Immunologic Lung Disease in the Critically Ill
Clinicopathologic Considerations during Mechanical Ventilation
The lungs of patients with immunologic diseases, in particular, interstitial lung diseases such as IPF and connective tissue lung diseases, exhibit restrictive physiology with decreased lung volumes, decreased parenchymal compliance, and a loss of functional capillary beds leading to a reduction in the diffusing capacity.1 In this manner the functionally smaller lungs (baby lungs) of these patients resemble the lungs of patients with the acute respiratory distress syndrome (ARDS). This concept is supported by computed tomography (CT) findings of heterogeneous disease involvement in the lungs of patients with both IPF and ARDS. This parallel can be used as a framework for managing tidal volumes during mechanical ventilation of patients with immunologic lung disease and respiratory failure. If large tidal volumes (or excessive inflation pressures) are used, relatively normal areas of lung will be overdistended, potentially exacerbating lung injury. Limiting tidal volumes to 6 mL/kg predicted body weight in patients with acute lung injury (ALI) or ARDS saves lives.2 Similar data are not available regarding safe parameters for ventilating patients with chronic restrictive lung diseases, but limiting tidal volumes to roughly 6 mL/kg (and raising the rate accordingly) carries little risk. Moreover, retrospective analysis of mechanically ventilated patients without ALI/ARDS suggests that large tidal volumes may produce ALI (odds ratio 1.3 for each milliliter above 6 mL/kg predicted body weight).3 Because there is little evidence that intrinsic positive end-expiratory pressure (PEEP) plays a physiologically important role in respiratory failure in patients with interstitial lung disease, rapid ventilatory rates are tolerated.1 This approach may allow ventilation without resorting to permissive hypercapnia, which may aggravate pulmonary hypertension.4,5 As in the ARDS lung, it seems likely that nonfunctional, diseased lung units exist alongside those with essentially normal function. However, unlike the acutely injured lung, atelectatic lung units available for recruitment through the use of elevated end-expiratory pressure are rare in conditions such as IPF. There is probably little clinical advantage to using high PEEP in patients with restrictive lung diseases; in fact, high levels of PEEP may be detrimental by overdistending the lung, as well as by contributing to cor pulmonale, as described later. In a cohort of ventilated patients with interstitial lung disease, high PEEP during the first 24 hours of mechanical ventilation was one of the independent determinants of death.6 Although it is likely that high PEEP served as a marker (rather than a cause) of severity in this study, high PEEP should nevertheless be applied cautiously to minimize harm.
Another clinicopathologic process to consider in the ventilatory management of the patient with immunologic lung disease is pulmonary hypertension. Pulmonary artery pressures are chronically elevated in advanced stages with lung fibrosis, and this pressure increases further with the increased cardiac output that accompanies exercise, fever, and hypercarbia.5,7 Pulmonary hypertension may eventually lead to cor pulmonale because of increased right ventricular afterload.8 Mechanical ventilation may interact adversely with pulmonary hypertension. Positive-pressure ventilation alone impairs right-sided heart function, and this effect is exaggerated by PEEP.9 PEEP increases afterload by increasing pulmonary vascular resistance.10 The resulting increase in wall tension decreases right ventricular perfusion, which leads to myocardial ischemia.10,11 Right ventricular ischemia may cause further dysfunction and dilation of the right side of the heart, in addition to diastolic dysfunction of the left ventricle (through ventricular interdependence), producing a cycle of progressively deteriorating circulatory function.8 Thus, it is important to minimize further increases in pulmonary artery pressure and to maintain adequate systemic pressure to preserve perfusion of the right ventricle. Additionally, adequate oxygenation is essential to prevent reflex increases in pulmonary artery pressure and to maintain peripheral oxygen delivery. Finally, hypercapnia, which tends to raise pulmonary artery pressures, should generally be avoided. Because predicting the degree of pulmonary hypertension clinically is difficult—and the need to avoid increases in pulmonary artery pressures is so important—we advocate liberal use of echocardiography. Other forms of monitoring, such as central venous saturation measurement or pulmonary artery catheterization, might also be useful. When acute-on-chronic cor pulmonale compromises the circulation, dobutamine or norepinephrine is often helpful.12 Inhaled nitric oxide or inhaled prostacyclin probably plays some role, at least to buy time in the critically impaired patient.13,14 The role of newer pulmonary vasodilators such as bosentan and sildenafil in patients with acute cor pulmonale is unclear.
Rescue therapies such as extracorporeal membrane oxygenation or pumpless extracorporeal lung assist devices may be of benefit in selected cases when used early.15,16 Data on noninvasive mechanical ventilation in immunologic lung disease is limited. Two small studies show that noninvasive ventilation can be used to avoid intubation and risk for ventilator-associated pneumonia with comparable or better short-term survival.17,18
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis is a disorder of unknown cause characterized by inflammation of the lower respiratory tract that usually leads to irreversible scarring. Current and former smokers are at increased risk, and there may be an inherited susceptibility to develop this disease.19–21 IPF most commonly presents as an outpatient illness with the insidious onset of exertional dyspnea and cough. On examination of the lungs, coarse crackles are found and clubbing of the fingers is characteristic.21 Chest radiographs may show a spectrum of findings from peripheral reticular densities to end-stage honeycombed lung.21 Alveolar infiltrates are unusual unless the patient has a concurrent lung cancer, pneumonia, or heart disease. The lung CT findings most closely associated with a pathologic diagnosis of IPF are lower-lung honeycombing and upper-lung irregular lines.22 The histologic examination of IPF reveals usual interstitial pneumonitis, which is characterized by inflammation, fibroblastic foci, areas of fibrosis, and remodeling of the lung parenchyma. The pulmonary fibrosis appears to follow collapse of involved alveoli.
Death in patients with IPF is most often directly attributable to progression of the underlying disease, even when the disease is only of moderate severity.21,23 Nevertheless, the clinician should seek treatable complicating conditions before making the difficult decision to withhold mechanical ventilatory support. Other causes of respiratory failure in IPF include infection, congestive heart failure, bronchogenic carcinoma, pulmonary embolism, and pneumothorax. Left ventricular failure is often found in association with IPF. These patients often have many of the risk factors (e.g., smoking, hyperlipidemia) that are associated with the development of atherosclerosis. For this reason, left ventricular failure may result from ischemic heart disease. Two other factors that may contribute to left ventricular failure in these individuals are systemic arterial hypertension and right ventricular failure. Hypoxemia may exacerbate these effects. A search for potentially treatable left ventricular failure should be considered in the deteriorating IPF patient.
Patients with IPF have about a 14-fold excess risk of developing lung cancer.24 These malignancies are difficult to detect on an already abnormal chest radiograph and often cause rapid deterioration in the IPF patient. Treatment options for malignancy are often limited by poor pulmonary reserve. However, the diagnosis of lung cancer may greatly alter therapeutic planning. Furthermore, relieving airway obstruction and postobstructive infection may significantly palliate dyspnea.
Pulmonary embolism can also cause rapid deterioration and respiratory failure in IPF patients. Ventilation/perfusion scans often reveal nonsegmental perfusion defects and inhomogeneous areas of poor ventilation as a result of the IPF alone, so the utility of these scans in diagnosing pulmonary emboli is limited.25 Pulmonary angiography or helical CT scanning should be considered if it can be performed safely.25 We recommend empiric long-term anticoagulation for individuals with advanced IPF and severe pulmonary hypertension who are suspected of having pulmonary embolism but for whom a diagnostic evaluation is not feasible.26
An important factor to consider in patients with advanced IPF who develop respiratory failure is that this disease is largely irreversible. Although many exciting new therapies are under investigation, current treatments are largely ineffective in reversing the decline in lung function.27–29 Most patients with IPF who are in respiratory failure do not respond to corticosteroid therapy.30 Cytotoxic therapy, such as cyclophosphamide, azathioprine, or cyclosporine, has not been shown to alter survival.31,32 Mortality rate after ICU admission is high, raising the question of appropriateness of mechanical ventilation in most cases, with the exception of perioperative support or as a bridge to lung transplantation.31,33–35 In a review of nine studies examining 135 patients with IPF ventilated in the ICU, the aggregate hospital mortality rate was 87% and the mortality rate within 3 months after discharge was 94%.36 If patients are young, have early disease, or may be diagnosed with interstitial lung disease other than IPF, an open lung biopsy should be considered to exclude alternative treatable diseases. Recently, lung transplantation has become a viable option for some patients with end-stage IPF. It is imperative that physicians caring for these patients familiarize themselves with the referral protocols and policies of their respective regional transplant centers.
Hamman-Rich Syndrome
Hamman-Rich syndrome, more recently called acute interstitial pneumonia (AIP), is a rapidly progressive interstitial pneumonia of unknown cause first described by Hamman and Rich.37 The mean age of patients is 50 to 60 years, with a broad range and perhaps an increased risk for men.37,38 The patients often describe a prodromal viral-like respiratory illness typically followed by subacute progressive dyspnea, fever, and nonproductive cough.
