Chapter 13 Immunity to Viruses
• Innate immune responses (mediated by anti-microbial peptides, type I interferons (IFNs), dendritic cells (DCs), natural killer (NK) cells, and macrophages) restrict the early stages of infection, delay spread of virus and promote the activation of adaptive responses. Innate defences are triggered following recognition of molecular ‘patterns’ characteristic of viral but not host components. Type I IFNs exert direct antiviral activity and also activate other innate and adaptive responses. NK cells are cytotoxic for virally infected cells. Macrophages act at three levels to destroy virus and virus-infected cells.
• As a viral infection proceeds, the adaptive (specific) immune response unfolds. Antibodies and complement can limit viral spread or reinfection. T cells mediate viral immunity in several ways – CD8+ T cells destroy virus-infected cells or cure them of infection; CD4+ T cells promote antibody and CD8+ T cell responses and are a major effector cell population in the response to some virus infections.
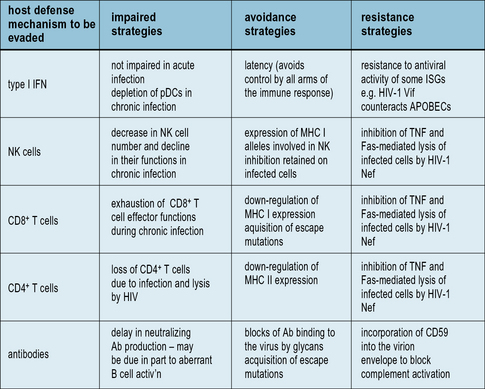
• Viruses have evolved strategies to evade the immune response. They may impair the host immune response at the induction and/or effector stages; avoid recognition by the immune response, e.g. via latency or antigenic variation; or resist control by immune effector mechanisms. Many viruses employ multiple strategies to prolong their replication in the host.
• Responses induced during viral infections can have pathological consequences. Damage can be mediated by antiviral responses (e.g. via the formation of immune complexes or T cell-induced damage to host tissues) or by autoimmune responses triggered during the course of infection.
Innate immune defenses against viruses
Type I interferons have critical antiviral and immunostimulatory roles
• type I (multiple subtypes of IFNα and one subtype of IFNβ);
• type III (IFNλ1, IFNλ2, and IFNλ3, also known as IL-29, IL-28a, and IL-28b).
Type I IFN production is triggered following recognition of molecular patterns characteristic of viral but not host components (Fig. 13.1). Host pattern-recognition receptors involved in detecting the presence of virus infections include:
• cytoplasmic pattern-recognition receptors expressed by almost all cells (e.g. the retinoic acid-inducible gene I (RIG-I)-like receptors, which recognize viral 5′-triphosphorylated ssRNA and dsRNA, and cytoplasmic DNA sensors);
• members of the Toll-like family of receptors (TLRs), which are expressed on the cell membrane or within endosomes/lysosomes of immune system cells and certain non-immune cells located at common sites of pathogen entry, e.g. epithelial cells (TLR3, TLR7, and TLR9, which recognize viral dsRNA, viral ssRNA, and DNA containing CpG motifs, respectively).
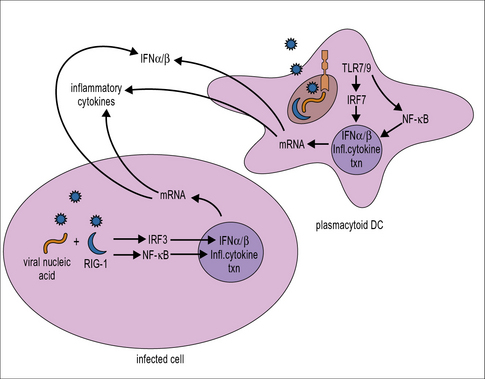
Triggering of pattern-recognition receptors initiates signaling along pathways that culminate in the activation of transcription factors including IFN regulatory factor (IRF)3 and NFκB, which translocate into the nucleus and activate the transcription of type I IFNs and inflammatory cytokines, respectively (see Fig. 13.1). Plasmacytoid DCs also have a unique signaling pathway for induction of type I IFN production in response to TLR7 or TLR9 ligation that involves the transcription factor IRF7.
