Chapter 15 Immunity to Protozoa and Worms
• Parasites stimulate a variety of immune defense mechanisms.
• Parasitic infections are often chronic and affect many people. They are generally host specific and most cause chronic infections. Many are spread by invertebrate vectors and have complicated life cycles. Their antigens are often stage specific.
• Innate immune responses are the first line of immune defense.
• T and B cells are pivotal in the development of immunity. Both CD4 and CD8 T cells are needed for protection from some parasites, and cytokines, chemokines, and their receptors have important roles.
• Effector cells such as macrophages, neutrophils, eosinophils, and platelets can kill both protozoa and worms. They secrete cytotoxic molecules such as reactive oxygen radicals and nitric oxide (NO•). All are more effective when activated by cytokines. Worm infections are usually associated with an increase in eosinophil number and circulating IgE, which are characteristic of TH2 responses. TH2 cells are necessary for the elimination of intestinal worms.
• Parasites have many different escape mechanisms to avoid being eliminated by the immune system. Some exploit the host response for their own development.
• Inflammatory responses can be a consequence of eliminating parasitic infections.
• Parasitic infections have immunopathological consequences. Parasitic infections are associated with pathology, which can include autoimmunity, splenomegaly, and hepatomegaly. Much immunopathology may be mediated by the adaptive immune response.
• Vaccines against human parasites are not yet routinely available.
Parasite infections
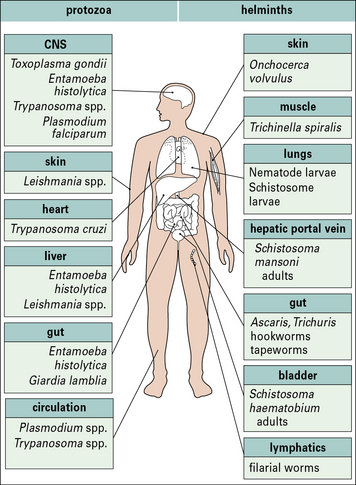
Parasitic infections typically stimulate a number of immune defense mechanisms, both antibody and cell mediated, and the responses that are most effective depend upon the particular parasite and the stage of infection. Some of the more important parasitic infections of humans (Fig. 15.1) affect the host in diverse ways. Parasitic protozoa may live:
• in the blood (e.g. African trypanosomes);
• within erythrocytes (e.g. Plasmodium spp.);
• in macrophages (e.g. Leishmania spp., Toxoplasma gondii);
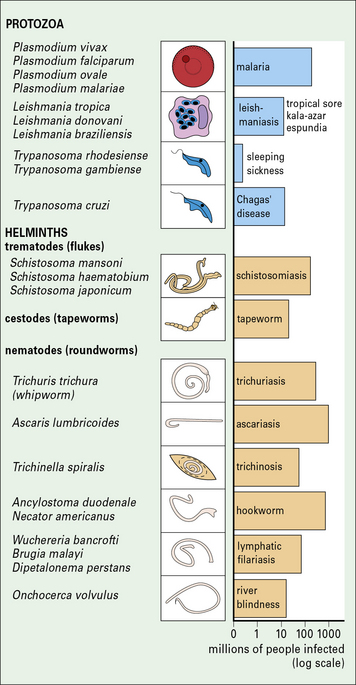
Fig. 15.1 Important parasitic infections of humans
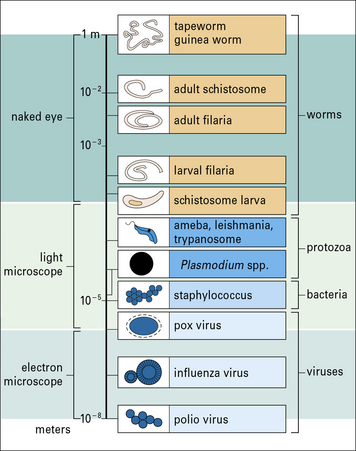
Important parasitic infections, including data from the World Health Organization (1993). Their sizes range from 1 m for the tapeworm to around 10− 5 m for Plasmodium spp. (cf. Fig. 15.w1).
Tapeworms and adult hookworms inhabit the gut, adult schistosomes live in blood vessels, and some filarial worms live in the lymphatics (Fig. 15.2). It is clear that there is widespread potential for damaging pathological reactions.

Parasitic infections are often chronic and affect many people
Parasitic infections present a major medical problem, especially in tropical countries (see Fig. 15.1), for example:
• malaria kills 1–2 million people every year;
• intestinal worms infect one-third of the world’s population – the severity of disease depends upon the worm burden, but in children even moderate intensities of infection may be associated with stunted growth and slow mental development.