AIP usually evolves over 1 to 3 months, and in some instances, it appears within 1 to 2 weeks after the onset of symptoms. Signs of right-sided heart failure may exist, and diffuse or basilar crackles may be found on auscultation of the lung. Diffuse, bilateral interstitial infiltrates are characteristic on chest radiograph. The findings on CT scan include diffuse, patchy alveolar ground glass infiltrates and pleural effusion in one third of the cases.39 Honeycombing may be present in subacute cases. Laboratory studies may show a leukocytosis with neutrophilia. Hypoxemia may be profound. Pulmonary function tests in patients without respiratory failure show a restrictive defect, generally without evidence of airway obstruction.38
For many years experts believed that this disease was simply a rapidly progressive form of IPF; now this disease is felt to be more related to ARDS. The pathologic features of AIP are characterized by diffuse, active fibrosis, with proliferating fibroblasts and minimal collagen. These findings appear acute and relatively uniform in age and resemble the organizing stage of diffuse alveolar damage as seen in ARDS.40
The prognosis of acute interstitial pneumonitis is poor, with only about a 40% short-term survival rate. Although the short-term mortality rate is similar to that for acute exacerbation of IPF, AIP survivors have near complete recovery of lung function in contrast to those with IPF.41,42 Supportive care may involve ventilatory support. Antibiotics to treat possible underlying infection and corticosteroids to treat inflammation have been used in many cases, but the efficacy of these treatments is not proved.38 In one small series, early efforts to exclude infection combined with lung-protective ventilation and high-dose corticosteroid therapy led to success in 8 of 10 patients.43 We recommend rigorous exclusion of an infectious cause including open lung biopsy, if necessary, before considering any immunosuppressive therapy.
Alveolar Hemorrhage Syndromes
Perhaps the most striking immunologically mediated lung diseases are those that present with alveolar hemorrhage. These disorders require prompt diagnosis and management. We limit our comments here to the disorders that most commonly present as alveolar hemorrhage: Good-pasture’s syndrome, Wegener’s granulomatosis (WG), microscopic polyangiitis (MPA), catastrophic antiphospholipid syndrome (CAPS), systemic lupus erythematosus (SLE), and idiopathic pulmonary hemosiderosis. Box 49.1 provides a more complete list of disorders that can lead to alveolar hemorrhage.
An essential goal of managing patients with alveolar hemorrhage is prompt diagnosis of the underlying disorder (Fig. 49.1). The first step is to document alveolar hemorrhage. The classic triad of hemoptysis, anemia, and diffuse infiltrates on chest radiograph strongly suggests alveolar hemorrhage, yet many patients with significant alveolar hemorrhage do not have hemoptysis.44 Consequently, the absence of hemoptysis does not exclude the presence of alveolar hemorrhage. Thus, diffuse pulmonary infiltrates, respiratory distress, and anemia associated with clinical evidence of glomerulonephritis or other conditions associated with vasculitis should arouse suspicion for alveolar hemorrhage, even in the absence of hemoptysis.
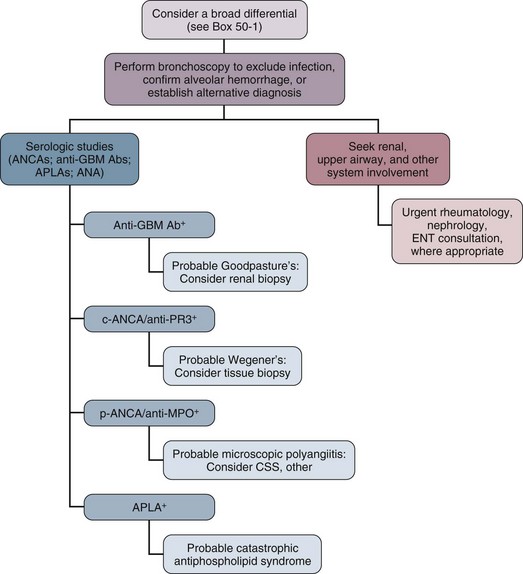
Before any specific therapy is instituted, it is important to document the presence of alveolar hemorrhage. Other processes that result in diffuse alveolar filling must be excluded, such as inflammatory exudate from infection, cardiogenic pulmonary edema, and ARDS. In addition, one should exclude hemorrhage from airway sources such as cancer, bronchitis, bronchiectasis, or excessive anticoagulation or an endogenous coagulation defect. Perhaps the most valuable test for documenting alveolar hemorrhage is bronchoscopy with BAL. Blood-tinged lavage fluid or frank blood in the airways is usually present.45 Another test that can be used involves staining alveolar macrophages retrieved by lavage for hemosiderin. Normal individuals will have few hemosiderin-laden macrophages in BAL, but the intensity of staining and percentage of cells staining positive have been found to be predictive of alveolar hemorrhage.46,47 We feel, however, that this test is of questionable clinical value. If patients have sufficient acute bleeding to cause infiltrates on chest radiograph, this should be seen easily in the lavage fluid. If the only evidence for hemorrhage is the presence of hemosiderin-laden macrophages, we would propose that the acute infiltrates are the result of another cause. Similarly, the negative predictive value of this test can be questioned because it may take up to 48 hours for intracellular hemosiderin accumulation after an acute bleed.48 Documentation of an elevated diffusion capacity for carbon monoxide (DLCO) is also a means of evaluating for alveolar hemorrhage but is not practical during active bleeding or critical illness.49,50 Bowley and coworkers demonstrated the usefulness of this measure as a sensitive index of recurrent alveolar hemorrhage in patients undergoing treatment.50
Treatment consists of supportive care including mechanical ventilation, prevention of infections and organ damage and immunosuppressive therapy directed at the underlying process. Rescue therapies such as recombinant factor VIIa and extracorporeal membrane oxygenation have been reported to be successful in anecdotal cases of refractory alveolar hemorrhage.51–55 Survival following alveolar hemorrhage due to immunologic lung disease tends to be better compared to patients with alveolar hemorrhage from thrombocytopenia or sepsis.56
Goodpasture’s Syndrome
Goodpasture’s syndrome accounts for 20% to 30% of the cases of alveolar hemorrhage.57 This disease is a classic pulmonary-renal syndrome with a high mortality rate from alveolar hemorrhage or renal failure if untreated. Anti–basement membrane antibody is a universal finding in this disease. Antibody deposition along the glomerular basement membrane (GBM) undoubtedly contributes to the renal pathologic examination of this disease; however, other cofactors, in addition to anti-GBM antibody, may be necessary for alveolar hemorrhage to develop. A higher incidence of alveolar hemorrhage has been reported in smokers with anti-GBM antibody disease.58 Experimental studies also showed that exposure to 100% oxygen in animals with circulating anti–basement membrane antibody resulted in alveolar hemorrhage, whereas unexposed animals did not develop lung disease.59 Genetic studies have shown a strong association with HLA-DRB alleles.60,61
A 2:1 male-female ratio exists with a median age of 21 years in patients with Goodpasture’s syndrome.62,63 Alveolar hemorrhage is the most common presentation of Goodpasture’s syndrome. Evidence of renal involvement is usually present; however, some patients may only have microscopic hematuria.57 Untreated, Goodpasture’s syndrome carries a mortality rate approaching 100%, the cause of death being equally divided between uremia and alveolar hemorrhage.63 With prompt dialysis, plasma exchange, and immunosuppression, however, the acute mortality rate of the disease is about 10%, with diffuse alveolar hemorrhage being the most common cause of early death.64
The evaluation of patients suspected of having Goodpasture’s syndrome should include confirmation of alveolar hemorrhage, evaluation for renal disease, and testing for anti–basement membrane antibody. Circulating anti–basement membrane antibody can be demonstrated in more than 95% of patients through radioimmunoassay or enzyme-linked immunosorbent assay (ELISA).65 Kidney biopsy should also be considered to confirm the diagnosis and to document the extent of glomerular loss. The characteristic glomerular lesion shows strong linear deposition of immunoglobulin G (IgG) along glomerular capillaries.57 Other histologic features include segmental, necrotizing, crescentic glomerulonephritis indistinguishable from that found in other forms of vasculitis. Walker and colleagues66 demonstrated that patients with greater than 85% crescents on biopsy were significantly less likely to regain renal function. Lung biopsy is rarely necessary and often nonspecific.