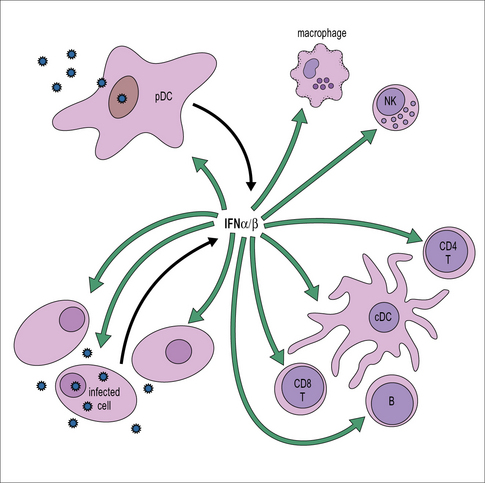
The IFN released acts on both the cell producing it and also neighboring cells where it establishes an antiviral state, enabling them to resist virus infection (Fig. 13.2).
• PKR disrupts virus infection by phosphorylating and inhibiting eukaryotic initiation factor (eIF)-2α, hence blocking the translation of viral mRNA and by initiating apoptosis via Bcl-2 and caspase-dependent mechanisms, killing the cell before virus can be released.
• 2′,5′-oligoadenylate synthetase specifically activates a latent endonuclease (RNaseL) that targets the degradation of viral RNA.
In addition to the direct inhibition of virus replication, IFNs also activate macrophages and NK cells and enhance their antiviral activity (see Fig. 13.2). In addition, they help to promote the activation of adaptive responses. They act on antigen presenting cells including conventional DCs to stimulate increased expression of MHC class I and II, along with components of the antigen processing machinery; and they also act directly on T and B cells to promote an antiviral response (see Fig. 13.2).
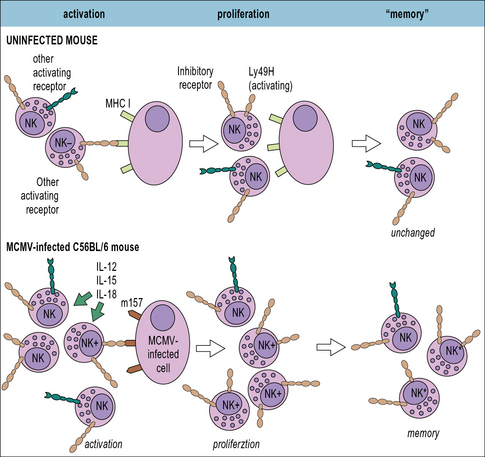
NK cells are cytotoxic for virally-infected cells
• Inhibitory NK receptors typically recognize ligands expressed on ‘normal’ host cells, such as MHC class I molecules. As discussed below, many viruses down-regulate MHC class I expression on the cells they infect to limit recognition by CD8+ T cells, but this helps to trigger NK cell activation.
• Activating NK receptors typically recognize host cell proteins that are up-regulated in response to stress or viral proteins, e.g. the natural cytotoxicity receptors NKp44 and NKp46 recognize certain viral glycoproteins including the influenza virus HA. Mice deficient in NKp46 are highly susceptible to influenza virus infection.
• NK cells can also be activated via antibody coating of the target cell which mediates crosslinking of the NK surface receptor FcγRIII. As discussed below, NK cells are one of the principal mediators of antibody-dependent cell-mediated cytotoxicity (ADCC).
Adaptive immune responses to viral infection
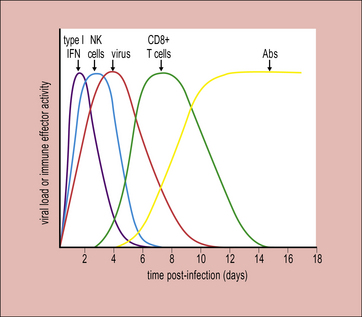
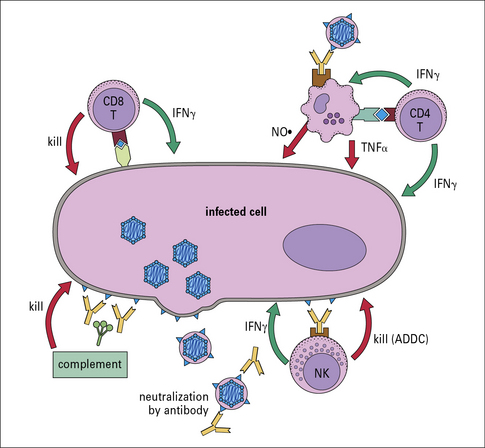
The adaptive immune response typically begins a few days after innate responses are activated (Fig. 13.3). T cells start to appear at sites of infection around 4 days after the initiation of viral expansion. In many virus infections, it is the action of CD8+ T cells that plays a key role in the resolution of infection. Antibodies are frequently induced slightly later, around day 6/7, and contribute to recovery from infection.