Anemia and malnutrition are also associated with parasitic disease.
There are some exceptions to this general rule, for example:
• the protozoan parasite T. gondii is not only able to invade and multiply in all nucleated mammalian cells, but can also infect immature mammalian erythrocytes, insect cell cultures, and the nucleated erythrocytes of birds and fish;
• similarly, the tapeworm of the pig can also infect humans.
Protozoan parasites and worms are considerably larger than bacteria and viruses (Fig. 15.w1), and have very different strategies for avoiding the host immune response.
Q. Apart from size, what is the basic biological difference between bacterial pathogens and parasites, and how would this affect the way they are recognized by the immune system?
A. Bacteria are prokaryotes, whereas parasites are eukaryotes. The plasma membrane and cell wall structures in bacteria are different, so they have distinct pathogen-associated molecular patterns (PAMPs, see Chapter 6). In addition, protein synthesis (initiated with formyl methionine) is different in prokaryotes.
Protozoa that are small enough to live inside human cells have evolved a special mode of entry:
• the merozoite, the invasive form of the blood stage of the malarial parasite, binds to certain receptors on the surface of the erythrocyte and uses a specialized organelle, the rhoptry, to enter the cell.
• Leishmania spp. parasites, which inhabit macrophages, use complement and Fc receptors to encourage the cells to engulf them.
Leishmania can also gain entry to the cell by using the mannose receptor (see Fig. 7.11) on the macrophage surface.
Immune defenses against parasites
Host resistance to parasite infection may be genetic
The resistance of individual hosts to infection varies, and may be controlled by a number of genes. These may be MHC, non-MHC, or other genes (Fig. 15.3).

Host defense depends upon a number of immunological mechanisms
In very general terms, humoral responses are important to eliminate extracellular parasites such as those that live in blood (Fig. 15.4), body fluids, or the gut.
Innate immune responses
Many different cells are involved in generating innate responses including phagocytic cells and NK cells. It is also becoming clear that early recognition of parasites by antigen-presenting cells (APCs), for example dendritic cells, determines the phenotype of the adaptive response (Fig. 15.5).
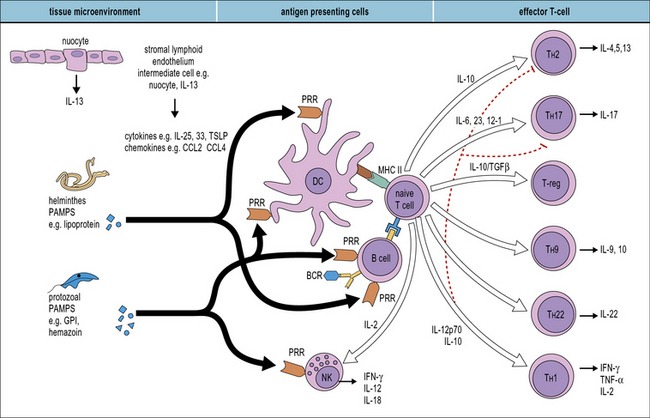
Q. Which groups of receptors and soluble molecules recognize PAMPs?
A. Toll-like receptors (see Fig. 6.20), the mannose receptor (see Fig. 7.11) and scavenger receptors (see Fig. 7.10) allow phagocytes to directly recognize pathogens. Ficolins, collectins, and pentraxins act as soluble opsonins by binding to pathogen surfaces (see Fig. 6.w3 ).
).
While major advances are being achieved in the area of microbial recognition by PRRs, a small but growing number of studies show that parasites also possess specific molecular patterns capable of engaging PRRs. Examples of some parasite PAMPs along with their receptors are given in Figure 15.6.
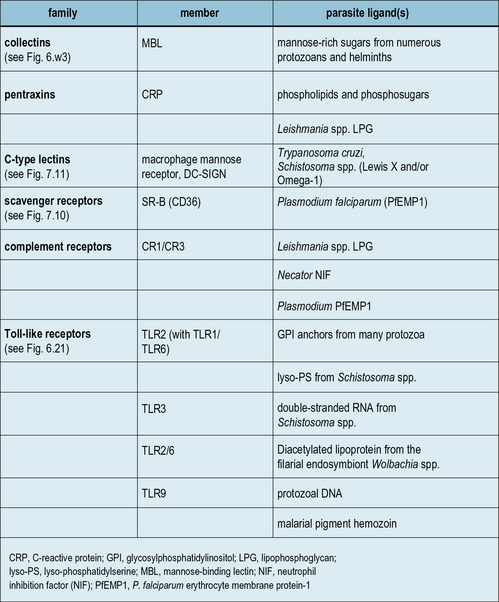
Toll-like receptors recognize parasite molecules
A few studies have examined the role of TLRs in immunity to parasites. For example:
• T. gondii binds TLR11 via profilin and associates with TLR3, TLR7, and TLR9 via the endoplasmic reticulum protein UNC93B1;
• TLR9 mediates innate immune activation by the malaria pigment hemozoin;
• lyso-phosphatidylserine (lyso-PS) from S. mansoni, glycophosphatidylinositol (GPI) anchors, and Tc52 from T. cruzi are capable of signaling through TLR2.