Treatment consists of dialysis, plasma exchange, and immunosuppressive therapy. Immunosuppression usually includes both cyclophosphamide and corticosteroids.67 Dialysis should be performed early to reverse uremic platelet dysfunction and to prevent fluid overload because both factors may perpetuate alveolar hemorrhage. Mechanical ventilation may be necessary to provide respiratory support, as well as to facilitate clearing blood from the airways. When mechanical ventilatory support is used, efforts should be made to select lung-protective tidal volumes and to minimize the fraction of inspired oxygen. We also aggressively treat possible respiratory infection because it may precipitate and perpetuate alveolar hemorrhage. If the patient has received drugs that impair platelet function, such as aspirin, we also administer platelets in cases of life-threatening hemorrhage. Alveolar hemorrhage generally responds within 1 to 3 days to this treatment.68 In refractory cases, there has been anecdotal response to mycophenolate or to anti-CD20 antibody. Once the patient has recovered, the importance of maintenance immunosuppression in preventing recurrent alveolar hemorrhage cannot be overemphasized.57,68,69
Wegener’s Granulomatosis
Another form of vasculitis that commonly presents as alveolar hemorrhage is WG. This disorder is characterized by a granulomatous vasculitis involving the upper and lower airways and is associated with rapidly progressive renal failure. WG represented 15% of the cases of alveolar hemorrhage in a series reported by Leatherman.57 The incidence of pulmonary hemorrhage in this disorder is reported to vary between 12% and 30%.70,71 Clinical findings that may suggest a diagnosis of WG include nodules visible on a chest radiograph and evidence of upper airway involvement including chronic otitis media, sinusitis, nasal septal perforation, and tracheal stenosis.72 Eye involvement with either proptosis or extraocular muscle entrapment may occur.72 Skin lesions may include petechiae, palpable purpura, ulcers, vesicles, papules, and subcutaneous nodules.70 Musculoskeletal findings include myalgias, arthralgias, and pauciarticular or migratory arthritis. Neurologic manifestations include sensorineural deafness, mononeuritis multiplex, and cranial nerve palsies.70,72 Subglottic tracheal stenosis or obstruction can occur in these patients and should be considered before endotracheal intubation is undertaken. In up to 10% of patients, tracheostomy may be required to manage the airway at some time during the course of their illness.72
Laboratory findings in WG include leukocytosis, anemia, thrombocytosis, and an elevated erythrocyte sedimentation rate. A valuable laboratory test is the antineutrophil cytoplasmic antibody (ANCA), which detects IgG directed at a variety of neutrophil and monocyte antigens. Clinically important ANCAs are of two types: antiproteinase 3 antibodies (anti-PR3) and antimyeloperoxidase antibodies (anti-MPO). When serum containing these antibodies is applied to neutrophils and stained by indirect immunofluorescence, anti-PR3 produces a cytoplasmic pattern of staining (c-ANCA), whereas anti-MPO produces a perinuclear or nuclear pattern (p-ANCA). Both anti-PR3 (seen almost exclusively in WG) and anti-MPO (which may be seen in pauci-immune rapidly progressive glomerulonephritis, Churg-Strauss syndrome, and MPA) can be measured more directly by ELISA. The sensitivity of ANCA has been reported to be 80% to 96% in patients with active generalized (e.g., having renal involvement) WG, and generally these patients have both c-ANCA and anti-PR3 positivity. In the ANCA-associated systemic vasculitides, which include WG, alveolar hemorrhage has been found in patients who have tested positive for either c-ANCA or p-ANCA (see “Microscopic Polyangiitis” later). More recently, the presence of an immunoglobulin M (IgM) isotype of ANCA has been strongly associated with alveolar hemorrhage; conversely, patients lacking IgM ANCA may have a low risk for alveolar hemorrhage.73 Frequently the diagnosis of WG relies on tissue examination. Biopsies of upper airway lesions are probably acceptable in nonemergent situations when diagnosis can be delayed. However, in the case of severe alveolar hemorrhage, there is frequently an emergent need for diagnosis so that effective treatment can be instituted. An open lung biopsy often provides the diagnosis. Potential infectious causes, especially mycobacterial and fungal pathogens, can also be excluded. Fauci and coworkers have developed a scheme of major and minor criteria for WG on the basis of histologic diagnosis. Three major pathologic manifestations were identified including parenchymal necrosis, vasculitis, and granulomatous inflammation74; however, in 18% of biopsies, less distinctive histologic features were the predominant findings. If definitive tissue biopsy cannot be obtained, a WG with alveolar hemorrhage diagnosis should be based on the histologic finding of a small vessel vasculitis or crescentic glomerulonephritis, together with compelling clinical evidence of WG consisting of cavitary pulmonary nodules or characteristic upper airway involvement.57 Moreover, the presence of a positive c-ANCA test can help to confirm the diagnosis.
The recommended treatment for alveolar hemorrhage resulting from WG includes high-dose corticosteroids and cyclophosphamide.57,75 We recommend that patients who are critically ill receive intravenous methylprednisolone at doses up to 1 g daily for the first 3 days in addition to cyclophosphamide at 3 to 5 mg/kg for 3 to 5 days. After this period, the prednisone dosage is reduced to l mg/kg/day and cyclophosphamide is continued at 1.5 mg/kg/day. Most patients respond favorably to this regimen, but the mortality rate remains substantial, often because of renal failure or sepsis. Plasma exchange or intravenous immunoglobulin may be useful in patients with life-threatening diffuse alveolar hemorrhage or when disease persists despite corticosteroids and cyclophosphamide.76 Other rescue therapies include rituxan,77 trimethoprim-sulfamethoxazole, anti–tumor necrosis factor antibodies, and antilymphocyte antibodies. The supportive measures described earlier for alveolar hemorrhage in Goodpasture’s syndrome also apply to this disease.
Microscopic Polyangiitis
Treatment requires high-dose corticosteroids and cyclophosphamide in the same doses as for WG. Intravenous immunoglobulin may be effective in difficult cases. Factor VIIa has also been tried, and the rescue therapies described earlier for WG may also be effective. For the patient with respiratory failure caused by alveolar hemorrhage, ventilator guidelines for ALI/ARDS should be followed. Once a patient survives the acute alveolar hemorrhage, there are reasonable prospects for long-term survival, although this depends to a large degree on whether renal function recovers. Pulmonary fibrosis is often present before, during, and after the initial diagnosis of MPA.78
Catastrophic Antiphospholipid Syndrome
The term antiphospholipid syndrome was coined to describe patients with systemic thrombosis or recurrent fetal loss having increased antiphospholipid antibodies in the circulation. A subset presenting with widespread vascular thrombosis and a fulminating clinical course, often involving respiratory failure, was subsequently called the catastrophic antiphospholipid syndrome (CAPS).79 This syndrome can occur in those without a recognized rheumatologic disease but also in those with SLE or other diseases.
CAPS generally has been considered a noninflammatory thrombotic disease leading to widespread ischemia and necrosis, producing multiorgan failure and death. More recently, however, cases of alveolar hemorrhage have been described in the setting of antiphospholipid syndrome in which thrombosis was not evident clinically or pathologically. These authors proposed that a nonthrombotic mechanism for pulmonary capillaritis and alveolar hemorrhage should be sought.80
Infections, trauma, procedures, drugs, and malignancy have been implicated as precipitating factors. Clinically there is often evidence of widespread arterial and venous occlusions. The renal, pulmonary, and central nervous systems are most often affected. Multiple pulmonary manifestations have been reported including pulmonary thromboembolism, pulmonary hypertension, and ALI in addition to diffuse alveolar hemorrhage. Serologic findings include elevated titers of anticardiolipin antibody or the lupus anticoagulant. Increased β2-glycoprotein I has been linked to CAPS.81
Treatment generally involves immunosuppression along the lines of treatment for other immune alveolar hemorrhage syndromes. Plasmapheresis may be effective.82 The role for anticoagulation in the acute setting of alveolar hemorrhage is uncertain, but we recommend first establishing control of bleeding with immunosuppressive therapy and plasmapheresis and then later instituting antithrombotic treatment.
Systemic Lupus Erythematosus
In patients with SLE, a wide variety of lung lesions are found. Histologic evidence of alveolar hemorrhage can be found in 40% of patients at autopsy.83 Massive alveolar hemorrhage is uncommon, being reported in only 5% of patients in one series of 99 patients with SLE and lung involvement.84 Although massive alveolar hemorrhage may be the presenting manifestation of SLE, this is uncommon.85,86 The clinical presentation of alveolar hemorrhage in SLE is similar to that in other alveolar hemorrhage syndromes; however, fever as high as 39° C to 40° C may be a prominent feature.84 At the time of alveolar hemorrhage, patients often manifest other typical clinical findings of SLE, particularly nephritis, highlighting the systemic nature of this disease.87
The cause of alveolar hemorrhage in SLE is not entirely clear. In some patients, antiphospholipid antibodies, as discussed earlier, may play a role.88 Pathologic studies of open lung biopsies have not always demonstrated immune complex deposition.83,86 Patients with SLE often show a microscopic angiitis on biopsy.85,87 Considerable clinical overlap between the alveolar hemorrhage syndrome and acute lupus pneumonitis exists.83,87 Because treatment of both lesions is similar, we recommend separating these two pulmonary manifestations on clinical grounds without the need for open lung biopsy, as long as infection has been adequately excluded.
Infection is the most important factor to exclude in diagnosing alveolar hemorrhage in the patient with SLE. At least half of the patients with SLE who present with infiltrates should be expected to have an infectious cause.83 Because many of these patients are receiving immunosuppressive therapy and because SLE is associated with impaired cellular immunity, the differential diagnosis for infectious agents is broad and includes bacterial, fungal, mycobacterial, viral, and parasitic pathogens. Bronchoscopy with lavage and transbronchial biopsies are the logical procedures used to search for an infectious cause. Open lung biopsy may be necessary in some cases.
Mortality rate from SLE-associated alveolar hemorrhage is variable in small series, ranging from about 20% to more than 85%.57,86,87 Many patients develop respiratory failure, and the need for mechanical ventilation is associated with an increased mortality risk.87 This high risk for respiratory failure is multifactorial, with contributions from alveolar hemorrhage, a high prevalence of underlying atelectasis, and diaphragmatic weakness.89,90 We recommend treating these patients with both high-dose corticosteroids and cyclophosphamide in a regimen similar to that used for WG.
Idiopathic Pulmonary Hemosiderosis
The diagnosis of idiopathic pulmonary hemosiderosis is, by definition, one of exclusion. The syndrome is typically an illness that presents in infancy or childhood.91 The disease is characterized by recurrent episodes of alveolar hemorrhage, although often these episodes may be subclinical.92 There have been familial clusters of cases.93,94 Some cases of unexplained alveolar hemorrhage with onset during adulthood have been reported. The specific treatment of this illness is not clear. Most patients do well during the acute episode with supportive care alone, but there may be a short-term benefit from corticosteroid therapy. Long-term corticosteroid treatment has been described in some cases.
