[level-membership-for-allergy-and-immunology-category]
Chapter 22 Immunity to Cancers
• The immune system can potentially survey the body for some types of developing tumor.
• Tumors can induce immunity. Mice, rats, hamsters, and frogs can be immunized against tumors. In most tumors of animals, tumor immunity elicited by immunization is specific (or strongest) to the individual tumor that was used to immunize.
• Tumor antigens have been characterized by three means – immunization-challenge experiments, T cell reactivity, and antibody reactivity. Immunization-challenge experiments have uncovered the immunogenicity of heat-shock protein-chaperoned antigenic peptides. Antigens identified by reactivity to T cells and antibodies include individual tumor-specific mutated antigens, cancer testes antigens, differentiation antigens, and viral tumor antigens.
• Vigorous anti-tumor immune responses are compromised by regulatory mechanisms. Tumors elicit immunity in their primary host, and such immunity is downregulated. Regulatory cells such as CD25+ CD4+ T cells and inhibition of activated anti-tumor T cells through T cell molecules such as CTLA-4 are involved in downregulation of tumor immunity.
Tumor immunity in the primary host
Nonetheless, amidst the barrage of experiments where MHC-mismatched tumors were transplanted into mice, were the experiments of Ludwik Gross, and later those of Prehn and Main, and of George and Eva Klein, who showed that, even when MHC-matched tumors were used to immunize mice, protection against subsequent tumor growth could be achieved (Fig. 22.1). These studies led to two principles, which have informed much of cancer immunology since and are discussed below.
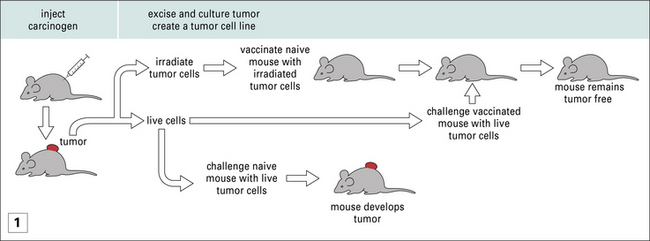
Cancers elicit protective immunity in the primary and syngeneic host
• The immunogenicity of tumors has provided the foundation stone for the idea of tumor-specific antigens. If one could immunize, then antigens must exist.
• Tumor immunity depends on many factors. The degree of tumor immunity depends upon the type of cancer and the method of its induction, or lack of induction. UV-induced cancers are highly immunogenic, methyl-cholanthrene-induced tumors less so, and spontaneous tumors even less so. Nonetheless, immunogenicity of tumors has been demonstrated in all model systems tested.
• The primary animal develops immunity to subsequent tumor challenge. Furthermore it is also immune to subsequent challenges with the same tumor (see Fig. 22.1).
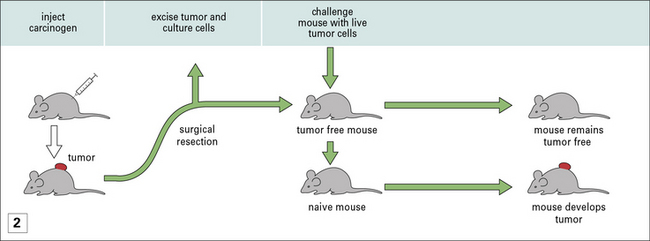
• Protective immunity is only seen in prophylactic immunization and not therapeutic immunization – once a mouse has been implanted with a tumor, immunization with irradiated cancer cells (derived from the growing tumor) does nothing to mitigate tumor growth (Fig. 22.2). Exploration of differences between prophylaxis and therapy has led to fundamental insights into tumor immunity.
Spontaneous tumors are also antigenically distinct
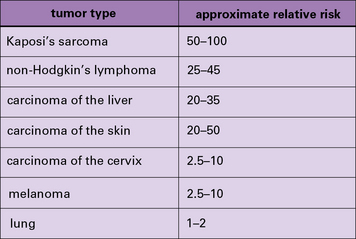
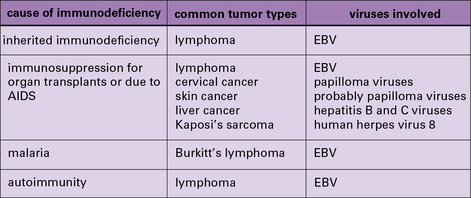
In addition to these experimental studies, several clinical observations point to the existence of tumor-protective immunity in humans. These include the increased relative risk of cancers in patients who are immunosuppressed because they are kidney transplant recipients (Fig. 22.3) or for a variety of other reasons (Fig. 22.4).