Interestingly, TLR2 triggering by these diverse parasite patterns leads to different immune outcomes: for S. mansoni, triggering leads to the development of fully mature dendritic cells capable of inducing a Treg response (see Chapter 11), characterized by elevated IL-10 levels; for T. cruzi, mature dendritic cells induce a TH1 response (see Chapter 7) with raised levels of IL-12. This dichotomous response could, in part, be explained by the cooperation between TLR2 and other TLRs. However, there has been difficulty in assigning definitive contributions of TLRs to activation by specific parasite proteins since samples are easily contaminated, e.g. with bacterial PAMPs.
Classical human PRRs also contribute to recognition of parasites
Classical PRRs also play important roles in the innate response to parasite infection (see Fig. 15.6) and include collectins (e.g. MBL), pentraxins (e.g. CRP), C-type lectins (e.g. macrophage mannose receptor, and scavenger receptors (e.g. CD36) – see Chapters 6 and 7. For example, MBL binds mannose-rich LPG from Leishmania, Plasmodium, trypanosomes, and schistosomes; and polymorphisms in the MBL gene are associated with increased susceptibility to severe malaria.
Complement receptors are archetypal PRRs
Complement receptors, in particular CR3, are archetypal PRRs involved in innate immune responses (see Fig. 15.6). They are truly multifunctional, being involved in phagocyte adhesion, recognition, migration, activation, and microbe elimination.
• CR3 offers a multiplicity of binding sites, enabling opsonic or non-opsonic binding;
• phagocytosis by CR3 alone does not generate an oxidative burst in phagocytic cells;
Adaptive immune responses to parasites
T and B cells are pivotal in the development of immunity
The T cell requirement is also demonstrable because nude (athymic) or T-deprived mice fail to clear otherwise non-lethal infections of protozoa such as T. cruzi or Plasmodium yoelii, and T cell-deprived rats fail to expel the intestinal worm Nippostrongylus brasiliensis (Fig. 15.7).
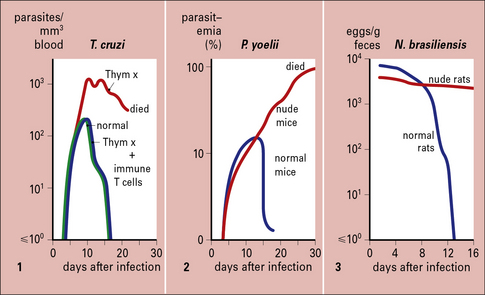
B cells also play key roles in regulating and controlling immunity to parasites. For example:
• B cells and antibodies are required for resistance to the parasitic gastrointestinal nematode Trichuris muris; and
Both CD4 and CD8 T cells are needed for protection from some parasites
• CD4+ T cells mediate immunity against blood-stage P. yoelii;
• CD8+ T cells protect against the liver stage of Plasmodium berghei.
The action of CD8+ T cells is twofold:
• they secrete IFNγ, which inhibits the multiplication of parasites within hepatocytes;
• they are able to kill infected hepatocytes, but not infected erythrocytes.
Regulatory T cells are able to modulate the extremes of both TH2 and TH1 responses.
Cytokines, chemokines, and their receptors have important roles
• the accumulation of macrophages in the granulomas that develop in the liver in schistosomiasis;
• the eosinophilia characteristic of helminth infections; and
• the recruitment of eosinophils and mast cells into the gut mucosa that occurs in worm infections of the gastrointestinal tract.
IL-10 and transforming growth factor-β (TGFβ), the regulatory cytokines (see Chapter 11), downregulate the proinflammatory response and thus minimize pathological damage.
T cell responses to protozoa depend on the species
For example, in mouse models, the induction of TH1 cells with concomitant upregulation of IFNγ and nitric oxide (NO•) is crucial for protection of mice from Leishmania. Strains of mice driving TH2 responses on infection, manifested by high levels of IL-4, IL-13, IL-10, and antibody, develop progressive and ultimately lethal disease (Fig. 15.8).