Cryptogenic Organizing Pneumonia
Cryptogenic organizing pneumonia (COP), also termed idiopathic bronchiolitis obliterans with organizing pneumonia (BOOP), frequently presents as an acute illness with respiratory failure. This disease often responds well to therapy without residual respiratory deficit, if timely diagnosis and treatment are undertaken. COP presents throughout adult life and shows no particular demographic associations.95 The presenting symptoms include cough, dyspnea, or both in more than two thirds of cases. Flulike symptoms are present in 14%, and patients usually present with subacute symptoms within 3 months.96,97 Examination of the lung reveals dry crackles in 50% to 75% of cases. Wheezing and finger clubbing are rarely seen. Up to 12% of patients can be expected to present with a normal physical examination.
The chest radiograph most often shows patchy alveolar infiltrates scattered throughout all lung fields. Interstitial infiltrates and nodular densities may be seen.97 Some reports suggest that interstitial densities on chest radiograph may be associated with a worse prognosis.98 High-resolution computed tomography (HRCT) of the chest shows predominantly subpleural or peribronchial areas of airspace consolidation, small nodules, or both in almost all cases.99 These changes are not pathognomonic for COP, but the HRCT images may be useful for directing biopsies to abnormal areas. The results of physiologic testing characteristically reveal a restrictive ventilatory defect with reduced lung volumes. Obstructive flow defects are seen only in smokers.97 The DLCO is frequently abnormal, out of proportion to the other pulmonary function tests.96 Resting and exercise-induced hypoxemia are almost always present.
BAL usually shows increased cellularity. An increased percentage of lymphocytes, neutrophils, or eosinophils may exist, but this finding does not help distinguish COP from other lung diseases.97 The diagnosis is difficult to make from a transbronchial lung biopsy because the tissue samples obtained are often not large enough.96 Transbronchial biopsy is, however, useful to rule out other disorders, especially infections. The gold standard for diagnosis is the open lung biopsy.96 Findings include patchy areas of intraluminal polyps of granulation tissue and constrictive bronchiolitis, organizing inflammation within the alveolar ducts, interstitial mononuclear cell infiltrate of variable density, alveolar space foam cells, and the absence of honeycombing or extensive interstitial fibrosis.100
COP may be associated with systemic diseases, certain inhalational exposures, or a drug reaction. A viral cause is hypothesized for at least a proportion of the cases of idiopathic COP.101 An association between COP and connective tissue diseases, especially rheumatoid arthritis (RA), exists.102 COP has also been reported in association with human immunodeficiency virus infection, radiation therapy, and smoking freebase cocaine.103–105 Thus, it appears that COP may appear secondary to a variety of pulmonary insults and may represent an aberrant healing process in the distal airspace.
The mortality rate of COP is about 5%.96 Typically, prednisone is used to treat this disease at a dose of 1 mg/kg/day. For the critically ill patient in the ICU, higher doses of parenteral corticosteroids can be used. Additional immunosuppression with cyclophosphamide is usually not required in COP and is associated with a high rate of complications.106,107 When respiratory failure develops, the ventilatory management of COP is similar to that in patients with IPF, except that chronic pulmonary hypertension and right ventricular failure are less of a problem.
Connective Tissue Diseases
Rheumatologic disorders affect the lungs in a variety of ways. Alveolar hemorrhage is discussed earlier. Interstitial fibrosis, vasculitis, pulmonary hypertension, and respiratory muscle weakness are mechanisms by which the patient with connective tissue disease may develop respiratory failure. In a recent study of 66 patients with connective tissue disease and respiratory failure, the most common underlying diagnoses were SLE, RA, and vasculitis.108 Pneumonia was the leading cause of respiratory failure, followed by pulmonary edema and alveolar hemorrhage. The hospital mortality rate in this cohort was 62%. This section focuses on those rheumatologic disorders most likely to be encountered in an ICU.
Lupus Pneumonitis
SLE is a systemic disorder characterized by widespread inflammation of serosal surfaces, skin, connective tissues, kidney, lung, and other organ systems. The characteristic finding of SLE is the presence of circulating autoantibodies, particularly antinuclear antibodies, and immune complexes. Pleuropulmonary involvement is common.109 For the purposes of this discussion, we define lupus pneumonitis as any acute presentation of respiratory disease and pulmonary infiltrates, associated with SLE, that is neither infection nor frank alveolar hemorrhage (see previous discussion). Matthay found acute presentation of lung disease in 11% of patients hospitalized for SLE.110 In 50% of these patients, lupus pneumonitis was the presenting manifestation of SLE, which is distinctly unusual in SLE-associated alveolar hemorrhage.87
The patients typically have dyspnea, cough, and pleuritic chest pain. Fever and tachypnea are also frequently present. The chest radiograph characteristically shows bilateral basilar or diffuse infiltrates, but unilateral infiltrates may be present and atelectasis may be a prominent feature. An accompanying pleural effusion is often present.109–111 Cyanosis and basilar rales are often found on physical examination. The arterial blood gas frequently shows severe hypoxemia. Histopathologic findings on open lung biopsy are variable and may include areas of desquamative or unusual interstitial pneumonia, COP, and microscopic alveolar hemorrhage. Pulmonary infarction is associated with anticardiolipin antibody, and focal atelectasis from respiratory muscle weakness can be seen.111–113 The rapidity of clinical deterioration can be alarming. The mortality rate for patients who present with the characteristic clinical features of lupus pneumonitis can be up to 50% despite treatment.110,113 The treatment of these patients usually includes high-dose corticosteroids. Cyclophosphamide and azathioprine have been used in cases of progressive disease.111–113 In patients with acute, severe neurologic lupus, cyclophosphamide was more effective than high-dose corticosteroids.114 We recommend the initial use of both cyclophosphamide and high-dose corticosteroids in a regimen similar to that used to treat vasculitis (see previous discussion). Before initiating immunosuppressive therapy, it is essential to exclude infection with bronchoscopy or open lung biopsy. Maintaining a high suspicion for infection in the patient who is unresponsive to immunosuppression or who shows clinical deterioration despite treatment is also important.
Rheumatoid Arthritis
RA is a disease of subacute and chronic inflammation characterized by erosive arthritis that is usually symmetrical, affecting mainly the peripheral joints. A positive rheumatoid factor is present in at least 75% of cases. RA, like SLE, has a variety of associated pleuropulmonary manifestations that can present during the course of illness.109 It is important for the intensivist to recognize the spectrum of lung disease associated with RA because many of the findings on chest radiograph can be ascribed to relatively benign disease.115 Moreover, even moderately severe chronic pulmonary disease may go undetected because it is obscured by musculoskeletal limitations. Two of the more common forms of rheumatoid involvement that present with severe lung disease are interstitial fibrosis and COP.
Interstitial fibrosis is a relatively common finding in patients who have RA. In the overwhelming majority of patients, this is an incidental finding and is asymptomatic.116 The clinical course of RA-associated interstitial lung disease is typically much more benign than that seen in IPF; however, a subset of patients presents with fulminant interstitial lung disease associated with RA.117 Like IPF, the physical examination frequently shows Velcro-like rales, and the chest radiograph in the more severe cases typically shows diffuse bilateral reticular or reticulonodular infiltrates.117
Patients with RA are generally admitted to the ICU with sepsis rather than complications of the arthritis itself.118 Most of these patients have been treated previously with corticosteroids or other immunosuppressive regimens. Airway management may be particularly challenging because many RA patients have limited mouth opening, atlantoaxial instability, or cricoarytenoid arthritis. Fiberoptic intubation is a necessary skill for safely managing patients with RA and ventilatory failure.
RA is the most common connective tissue disease to present with COP. This illness typically presents with a subacute onset of dyspnea. The presentation and pathologic features of COP associated with RA are indistinguishable from those of idiopathic COP.119 Diagnosis usually requires an open lung biopsy to define the histologic features. In a few patients the diagnosis is made by transbronchial biopsy. Bronchoscopy should be undertaken before immunosuppressive therapy to rule out infection. Treatment of this disorder is identical to the treatment of COP, but the prognosis for RA-associated COP appears to be worse.120 For this reason, we consider cyclophosphamide earlier to treat this disease when it does not respond rapidly to corticosteroids.
Progressive Systemic Sclerosis
Progressive systemic sclerosis (PSS) and the related disorder, the CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasis) syndrome, are disorders characterized by fibrosing inflammation of the skin with variable visceral involvement. Patients with PSS and CREST develop interstitial lung disease that histopathologically resembles the lung fibrosis associated with RA and IPF.121 The prevalence of pulmonary fibrosis detected by chest radiograph is approximately 36% in PSS and 20% in CREST.122 The clinical presentation is indistinguishable from other secondary causes of pulmonary fibrosis. Many of these patients also have chronic aspiration resulting from esophageal dysfunction, which can precipitate and exacerbate pulmonary inflammation and fibrosis.123
The diagnosis of lung disease in PSS and CREST usually does not require an open lung biopsy. Pulmonary function tests characteristically reveal a restrictive ventilatory defect with low lung volumes.124 The detection of circulating autoantibodies may be helpful in diagnosing these diseases.109 Anticentromere antibody presence is associated with a lower incidence of pulmonary fibrosis in CREST.125,126 Anti-SCL-70 antibody presence is associated with a higher incidence of pulmonary fibrosis.127 Bronchoscopy can be used to evaluate for an infectious process and to look for vegetable matter or lipid-laden macrophages, which may suggest chronic aspiration. Treatment of interstitial lung disease associated with PSS or CREST with cyclophosphamide has been shown to be effective in the National Institutes of Health Scleroderma Lung Study by improving physiology, relieving dyspnea, and enhancing quality of life. Nevertheless, the prognosis remains poor. The role of steroids is unproved.
