[level-membership-for-basic-science-category]
CHAPTER 13 Immunogenetics
Innate Immunity
Cell-Mediated Innate Immunity
Phagocytosis
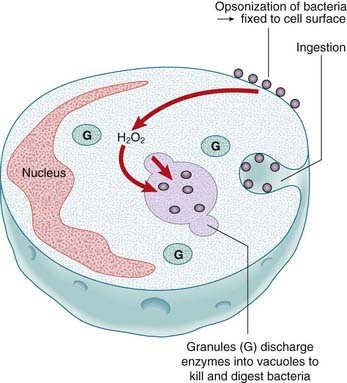
Two major cell types go on the offensive when a foreign microorganism invades—macrophages and neutrophils. Macrophages are the mature form of circulating monocytes that migrate into tissues and occur primarily around the basement membrane of blood vessels in connective tissue, lung, liver, and the lining of the sinusoids of the spleen and the medullary sinuses of the lymph nodes. They are believed to play a key role in the orchestration of both the innate and adaptive responses, and can recognise invading microorganisms through surface receptors able to distinguishing between self and pathogen. Recognition of the foreign material leads to phagocytosis by the macrophage, followed rapidly by neutrophils recruited from the circulation during the inflammatory process. The activation of the macrophage triggers the inflammatory process through the release of inflammatory mediators. The invading organism is destroyed by fusion with intracellular granules of the phagocyte and exposure to the action of hydrogen peroxide, hydroxyl radicals, and nitrous oxide (Figure 13.1).
The Toll-like Receptor Pathway
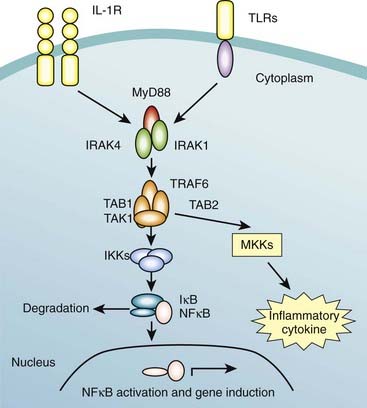
There are 10 TLRs in man, each receptor being responsible for recognition of a specific set of pathogen-associated molecular patterns. TLR2 has been well characterised and has an essential role in the detection of invading pathogens, recognizing peptidoglycans and lipoproteins associated with gram-positive bacteria, as well as a host of other microbial and endogenous ligands. TLR2’s primary function is therefore lipoprotein-mediated signaling, and activation of the pathway by recognition of its ligand results in activation of the transcription factor NF-κB, which in turn results in the increased expression of co-stimulatory molecules and inflammatory cytokines (Figure 13.3). These cytokines help mediate migration of dendritic cells from infected tissue to lymph nodes, where they may encounter and activate leukocytes involved in the adaptive immune response. The signaling pathways used by TLRs share many of the same proteins as the interleukin-1 receptor (IL-1R) pathway (Figure 13.2). Activation of TLR leads to recruitment of the MyD88 (this is sometimes known as the MyD88-dependent pathway) which mediates the interaction between IL-1R associated kinases 1 and 4 (IRAK1 and IRAK4).
The activation of the Toll pathway has several important effects in inducing innate immunity. These effects include the production of cytokines and chemokines, including IL-1, IL-6, and TNF-α (tumor necrosis factor-alpha), which have local effects in containing infection and systemic effects with the generation of fever and induction of acute phase responses, including production of C-reactive protein. One important medical condition related to the Toll pathway is septic shock, as activation of the Toll pathway by certain ligands induces systemic release of TNF-α. There are also important health-related consequences that result from TLR2 deficiency or mutation. TLR2 deficient mice are susceptible to infection by Gram-positive bacteria as well as meningitis from Streptococcus pneumoniae.
Humoral Innate Immunity
Complement
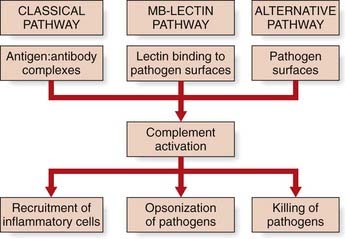
The complement system is a complex of 20 or so plasma proteins that cooperate to attack extracellular pathogens. Although the critical role of the system is to opsonize pathogens, it also recruits inflammatory cells and kills pathogens directly through membrane attack complexes. The complement system can be activated through three pathways: the classical pathway, the alternative pathway, and the mannose-binding lectin (MBL) pathway (see Figure 13.3).