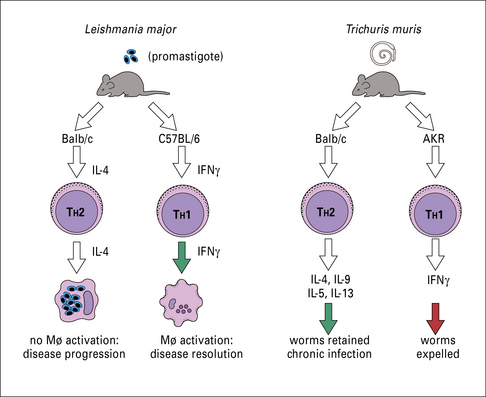
The importance of TH1 cells for protection from toxoplasmosis is also evident in murine models.
Q. What effects will IFNγ and TNFα have on cerebral blood vessels?
A. These cytokines cause an increase in adhesion molecules (ICAM-1, VCAM-1) and synthesis of inflammatory chemokines (CCL2, CXCL10) producing leukocyte migration into the brain (see Chapter 6). They also cause an increase in the permeability of the vessels so that large serum molecules enter the CNS, and ionic equilibria are disturbed. This is referred to as a breakdown in the blood–brain barrier.
The immune response to worms depends upon TH2-secreted cytokines
IgE and eosinophilia are the hallmarks of the immune response to worm infections, and depend upon cytokines secreted by TH2 cells (see Fig. 15.8).
The mechanisms of induction of TH2 responses are less well understood than TH1 responses.
• one hypothesis, the default hypothesis, suggests that unless the triggers for TH1 responses are received (including high IL-12), TH2 responses occur;
• more recent evidence, however, suggests that specific signals induce the T cell to make TH2 cytokines, probably including cell–cell interactions.
• in mice infected with S. mansoni, IL-5-producing TH2 cells predominate;
• in mice that have been immunized, IgE levels and eosinophil numbers are low and TH1 cells predominate.
IFNγ activates effector cells that destroy lung stage larvae, via the production of NO•.
• animals normally resistant to infection develop persistent infections in IL-4 and/or IL-13 knockout mice;
• conversely, susceptible mice expel the worms if IL-4 activity is promoted by administration of neutralizing antibody against IFNγ.
Some worm infections deviate the immune response
The role of IFNγ in promoting chronic infection is again shown by the administration of IL-12 to mice soon after infection with the intestinal worm Nippostrongylus braziliensis (Fig. 15.w2  ).
).
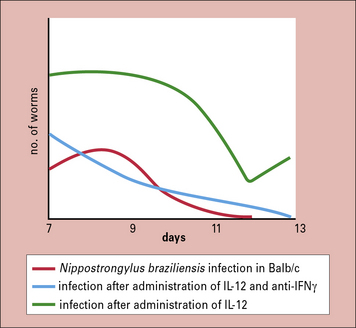
N. braziliensis stimulates IFNγ production, which delays expulsion of the worms.
Parasites induce non-specific and specific antibody production
Levels of total immunoglobulins are raised:
The relative importance of antibody-dependent and antibody-independent responses varies with the infection and host (Fig. 15.9).
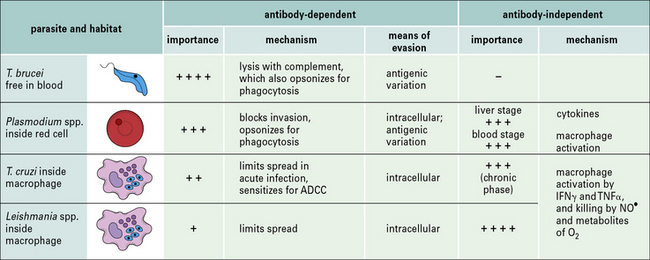
Fig. 15.9 Relative importance of antibody-dependent and antibody-independent responses in protozoal infections
The mechanisms by which specific antibody can control parasitic infections and its effects are summarized in Figure 15.10. Antibody:
• can act directly on protozoa to damage them, either by itself or by activating the complement system (Fig. 15.11);
• can neutralize a parasite directly by blocking its attachment to a new host cell, as with Plasmodium spp., whose merozoites enter red blood cells through a special receptor – their entry is inhibited by specific antibody (Fig. 15.w3  );
);
• may prevent spread (e.g. in the acute phase of infection by T. cruzi);
• can enhance phagocytosis by macrophages – phagocytosis is increased even more by the addition of complement; these effects are mediated by Fc and C3 receptors on macrophages, which may increase in number as a result of macrophage activation;
• is involved in antibody-dependent cell-mediated cytotoxicity (ADCC), for example in infections caused by T. cruzi, T. spiralis, S. mansoni, and filarial worms – cytotoxic cells such as macrophages, neutrophils, and eosinophils adhere to antibody-coated worms by means of their Fc and C3 receptors and degranulate, spilling their toxic contents onto the worm (see Fig. 15.13).