Pulmonary hypertension is another common manifestation of lung involvement in PSS and the CREST syndrome. This can occur without evidence of other lung disease, but it is often associated with interstitial disease. When accompanying interstitial lung disease, pulmonary hypertension is often more prominent than one would expect from the degree of interstitial lung disease alone. The prevalence of pulmonary hypertension in PSS has been found to be about 33%. In CREST the prevalence of pulmonary hypertension is at least as high.128 The cause of pulmonary hypertension in these disorders is not well understood. Some experts have speculated that early in the course of disease there is a period of vascular reactivity associated with Raynaud’s phenomenon.129 This period is hypothesized to be followed by a period of increased pulmonary pressures associated with local hypoxia.130 Finally, there is vascular remodeling with intimal thickening and loss of capillary beds.131
The patient with pulmonary hypertension may present with exertional dyspnea or impending respiratory failure, but pulmonary hypertension may also be asymptomatic.128 Physical findings include those features commonly associated with PSS or CREST. Findings suggestive of cor pulmonale may be present, including jugular venous distention with prominent “a” waves, loud or palpable S2, left parasternal lift, and an S4 gallop that increases with inspiration. Although approximately 88% specific, the physical examination is only about 63% sensitive to identifying definite pulmonary hypertension in PSS.128 The single best marker of underlying pulmonary hypertension is a low DLCO. When the DLCO is below 40% to 55% of predicted normal values, pulmonary hypertension is likely to be present.128,132 The sensitivity of this finding, irrespective of the presence of interstitial lung disease, is about 87% with a specificity of 88%.128,132 The electrocardiogram may show right bundle branch block, right ventricular hypertrophy, or right atrial enlargement. Echocardiography is highly specific for pulmonary hypertension if a Doppler gradient analysis of tricuspid regurgitation suggests pulmonary hypertension.133 The gold standard has been pulmonary artery catheterization with documentation of an elevated pulmonary artery pressure and a normal pulmonary capillary wedge pressure.
Early intervention with vasodilating agents may alter the course of pulmonary hypertension by preventing progression that is dependent on high pulmonary artery pressures or by ameliorating angiogenesis or fibrosis. Bosentan, the dual endothelin receptor inhibitor, has been found effective in clinical trials of subjects with pulmonary hypertension including those with scleroderma.134 Other treatments such as prostanoids, endothelin receptor blockers, and sildenafil may play a role in chronic management of the patient with pulmonary hypertension. In the acute ICU setting, treatment of acute-on-chronic cor pulmonale generally involves seeking treatable precipitants, infusing rapidly acting vasoactive drugs such as dobutamine, and giving short-acting pulmonary vasodilators such as inhaled nitric oxide or inhaled prostacyclin. In mechanically ventilated patients, tidal volumes should be limited to reduce the potential for superimposing ALI.
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis, in the majority of cases, does not result in an illness that requires critical care management. The cases that do present acutely are important to identify because these patients respond well to treatment. Furthermore, identifying an inciting exposure can prevent serious relapse or progression to chronic lung disease. Acute and subacute hypersensitivity pneumonitis are the most likely forms of this illness to result in admission to the ICU. The cause of hypersensitivity pneumonitis involves exposure to an airborne agent (Table 49.1).135 Associated symptoms include malaise, myalgia, fever, nonproductive cough, and dyspnea.136 The patient’s history may reveal an onset of symptoms within 4 to 6 hours of the exposure to a previously sensitized antigen.137 The physical examination frequently reveals diffuse basilar lung crackles. The chest radiograph findings vary from normal to nodular or diffuse fluffy infiltrates. A predilection for involvement of the lung bases exists.138 This is in contrast to the upper lung zone predominance seen in chronic hypersensitivity pneumonitis. The HRCT scan in the acute phase shows diffuse airspace consolidation that evolves to a fine nodular or reticulonodular pattern over the course of days to weeks. Laboratory studies generally show a leukocytosis with a leftward shift in neutrophils. Eosinophilia is variably present, usually at low levels.136 A polyclonal gammopathy may be present. Specific serum precipitins should be interpreted only as evidence of exposure, not as definitive evidence of disease. Rheumatoid factor may be present in as high as 50% of cases.136,137 Pulmonary function tests usually show restrictive defects with maintenance of expiratory flow rates.139
Table 49.1
Hypersensitivity Pneumonitis (HSP) (Extrinsic Allergic Alveolitis): Reported Associations
| Disease | Source of Particles |
| Farmer’s lung | “Moldy” hay, grain, silage |
| Bird fancier’s, breeder’s, or handler’s lung | Avian droppings or feathers |
| Humidifier or air conditioner lung | Contaminated water in humidification and air conditioning systems |
| Chemical worker’s lung | Polyurethane foam, varnishes, lacquer |
| Bagassosis | “Moldy” bagasse (sugar cane) |
| Malt worker’s lung | Moldy barley |
| Mushroom worker’s lung | Mushroom compost |
| Sequoiosis | Redwood sawdust |
| Maple bark disease | Maple bark |
| Woodworker’s lung | Oak, cedar, mahogany dusts; pine and spruce pulp |
| Cheese washer’s lung | Moldy cheese |
| Suberosis | Cork dust |
| Sauna taker’s lung | Contaminated sauna water |
| Pituitary snuff taker’s lung | Heterologous pituitary snuff |
| Coffee worker’s lung | Coffee beans |
| Miller’s lung | Infested wheat flour |
| Fish meal worker’s lung | Fish meal |
| Furrier’s lung | Animal pelts |
| Lycoperdonosis | Lycoperdon puffballs |
| Compost lung | Compost |
| Wood trimmer’s disease | Contaminated wood trimmings |
| Thatched roof disease | Dried grasses and leaves |
| Streptomyces albus HSP | Contaminated fertilizer |
| Cephalosporium HSP | Contaminated basement (sewage) |
| Detergent worker’s disease | Detergent |
| Japanese summer house HSP | House dust? Bird droppings |
| Potato riddler’s lung | “Moldy” hay around potatoes |
| Tobacco worker’s disease | Mold on tobacco |
| Hot tub lung | Mold on ceiling |
| Winegrower’s lung | Mold on grapes |
| Laboratory worker’s HSP | Laboratory rat |
| Tapwater lung | Contaminated tapwater |
| Pauli’s HSP | Laboratory reagent |
| Woodman’s disease | Oak and maple trees |
Adapted from Richerson HB, Bernstein IL, Fink JN, et al: Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol 1989;84:839.
BAL nearly always shows increased cellularity.140 In the acute phase of illness, within 24 to 48 hours of the onset of symptoms, BAL typically shows a predominance of neutrophils; as the illness progresses, BAL shows a predominance of lymphocytes, up to as high as 80%.141–143 Most of the lymphocytes are suppressor/cytotoxic (CD8, suppressor cytotoxic; CD4, helper) T cells. The presence of many foamy macrophages in the BAL is also highly suggestive of hypersensitivity pneumonitis. Histopathologic examination of transbronchial biopsies or open lung biopsy shows an inflammatory process involving both the airspaces and the interstitium. A mononuclear cell infiltration with many lymphocytes exists. Foamy histiocytes and plasma cells can frequently be seen. Interstitial, often poorly formed, noncaseating granulomas may be present.135,139
The differential diagnosis of acute hypersensitivity pneumonitis should include other causes of interstitial pneumonitis such as COP or AIP (Hamman-Rich syndrome). Organic dust toxic syndrome also occurs under similar environmental exposures as hypersensitivity pneumonitis but represents an acute response to inhaled bacterial and fungal cell wall products.144 This illness tends to be more acute, resolves spontaneously, and often appears in case clusters because the response is not a specific allergic hypersensitivity. Atypical community-acquired pneumonia should also be considered in the critically ill patient. BAL and transbronchial biopsies are helpful in evaluating for an infectious cause. After the diagnosis of hypersensitivity pneumonitis is made, treatment usually includes corticosteroids and environmental counseling to avoid repeated exposure.