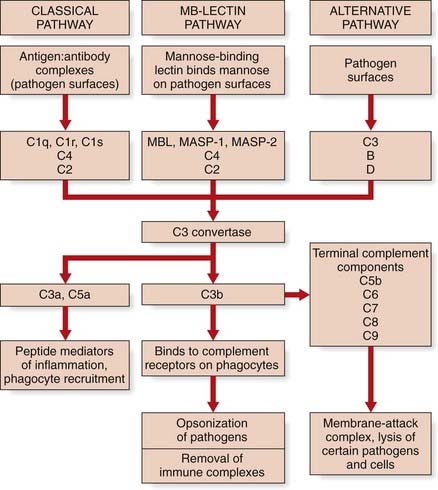
Complement nomenclature, like much else in immunology, can be confusing. Each component is designated by the letter C, followed by a number. But they were numbered in order of their discovery rather than the sequence of reactions. The reaction sequence is C1, C4, C3, C5, C6, C7, C8, and C9. The product of each cleavage reaction is designated by letters, the larger fragment being ‘b’ (b = big), and the smaller fragment ‘a’. In the lectin pathway, MBL in the blood binds another protein, a serine protease called MASP (MBL-associated serine protease). When MBL binds to its target (for example, mannose on the surface of a bacterium), the MASP protein functions like a convertase to clip C3 into C3a and C3b. C3 is abundant in the blood, so this happens very efficiently. The other two complement pathways also converge toward C3 convertase, which cleaves C3. C3a mediates inflammation while C3b binds to the pathogen surface, coating it and acting as an opsonin. The effector roles of the major complement proteins can be summarized according to function as follows (Figure 13.4):
Specific Acquired Immunity
Humoral Specific Acquired Immunity
The main mediators of humoral specific acquired immunity are immunoglobulins or antibodies. Antibodies are able to recognize and bind to surface antigens of infecting microorganisms, leading to the activation of phagocytes and the initiation of the classic pathway of complement, resulting in the generation of the MAC (see Figure 13.4) and availability of other complement effector functions. Exposure to a specific antigen results in the clonal proliferation of a small lymphocyte derived from the bone marrow (hence ‘B’ lymphocytes), resulting in mature antibody-producing cells or plasma cells.
Immunoglobulins
Immunoglobulin structure
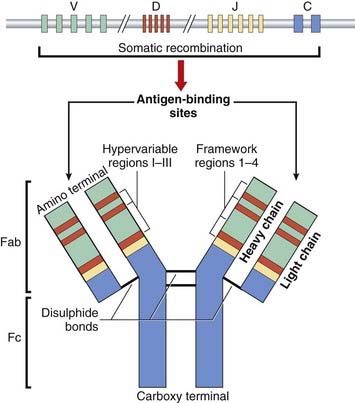
The immunoglobulin molecule is made up of four polypeptide chains—two ‘light’ (L) and two ‘heavy’ (H)—of approximately 220 and 440 amino acids in length, respectively. They are held together in a Y-shape by disulfide bonds and non-covalent interactions. Each Fab fragment is composed of L chains linked to the amino-terminal portion of the H chains, whereas each Fc fragment is composed only of the carboxy-terminal portion of the H chains (Figure 13.5).
Immunoglobulin isotypes, subclasses, and idiotypes
There are five different types of heavy chain, designated respectively as γ, µ, α, δ, and ε, one each for the five major antibody classes—the isotypes: IgG, IgM, IgA, IgD, and IgE, respectively. The L chains are of two types—kappa (κ) or lambda (λ), and these occur in all five classes of antibody, but only one type occurs in each individual antibody. Thus, the molecular formula for IgG is λ2γ2 or κ2γ2. The characteristics of the various classes of antibody are outlined in Table 13.1. In addition, there are four IgG subclasses—IgG1, IgG2, IgG3, and IgG4—and two IgA subclasses—IgA1 and IgA2—that differ in their amino acid sequence and interchain disulfide bonds. Individual antibody molecules that recognize specific antigens are known as idiotypes.

Immunoglobulin allotypes
The five immunoglobulin classes occur in all normal individuals, but allelic variants, or what are known as antibody allotypes of these classes, have also been identified. These are the Gm system associated with the heavy chain of IgG, the Am system associated with the IgA heavy chain, the Km and Inv systems associated with the κ light chain, the Oz system for the λ light chain and the Em allotype for the IgE heavy chain. The Gm and Km systems are independent of each other and are polymorphic (p. 135), the frequencies of the different alleles varying in different ethnic groups.
Generation of Antibody Diversity
Multiple myeloma
People with multiple myeloma make a single or monoclonal antibody species in large abundance, which in a proportion of patients is detected in their urine. This is known as Bence Jones protein and consists of antibody L chains. The amino-terminal ends of this protein molecule in different patients are quite variable in sequence, whereas the carboxy-terminal ends are relatively constant. These are called the variable, or V, and constant, or C, regions, respectively. However, the V regions of different myeloma proteins show four regions that vary little from one antibody to another, known as framework regions (FR 1–4), and three markedly variable regions interspersed between these, known as hypervariable regions (HV I–III) (see Figure 13.5).
DNA studies of antibody diversity
There are estimated to be some 60 different DNA segments coding for the V region of the H-chain, 40 for the V region of the κ L-chain, and 30 for the λ L-chain V region. Six functional DNA segments code for the J region of the H-chain, five for the J region of the κ L-chain, and four for the J region of the λ L-chain. A single DNA segment codes for the C region of the κ L-chain, seven for the C region of the λ L-chain and 11 functional DNA segments code for the C region of the different classes of H-chain. There are also 27 functional DNA segments coding for the D region of the H-chain (Figure 13.6).
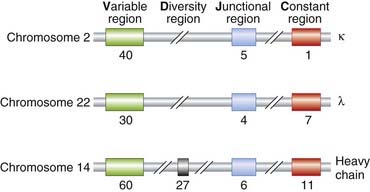
FIGURE 13.6 Estimated number of the various DNA segments coding for the κ, λ, and various heavy chains.