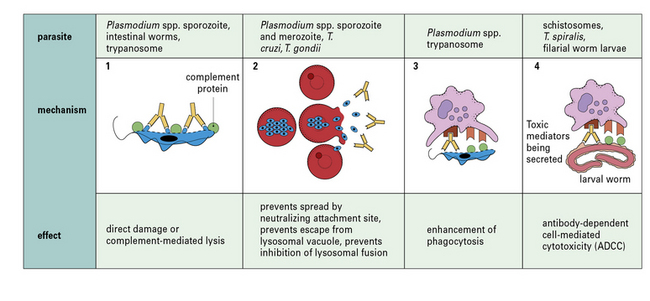
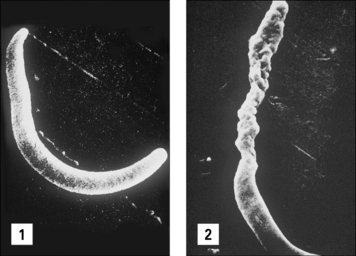
Fig. 15.11 Direct effect of specific antibody on sporozoites of malaria parasites
(Courtesy of Dr R Nussenzweig.)
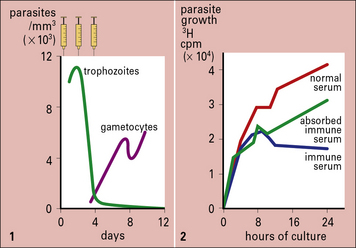
Q. What roles does IgE have in immune defense?
A. IgE mediates inflammation by binding to mast cells and basophils, sensitizing them to parasite antigens. Additionally it can act as an opsonin for eosinophils (see Chapter 3). Polyclonal activation of IgE production can inhibit cross-linking of specific IgE on mast cells by ‘crowding out’.
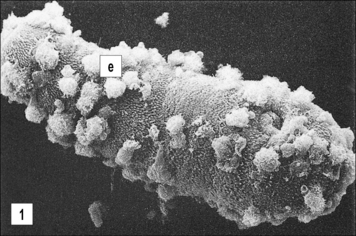
In many infections it is difficult to distinguish between cell-mediated and antibody-mediated responses because both can act in concert against the parasite. This is illustrated in Figure 15.12, which summarizes the immune reaction that can be mounted against schistosome larvae.
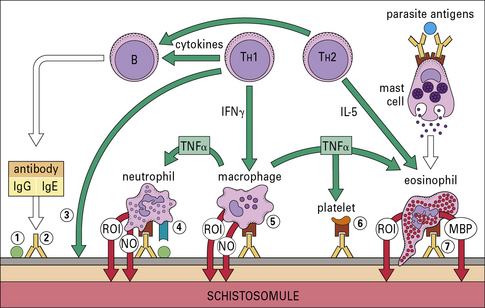
Immune effector cells
• the cercariae of S. mansoni enter through the skin – experimental depletion of macrophages, neutrophils, and eosinophils from the skin of mice increases their susceptibility to infection;
• trypanosomes and malarial parasites entering the blood are removed from the circulation by phagocytic cells in the skin, spleen and liver;
• comparison of strains of mice with various immunological defects for their resistance to infection by Trypanosoma rhodesiense shows that the African trypanosomes are destroyed by macrophages, and later in infection, when opsonized with antibodies and complement C3b, they are taken up by macrophages in the liver more quickly still.
• some of these molecules – IL-1, IL-12, TNFα, and the colony stimulating factors (CSFs) – enhance immunity by activating other cells or stimulating their proliferation;
• others, like IL-10, prostaglandins, and TGFβ, may be anti-inflammatory and immunosuppressive.
Macrophages can kill extracellular parasites
• act as killer cells through ADCC – specific IgG and IgE, for instance, enhance their ability to kill schistosomules;
• secrete cytokines, such as TNFα and IL-1, which interact with other types of cell, for example rendering hepatocytes resistant to malarial parasites.
Nitric oxide, a product of L-arginine metabolism, is another potent toxin. Its synthesis by macrophages in mouse experimental systems is induced by the cytokines IFNγ and TNFα and is greatly increased when they act synergistically. NO• can also be produced by endothelial cells. It contributes to host resistance in leishmaniasis, schistosomiasis, and malaria, and is probably important in the control of most parasitic infections (see Fig. 15.13). For instance, the innate resistance to infection by T. gondii that is lost in immunocompromised individuals appears to be due to the inhibition of parasite multiplication by such an oxygen-independent mechanism.