Drug-Induced Respiratory Failure
Drug-induced interstitial lung disease is a common complication of a variety of drugs including antibiotics and novel molecular targeted agents. Pathologic examination can include interstitial pneumonitis (interferons, methotrexate), acute eosinophilic pneumonia (SSRIs, sulfamides), alveolar hemorrhage (abciximab, allopurinol, retinoic acid), BOOP (minocycline, nitrofurantoin), and diffuse alveolar damage (bleomycin, cyclophosphamide, gemcitabine). Diagnosis is challenging as there are no pathognomonic findings in lung biopsy. Bronchoscopy might be helpful when there is eosinophilic predominance in the lavage and a clinical correlation. We recommend empirically withholding the offending agent when a patient presents with any of abovementioned forms of lung disease unless a compelling alternative diagnosis such as an infection has been identified. This complication often resolves when the offending agent is discontinued, but steroids are commonly used in the ICU setting to hasten recovery. The pneumotox.com website is a comprehensive resource that has an ever-evolving list of drugs that have been implicated in lung disease.145
Summary
Patients with immunologic lung diseases can present with fulminant respiratory failure requiring care in an ICU. These conditions require a high index of suspicion because they may mimic many atypical pneumonia syndromes.146 An efficient management strategy must include a rapid diagnosis; aggressive supportive care; and, often, therapy with immunosuppressive agents. Patients with an established diagnosis may already have received potent corticosteroids or cytotoxic therapy and are at great risk of opportunistic infection that can mimic a flare of their underlying immunologic lung disease.107,147 In addition, although in many cases immunomodulatory therapies have greatly altered the course of these diseases, treatment remains nonspecific and involves considerable toxicity. Indeed, in some series up to half of disease-related deaths can be attributed to treatment toxicity including infections and secondary malignancies.70 Supportive management generally entails lung-protective ventilation and, in appropriate patients, surveillance for pulmonary hypertension.
References
1. Nava, S, Rubini, F. Lung and chest wall mechanics in ventilated patients with end stage idiopathic pulmonary fibrosis. Thorax. 1999; 54(5):390–395.
2. Brower, RG. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342(18):1301–1308.
3. Gajic, O, Dara, SI, Mendez, JL, et al. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit Care Med. 2004; 32(9):1817–1824.
4. Carvalho, CR, Barbas, CS, Medeiros, DM, et al. Temporal hemodynamic effects of permissive hypercapnia associated with ideal PEEP in ARDS. Am J Respir Crit Care Med. 1997; 156(5):1458–1466.
5. Thorens, JB, Jolliet, P, Ritz, M, et al. Effects of rapid permissive hypercapnia on hemodynamics, gas exchange, and oxygen transport and consumption during mechanical ventilation for the acute respiratory distress syndrome. Intensive Care Med. 1996; 22(3):182–191.
6. Fernandez-Perez, ER, Yilmaz, M, Jenad, H, et al. Ventilator settings and outcome of respiratory failure in chronic interstitial lung disease. Chest. 2008; 133(5):1113–1119.
7. Nadrous, HF, Pellikka, PA, Krowka, MJ, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005; 128(4):2393–2399.
8. Schulman, DS, Biondi, JW, Matthay, RA, et al. Differing responses in right and left-ventricular filling, loading and volumes during positive end-expiratory pressure. Am J Cardiol. 1989; 64(12):772–777.
9. Biondi, JW, Schulman, DS, Matthay, RA. Effects of mechanical ventilation on right and left-ventricular function. Clin Chest Med. 1988; 9(1):55–71.
10. Schulman, DS, Biondi, JW, Matthay, RA, et al. Effect of positive end-expiratory pressure on right ventricular performance—Importance of baseline right ventricular function. Am J Med. 1988; 84(1):57–67.
11. Mebazaa, A, Karpati, P, Renaud, E, et al. Acute right ventricular failure—From pathophysiology to new treatments. Intensive Care Med. 2004; 30(2):185–196.
12. Prewitt, RM, Ghignone, M. Treatment of right ventricular dysfunction in acute respiratory-failure. Crit Care Med. 1983; 11(5):346–352.
13. Theodoraki, K, Rellia, P, Thanopoulos, A, et al. Inhaled iloprost controls pulmonary hypertension after cardiopulmonary bypass. Can J Anaesth. 2002; 49(9):963–967.
14. Bhorade, S, Christenson, J, O’Connor, M, et al. Response to inhaled nitric oxide in patients with acute right heart syndrome. Am J Respir Crit Care Med. 1999; 159(2):571–579.
15. Petzoldt, M, Braune, S, Bittmann, I, Kluge, S. Rescue therapy with a pumpless extracorporeal lung assist device in a patient with acute interstitial lung disease and severe refractory hypercapnia. Respir Care. 2012; 57(2):293–297.
16. Zulian, F, Martinez Toledo, MM, Amigoni, A, et al. Successful use of extracorporeal membrane oxygenation for severe interstitial lung disease in a child with dermatomyositis. Intensive Care Med. 2007; 33(9):1663–1666.
17. Tomii, K, Tachikawa, R, Chin, K, et al. Role of non-invasive ventilation in managing life-threatening acute exacerbation of interstitial pneumonia. Intern Med. 2010; 49(14):1341–1347.
18. Yokoyama, T, Kondoh, Y, Taniguchi, H, et al. Noninvasive ventilation in acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2010; 49(15):1509–1514.
19. Carrington, CB, Gaensler, EA, Coutu, RE, et al. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med. 1978; 298(15):801–809.
20. Bitterman, PB, Rennard, SI, Keogh, BA, et al. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members. N Engl J Med. 1986; 314(21):1343–1347.
21. Turner-Warwick, M, Burrows, B, Johnson, A. Cryptogenic fibrosing alveolitis—Clinical features and their influence on survival. Thorax. 1980; 35(3):171–180.
22. Hunninghake, GW, Lynch, DA, Galvin, JR. Radiologic findings are strongly associated with a pathological diagnosis of usual interstitial pneumonia. Chest. 2003; 124:1215–1223.
23. Martinez, FJ, Safrin, S, Weycker, D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005; 142(12):963–967.
24. Turner-Warwick, M, Lebowitz, M, Burrows, B, et al. Cryptogenic fibrosing alveolitis and lung cancer. Thorax. 1980; 35(7):496–499.
25. Pochis, WT, Krasnow, AZ, Collier, BD, et al. Idiopathic pulmonary fibrosis—A rare cause of scintigraphic ventilation-perfusion mismatch. Clin Nucl Med. 1990; 15(5):321–323.
26. Kubo, H, Nakayama, K, Yanai, M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005; 128(3):1475–1482.
27. Collard, HR, Ryu, JH, Douglas, WW, et al. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest. 2004; 125(6):2169–2174.
28. Hunninghake, GW. Antioxidant therapy for idiopathic pulmonary fibrosis. N Engl J Med. 2005; 353(21):2285–2287.
29. Raghu, G, Brown, KK, Bradford, WZ, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004; 350(2):125–133.
30. Parambil, JG, Myers, JL, Ryu, JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005; 128(5):3310–3315.
31. Fumeaux, T, Rothmeier, C, Jolliet, P. Outcome of mechanical ventilation for acute respiratory failure in patients with pulmonary fibrosis. Intensive Care Med. 2001; 27(12):1868–1874.
32. Al-Hameed, FM, Sharma, S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J. 2004; 11(2):117–122.
33. Huie, TJ, Olson, AL, Cosgrove, GP, et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: Aetiology and outcomes. Respirology. 2010; 15(6):909–917.
34. Rangappa, P, Moran, JL. Outcomes of patients admitted to the intensive care unit with idiopathic pulmonary fibrosis. Crit Care Resuscitation. 2009; 11(2):102–109.
35. Stern, JB, Mal, H, Groussard, O, et al. Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure. Chest. 2001; 120(1):213–219.
36. Mallick, S. Outcome of patients with idiopathic pulmonary fibrosis (IPF) ventilated in intensive care unit. Respir Med. 2008; 102(10):1355–1359.
37. Hamman, L, Rich, AR. Acute diffuse interstitial fibrosis of the lungs. Bull Johns Hopkins Hosp. 1944; 74:177–212.
38. Olson, J, Colby, TV, Elliott, CG. Hamman-Rich syndrome revisited. Mayo Clin Proc. 1990; 65(12):1538–1548.
39. Johkoh, T, Müller, NL, Taniguchi, H, et al. Acute interstitial pneumonia: Thin-section CT findings in 36 patients. Radiology. 1999; 211(3):859–863.
40. Katzenstein, ALA, Myers, JL. Idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998; 157:1301.
41. Quefatieh, A, Stone, CH, DiGiovine, B, et al. Low hospital mortality in patients with acute interstitial pneumonia. Chest. 2003; 124(2):554–559.
42. Bonaccorsi, A, Cancellieri, A, Chilosi, M, et al. Acute interstitial pneumonia: Report of a series. Eur Respir J. 2003; 21(1):187–191.
43. Suh, GY, Kang, EH, Chung, MP, et al. Early intervention can improve clinical outcome of acute interstitial pneumonia. Chest. 2006; 129(3):753–761.
44. Bradley, JD. The pulmonary hemorrhage syndromes. Clin Chest Med. 1982; 3(3):593–605.
45. Robbins, RA, Linder, J, Stahl, MG, et al. Diffuse alveolar hemorrhage in autologous bone-marrow transplant recipients. Am J Med. 1989; 87(5):511–518.
46. Perez-Arellano, JL, Losa, GJ, Garcia, MM, et al. Hemosiderin-laden macrophages in bronchoalveolar lavage fluid. Acta Cytol. 1992; 36(1):26–30.
47. Kahn, FW, Jones, JM, England, DM. Diagnosis of pulmonary hemorrhage in the immunocompromised host. Am Rev Respir Dis. 1987; 136(1):155–160.
48. Sherman, JM, Winnie, G, Thomassen, MJ, et al. Time course of hemosiderin production and clearance by human pulmonary macrophages. Chest. 1984; 86(3):409–411.
49. Ewan, PW, Jones, HA, Rhodes, CG, et al. Detection of intrapulmonary hemorrhage with carbon monoxide uptake. Application in Goodpasture’s syndrome. N Engl J Med. 1976; 295(25):1391–1396.
