (RAR) related orphan nuclear receptor RORγt expression and therefore, TH17 differentiation. The transcription factor IRF4 is necessary for the down-regulation of TGF-β-induced FoxP3 expression in response to IL-6 (Fig. 19.7).

Treg effector functions
Tregs can act on a number of different target cells including effector T cells and dendritic cells but also on numerous other cell types including B cells, macrophages, NK cells, NKT cells, mast cells, osteoblasts and osteoclasts. They produce or consume cytokines to modulate their target cells and Tregs are also capable of lysing target cells (Fig. 19.8).
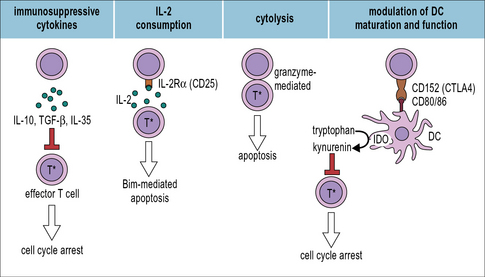
Fig 19.8 Treg effector mechanisms
(Adapted from Shevach EM, Immunity 2009;30:636 and Vignali DAA et al. Nature Reviews Immunology 2008;8:523.)
Tregs secrete immunosuppressive cytokines
One possible mechanism of immunosuppression by Tregs would be the secretion of immunosuppressive cytokines (see Fig. 19.8). Indeed, three inhibitory cytokines, IL-10, TGF-β, and IL-35 are produced by Tregs and important for Treg development or effector function. Importantly, IL-10 and TGF-β are not exclusively produced by Tregs but can be produced by many different cell types. Moreover, most in vitro studies found IL-10 or TGF-β produced by Treg non-essential for Treg mediated suppression.
Modulation of DC maturation and function
Treg cells can induce DCs to express indoleamine 2,3-dioxygenase (IDO), which depletes tryptophan resulting in the suppression of effector T cell responses. This interaction also depends on the interaction between CD152 and CD80/86 (see Fig. 19.8).
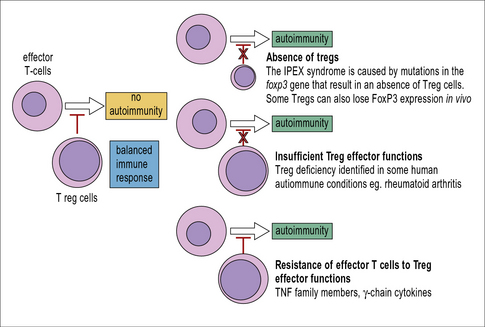
Can loss of Treg function explain autoimmune disease?

However, except for the IPEX syndrome (Fig. 19.9), in which FoxP3+ Tregs are completely absent, there is currently no convincing evidence that reduced numbers of Tregs would cause autoimmune disease. In contrast, Tregs are frequently found in increased numbers at the site of autoimmune lesions.
T cell anergy
The induction of anergy is an active process
B cell tolerance
B lymphocytes and plasma cells that produce antibodies that recognize self-antigens, so called auto-antibodies, pose a threat to the organism. Grave’s disease, which is clinically characterized by overshooting production of thyroid hormones, is caused by autoantibodies that act as agonists for the receptor for thyroid stimulating hormone. Blistering skin diseases are caused by autoantibodies that recognize adhesion molecules in the epidermis. In addition to such organ-specific diseases autoantibodies can cause systemic autoimmunity as exemplified by the multi-organ autoimmune disease systemic lupus erythematosus (see Chapter 20).
• first, B cells mature in the bone marrow and in mammals there is no B-cell equivalent of thymic repertoire selection in T cells;
• second, mature B cells change their receptors by random mutation in a process called affinity maturation. While affinity maturation serves to generate antibodies with increased affinity for harmful antigens these random mutations could also generate antibodies with high affinity for self antigens. The immune system must have developed mechanisms to prevent the generation of such high-affinity autoantibodies. Indeed, even after immunisation with foreign antigens that closely resemble self antigens, high affinity autoantibodies are usually not produced;
• third, the production of high affinity, class-switched antibodies depends on T cell help. Thus, B cell tolerance is taken care of in large part, if not completely, by T cell tolerance of self-antigens.
Altogether, less than 10% of the mature follicular B cells have been found to be autoreactive. B cell tolerance to autoantigens is established by clonal deletion, receptor editing, the induction of anergy, and the B cells’ dependence on T cell help (Fig. 19.10).
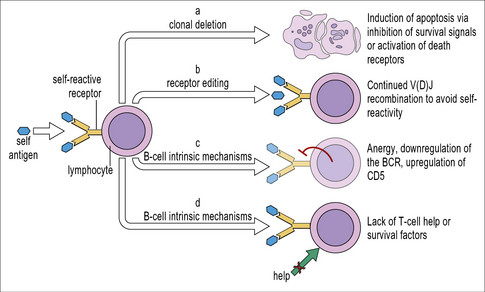
B cells undergo negative selection in the bone marrow
Clonal deletion of B lymphocytes was first directly demonstrated in mice that expressed a transgenic BCR specific for a foreign antigen (hen egg lysosyme, HEL). These mice were bred with another strain of transgenic mice that expressed HEL. The F1 mice expressed both HEL and BCRs that recognized HEL. The HEL-specific B-cells were deleted (negatively selected) in the bone marrow of the F1 mice (see Fig. 19.10(1)). When HEL-specific mature B cells from mice that did not express the HEL-transgene were adoptively transferred into mice that expressed HEL, these B cells were also deleted in the recipient mice. Apoptosis of mature germinal centre B cells occurs rapidly, within 4–8 hours of encountering the self-antigen. When the BCR transgenic mice were also Bim-deficient, they survived in the recipient mice. Similar to T cells, the pro-apoptotic factor Bim is important for BCR-induced apoptosis. This is further illustrated by the fact that Bim-deficient mice spontaneously produce autoantibodies against DNA.