Characterization of tumor antigens
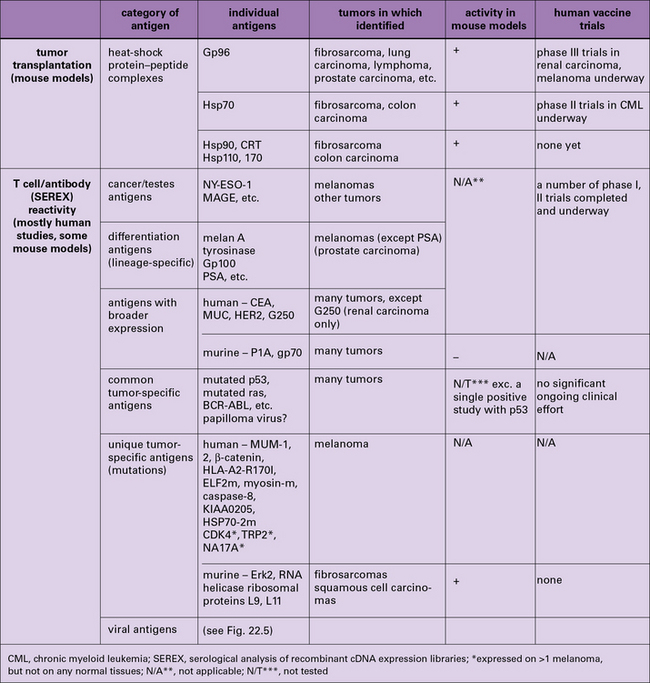
The two broad approaches to the identification of tumor antigens are shown in Figure 22.5 and discussed individually below. Not surprisingly, the approaches have yielded results that are not fully concordant. These differences have helped highlight a fascinating interplay between immunity and tolerance to tumors, discussed below.
Tumor-specific antigens defined by immunization all belong to the family of HSPs
• could elicit protective immunity;
• were of the HSP90 (gp96 and HSP90), HSP70 (HSP110 and HSP/c70), calreticulin, and HSP170 (also known as grp170) families.
HSPs must be isolated directly from tumors to be immunologically active
Two aspects of HSP-elicited tumor immunity are notable.
• First, HSPs are present in normal tissues as well as tumors, and normal tissue-derived HSPs do not elicit rejection. They must be isolated from tumors to be immunologically active.
• Second, HSPs elicit immunity specifically against the individual tumors from which they are isolated.
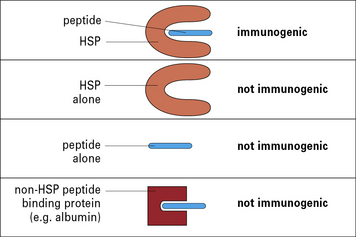
This conundrum was resolved by the demonstration that the HSP molecules chaperone peptides in a peptide-binding pocket, much as the MHC molecules do, although the structural details of the pockets in HSP and MHC differ (Fig. 22.6). The specificity of immunogenicity derives from the peptides rather than the HSP itself – dissociation of HSP-associated peptides from HSPs abrogate the tumor rejection activity.