Activation of macrophages is a feature of early infection
Neutrophils can kill large and small parasites
Neutrophils can be activated by cytokines, such as IL-8, IFNγ, TNFα, and GM-CSF.
Eosinophils are characteristically associated with worm infections
• the eosinophil evolved specifically as a defense against the tissue stages of parasites that are too large to be phagocytosed;
• the IgE-dependent mast cell reaction has evolved primarily to localize eosinophils near the parasite and enhance their antiparasitic functions.
Eosinophils can kill helminths by oxygen-dependent and independent mechanisms
Eosinophils are less phagocytic than neutrophils. They degranulate in response to perturbation of their surface membrane and their activities are enhanced by cytokines such as TNFα and GM-CSF. Most of their activities, however, are controlled by antigen-specific mechanisms. Thus their binding in vitro to the larvae of worms (e.g. S. mansoni and T. spiralis) increases the release of their granular contents onto the surface of the worms (Fig. 15.13).
Mast cells control gastrointestinal helminths
In the case of T. spiralis and Heligmosomoides polygyrus there is good evidence to suggest the involvement of mucosal mast cells (Fig. 15.14)
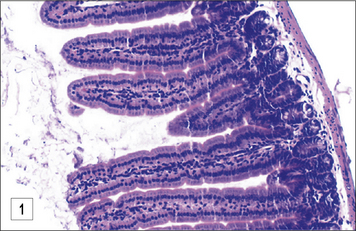
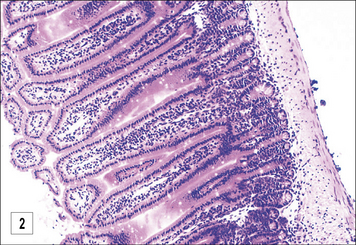
Parasite escape mechanisms
Despite their protective role in the immune response to many different parasites:
Parasites can resist destruction by complement
In the case of Leishmania, resistance correlates with virulence:
• L. tropica, which is easily killed by complement, causes a localized self-healing infection in the skin; whereas
• L. donovani, which is ten times more resistant to complement, becomes disseminated throughout the viscera, causing a disease that is often fatal.
The mechanisms whereby parasites can resist the effect of complement differ:
• the lipophosphoglycan (LPG) surface coat of L. major activates complement, but the complex is then shed so the parasite avoids lysis;
• the trypomastigotes of T. cruzi bear a surface glycoprotein that has activity resembling the decay accelerating factor (DAF) that limits the complement reaction. The resistance that schistosomules acquire as they mature is also correlated with the appearance of a surface molecule similar to DAF.
Intracellular parasites can avoid being killed by oxygen metabolites and lysosomal enzymes
Intracellular parasites that live inside macrophages have evolved different ways of avoiding being killed by oxygen metabolites and lysosomal enzymes (Fig. 15.15):
• T. gondii penetrates the macrophage by a non-phagocytic pathway and so avoids triggering the oxidative burst;
• Leishmania spp. can enter by binding to complement receptors – another way of avoiding the respiratory burst.
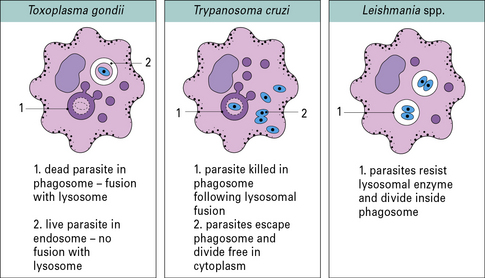
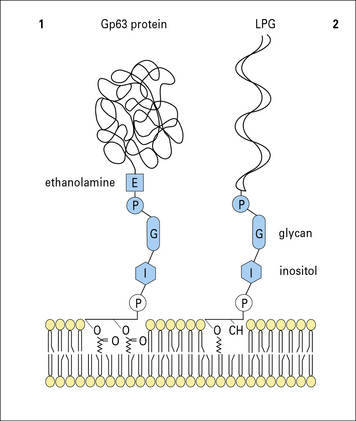
It can be demonstrated that the vacuole in which Leishmania organisms survive is lyosomal in nature (Fig. 15.16), but the parasites have evolved mechanisms that protect it against enzymatic attack. The LPG surface coat acts as a scavenger of oxygen metabolites and affords protection against enzymatic attack, but a glycoprotein, Gp63 (Fig 15.w4  ) inhibits the action of the macrophage’s lysosomal enzymes.