Antibodies and complement can limit viral spread or reinfection
Antibodies can neutralize the infectivity of viruses
Antibodies may be generated against any viral protein in the infected cell.
Defense against free virus particles involves neutralization of infectivity, which can occur in various ways (Fig. 13.4). Such mechanisms are likely to operate in vivo because injection of neutralizing monoclonal antibodies is highly effective at inhibiting virus replication. The presence of circulating virus-neutralizing antibodies is an important factor in the prevention of reinfection. Passively-administered monoclonal antibodies have been used therapeutically to inhibit respiratory syncytial virus and influenza virus infections.
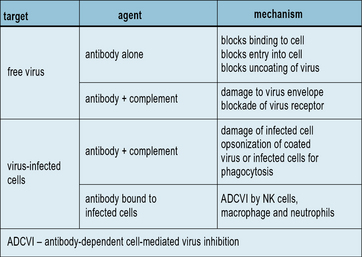
Antibodies mobilize complement and/or effector cells to destroy virus-infected cells
Antibodies are also effective in mediating the destruction of virus-infected cells. This can occur by antibody-mediated activation of the complement system, leading to the assembly of the membrane attack complex and lysis of the infected cell (see Chapter 4). This process requires a high density of viral antigens on the membrane (about 5 × 106/cell) to be effective. In contrast, ADCC mediated by NK cells requires as few as 103 IgG molecules in order to activate NK cell binding to and lysis of the infected cell.
T cells mediate viral immunity in several ways
T cells exhibit a variety of functions in antiviral immunity:
• CD8+ T cells are important effector cells that play a key role in the control of established virus infections;
• most of the antibody response is T-dependent, requiring the presence of helper CD4+ T cells for class switching and affinity maturation;
• CD4+ T cells also help in the induction of CD8+ T cell responses and in the recruitment and activation of macrophages at sites of virus infection;
• memory CD8+ T cells are effective in combating reinfection with viruses such as influenza virus and respiratory syncytial virus – however, even memory T cells need time to develop a response when infection is re-encountered, so antibodies typically assume a more dominant role in protection against reinfection by neutralizing incoming virus, containing the infection and preventing spread to other tissues.
CD8+ T cells target virus-infected cells
• kill infected cells through the release of perforin, granzymes, and other cytolytic proteins;
• trigger the death of infected cells through binding of soluble factors (e.g. TNFα) or ligands they express (e.g. FasL) to cell-surface receptors (such as Fas) that signal the cell to undergo apoptosis, i.e. effectively ‘commit suicide’; and
• produce soluble factors such as IFNγ and/or TNFα that can ‘cure’ infection with some viruses (e.g. hepatitis B virus) without death of the cell. This can result in eradication of virus from not only the target cell with which the CD8+ T cell is interacting, but also from neighboring cells.
• the adoptive transfer of specific T cell subpopulations or T cell clones to infected animals;
• depletion of T cell populations in vivo using monoclonal antibodies to CD4 or CD8; and
• creation of ‘gene knockout’ mice, in which genes encoding cell surface receptors (e.g. CD8, CD4), signal transduction molecules (e.g. signal transducer and activator of transcription (STAT)) or transcription factors (e.g. T-bet) are removed from the germline.
• correlative studies addressing the relationship between the magnitude or functional efficacy of antigen-specific T cell responses and the efficiency of control of virus replication in different infected individuals;
• assessment of control of virus replication in patients with defects in selected immune functions (e.g. DiGeorge syndrome patients lacking a thymus); and
• finding of evidence that viruses have evolved strategies to escape recognition by host T cells (this would not happen unless T cells were exerting selective pressure on virus replication).