The genomic regions in question also contain a large number of unexpressed DNA sequences or pseudogenes (p. 17). Although the coding DNA segments for the various regions of the antibody molecule can be referred to as ‘genes’, use of this term in regard to antibodies has deliberately been avoided because they could be considered an exception to the general rule of ‘one gene–one enzyme (or protein)’ (p. 167).
Antibody gene rearrangement
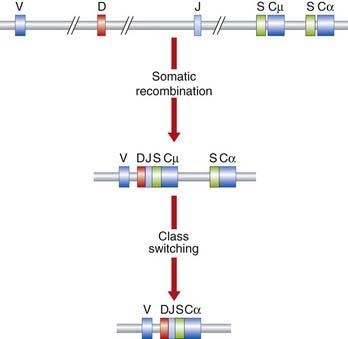
The genes for the κ and λ L-chains and the H-chains are located on chromosomes 2, 22, and 14, respectively. Only one of each of the relevant types of DNA segment is expressed in any single antibody molecule. The DNA coding segments for the various portions of the antibody chains on these chromosomes are separated by DNA that is non-coding. Somatic recombinational events involved in antibody production involve short conserved recombination signal sequences that flank each germline DNA segment (Figure 13.7). Further diversity occurs by variable mRNA splicing at the V–J junction in RNA processing and by somatic mutation of the antibody genes. These mechanisms readily account for the antibody diversity seen in nature, even though it is still not entirely clear how particular DNA segments are selected to produce an antibody to a specific antigen.
Class Switching of Antibodies
There is a normal switch of antibody class produced by B cells on continued, or further, exposure to antigen—from IgM, the initial class of antibody produced in response to exposure to an antigen, to IgA or IgG. This class switching involves retention of the specificity of the antibody to the same antigen. Analysis of class switching in a population of cells derived from a single B cell has shown that both classes of antibody have the same antigen-binding sites, having the same V region but differing only in their C region. Class switching occurs by a somatic recombination event that involves DNA segments designated S (for switching) that lead to looping out and deletion of the intervening DNA. The result is to eliminate the DNA segment coding for the C region of the H-chain of the IgM molecule, and to bring the gene segment encoding the C region of the new class of H-chain adjacent to the segment encoding the V region (see Figure 13.7).
The Immunoglobulin Gene Superfamily
Several other molecules involved in the immune response have been shown to have structural and DNA sequence homology to the immunoglobulins. This involves a 110–amino acid sequence characterized by a centrally placed disulfide bridge that stabilizes a series of antiparallel β strands into an ‘antibody fold’. This group of structurally similar molecules has been called the immunoglobulin superfamily (p. 16). It consists of eight multigene families that, in addition to the κ and λ L-chains and different classes of H-chain, include the chains of the T-cell receptor (p. 16), the class I and II MHC, or human leukocyte antigens (HLA) (p. 200). Other molecules in this group include the T-cell CD4 and CD8 cell surface receptors, which cooperate with T-cell receptors in antigen recognition, and the intercellular adhesion molecules-1, -2, and -3, which are involved in leukocyte-endothelial adhesion and extravasation, T-cell activation, and T-cell homing.
Antibody Engineering
In the 1970s it was understood that the B-cell cancer multiple myeloma produced a single type of antibody—a paraprotein. The structure of antibodies was studied from this but it was not possible to produce identical antibodies specific to a given antigen. Myeloma cells cannot grow because they lack hypoxanthine-guanine-phosphoribosyl transferase, which is necessary for DNA replication. Typically, mAb are made by fusing myeloma cells with spleen cells from a mouse (or rabbit) that has been immunized with the desired antigen. They are then grown in medium which is selective for these hybrids—the spleen cell partner supplies hypoxanthine-guanine-phosphoribosyl transferase and the myeloma has immortal properties because it is a cancer cell. The cell mixture is diluted and clones grown from single parent cells. The antibodies secreted by different clones are assayed for their ability to bind to the antigen in question, with the healthiest clone selected for future use. The hybrids can also be injected into the peritoneal cavity of mice to produce tumors containing antibody-rich ascitic fluid, and the mAb then has to be extracted and purified.
Cell-Mediated Specific Acquired Immunity
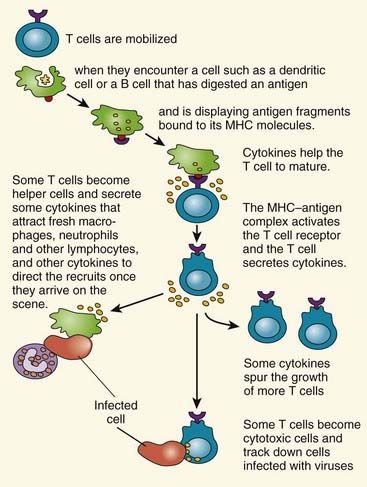
Certain microorganisms, viruses, and parasites live inside host cells. As a result, a separate form of specific acquired immunity has developed to combat intracellular infections involving lymphocytes differentiated and mature in the thymus—hence T cells. T lymphocytes have specialized receptors on the cell surface, known as T-cell surface antigen receptors, which in conjunction with the MHC on the cell surface of the infected cell result in the involvement of different subsets of T cells, each with a distinct function—T helper cells and cytotoxic T cells. The battle against intracellular infections is a cooperative, coordinated response from these separate components of the immune system, leading to death of the infected cell (Figure 13.8).