) inhibits the action of the macrophage’s lysosomal enzymes.
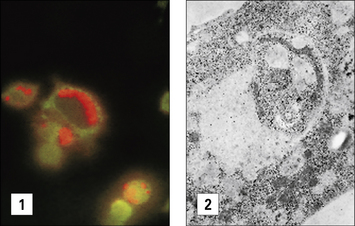
These escape mechanisms, however, are less efficient in the immune host.
Q. Why would reduction in MHC molecule expression be less effective in an immune host?
A. In immune hosts, the release of IFNγ enhances MHC molecule expression by APCs. In addition the level of stimulation required by a primed T cell is less than for a naive T cell, due in part to its enhanced level of receptors for co-stimulatory signals (see Chapter 8).
Parasites can disguise themselves
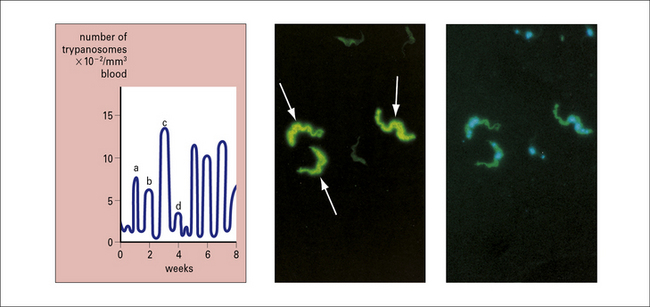
African trypanosomes and malaria undergo antigenic variation
The molecule that forms the surface coat of the African trypanosome, the variable surface glycoprotein (VSG), changes to protect the underlying surface membrane from the host’s defense mechanisms. New populations of parasites are antigenically distinct from previous ones (Fig. 15.17).
Several antigens of malarial parasites also undergo antigenic variation.
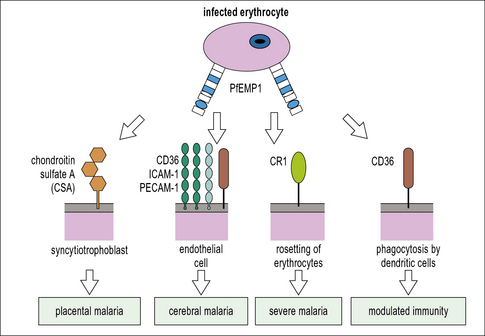
For example, the P. falciparum erythrocyte membrane protein-1 (PfEMP1) is extremely polymorphic and variable between different strains of the parasite because it is perpetually exposed to the immune system by its location on the red cell membrane. PfEMP1 can bind numerous host immune proteins, but particularly scavenger receptors, e.g. CD36 and scavenging antibodies that eliminate apoptotic, or damaged cells, e.g. natural IgM (see Fig. 15.20).
Some extracellular parasites hide from or resist immune attack
There are numerous examples of simple, physical, protective strategies in parasites:
• nematodes have a thick extracellular cuticle, which protects them from the toxic effects of an immune response;
• the tegument of schistosomes thickens during maturation to offer similar protection;
• the loose surface coat of many nematodes may slough off under immune attack;
• tapeworms actually prevent attack by secreting an elastase inhibitor, which stops them attracting neutrophils.
Most parasites interfere with immune responses for their benefit
Parasites produce molecules that interfere with host immune function
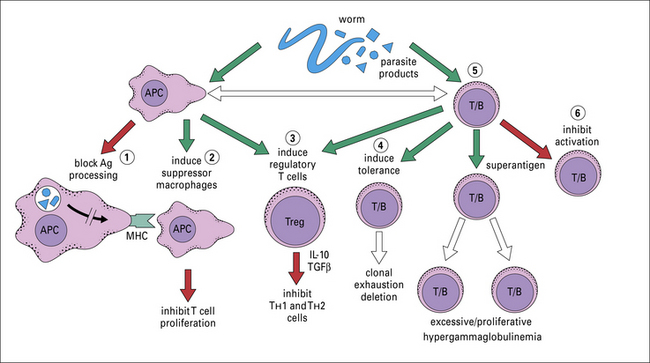
Parasites produce molecules that can affect the phenotype of the adaptive response, which may be to their own advantage (Figs 15.18 and 15.19).