50. Bowley, NB, Hughes, JM, Steiner, RE. The chest x-ray in pulmonary capillary haemorrhage: Correlation with carbon monoxide uptake. Clin Radiol. 1979; 30(4):413–417.
51. Barnes, SL, Naughton, M, Douglass, J, Murphy, D. Extracorporeal membrane oxygenation with plasma exchange in a patient with alveolar haemorrhage secondary to Wegener’s granulomatosis. Intern Med J. 2012; 42(3):341–342.
52. Ahmed, SH, Aziz, T, Cochran, J, Highland, K. Use of extracorporeal membrane oxygenation in a patient with diffuse alveolar hemorrhage. Chest. 2004; 126(1):305–309.
53. Heslet, L, Nielsen, JD, Levi, M, Sengeløv, H, et al. Successful pulmonary administration of activated recombinant factor VII in diffuse alveolar hemorrhage. Crit Care. 2006; 10(6):R177.
54. Betensley, AD, Yankaskas, JR. Factor VIIa for alveolar hemorrhage in microscopic polyangiitis. Am J Respir Crit Care Med. 2002; 166(9):1291–1292.
55. Sun, LC, Tseng, YR, Huang, SC, et al. Extracorporeal membrane oxygenation to rescue profound pulmonary hemorrhage due to idiopathic pulmonary hemosiderosis in a child. Pediatr Pulmonol. 2006; 41(9):900–903.
56. Rabe, C, Appenrodt, B, Hoff, C, et al. Severe respiratory failure due to diffuse alveolar hemorrhage: Clinical characteristics and outcome of intensive care. J Crit Care. 2010; 25(2):230–235.
57. Leatherman, JW, Davies, SF, Hoidal, JR. Alveolar hemorrhage syndromes: Diffuse microvascular lung hemorrhage in immune and idiopathic disorders. Medicine. 1984; 63(6):343–361.
58. Donaghy, M, Rees, AJ. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. Lancet. 1983; 2(8364):1390–1393.
59. Jennings, L, Rohold, JA, Pressman, D, et al. Experimental anti-alveolar basement membrane antibody-mediated pneumonitis. I. The role of increased permeability of the alveolar capillary wall induced by oxygen. J Immunol. 1981; 127(1):129–134.
60. Dunckley, H, Chapman, JR, Burke, J, et al. HLA-DR and -DQ genotyping in anti-GBM disease. Disease markers.. 1991; 9(5):249–256.
61. Rees, A, Peters, DK, Compston, DAS. Strong association between HLA-DRw2 and antibody-mediated Goodpasture’s syndrome. Lancet. 1978; I:966–968.
62. Young, KR, Jr. Pulmonary-renal syndromes. Clin Chest Med. 1989; 10(4):655–675.
63. Benoit, FL, Rulon, DB, Theil, GB, et al. Goodpasture’s syndrome: A clinicopathologic entity. Am J Med. 1964; 37:424–444.
64. Chan, AL, Louie, S, Leslie, KO, et al. Cutting edge issues in Goodpasture’s disease. Clin Rev Allergy Immunol. 2011; 41(2):151–162.
65. Fish, AJ, Kieppel, M, Jeraj, K, et al. Enzyme immunoassay of anti-glomerular basement membrane antibodies. J Lab Clin Med. 1985; 105(6):700–705.
66. Walker, RG, Scheinkestel, C, Becker, GJ, et al. Clinical and morphological aspects of the management of crescentic anti-glomerular basement membrane antibody (anti-GBM) nephritis/Goodpasture’s syndrome. Q J Med. 1985; 54(213):75–89.
67. Levy, JB, Turner, AN, Rees, AJ, et al. Long-term outcome of anti-glomerular basement membrane antibody disease treated with plasma exchange and immunosuppression. Ann Intern Med. 2001; 134(11):1033–1042.
68. Johnson, JP, Moore, J, Austin, HA, et al. Therapy of anti-glomerular basement membrane antibody disease: Analysis of prognostic significance of clinical, pathologic and treatment factors. Medicine. 1985; 64(4):219–227.
69. Lockwood, CM, Rees, M, Pearson, TA, et al. Immunosuppression and plasma-exchange in the treatment of Goodpasture’s syndrome. Lancet. 1976; 1(7962):711–715.
70. Hoffman, GS, Kerr, GS, Leavitt, MD, et al. Wegener’s granulomatosis—An analysis of 158 patients. Ann Intern Med. 1992; 116(6):488–498.
71. Misset, B, Glotz, D, Escudier, B, et al. Wegener’s granulomatosis presenting as diffuse pulmonary hemorrhage. Intensive Care Med. 1991; 17(2):118–120.
72. Mcdonald, TJ, Deremee, RA. Wegener’s granulomatosis. Laryngoscope. 1983; 93(2):220–231.
73. Esnault, VLM, Soleimani, B, Keogan, MT, et al. Association of IgM with IgG ANCA in patients presenting with pulmonary hemorrhage. Kidney Int. 1992; 41(5):1304–1310.
74. Travis, WD, Hoffman, GS, Leavitt, RY, et al. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol. 1991; 15(4):315–333.
75. Jantz, MA, Sahn, SA. Corticosteroids in acute respiratory failure. Am J Respir Crit Care Med. 1999; 160(4):1079–1100.
76. Frankel, SK, Cosgrove, GP, Fischer, A, et al. Update in the diagnosis and management of pulmonary vasculitis. Chest. 2006; 129(2):452–465.
77. Stone, JH, Merkel, PA, Spiera, R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010; 363(3):221–232.
78. Tzelepis, GE, Kokosi, M, Tzioufas, A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J. 2010; 36(1):116–121.
79. Asherson, RA. The catastrophic antiphospholipid syndrome. J Rheumatol. 1992; 19(4):508–512.
80. Deane, K, West, S. Antiphospholipid antibodies as a cause of pulmonary capillaritis and diffuse alveolar hemorrhage. J Invest Med. 2004; 52(1):S127.
81. Abinader, A, Hanly, AJ, Lozada, CJ. Catastrophic antiphospholipid syndrome associated with anti-beta-2-glycoprotein I IgA. Rheumatology. 1999; 38(1):84–85.
82. Waterer, GW, Latham, B, Waring, JA, et al. Pulmonary capillaritis associated with the antiphospholipid antibody syndrome and rapid response to plasmapheresis. Respirology. 1999; 4(4):405–408.
83. Miller, LR, Greenberg, SD, McLarty, JW. Lupus lung. Chest. 1985; 88(2):265–269.
84. Onomura, K, Nakata, H, Tanaka, Y, et al. Pulmonary hemorrhage in patients with systemic lupus erythematosus. J Thorac Imaging. 1991; 6(2):57–61.
85. Myers, JL, Katzenstein, ALA. Microangiitis in lupus-induced pulmonary hemorrhage. Am J Clin Pathol. 1986; 85(5):552–556.
86. Mintz, G, Galindo, LF, Fernandez, DJ, et al. Acute massive pulmonary hemorrhage in systemic lupus erythematosus. J Rheumatol. 1978; 5(1):39–50.
87. Zamora, MR, Warner, ML, Tuder, R, et al. Diffuse alveolar hemorrhage and systemic lupus erythematosus—Clinical presentation, histology, survival, and outcome. Medicine. 1997; 76(3):192–202.
88. Nguyen, VA, Gotwald, T, Prior, C, et al. Acute pulmonary edema, capillaritis and alveolar hemorrhage: Pulmonary manifestations coexistent in antiphospholipid syndrome and systemic lupus erythematosus? Lupus. 2005; 14(7):557–560.
89. Thompson, PJ, Dhillon, DP, Ledingham, J, et al. Shrinking lungs, diaphragmatic dysfunction, and systemic lupus erythematosus. Am Rev Respir Dis. 1985; 132(4):926–928.
90. Gibson, GJ, Edmonds, JP, Hughes, GRV. Diaphragm function and lung involvement in systemic lupus erythematosus. Am J Med. 1977; 63(6):926–932.
91. Morgan, PG, Turner-Warwick, M. Pulmonary haemosiderosis and pulmonary haemorrhage. Br J Dis Chest. 1981; 75(3):225–242.
92. Cutz, E. Idiopathic pulmonary hemosiderosis and related disorders in infancy and childhood. Perspect Pediatr Pathol. 1987; 11:47–81.
93. Beckerman, RC, Taussig, LM, Pinnas, JL. Familial idiopathic pulmonary hemosiderosis. Am J Dis Child. 1979; 133(6):609–611.
94. Thaell, JF, Greipp, PR, Stubbs, SE, et al. Idiopathic pulmonary hemosiderosis: Two cases in a family. Mayo Clin Proc. 1978; 53(2):113–118.
95. Epler, GR. Bronchiolitis obliterans organizing pneumonia: Definition and clinical features. Chest. 1992; 102(1 Suppl):2S–6S.
96. Epler, GR, Colby, TV, McLoud, TC, et al. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985; 312(3):152–158.
97. King, TE, Jr., Mortenson, RL. Cryptogenic organizing pneumonitis. The North American experience. Chest. 1992; 102(1 Suppl):8S–13S.
98. Cordier, JF, Loire, R, Brune, J. Idiopathic bronchiolitis obliterans organizing pneumonia. Definition of characteristic clinical profiles in a series of 16 patients. Chest. 1989; 96(5):999–1004.