The Major Histocompatibility Complex
The MHC plays a central role in the immune system. Its role is to bind antigen peptides processed intracellularly and present this material on the cell surface, with co-stimulatory molecules, where it can be recognised by T cells. MHC molecules occur in three classes: class I occur on virtually all cells and are responsible for presenting cytotoxic T cells; class II occur on B cells and macrophages and are involved in signaling T-helper cells to present further B cells and macrophages; and the non-classic class III molecules include a number of other proteins with a variety of other immunological functions. The latter include inflammatory mediators such as the TNF, heat-shock proteins, and the various components of complement.
Structural analysis of class I and II MHC molecules reveal them to be heterodimers with homology to immunoglobulin. The genes coding for the class I (A, B, C, E, F, and G), class II (DR, DQ, and DP) and class III MHC molecules, or what is also known as the human leukocyte antigen (HLA) system (p. 200), are located on chromosome 6.
Transplantation Genetics
Organ transplantation has become routine in clinical medicine and, with the exception of corneal and bone grafts, success depends on the degree of antigenic similarity between donor and recipient. The closer the similarity, the greater the likelihood that the transplanted organ or tissue (the homograft), will be accepted rather than rejected. Homograft rejection does not occur between identical twins or between non-identical twins where there has been mixing of the placental circulations before birth (p. 50). In all other instances, the antigenic similarity of donor and recipient has to be assessed by testing them with suitable antisera or monoclonal antibodies for antigens on donor and recipient tissues. These were originally known as transplantation antigens but are now known to be a result of the MHC. As a general rule, a recipient will reject a graft from any person who has antigens that the recipient lacks. HLA typing of an individual is carried out using PCR-based techniques (p. 56).
The HLA system is highly polymorphic (Table 13.2). A virtually infinite number of phenotypes resulting from different combinations of the various alleles at these loci is theoretically possible. Two unrelated individuals are therefore very unlikely to have identical HLA phenotypes. The close linkage of the HLA loci means that they tend to be inherited en bloc, the term haplotype being used to indicate the particular HLA alleles that an individual carries on each of the two copies of chromosome 6. Thus, any individual will have a 25% chance of having identical HLA antigens with a sibling, as there are only four possible combinations of the two paternal haplotypes (say P and Q) and the two maternal haplotypes (say R and S), i.e., PR, PS, QR and QS. The siblings of a particular recipient are more likely to be antigenically similar than either of his or her parents, and the latter more than a non-relative. Therefore, a sibling is frequently selected as a potential donor.
| HLA Locus | Number of Alleles |
|---|---|
| A | 57 |
| B | 111 |
| C | 34 |
| D | 228 |
HLA, human leukocyte antigen.
Although recombination occurs within the HLA region, certain alleles tend to occur together more frequently than would be expected by chance, i.e. they tend to exhibit linkage disequilibrium (p. 138). An example is the association of the HLA antigens A1 and B8 in populations of western European origin.
H-Y antigen
In a number of different animal species, it was noted that tissue grafts from males were rejected by females of the same inbred strain. These incompatibilities were found to be due to a histocompatibility antigen known as H-Y. However, H-Y seems to play little part in transplantation in humans. The H-Y antigen (not the same as the SRY gene, see p. 92) is important for testicular differentiation and function but its expression does not correlate with the presence or absence of testicular tissue.
HLA Polymorphisms and Disease Associations
The association of certain diseases with certain HLA types (Table 13.3) should shed light on the pathogenesis of the disease, but in reality this not well understood. The best documented is between ankylosing spondylitis and HLA-B27. Narcolepsy, a condition of unknown etiology characterized by a periodic uncontrollable tendency to fall asleep, is almost invariably associated with HLA-DR2. The possession of a particular HLA antigen does not mean that an individual will necessarily develop the associated disease, only that the relative risk of being affected is greater than the general population (p. 384). In a family, the risks to first-degree relatives of those affected are low, usually no more than 5%.
| Disease | HLA |
|---|---|
| Ankylosing spondylitis | B27 |
| Celiac disease | DR4 |
| 21-Hydroxylase deficiency | A3/Bw47/DR7 |
| Hemochromatosis | A3 |
| Insulin-dependent diabetes (type 1) | DR3/4 |
| Myasthenia gravis | B8 |
| Narcolepsy | DR2 |
| Rheumatoid arthritis | DR4 |
| Systemic lupus erythematosus | DR2/DR3 |
| Thyrotoxicosis (Graves disease) | DR3 |
HLA, human leukocyte antigen.
Explanations for the various HLA-associated disease susceptibilities include close linkage to a susceptibility gene near the HLA complex, cross-reactivity of antibodies to environmental antigens or pathogens with specific HLA antigens, and abnormal recognition of ‘self’ antigens through defects in T-cell receptors or antigen processing. These conditions are known as autoimmune diseases. An example of close linkage is congenital adrenal hyperplasia from a 21-hydroxylase deficiency (p. 174) from mutated CYP21, which lies within the HLA major histocompatibility locus. This form of congenital adrenal hyperplasia is strongly associated HLA-A3/Bw47/DR7 in northern European populations. Non-classical 21-hydroxylase deficiency is associated with HLA-B14/DR1, and HLA-A1/B8/DR3 is negatively associated with 21-hydroxylase deficiency.