HSPs can chaperone many different types of antigen
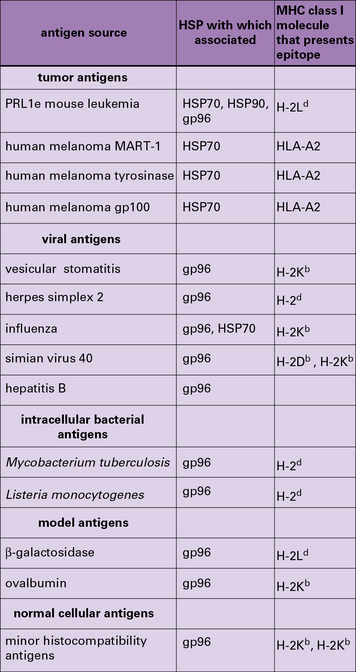
HSP-chaperoned peptide pools contain cytotoxic T lymphocyte (CTL) epitopes and CTL epitope precursors for any antigens that the cell expresses (Fig. 22.w1). This evidence comes from mouse and human tumors, normal tissues, and virus-infected cells.
HSP molecules bind to APCs and target peptides with high efficiency
The HSP molecule itself plays at least two crucial roles other than chaperoning peptides:
• HSPs bind antigen-presenting cells (APCs) such as macrophages and dendritic cells (DCs) through HSP receptors such as CD91 and thus target the peptides chaperoned by them into the APCs with high efficiency;
• further, the HSP-chaperoned peptides, once introduced into the APC, follow the endogenous as well as the exogenous pathway of antigen presentation and are processed and re-presented by the MHC I and MHC II molecules of the APCs (Fig. 22.7).
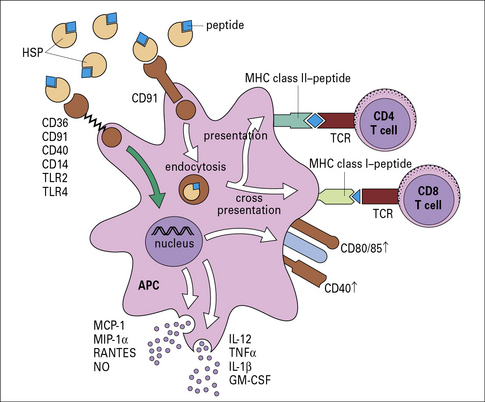
Fig. 22.7 Mechanism of specific immunogenicity of HSP-peptide complexes
(Redrawn from Srivastava P. Nat Rev Immunol 2002;2:185–194. Copyright 2002, Nature Reviews Immunology.)
‘Tumor-specific antigens’ recognized by T cells show a wide spectrum of specificity
The tumor antigens identified as T cell epitopes fall into the following categories (see Fig. 22.5).
Unique tumor-specific antigens have been characterized in melanomas
Individually tumor-specific or unique antigens presented by MHC class I or class II molecules have been characterized in human melanomas (see Fig. 22.5).
T cell epitopes of viral antigens have been identified
T cell epitopes of viral antigens of virus-induced tumors have been identified such as:
• the T antigen of SV40 and the polyoma viruses;
• the E6 and E7 antigens of the human papilloma viruses that cause cervical cancer; and
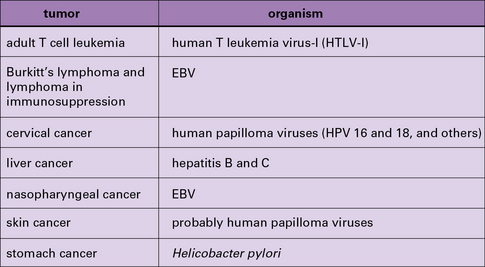
Some of these, such as the HPV antigens, are used for prophylactic vaccination (see Chapter 18) and are being tested for clinical activity in human tumors.
‘Tumor-specific antigens’ defined by antibodies are rarely tumor specific
The search for antibodies that discriminate between cancer cells and normal cells has a long pedigree and has been carried out with the whole range of tools starting from antisera to panning antibody libraries. This search has been largely unsuccessful and rarely have tumor-specific antibodies been generated. Most anti-tumor antibodies, like anti-tumor T cells, happen to recognize CT antigens, differentiation antigens, and even more broadly distributed common antigens (see Fig. 22.5).
• do not recognize tumor-specific antigens;
• are actually used as pharmacological rather than immunological reagents.