CD4+ T cells are a major effector cell population in the response to some virus infections
CD4+ T cells have also been identified as a major effector cell population in the immune response to some virus infections. A good example is in HSV-1 infection of epithelial surfaces. Here, CD4+ T cells participate in a delayed-type hypersensitivity response (see Chapter 26) that results in accelerated clearance of virus. They produce cytokines such as IFNγ and TNFα, which mediate direct antiviral effects and also help to activate macrophages at the site of infection. Macrophages play an important role in inhibiting virus infection, probably through the generation and action of nitric oxide (Fig. 13.5).
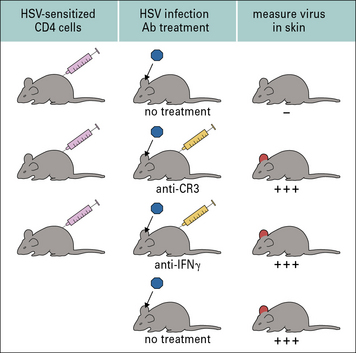
In measles virus and Epstein–Barr virus (EBV) infections, CD4+ CTLs are generated that recognize and kill MHC class II-positive cells infected with the virus using the cytolytic mechanisms also employed by CD8+ CTLs. This suggests that measles virus and EBV peptides are generated by normal pathways of antigen presentation (i.e. following phagocytosis and degradation, see Chapter 8). However, other pathways have been implicated in which some measles proteins/peptides enter class II vesicles from the cytosol.
A summary of antiviral defense mechanisms is illustrated in Figure 13.6.
Virus strategies to evade host immune responses
Viral immune evasion strategies can be categorized into mechanisms for:
Some viruses employ multiple strategies in each category to promote their persistence in vivo: human immunodeficiency virus type 1 (HIV-1) provides a particularly good example of this (Fig. 13.7).
Viruses can impair the host immune response
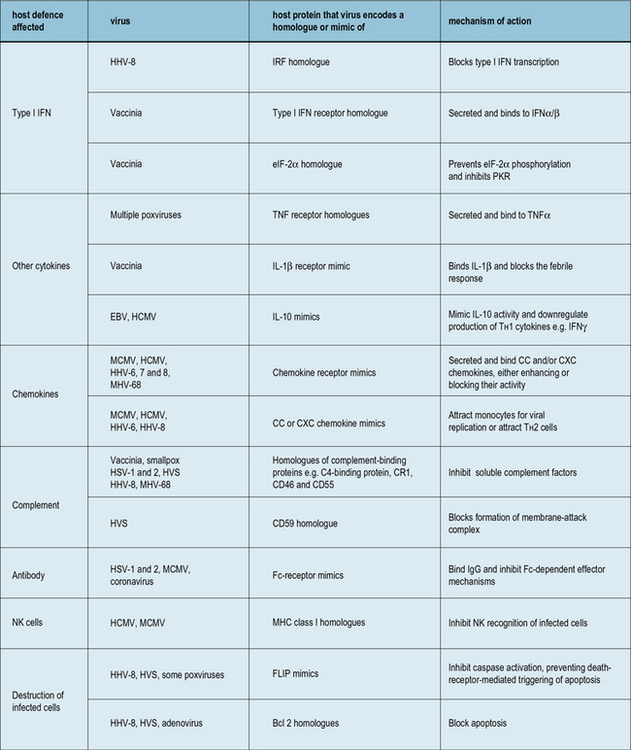
• chemokine homologues (e.g. CCL3);
• chemokine receptor homologues; and
• chemokine-binding proteins, which have powerful effects on delaying or inhibiting cell migration during inflammation.
• Some persistent virus infections are transmitted from mother to offspring in utero (e.g. lymphocytic choriomeningitis virus (LCMV) infection of mice and hepatitis B virus infection in humans). This can result in tolerization of virus-specific T cells as self-tolerance is established in the developing host immune system, leading to failure to mount a virus-specific T cell (or an effective antibody) response to the virus.
• Virus-specific CD8+ T cell responses in immunologically-mature hosts may be impaired by processes termed ‘exhaustion’ or ‘stunning’ during which virus-specific T cells are rendered increasingly functionally-defective in the face of high-level ongoing virus replication and may eventually be driven to undergo apoptotic death.