Receptor editing allows potentially self-reactive B cells to avoid negative selection
Death by apoptosis is not the only possible outcome for immature B cells in the bone marrow when the strength of signals received through the BCR exceeds a certain threshold. Internalization of the BCR is an important early step in the BCR-induced apoptosis program. This has a number of consequences relevant for BCR-induced apoptosis including a reduced expression of receptors for the cytokine B-cell-activating factor (BAFF) which is necessary to sustain B-cell survival. In contrast, expression of the genes that encode the key enzymes for V(D)J recombination, RAG1 and RAG2, continues. This offers B cells the chance to rearrange their VH or VL regions to replace the autoreactive one. This process is called BCR editing (see Fig. 19.10(2)). Approximately two days remain for the B cell to rearrange a less autoreactive receptor. If it fails to do so, it will undergo apoptosis, either in the bone marrow or upon arrival in the spleen. Single-cell PCR analyses reveal secondary rearrangements in roughly two thirds of the immature B cells in the bone marrow. As the frequency of cells with secondary rearrangements is only about 50% of all B cells in the spleen, receptor editing does not always result in a useful, non-autoreactive BCR.
B cell anergy can be induced by self antigens
B cells can also become anergic upon recognizing a tolerizing self-antigen (see Fig. 19.10(3)). Anergic B cells lack the capacity to proliferate and produce antibodies in response to BCR signaling. In anergic B cells signaling via the BCR is uncoupled from NFκB, thus preventing B cell proliferation. At the same time BCR signaling still prevents apoptosis. Similar to other immature and naive B cells, anergic B cells have a very limited life span if they do not receive appropriate signals. The anergic state can be reversed if the B cell receives signaling via the BCR simultaneously with synergistic signals from another receptor. Such simultaneous signaling is likely to occur when the B cell encounters microbial rather than self antigens. One example is signaling both via the BCR and toll like receptors (TLRs). This would typically be the case when a B cell encounters a pathogen, e.g. gram-negative bacteria possessing LPS which triggers signaling via TLR4.
B cell tolerance due to lack of T cell help
Perhaps the main mechanism to ensure B cell tolerance is the B cells’ dependence on T cell help for high affinity isotype switched antibody production (see Fig. 19.10(4)). BCR signaling results in changes in gene expression that facilitate antigen presentation to T cells:
• enhanced CCR7 expression enables the B cells to migrate from the follicle towards the T cell zone in the secondary lymphatic organs;
• induction of CD86 increases the number of ligands for the co-stimulatory receptor CD28 on T cells;
• productive antigen-presentation to T cells results in the increased secretion of T-cell cytokines such as IL-4 and IL-21 that prevent B-cell apoptosis and support B-cell proliferation.
Critical Thinking: Immunological tolerance (see Click here for explanations)
1. Figure 19.10 depicts four mechanisms to establish B cell tolerance. Compare these with the mechanisms used to establish T cell tolerance and explain similarities and differences.
2. The transcriptional regulator AIRE (autoimmune regulator) is considered to be critical for the establishment of self tolerance. Explain the experimental and clinical evidence on which this statement is based.
3. T-cell precursors undergo positive selection processes in the thymus. Explain ‘positive selection’. There is no positive selection for B cells. Can you speculate why positive selection is required for T-cells but not for B-cells?
4. Explain how dendritic cells contribute to peripheral T cell tolerance.
5. Pathogens with exclusively peripheral-tissue tropism (such as papillomavirus) can evade immune responses. Can you explain which tolerance-mechanism is subverted by such pathogens?
6. Mice that express a transgene-encoded TCR which is specific for a self antigen usually do not develop autoimmune disease. How could you distinguish in an in vitro assay if the transgenic T cells were ignorant or anergic?
7. Patients with autoimmune diseases often have increased numbers of regulatory T cells (Tregs) in diseased tissue – speculate about possible explanations for this seemingly paradoxical finding.
Germain R.N. Special regulatory T-cell review: A rose by any other name: from suppressor T cells to Tregs, approbation to unbridled enthusiasm. Immunology. 2008;123:20–27.
Goldrath A.W., Bevan M.J. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262.
Goodnow C.C., Vinuesa C.G., Randall K.L., et al. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688.
Kamradt T., Mitchison N.A. Tolerance and autoimmunity. N Engl J Med. 2001;344:655–664.
Klein L., Hinterberger M., Wirnsberger G., Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol. 2009;9:833–844.
Matzinger P., Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol. 2011;11:221–230.
Pulendran B., Tang H., Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11:647–655.
Saibil S.D., Deenick E.K., Ohashi P.S. The sound of silence: modulating anergy in T lymphocytes. Curr Opin Immunol. 2007;19:658–664.
Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787.
Schwartz R.H. T cell anergy. Annu Rev Immunol. 2003;21:305–334.
Shevach E.M. Mechanisms of FoxP3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645.
von Boehmer H., Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol. 2010;11:14–20.