Antibodies used in the diagnosis of cancer may not be tumor specific
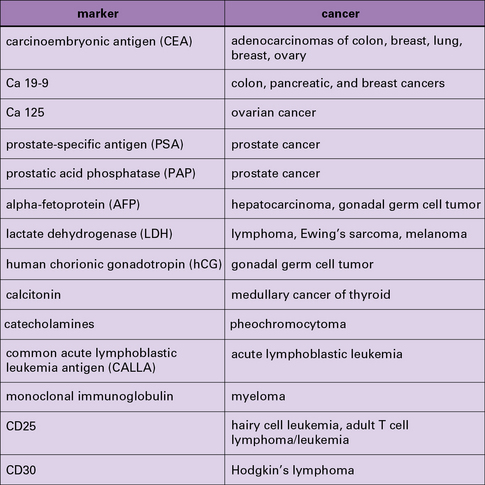
As serum antibodies are technically easy to measure, they have always attracted the attention of diagnosticians (Fig. 22.w2  ). Thus, antibody to carcinoembryonic antigen (CEA) is often used as a marker for progression or status of certain carcinomas; however, CEA or other such antigens are not tumor-specific antigens and diagnostic markers need not necessarily have the specificity required of a ‘tumor-specific
). Thus, antibody to carcinoembryonic antigen (CEA) is often used as a marker for progression or status of certain carcinomas; however, CEA or other such antigens are not tumor-specific antigens and diagnostic markers need not necessarily have the specificity required of a ‘tumor-specific  antigen’.
antigen’.
Anti-tumor immune responses
Successful tumor immunity is rare in patients who have cancer
Not surprisingly, the pathways to elicitation of immune response to tumors are straightforward (Fig. 22.9). The tumor inoculum (with its antigenic load) is taken up by the APCs at the site of immunization and is cross-presented by them to the naive CD8 cells in the draining lymph nodes. Both responses are generally necessary in the mouse models tested and both responses have been shown to be present in the cancer patients studied.
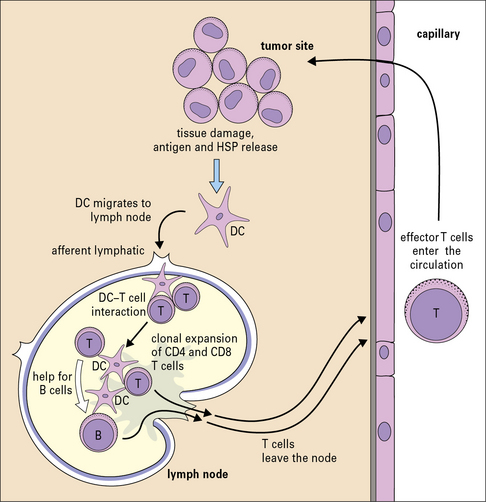
Antibodies have not generally been shown to be protective in the natural setting.
Despite an immune response, tumors continue to grow
Despite clear evidence for the existence of tumor-specific antigens and the immune response elicited by them, tumors generally continue to grow. In this regard, Ehrlich made a curious observation that remains at the center of cancer immunity. He noted that animals with already growing tumors were strangely resistant to a second tumor challenge even as the first tumor kept growing (Fig. 22.10). This phenomenon, termed concomitant immunity as early as 1908, remained relatively unexamined until recently.
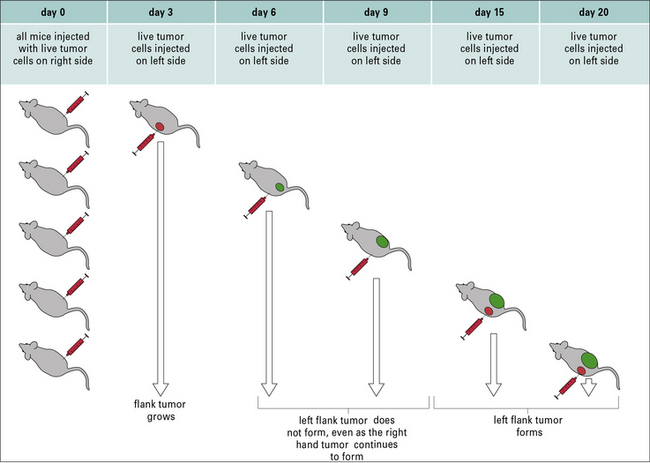
Concomitant immunity shows two aspects of tumor immunity
Concomitant immunity shows two aspects of tumor immunity:
• first, a growing tumor elicits in the primary host a tumor-protective immune response;
• second, although this response is sufficient to eliminate a nascent tumor, it fails to eliminate the tumor that elicited the response.