Viral strategies for avoidance of recognition by host immune defenses
Strategies viruses use to avoid recognition by host immune defenses include:
• infection of ‘immune privileged’ sites;
• rendering infected cells less visible to host effector cells; and
Another strategy that viruses use to avoid recognition by host immune defences is to replicate in ‘immune privileged’ sites, i.e. parts of the body to which adaptive responses have limited access and where there may also be an ‘immune-suppressive’ environment, such as in the brain (see Chapter 12). A surprising number of different viruses persist in the brain.
Viruses avoid recognition by T cells by reducing MHC expression on infected cells
MHC class I expression can be disrupted by:
• downregulating MHC class I synthesis (e.g. HIV-1);
• reducing the generation of epitope peptides in the cytoplasm (e.g. EBV);
• blocking peptide uptake into the endoplasmic reticulum (e.g. HSV-1);
• preventing maturation, assembly and migration of the trimolecular MHC complex (e.g. human cytomegalovirus [HCMV]); and/or
• recycling of MHC class I molecules from the cell surface (e.g. HIV-1).
Mutation of viral target antigen allows escape from recognition by antibodies or T cells
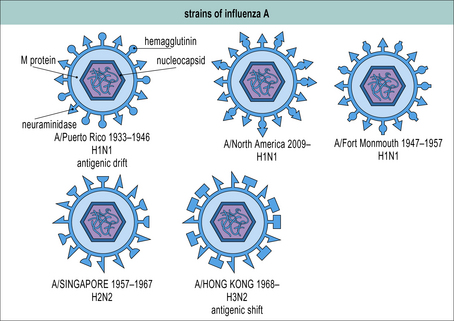
Antigenic variation involves a virus acquiring sequence changes (mutations) in sites on proteins that are normally targeted by antibody or T cells so that these sites are no longer recognized. Antigenic variation can promote virus persistence within a given host, e.g. during HIV-1 infection mutations are frequently selected for in and around the epitopes recognized by the initial T cell responses and neutralizing antibody responses, which confer escape from recognition by these responses and enable enhanced virus replication. It can also promote virus persistence at the population level, as exemplified by the antigenic shift and drift seen with influenza virus (Fig. 13.8). Humoral immunity to influenza virus provides protection against re-infection only until a new virus strain emerges, making effective longlasting vaccines difficult to produce.
Viral strategies for resisting control by immune effector mechanisms
• the antiviral activity of type I IFNs and other cytokines;
• the lytic mechanisms by which NK cells and CD8+ T cells destroy infected cells; and
• production of soluble IFN receptors (e.g. pox viruses);
• interference with IFN signaling (e.g. paramyxoviruses); and
• disruption of intracellular IFN-induced defences such as PKR or 2′,5′-oligoadenylate synthetase activity (e.g. adenovirus and herpesviruses).
Examples of virus-encoded homologues or mimics of the host defense system are shown in Figure 13.9.
Pathological consequences of immune responses induced by viral infections
Excessive cytokine production and immune activation can be pathological
• Infection with severe acute respiratory syndrome (SARS) virus and the highly pathogenic influenza viruses (H5N1), if not rapidly controlled by the early innate response, can be associated with hypercytokinemia (cytokine storms) which drive an aggressive inflammatory response that can result in massive tissue damage (pneumonia) leading to death.
• Activated CD4+ T cells constitute the main cellular sites for HIV replication. The virus triggers an intense cytokine storm associated with extensive immune activation during acute infection, which helps to fuel virus replication by providing a large pool of activated CD4+ target cells. A key difference between non-pathogenic simian immunodeficiency virus (SIV) infections of non-human primates and pathogenic SIV infection or HIV infection is that in non-pathogenic infections immune activation is rapidly down-modulated after the acute phase of infection, whilst a constant state of immune activation is maintained in pathogenic infections. This helps to drive ongoing virus replication and CD4+ T cell loss, ultimately leading to the development of AIDS.
