[level-membership-for-allergy-and-immunology-category]
Chapter 14 Immunity to Bacteria and Fungi
• Mechanisms of protection from bacteria can be deduced from their structure and pathogenicity. There are four main types of bacterial cell wall and pathogenicity varies between two extreme patterns. Non-specific, phylogenetically ancient recognition pathways for conserved bacterial structures trigger protective innate immune responses and guide the development of adaptive immunity.
• Lymphocyte-independent bacterial recognition pathways have several consequences. Complement is activated via the alternative pathway. Release of proinflammatory cytokines and chemokines increases the adhesive properties of the vascular endothelium and promotes neutrophil and macrophage recruitment. Pathogen recognition generates signals then regulate the lymphocyte-mediated response.
• Antibody provides an antigen-specific protective mechanism. Neutralizing antibody may be all that is needed for protection if the organism is pathogenic only because of a single toxin or adhesion molecule. Opsonizing antibody responses are particularly important for resistance to extracellular bacterial pathogens. Complement can kill some bacteria, particularly those with an exposed outer lipid bilayer, such as Gram-negative bacteria.
• Ultimately most bacteria are killed by phagocytes following a multistage process of chemotaxis, attachment, uptake, and killing. Macrophage killing can be enhanced on activation. Optimal activation of macrophages is dependent on TH1 CD4 T cells, whereas neutrophil responses are promoted by TH17 CD4 T cells. Persistent macrophage recruitment and activation can result in granuloma formation, which is a hallmark of cell-mediated immunity to intracellular bacteria.
• Successful pathogens have evolved mechanisms to avoid phagocyte-mediated killing and have evolved a startling diversity of mechanisms for avoiding other aspects of innate and adaptive immunity.
• Infected cells can be killed by CTLs. Other T cell populations and some tissue cells can contribute to antibacterial immunity.
• The response to bacteria can result in immunological tissue damage. Excessive release of cytokines caused by microorganisms can result in immunopathological syndromes, such as endotoxin shock and the Schwartzman reaction.
• Fungi can cause life-threatening infections. Immunity to fungi is predominantly cell mediated and shares many similarities with immunity to bacteria.
Innate recognition of bacterial components
The immune defense mechanisms elicited against pathogenic bacteria are determined by their:
• mechanism(s) of pathogenicity; and
• whether they are predominantly extracellular or also have the ability to survive inside mammalian cells.
There are four main types of bacterial cell wall
The four main types of bacterial cell wall (Fig. 14.1) belong to the following groups.
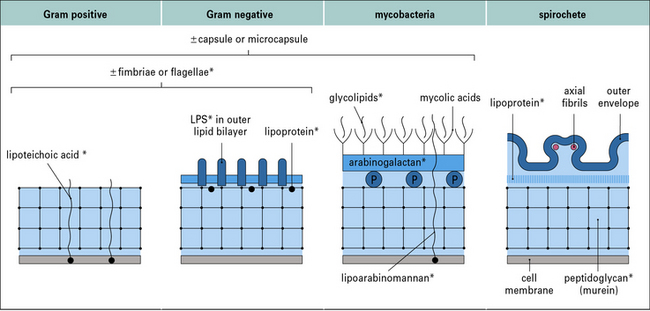
Pathogenicity varies between two extreme patterns
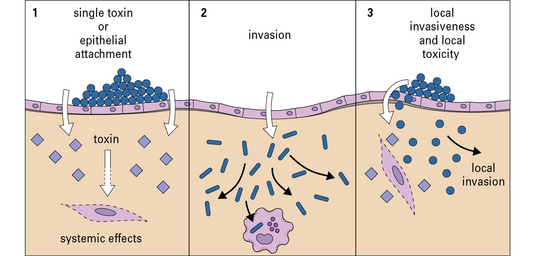
The first lines of defense do not depend on antigen recognition
The second line of defense is mediated by recognition of bacterial components
If organisms do enter the tissues, they can be combated initially by further elements of the innate immune system. Numerous bacterial components are recognized in ways that do not rely on the antigen-specific receptors of either B cells or T cells. These types of recognition are phylogenetically ancient ‘broad-spectrum’ mechanisms that evolved before antigen-specific T cells and immunoglobulins, allowing protective responses to be triggered by common microbial components bearing so-called ‘pathogen-associated molecular patterns’ (PAMPs), recognized by ‘pattern recognition molecules’ of the innate immune system (see Chapter 6).