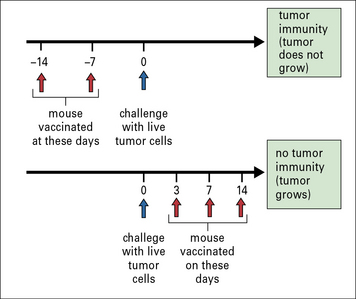
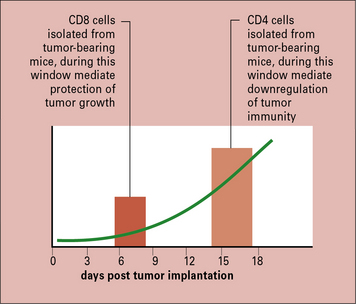
It was shown that concomitant immunity was tumor specific and operational only within a narrow window of 7–10 days after tumor implantation; if the second tumor was implanted beyond this time, it was not rejected. The lack of immunity beyond the narrow window was attributable to a new population of suppressor T cells that appeared at that time (Fig. 22.11). Similar to the phenomenon of concomitant immunity, it was also noted that mice in the process of rejecting an allograft were unable to concomitantly reject a growing tumor bearing the same alloantigens as the allograft.
Immunization is effective prophylactically but rarely as therapy
Prophylaxis is relatively effective, even if begun as late as 3 days before tumor implantation, but therapy is ineffective even if begun as early as 2 days after tumor implantation (see Fig. 22.2).
The naive state and tumor-bearing state are essentially different
What are the mechanisms behind this change of status? They are exactly the same as envisaged for the mechanisms for the initiation and maintenance of peripheral tolerance. None of the explanations is fully satisfactory by itself, but each perhaps contributes to the final state of tolerance in some measure. Immune unresponsiveness per se is addressed in detail in Chapter 19. Aspects that are of specific relevance to tumor immunity include:
• inhibitory cytokines including TGFβ and IL-10;
• inhibition of T cell activity through CTLA-4 and PD1;
• down-regulation of the immune response through regulatory T cells;
• resistance to recognition by down-regulation of MHC class I molecules; and
Downregulation of MHC I molecule expression may result in resistance to recognition and lysis
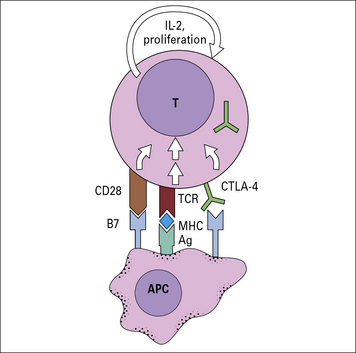
T cell activity is inhibited through CTLA-4
CTLA-4 inhibits the T cell by raising the stimulatory threshold or by inhibiting the proliferative drive of T cells (Fig. 22.12). The biological role of CTLA-4 appears to lie in limiting the T cell response to foreign antigens as well as to autoantigens.
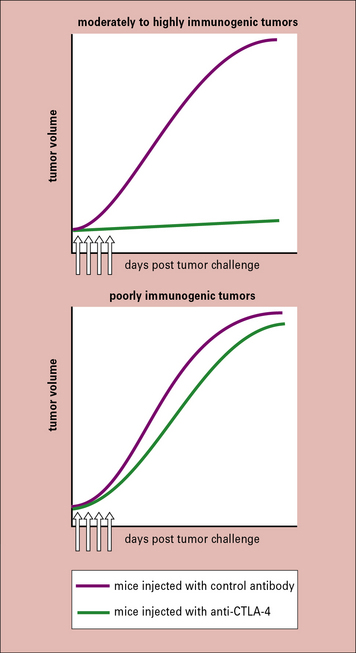
Administering antibodies to CTLA-4 (that inhibit CTLA-4:B7 interactions) to mice bearing a broad array of tumors inhibited tumor growth, even when the antibody was administered after the tumors were visible and palpable (Fig. 22.w3  ). Such activity was generally seen only against the more immunogenic tumors and not against a poorly immunogenic melanoma (e.g. B16). In that instance, combination of anti-CTLA-4 antibody with a vaccine consisting of irradiated melanoma cells that were also transfected with the cytokine granulocyte–macrophage colony stimulating factor (GM-CSF), resulted in a stronger anti-tumor response than by anti-CTLA-4 antibody or the vaccine alone. Clinical development of this idea is discussed below.