Inherited Immunodeficiency Disorders
Primary Inherited Disorders of Immunity
Disorders of Innate Immunity
Disorders of innate humoral immunity
A variety of defects of complement can lead to disordered innate immunity.
The clinical effects of MBL deficiency have been described previously. Defects of the third component of complement, C3, lead to abnormalities of opsonization of bacteria, resulting in difficulties in combating pyogenic infections. Defects in the later components of complement—those involved in the formation of the MAC (p. 195)—also result in susceptibility to bacterial infection, though in particular Neisseria (meningococcal infections). This includes deficiency of properdin (factor P), a plasma protein active in the alternative complement pathway.
Defects in NFκB signaling
Since 2000, mutations have occasionally been found in the X-linked IKK-gamma gene, part of the TLR pathway (p. 193), in children demonstrating failure to thrive, recurrent digestive tract infections, often with intractable diarrhea, and recurrent ulcerations, respiratory tract infections with bronchiectasis, and recurrent skin infections, presenting in infancy, suggesting susceptibility to various gram-positive and gram-negative bacteria. Sparse scalp hair is sometimes a feature and in older children oligodontia and conical-shaped maxillary lateral incisors have been noted. Survival ranged from 9 months to 17 years in one study. IgG is low and IgM usually high. Interestingly, IKKg is the same as NEMO, the gene that causes X-linked dominant incontinentia pigmenti (p. 117–118). However, in this condition of the immune system mutations occur in exon 10 of the gene.
IRAK4 is another component of the TLR pathway and deficiency leads to recurrent infections, mainly from gram-positive microorganisms, though also fungi. There is a reduced inflammatory response. Infections begin early in life but become less frequent with age, some patients requiring no treatment by late childhood. It follows autosomal recessive inheritance.
Disorders of innate cell-mediated immunity
Chronic granulomatous disease. Chronic granulomatous disease (CGD) is the best known example of a disorder of phagocytic function, and follows either an X-linked or an autosomal recessive inheritance. It results from an inability of phagocytes to kill ingested microbes, because of any of several defects in the NADPH oxidase enzyme complex which generates the so-called microbicidal ‘respiratory burst’ (see Figure 13.1). Hypergammaglobulinemia may be present. CGD is therefore associated with recurrent bacterial or fungal infections, and may present as suppurative lymphadenitis, hepatosplenomegaly, pulmonary infiltrates, and/or eczematoid dermatitis. Childhood mortality was high until the advent of supportive treatment and prophylactic antibiotics. Bone marrow transplant has been successful, as well as transplantation of peripheral blood stem cells from an HLA-identical sibling. The X-linked gene mutated in CGD, CYBB, was the first human disease gene cloned by positional cloning (p. 75).
Disorders of Specific Acquired Immunity
Disorders of humoral acquired immunity
Bruton-type agammaglobulinemia. Boys with this X-linked immunodeficiency usually develop multiple recurrent bacterial infections of the respiratory tract and skin after the first few months of life, having been protected initially by placentally transferred maternal IgG. Features similar to rheumatoid arthritis develop in many and they are not prone to viral infection. Treatment of life-threatening infections with antibiotics and the use of prophylactic intravenous immunoglobulins have improved survival prospects, but children with this disorder can still die from respiratory failure through complications of repeated lung infections. The diagnosis of this type of immunodeficiency is confirmed by demonstration of immunoglobulin deficiency and absence of B lymphocytes. The disorder has been shown to result from mutations in a tyrosine kinase specific to B cells (Btk) that result in loss of the signal for B cells to differentiate to mature antibody-producing plasma cells. A rarer, autosomal recessive, form of agammaglobulinemia shows marked depression of the circulating lymphocytes, and lymphocytes are absent from the lymphoid tissue.
Disorders of cell-mediated specific acquired immunity
T-B-SCID includes adenosine deaminase deficiency, which accounts for approximately 15% of all SCID and one-third of autosomal recessive SCID. The phenotypic spectrum is variable, the most severe being SCID presenting in infancy and usually resulting in early death. Ten to 15% of patients have a ‘delayed’ clinical onset by age 6 to 24 months, and a smaller percentage of patients have ‘later’ onset, diagnosed from ages 4 years to adulthood, showing less severe infections and gradual immunologic deterioration. The immune system is affected through the accumulation of purine degradation products that are selectively toxic to T cells. Rare forms of B-cell negative SCID include mutated RAG1/RAG2 (recombination activating genes), which are normally responsible for VDJ recombinations (p. 199) that lead to mature immunoglobulin chains and T-cell receptors. In addition, cases occur due to mutation in the Artemis gene (DNA cross-link repair protein 1c—DCLRE1C). The latter forms are both sensitive to ionizing radiation. Lastly, reticular dysgenesis is a rare and very severe form of SCID characterized by congenital agranulocytosis, lymphopenia, and lymphoid and thymic hypoplasia with absent cellular and humoral immunity functions. It is due to mutation in the mitochondrial Adenylate kinase-2 gene (AK2).