Pathological consequences of antiviral antibody production
Antiviral antibodies can form immune complexes that cause tissue damage
Immune complexes may arise in body fluids or on cell surfaces and are most common during persistent or chronic infections (e.g. with hepatitis B virus). Antibody is ineffective (non-neutralizing) in the presence of large amounts of the viral antigen; instead, immune complexes form and are deposited in the kidney or in blood vessels, where they evoke inflammatory responses leading to tissue damage (e.g. glomerulonephritis, see Chapter 25).
Virus-specific T cell responses can cause severe tissue damage
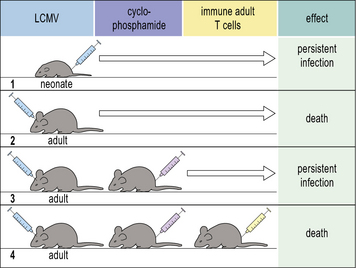
In any virus infection some tissue damage is likely to arise from the activity of infiltrating T cells. However, in some situations this damage may be considerable, resulting in the death of the host. A classic illustration of this is the CD8+ T cell response to LCMV in the central nervous system (Fig. 13.10). Removal of T cells protects LCMV-infected mice from death, indicating that they, rather than the virus, are mediating damage to the brain.
Viral infection may provoke autoimmunity
Viruses may trigger autoimmune disease in a number of ways, including:
• Virus-induced damage – during the course of some virus infections, tissues become damaged, provoking an inflammatory response during which ‘hidden’ antigens become exposed and can be processed and presented to the immune system. Examples of this include Theiler’s virus (a murine picornavirus) and murine hepatitis virus infection of the nervous system, in which the constituents of myelin (the insulating material of axons) become targets for antibody and T cells.
• Molecular mimicry – a sequence in a viral protein that is homologous to a ‘self’ protein can become recognized, leading to a breakdown in immunological tolerance to cryptic self antigens in the consequent attack on host tissues by the immune system (see Chapter 19). A good example is coxsackie B virus-induced myocarditis. Patients with inflammatory cardiomyopathy have antibodies that cross-react with peptides derived from coxsackie B3 protein and with peptides derived from cellular adenine nucleotide translocator.
Critical thinking: Virus–immune system interactions (see p. 436 for explanations)
1. How do you explain the protection achieved by the non-neutralizing monoclonal antibodies?
2. What experiments would you propose to support some of your conclusions?
3. Why might memory CD8+ T cells be unable to prevent the establishment of HIV infection?
4. Would T cell responses directed against epitopes in any of the HIV proteins be likely to control viral replication equally well?
5. How could viral escape from vaccine-elicited T cell responses be minimized?
Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol. 2003;3:36–50.
Goulder P.J., Watkins D.I. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol. 2004;4:630–640.
Guidotti L.G., Chisari F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91.
Hansen T.H., Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol. 2009;9:503–513.
Kawai T., Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137.
Klenerman P., Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–879.
Klotman M.E., Chang T.L. Defensins in antiviral immunity. Nat Rev Immun.. 2006;6:247–256.
Koelle D.M., Corey L. Herpes simplex: insights on pathogenesis and possible vaccines. Annu Rev Med. 2008;59:381–395.
Kohlmeier J.E., Woodland D.L. Immunity to respiratory viruses. Annu Rev Immunol. 2009;27:61–82.
Lanier L. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. 2008;8:259–268.
McMichael A.J., Borrow P., Tomaras G.D., et al. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10:11–23.
Paiardini M., Pandrea I., Apetrei C., Silvestri G. Lessons learned from the natural hosts of HIV-related viruses. Annu Rev Med. 2009;60:485–495.
Peiris J.S., Hui K.P., Yen H.L. Host response to influenza virus: protection versus immunopathology. Curr Opin Immunol. 2010;22:475–481.
Powers C., DeFilippis V., Malouli D., Früh K. Cytomegalovirus immune evasion. Curr Top Microbiol Immunol. 2008;325:333–359.
Randall R.E., Goodbourn S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47.
Reading S.A., Dimmock N.J. Neutralization of animal virus infectivity by antibody. Arch Virol. 2007;152:1047–1059.
Rehermann B., Hepatitis C. virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest. 2009;119:1745–1754.
Sadler A.J., Williams B.R.G. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568.
Tirado S.M., Yoon K.J. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003;16:69–86.
Tortorella D., Gewurz B.E., Furman M.H., et al. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926.