). Such activity was generally seen only against the more immunogenic tumors and not against a poorly immunogenic melanoma (e.g. B16). In that instance, combination of anti-CTLA-4 antibody with a vaccine consisting of irradiated melanoma cells that were also transfected with the cytokine granulocyte–macrophage colony stimulating factor (GM-CSF), resulted in a stronger anti-tumor response than by anti-CTLA-4 antibody or the vaccine alone. Clinical development of this idea is discussed below.
Abrogation of CD25+ cells leads to protective tumor immunity
The notion that progressive tumor growth results in the generation of inhibitory influences on the anti-tumor immune response is further supported by the recent work on the CD25+ CD4+ Treg cells (see Chapter 11). These cells have been shown to suppress CD8+ T cell responses in general, including autoimmune responses.
Immunotherapy for human cancer
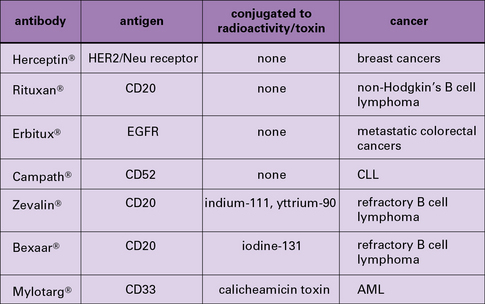
Antibodies have been used successfully
Vaccination can be used to treat cancer
Although the term vaccination is typically used to indicate prophylactic vaccination, cancer researchers use it to indicate the treatment of someone who already has cancer, with agents that stimulate anti-cancer immune response. Several vaccination approaches are currently being pursued (Fig. 22.13).
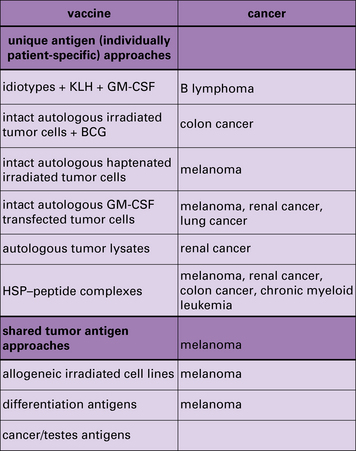
The idiotypes of B lymphomas have been used as vaccines

Immunization with defined MHC I epitope does not lead to clinical benefit
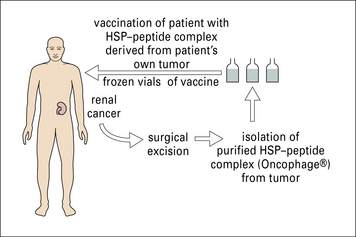
Immunization with HSP–peptide complexes demonstrates clinical benefit
The role of HSP–peptide complexes in eliciting tumor immunity has been discussed earlier.
In these trials, surgically obtained tumor specimens (or leukemia cells obtained by leukopheresis) from a given patient are used as the starting material for preparation of gp96–peptide or hsp70–peptide complexes specifically for that patient (Fig. 22.14).
Adoptive immunotherapy using T cells: the clinical benefits
Adoptive immunotherapy using T cells has a successful pedigree in murine models of cancer (Fig. 22.15). Clinical experience with bone marrow transplant recipients also provides a strong rationale for the approach.
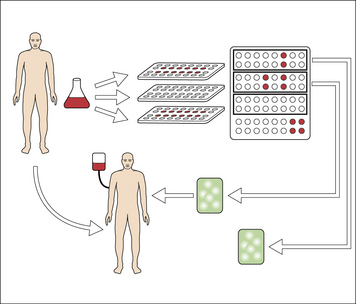
Fig. 22.15 Adoptive immunotherapy with T cells
(Redrawn from Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer 2003;3:666–675. Copyright 2002, Nature Reviews Cancer, Macmillan Magazines Ltd.)