Secondary or Associated Immunodeficiency
DiGeorge/Sedláčková Syndrome
Children with the DiGeorge syndrome (also well described by Sedláčková, 10 years earlier than DiGeorge) present with recurrent viral illnesses and are found to have abnormal cellular immunity as characterized by reduced numbers of T lymphocytes, as well as abnormal antibody production. This has been found to be associated with partial absence of the thymus gland, leading to defects in cell-mediated immunity and T cell–dependent antibody production. Usually these defects are relatively mild and improve with age, as the immune system matures, but occasionally the immune deficiency is very severe because no T cells are produced and bone marrow transplantation is indicated. It is important for all patients diagnosed to be investigated by taking a full blood count with differential CD3, CD4, and CD8 counts, and immunoglobulins. The levels of diphtheria and tetanus antibodies can indicate the ability of the immune system to respond. These patients usually also have a number of characteristic congenital abnormalities, which can include heart disease and absent parathyroid glands. The latter finding can result in affected individuals presenting in the newborn period with tetany due to low serum calcium levels secondary to low parathyroid hormone levels. This syndrome has been recognized to be part of the spectrum of phenotypes caused by abnormalities of the third and fourth pharyngeal pouches (p. 95) as a consequence of a microdeletion of chromosome band 22q11.2 (p. 282).
Ataxia Telangiectasia
Ataxia telangiectasia is an autosomal recessive disorder in which children present in early childhood with signs of cerebellar ataxia, dilated blood vessels on the sclerae of the eyes, ears, and face (oculocutaneous telangiectasia), and a susceptibility to sinus and pulmonary infections. Low serum IgA levels occur and a hypoplastic thymus as a result of a defect in the cellular response to DNA damage. The diagnosis is made by the demonstration of low or absent serum IgA and IgG as well as characteristic chromosome abnormalities on culture of peripheral blood lymphocytes—a form of chromosome instability (p. 288). Patients have an increased risk of developing leukemia or lymphoid malignancies.
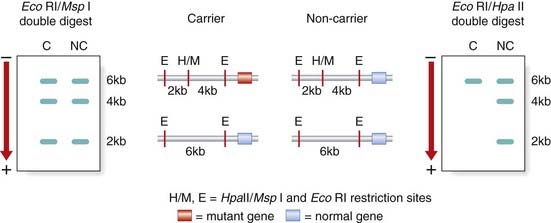
Carrier Tests for X-Linked Immunodeficiencies
Before it was possible to sequence the genes responsible for Wiskott-Aldrich syndrome, Bruton-type hypogammaglobulinemia, and X-linked SCID, the availability of closely linked DNA markers allowed female carrier testing by studies of the pattern of X-inactivation (p. 103) in the lymphocytes of females at risk. A female relative of a sporadically affected male with an X-linked immunodeficiency would be confirmed as a carrier by the demonstration of a non-random pattern of X-inactivation in the T-lymphocyte population, indicating that all her peripheral blood T lymphocytes had the same chromosome inactivated (Figure 13.9).
Blood Groups
Blood groups reflect the antigenic determinants on red cells and were one of the first areas in which an understanding of basic biology led to significant advances in clinical medicine. Our knowledge of the ABO and Rhesus blood groups has resulted in safe blood transfusion and the prevention of Rhesus hemolytic disease of the newborn.
The ABO Blood Groups
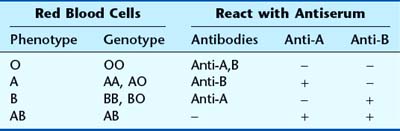
The ABO blood groups were discovered by Landsteiner early in the twentieth century. In some cases blood transfusion resulted in rapid hemolysis because of incompatibility. Four major ABO blood groups were discovered: A, B, AB, and O. Those with blood group A possess the antigen A on the surface of their red blood cells, blood group B has antigen B, AB has both antigens, and those with blood group O have neither. People of blood group A have naturally occurring anti-B antibodies, and blood group B have anti-A, whereas blood group O have both. The alleles at the ABO blood group locus are inherited in a co-dominant manner but are both dominant to the gene for the O antigen. There are, therefore, six possible genotypes (Table 13.4).
Rhesus Blood Group
Rhesus Hemolytic Disease of the Newborn
To avoid sensitizing an Rh-negative woman, Rh-compatible blood must always be used in any blood transfusion. Furthermore, the development of sensitization, and therefore Rh incompatibility after delivery, can be prevented by giving the mother an injection of Rh antibodies—anti-D—so that fetal cells in the maternal circulation are destroyed before the mother can become sensitized.
Other Blood Groups
There are approximately a further 12 ‘common’ blood group systems of clinical importance in humans, including Duffy, Lewis, MN, and S. These are usually of concern only when cross-matching blood for persons who, because of repeated transfusions, have developed antibodies to one of these other blood group antigens. Until the advent of DNA fingerprinting (p. 69), they were used in linkage studies (p. 137) and paternity testing (p. 270).
Bell JI, Todd JA, McDevitt HO. The molecular basis of HLA–disease association. Adv Hum Genet. 1989;18:1-41.
Good review of the HLA–disease associations.