99. Muller, NL, Staples, CA, Miller, RR. Bronchiolitis obliterans organizing pneumonia: CT features in 14 patients. Am J Roentgenol. 1990; 154(5):983–987.
100. Colby, TV. Pathologic aspects of bronchiolitis obliterans organizing pneumonia. Chest. 1992; 102(1 Suppl):38S–43S.
101. Marinopoulos, GC, Huddle, KR, Wainwright, H. Obliterative bronchiolitis: Virus induced? Chest. 1991; 99(1):243–245.
102. Rees, JH, Woodhead, MA, Sheppard, MN, et al. Rheumatoid arthritis and cryptogenic organizing pneumonitis. Respir Med. 1991; 85(3):243–246.
103. Allen, JN, Wewers, MD. HIV-associated bronchiolitis obliterans organizing pneumonia. Chest. 1989; 96(1):197–198.
104. Kaufman, J, Komorowski, R. Bronchiolitis obliterans. A new clinical-pathologic complication of irradiation pneumonitis. Chest. 1990; 97(5):1243–1244.
105. Patel, RC, Dutta, D, Schonfeld, SA. Free-base cocaine use associated with bronchiolitis obliterans organizing pneumonia. Ann Intern Med. 1987; 107(2):186–187.
106. McLoud, TC, Epler, GR, Colby, TV, et al. Bronchiolitis obliterans. Radiology. 1986; 159(1):1–8.
107. Sen, RP, Walsh, TE, Fisher, W, et al. Pulmonary complications of combination therapy with cyclophosphamide and prednisone. Chest. 1991; 99(1):143–146.
108. Lee, J, Yim, JJ, Yang, SC, et al. Outcome of patients with connective tissue disease requiring intensive care for respiratory failure. Rheumatol Int. 2012; 32(11):3353–3358.
109. Hunninghake, GW, Fauci, AS. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis. 1979; 119(3):471–503.
110. Matthay, RA, Schwarz, MI, Petty, TH, et al. Pulmonary manifestations of systemic lupus erythematosus: Review of twelve cases of acute lupus pneumonitis. Medicine. 1975; 54(5):397–409.
111. Pines, A, Kaplinsky, N, Olchovsky, D, et al. Pleuro-pulmonary manifestations of systemic lupus erythematosus: Clinical features of its subgroups—Prognostic and therapeutic implications. Chest. 1985; 88(1):129–135.
112. Gammon, RB, Bridges, TA, al-Nezir, H, et al. Bronchiolitis obliterans organizing pneumonia associated with systemic lupus erythematosus. Chest. 1992; 102(4):1171–1174.
113. Pertschuk, LP, Moccia, LF, Rosen, Y, et al. Acute pulmonary complications in systemic lupus erythematosus—Immunofluorescence and light microscopic study. Am J Clin Pathol. 1977; 68(5):553–557.
114. Barile-Fabris, L, Ariza-Andraca, R, Olgun-Ortega, L, et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann Rheum Dis. 2005; 64(4):620–625.
115. Helmers, R, Galvin, J, Hunninghake, GW. Pulmonary manifestations associated with rheumatoid arthritis. Chest. 1991; 100(1):235–238.
116. Salorinne, Y. Single-breath pulmonary diffusing-capacity. Reference values and application in connective tissue diseases and in various lung diseases—Introduction. Scand J Respir Dis. 1976; 96:1.
117. Hakala, M. Poor prognosis in patients with rheumatoid-arthritis hospitalized for interstitial lung fibrosis. Chest. 1988; 93(1):114–118.
118. Dedhia, HV, DiBartolomeo, A. Rheumatoid arthritis. Crit Care Clin. 2002; 18(4):841.
119. Vanthiel, RJ, van der Burg, S, Groote, AD, et al. Bronchiolitis obliterans organizing pneumonia and rheumatoid arthritis. Eur Respir J. 1991; 4(7):905–911.
120. Geddes, DM, Corrin, B, Brewerton, DA, et al. Progressive airway obliteration in adults and its association with rheumatoid disease. Q J Med. 1977; 46(184):427–444.
121. Harrison, NK, Myers, AR, Corrin, B, et al. Structural features of interstitial lung disease in systemic sclerosis. Am Rev Respir Dis. 1991; 144(3):706–713.
122. Alton, E, Turner-Warwick, M. Lung involvement in scleroderma. In: Jayson M, Black CM, eds. Systemic Sclerosis. New York: John Wiley, 1988.
123. Johnson, DA, Drane, WE, Curran, J, et al. Pulmonary disease in progressive systemic sclerosis. A complication of gastroesophageal reflux and occult aspiration? Arch Intern Med. 1989; 149(3):589–593.
124. Bagg, LR, Hughes, DT. Serial pulmonary function tests in progressive systemic sclerosis. Thorax. 1979; 34(2):224–228.
125. Owens, GR, Fino, GJ, Herbert, DL, et al. Pulmonary function in progressive systemic sclerosis. Comparison of CREST syndrome variant with diffuse scleroderma. Chest. 1983; 84(5):546–550.
126. Steen, VD, Ziegler, GL, Rodnan, GP, et al. Clinical and laboratory associations of anticentromere antibody in patients with progressive systemic sclerosis. Arthritis Rheum. 1984; 27(2):125–131.
127. Manoussakis, MN, Constantopoulos, SH, Gharavi, AE, et al. Pulmonary involvement in systemic sclerosis. Association with anti-Scl 70 antibody and digital pitting. Chest. 1987; 92(3):509–513.
128. Ungerer, RG, Tashkin, DP, Furst, D, et al. Prevalence and clinical correlates of pulmonary arterial hypertension in progressive systemic sclerosis. Am J Med. 1983; 75(1):65–74.
129. Ohar, JM, Robichaud, AM, Fowler, AA, et al. Increased pulmonary artery pressure in association with Raynaud’s phenomenon. Am J Med. 1986; 81(2):361–362.
130. Morgan, JM, Griffiths, M, du Bois, RM, et al. Hypoxic pulmonary vasoconstriction in systemic sclerosis and primary pulmonary hypertension. Chest. 1991; 99(3):551–556.
131. Salerni, R, Rodnan, GP, Leon, DF, et al. Pulmonary hypertension in the CREST syndrome variant of progressive systemic sclerosis (scleroderma). Ann Intern Med. 1977; 86(4):394–399.
132. Steen, VD, Graham, G, Conte, C, et al. Isolated diffusing capacity reduction in systemic sclerosis. Arthritis Rheum. 1992; 35(7):765–770.
133. Murata, I, Kihara, H, Shinohara, S, et al. Echocardiographic evaluation of pulmonary arterial hypertension in patients with progressive systemic sclerosis and related syndromes. Jpn Circ J. 1992; 56(10):983–991.
134. Rubin, LJ, Badesch, DB, Barst, RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002; 346(12):896–903.
135. Costabel, U. The alveolitis of hypersensitivity pneumonitis. Eur Respir J. 1988; 1(1):5–9.
136. Fink, JN. Hypersensitivity pneumonitis. In: Lynch JP, III., DeRemee RA, eds. Immunologically Mediated Pulmonary Diseases. Philadelphia: JB Lippincott, 1991.
137. Salvaggio, JE, Robert, A. Cooke memorial lecture. Hypersensitivity pneumonitis. J Allergy Clin Immunol. 1987; 79:558.
138. Gurney, JW. Hypersensitivity pneumonitis. Radiol Clin North Am. 1992; 30(6):1219–1230.
139. Richerson, HB, Bernstein, IL, Fink, JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989; 84(5 Pt 2):839–844.
140. Semenzato, G. Current concepts on bronchoalveolar lavage cells in extrinsic allergic alveolitis. Respiration. 1988; 54(Suppl 1):59–65.
141. Salmeron, S, Brochard, L, Rain, B, et al. Early neutrophil alveolitis after rechallenge in drug induced alveolitis. Thorax. 1988; 43(8):647–648.
142. Semenzato, G, Chilosi, M, Ossi, E, et al. Bronchoalveolar lavage and lung histology. Comparative analysis of inflammatory and immunocompetent cells in patients with sarcoidosis and hypersensitivity pneumonitis. Am Rev Respir Dis. 1985; 132(2):400–404.
143. Fournier, E, Tonnel, AB, Gosset, P, et al. Early neutrophil alveolitis after antigen inhalation in hypersensitivity pneumonitis. Chest. 1985; 88(4):563–566.
144. Von Essen, S, Robbins, RA, Thompson, AB, et al. Organic dust toxic syndrome: An acute febrile reaction to organic dust exposure distinct from hypersensitivity pneumonitis. J Toxicol. 1990; 28(4):389–420.
145. Chaiyakunapruk, N, Somkrua, R, Hutubessy, R, et al, Cost effectiveness of pediatric pneumococcal conjugate vaccines: A comparative assessment of decision-making tools. BMC 2011; 9:53. http://www.pneumotox.com
146. Gross, TJ, Chavis, AD, Lynch, JP. Noninfectious pulmonary diseases masquerading as community-acquired pneumonia. Clin Chest Med. 1991; 12(2):363–393.
147. Weiss, DJ, Greenfield, JW, Jr., O’Rourke, KS, McCune, WJ. Systemic cytomegalovirus infection mimicking an exacerbation of Wegener’s granulomatosis. J Rheumatol. 1993; 20(1):155–157.