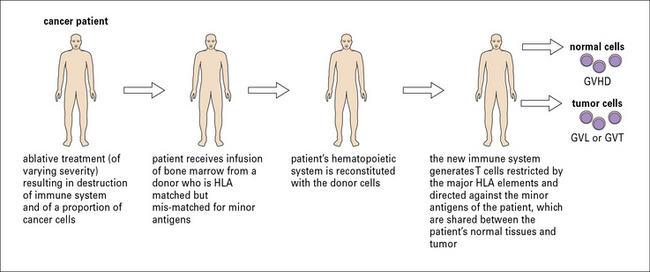
Interestingly, patients who develop GVHD also have a lower cancer relapse or graft versus tumor (GVT) incidence. The clinical experience with GVHD and GVT has long remained a compelling piece of evidence for the premise that T cells can eliminate human cancers in vivo (Fig. 22.16).
Inhibition of downregulation of immune modulation is clinically valuable
Critical thinking: Immunity to cancers (see p. 441 for explanations)
1. Animals can be easily immunized against cancers. Why then do the same cancers grow progressively and kill their hosts?
2. Heat-shock proteins do not differ between tumors and normal tissues. Yet, HSP preparations isolated from tumors elicit tumor immunity, whereas similar preparations from normal tissues do not. Explain the mechanistic basis of this phenomenon.
3. How have tumor antigens been defined? How many classes of tumor antigen have been defined thus far?
4. Explain the phenomenon of concomitant immunity.
5. Discuss the evidence that the immune response to tumors is downregulated in tumor-bearing mice and in patients with cancer.
Belli F., Testori A., Rivoltini L., et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96–peptide complexes: clinical and immunologic findings. J Clin Oncol. 2002;20:4169–4180.
Coulie P.G., Karanikas V., Lurquin C., et al. Cytolytic T-cell responses of cancer patients vaccinated with a MAGE antigen. Immunol Rev. 2002;188:33–42.
Egen J.G., Kuhns M.S., Allison J.P. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat Immunol. 2002;3:611–618.
Fehervari Z., Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest. 2004;114:1209–1217.
Ho W.Y., Blattman J.N., Dossett M.L., et al. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437.
Klein G. The strange road to the tumor-specific transplantation antigens (TSTAs). Cancer Immun. 2001;1:6.
North R.J. Down-regulation of the antitumor immune response. Adv Cancer Res. 1985;45:1–43.
Scanlan M.J., Gure A.O., Jungbluth A.A., et al. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32.
Srivastava P.K. Do human cancers express shared protective antigens? or the necessity of remembrance of things past. Semin Immunol. 1996;8:295–302.
Srivastava P.K. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425.
Van Der Bruggen P., Zhang Y., Chaux P., et al. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64.
Wick M., Dubey P., Koeppen H., et al. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J Exp Med. 1997;186:229–238.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 22 Immunity to Cancers
• The immune system can potentially survey the body for some types of developing tumor.
• Tumors can induce immunity. Mice, rats, hamsters, and frogs can be immunized against tumors. In most tumors of animals, tumor immunity elicited by immunization is specific (or strongest) to the individual tumor that was used to immunize.
• Tumor antigens have been characterized by three means – immunization-challenge experiments, T cell reactivity, and antibody reactivity. Immunization-challenge experiments have uncovered the immunogenicity of heat-shock protein-chaperoned antigenic peptides. Antigens identified by reactivity to T cells and antibodies include individual tumor-specific mutated antigens, cancer testes antigens, differentiation antigens, and viral tumor antigens.
• Vigorous anti-tumor immune responses are compromised by regulatory mechanisms. Tumors elicit immunity in their primary host, and such immunity is downregulated. Regulatory cells such as CD25+ CD4+ T cells and inhibition of activated anti-tumor T cells through T cell molecules such as CTLA-4 are involved in downregulation of tumor immunity.