Dreyer WJ, Bennet JC. The molecular basis of antibody formation: a paradox. Proc Natl Acad Sci. 1965;54:864-869.
The proposal of the generation of antibody diversity.
Hunkapiller T, Hood L. Diversity of the immunoglobulin gene superfamily. Adv Immunol. 1989;44:1-63.
Good review of the structure of the immunoglobulin gene superfamily.
Lachmann PJ, Peters K, Rosen FS, Walport MJ. Clinical aspects of immunology, 5 edn. Oxford: Blackwell; 1993.
A comprehensive three-volume multiauthor text covering both basic and clinical immunology.
Murphy KM, Travers P, Walport M. Janeway’s immunobiology, 7 edn. Oxford: Garland Science; 2007.
Good, well-illustrated, textbook of the biology of immunology.
Roitt I. Essential immunology, 9th edn. Oxford: Blackwell; 1997.
Excellent basic immunology textbook.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 13 Immunogenetics
Innate Immunity
Cell-Mediated Innate Immunity
Phagocytosis
Two major cell types go on the offensive when a foreign microorganism invades—macrophages and neutrophils. Macrophages are the mature form of circulating monocytes that migrate into tissues and occur primarily around the basement membrane of blood vessels in connective tissue, lung, liver, and the lining of the sinusoids of the spleen and the medullary sinuses of the lymph nodes. They are believed to play a key role in the orchestration of both the innate and adaptive responses, and can recognise invading microorganisms through surface receptors able to distinguishing between self and pathogen. Recognition of the foreign material leads to phagocytosis by the macrophage, followed rapidly by neutrophils recruited from the circulation during the inflammatory process. The activation of the macrophage triggers the inflammatory process through the release of inflammatory mediators. The invading organism is destroyed by fusion with intracellular granules of the phagocyte and exposure to the action of hydrogen peroxide, hydroxyl radicals, and nitrous oxide (Figure 13.1).
The Toll-like Receptor Pathway
There are 10 TLRs in man, each receptor being responsible for recognition of a specific set of pathogen-associated molecular patterns. TLR2 has been well characterised and has an essential role in the detection of invading pathogens, recognizing peptidoglycans and lipoproteins associated with gram-positive bacteria, as well as a host of other microbial and endogenous ligands. TLR2’s primary function is therefore lipoprotein-mediated signaling, and activation of the pathway by recognition of its ligand results in activation of the transcription factor NF-κB, which in turn results in the increased expression of co-stimulatory molecules and inflammatory cytokines (Figure 13.3). These cytokines help mediate migration of dendritic cells from infected tissue to lymph nodes, where they may encounter and activate leukocytes involved in the adaptive immune response. The signaling pathways used by TLRs share many of the same proteins as the interleukin-1 receptor (IL-1R) pathway (Figure 13.2). Activation of TLR leads to recruitment of the MyD88 (this is sometimes known as the MyD88-dependent pathway) which mediates the interaction between IL-1R associated kinases 1 and 4 (IRAK1 and IRAK4).
The activation of the Toll pathway has several important effects in inducing innate immunity. These effects include the production of cytokines and chemokines, including IL-1, IL-6, and TNF-α (tumor necrosis factor-alpha), which have local effects in containing infection and systemic effects with the generation of fever and induction of acute phase responses, including production of C-reactive protein. One important medical condition related to the Toll pathway is septic shock, as activation of the Toll pathway by certain ligands induces systemic release of TNF-α. There are also important health-related consequences that result from TLR2 deficiency or mutation. TLR2 deficient mice are susceptible to infection by Gram-positive bacteria as well as meningitis from Streptococcus pneumoniae.
Humoral Innate Immunity
Complement
The complement system is a complex of 20 or so plasma proteins that cooperate to attack extracellular pathogens. Although the critical role of the system is to opsonize pathogens, it also recruits inflammatory cells and kills pathogens directly through membrane attack complexes. The complement system can be activated through three pathways: the classical pathway, the alternative pathway, and the mannose-binding lectin (MBL) pathway (see Figure 13.3).
Complement nomenclature, like much else in immunology, can be confusing. Each component is designated by the letter C, followed by a number. But they were numbered in order of their discovery rather than the sequence of reactions. The reaction sequence is C1, C4, C3, C5, C6, C7, C8, and C9. The product of each cleavage reaction is designated by letters, the larger fragment being ‘b’ (b = big), and the smaller fragment ‘a’. In the lectin pathway, MBL in the blood binds another protein, a serine protease called MASP (MBL-associated serine protease). When MBL binds to its target (for example, mannose on the surface of a bacterium), the MASP protein functions like a convertase to clip C3 into C3a and C3b. C3 is abundant in the blood, so this happens very efficiently. The other two complement pathways also converge toward C3 convertase, which cleaves C3. C3a mediates inflammation while C3b binds to the pathogen surface, coating it and acting as an opsonin. The effector roles of the major complement proteins can be summarized according to function as follows (Figure 13.4):
Specific Acquired Immunity
Humoral Specific Acquired Immunity
The main mediators of humoral specific acquired immunity are immunoglobulins or antibodies. Antibodies are able to recognize and bind to surface antigens of infecting microorganisms, leading to the activation of phagocytes and the initiation of the classic pathway of complement, resulting in the generation of the MAC (see Figure 13.4) and availability of other complement effector functions. Exposure to a specific antigen results in the clonal proliferation of a small lymphocyte derived from the bone marrow (hence ‘B’ lymphocytes), resulting in mature antibody-producing cells or plasma cells.