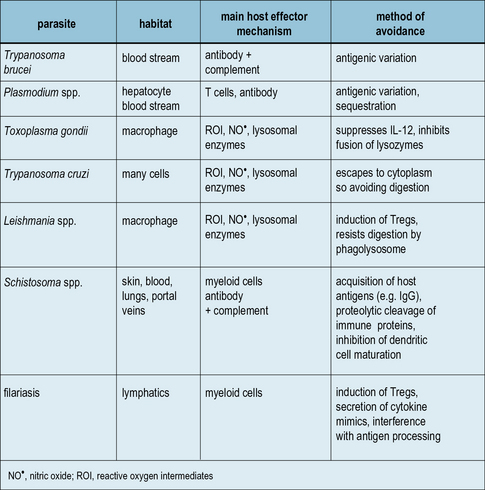
Fig. 15.18 Some mechanisms by which parasites avoid host immunity
A summary of the various methods that parasites have evolved to avoid host defense mechanisms.
Many of the surface antigens that are shed are soluble forms of molecules inserted into the parasite membrane by a GPI anchor, including the VSG of T. brucei, the LPG or ‘excreted factor’ of Leishmania (see Fig. 15.w4 ) and several surface antigens of schistosomules. These are released by endogenous phosphatidylinositol-specific phospholipases.
) and several surface antigens of schistosomules. These are released by endogenous phosphatidylinositol-specific phospholipases.
Some parasites suppress inflammation or immune responses
The ability of parasites to suppress hyperactive immune responses is believed to be due to the induction of regulatory T cells (see Fig. 15.19). Understanding how regulatory T cells, also known as suppressor T cells, are induced and how they dampen immune responses is the focus of intensive research. It seems to be the dendritic cell that polarizes the T cell towards a regulator phenotype after exposure to parasite extracts.
Immunopathological consequences of parasite infections
A single parasite protein may produce multiple pathological effects, as seen with PfEMP1, coded by the var genes, and expressed on the surface of infected erythrocytes (Fig. 15.20).

Vaccines against human parasites
Some vaccines composed of attenuated living parasites have proved successful in veterinary practice. However, so far there is none in use against human parasites, though much effort has been directed towards the development of subunit vaccines against malarial parasites and schistosomes in particular. Some clinical trials of vaccines against malaria, based on combinations of putatively protective antigens are in progress (see Chapter 18). There is an urgent need to identify the key antigens involved and develop non-toxic adjuvants for their effective delivery to the immune system.
Parasite genome sequencing ventures and analysis of whole parasite proteomes are highlighting novel targets for both drug and vaccine design. These modern technologies are providing startling insights into how parasite and host immune systems interact. For example, the malaria protein PfEMP1 encoded by var genes, known to be expressed in the red blood cell stage within the human host and implicated in immune evasion, has been identified in the mosquito sporozoite stage, indicating that it may have several alternative functions (see Fig. 15.20).
Critical thinking: Immunity to protozoa and helminths (see p. 437 for explanations)
1. In general, protozoa and helminths adopt different strategies for survival and for transmission to the subsequent host. How do they differ?
2. Many parasites have evolved to live in host cells. Consider the advantages and disadvantages to this mode of existence. Consider the different cell types and how parasites have to adapt this environment to their advantage. In particular Toxoplasma gondii, Trypanosoma cruzi, and Leishmania spp. have adapted to live in the macrophage and can escape destruction by lysosomal enzymes, but the way in which they do this differs. How have these adaptations helped parasite survival?
3. Extracellular parasites have evolved sophisticated mechanisms to avoid the immune response. Consider examples of how they do this.
Anthony R.M., Rutitzky L.I., Urban J.F., Jr., et al. Protective immune mechanisms in helminth infection. Nat Rev Immunol. 2007;7:975–987.
Couper K.N., Blount D.G., Riley E.M. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777.
Czajkowsky D.M., Salanti A., Ditlev S.B., et al. IgM, Fc mu Rs, and malarial immune evasion. J Immunol. 2010;184:4597–4603.
Erb K.J. Helminths, allergic disorders and IgE-mediated immune responses: where do we stand? Eur J Immunol. 2007;37:1170–1173.
Langhorne J., Ndungu F.M., Sponaas A.M., Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. 2008;9:725–732.
Martinez F.O., Helming L., Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483.
Paul W.E., Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235.
Pleass R.J., Behnke J.M. B-cells get the T-cells but antibodies get the worms. Trends Parasitol. 2009;25:443–446.
Soong L. Modulation of dendritic cell function by Leishmania parasites. J Immunol. 2008;180:4355–4360.
Voehringer D. The role of basophils in helminth infection. Trends Parasitol. 2009;25:551–556.
Various news groups on the www (though not all dedicated to immunology) can be accessed by exploring keywords
Several discussion groups operate through the bionet, for example http://www.bio.net/archives.html (a parasitology mail newsgroup) and http://www.parasitology.org.uk (British Society for Parasitology)