Tumor immunity in the primary host
Nonetheless, amidst the barrage of experiments where MHC-mismatched tumors were transplanted into mice, were the experiments of Ludwik Gross, and later those of Prehn and Main, and of George and Eva Klein, who showed that, even when MHC-matched tumors were used to immunize mice, protection against subsequent tumor growth could be achieved (Fig. 22.1). These studies led to two principles, which have informed much of cancer immunology since and are discussed below.
Cancers elicit protective immunity in the primary and syngeneic host
• The immunogenicity of tumors has provided the foundation stone for the idea of tumor-specific antigens. If one could immunize, then antigens must exist.
• Tumor immunity depends on many factors. The degree of tumor immunity depends upon the type of cancer and the method of its induction, or lack of induction. UV-induced cancers are highly immunogenic, methyl-cholanthrene-induced tumors less so, and spontaneous tumors even less so. Nonetheless, immunogenicity of tumors has been demonstrated in all model systems tested.
• The primary animal develops immunity to subsequent tumor challenge. Furthermore it is also immune to subsequent challenges with the same tumor (see Fig. 22.1).
• Protective immunity is only seen in prophylactic immunization and not therapeutic immunization – once a mouse has been implanted with a tumor, immunization with irradiated cancer cells (derived from the growing tumor) does nothing to mitigate tumor growth (Fig. 22.2). Exploration of differences between prophylaxis and therapy has led to fundamental insights into tumor immunity.
Spontaneous tumors are also antigenically distinct
In addition to these experimental studies, several clinical observations point to the existence of tumor-protective immunity in humans. These include the increased relative risk of cancers in patients who are immunosuppressed because they are kidney transplant recipients (Fig. 22.3) or for a variety of other reasons (Fig. 22.4).
Characterization of tumor antigens
The two broad approaches to the identification of tumor antigens are shown in Figure 22.5 and discussed individually below. Not surprisingly, the approaches have yielded results that are not fully concordant. These differences have helped highlight a fascinating interplay between immunity and tolerance to tumors, discussed below.
Tumor-specific antigens defined by immunization all belong to the family of HSPs
• could elicit protective immunity;
• were of the HSP90 (gp96 and HSP90), HSP70 (HSP110 and HSP/c70), calreticulin, and HSP170 (also known as grp170) families.
HSPs must be isolated directly from tumors to be immunologically active
Two aspects of HSP-elicited tumor immunity are notable.
• First, HSPs are present in normal tissues as well as tumors, and normal tissue-derived HSPs do not elicit rejection. They must be isolated from tumors to be immunologically active.
• Second, HSPs elicit immunity specifically against the individual tumors from which they are isolated.
This conundrum was resolved by the demonstration that the HSP molecules chaperone peptides in a peptide-binding pocket, much as the MHC molecules do, although the structural details of the pockets in HSP and MHC differ (Fig. 22.6). The specificity of immunogenicity derives from the peptides rather than the HSP itself – dissociation of HSP-associated peptides from HSPs abrogate the tumor rejection activity.
HSPs can chaperone many different types of antigen
HSP-chaperoned peptide pools contain cytotoxic T lymphocyte (CTL) epitopes and CTL epitope precursors for any antigens that the cell expresses (Fig. 22.w1). This evidence comes from mouse and human tumors, normal tissues, and virus-infected cells.
HSP molecules bind to APCs and target peptides with high efficiency
The HSP molecule itself plays at least two crucial roles other than chaperoning peptides:
• HSPs bind antigen-presenting cells (APCs) such as macrophages and dendritic cells (DCs) through HSP receptors such as CD91 and thus target the peptides chaperoned by them into the APCs with high efficiency;
• further, the HSP-chaperoned peptides, once introduced into the APC, follow the endogenous as well as the exogenous pathway of antigen presentation and are processed and re-presented by the MHC I and MHC II molecules of the APCs (Fig. 22.7).
Fig. 22.7 Mechanism of specific immunogenicity of HSP-peptide complexes
(Redrawn from Srivastava P. Nat Rev Immunol 2002;2:185–194. Copyright 2002, Nature Reviews Immunology.)