Immunoglobulins
Immunoglobulin structure
The immunoglobulin molecule is made up of four polypeptide chains—two ‘light’ (L) and two ‘heavy’ (H)—of approximately 220 and 440 amino acids in length, respectively. They are held together in a Y-shape by disulfide bonds and non-covalent interactions. Each Fab fragment is composed of L chains linked to the amino-terminal portion of the H chains, whereas each Fc fragment is composed only of the carboxy-terminal portion of the H chains (Figure 13.5).
Immunoglobulin isotypes, subclasses, and idiotypes
There are five different types of heavy chain, designated respectively as γ, µ, α, δ, and ε, one each for the five major antibody classes—the isotypes: IgG, IgM, IgA, IgD, and IgE, respectively. The L chains are of two types—kappa (κ) or lambda (λ), and these occur in all five classes of antibody, but only one type occurs in each individual antibody. Thus, the molecular formula for IgG is λ2γ2 or κ2γ2. The characteristics of the various classes of antibody are outlined in Table 13.1. In addition, there are four IgG subclasses—IgG1, IgG2, IgG3, and IgG4—and two IgA subclasses—IgA1 and IgA2—that differ in their amino acid sequence and interchain disulfide bonds. Individual antibody molecules that recognize specific antigens are known as idiotypes.
Immunoglobulin allotypes
The five immunoglobulin classes occur in all normal individuals, but allelic variants, or what are known as antibody allotypes of these classes, have also been identified. These are the Gm system associated with the heavy chain of IgG, the Am system associated with the IgA heavy chain, the Km and Inv systems associated with the κ light chain, the Oz system for the λ light chain and the Em allotype for the IgE heavy chain. The Gm and Km systems are independent of each other and are polymorphic (p. 135), the frequencies of the different alleles varying in different ethnic groups.
Generation of Antibody Diversity
Multiple myeloma
People with multiple myeloma make a single or monoclonal antibody species in large abundance, which in a proportion of patients is detected in their urine. This is known as Bence Jones protein and consists of antibody L chains. The amino-terminal ends of this protein molecule in different patients are quite variable in sequence, whereas the carboxy-terminal ends are relatively constant. These are called the variable, or V, and constant, or C, regions, respectively. However, the V regions of different myeloma proteins show four regions that vary little from one antibody to another, known as framework regions (FR 1–4), and three markedly variable regions interspersed between these, known as hypervariable regions (HV I–III) (see Figure 13.5).
DNA studies of antibody diversity
There are estimated to be some 60 different DNA segments coding for the V region of the H-chain, 40 for the V region of the κ L-chain, and 30 for the λ L-chain V region. Six functional DNA segments code for the J region of the H-chain, five for the J region of the κ L-chain, and four for the J region of the λ L-chain. A single DNA segment codes for the C region of the κ L-chain, seven for the C region of the λ L-chain and 11 functional DNA segments code for the C region of the different classes of H-chain. There are also 27 functional DNA segments coding for the D region of the H-chain (Figure 13.6).
FIGURE 13.6 Estimated number of the various DNA segments coding for the κ, λ, and various heavy chains.
The genomic regions in question also contain a large number of unexpressed DNA sequences or pseudogenes (p. 17). Although the coding DNA segments for the various regions of the antibody molecule can be referred to as ‘genes’, use of this term in regard to antibodies has deliberately been avoided because they could be considered an exception to the general rule of ‘one gene–one enzyme (or protein)’ (p. 167).
Antibody gene rearrangement
The genes for the κ and λ L-chains and the H-chains are located on chromosomes 2, 22, and 14, respectively. Only one of each of the relevant types of DNA segment is expressed in any single antibody molecule. The DNA coding segments for the various portions of the antibody chains on these chromosomes are separated by DNA that is non-coding. Somatic recombinational events involved in antibody production involve short conserved recombination signal sequences that flank each germline DNA segment (Figure 13.7). Further diversity occurs by variable mRNA splicing at the V–J junction in RNA processing and by somatic mutation of the antibody genes. These mechanisms readily account for the antibody diversity seen in nature, even though it is still not entirely clear how particular DNA segments are selected to produce an antibody to a specific antigen.
Class Switching of Antibodies
There is a normal switch of antibody class produced by B cells on continued, or further, exposure to antigen—from IgM, the initial class of antibody produced in response to exposure to an antigen, to IgA or IgG. This class switching involves retention of the specificity of the antibody to the same antigen. Analysis of class switching in a population of cells derived from a single B cell has shown that both classes of antibody have the same antigen-binding sites, having the same V region but differing only in their C region. Class switching occurs by a somatic recombination event that involves DNA segments designated S (for switching) that lead to looping out and deletion of the intervening DNA. The result is to eliminate the DNA segment coding for the C region of the H-chain of the IgM molecule, and to bring the gene segment encoding the C region of the new class of H-chain adjacent to the segment encoding the V region (see Figure 13.7).