Q. List some examples of soluble molecules, cell surface receptors, and intracellular molecules that recognize PAMPs
A. Collectins and ficolins (see Fig. 6.w3) , the Toll-like receptors (see Fig. 6.21), the mannose receptor (see Fig. 7.11), and the NOD-like receptor proteins (see Fig. 7.13) all recognize PAMPs.
, the Toll-like receptors (see Fig. 6.21), the mannose receptor (see Fig. 7.11), and the NOD-like receptor proteins (see Fig. 7.13) all recognize PAMPs.
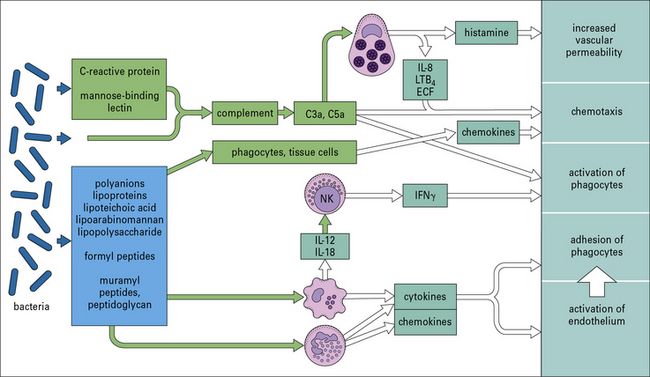
Many organisms, such as non-pathogenic cocci, are probably removed from the tissues as a consequence of these pathways, without the need for a specific adaptive immune reaction. Figure 14.3 shows some of the microbial components involved and the host responses that are triggered.

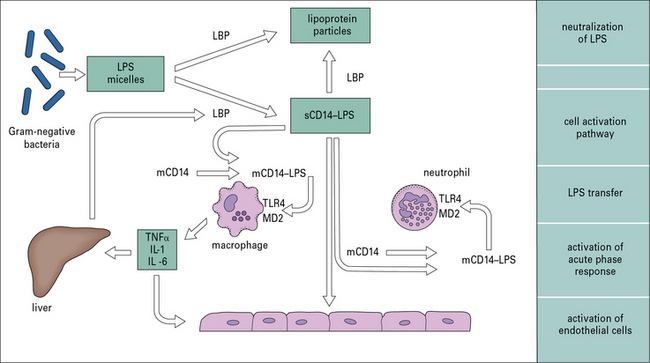
LPS is the dominant activator of innate immunity in Gram-negative bacterial infection
Recognition of LPS is a complex process involving molecules that bind LPS and pass it on to cell membrane-associated receptors on leukocytes, and endothelial and other cells, which initiate this proinflammatory cascade (Fig. 14.4).
 ).
).Lymphocyte-independent effector systems
Complement is activated via the alternative pathway
Perhaps more importantly, complement activation releases C5a, which attracts and activates neutrophils and causes degranulation of mast cells (see Chapter 3). The consequent release of histamine and leukotriene (LTB4) contributes to further increases in vascular permeability (see Fig. 14.3).
Release of proinflammatory cytokines increases the adhesive properties of the vascular endothelium
The rapid release of cytokines such as TNF and IL-1 (see Fig. 14.4) from macrophages increases the adhesive properties of the vascular endothelium and facilitates the passage of more phagocytes into inflamed tissue. Combined with the release of chemokines such as CCL2, CCL3, and CXCL8 (see Chapter 6), this directs the recruitment of different leukocyte populations.
Pathogen recognition generates signals that regulate the lymphocyte-mediated response
• display of MHC molecule–peptide complexes;
• expression of co-stimulatory molecules (such as CD40, CD80, and CD86); and
Some of this DC activation occurs secondary to their production of cytokines such as type I IFN.
Immunologists have made use of these effects for many decades (even without knowing their true molecular basis) in the use of adjuvants in vaccination. ‘Adjuvant’ is derived from the Latin adjuvare, to help. When given experimentally, soluble antigens evoke stronger T and B cell-mediated responses if they are mixed with bacterial components that act as adjuvants. Components with this property are indicated in Figure 14.1. This effect probably reflects that the antigen-specific immune response evolved in a tissue environment that already contained these pharmacologically active bacterial components.
Antibody dependent anti-bacterial defenses
• it neutralizes diphtheria toxin by blocking the attachment of the binding portion of the molecule to its target cells;
• similarly it may block locally acting toxins or extracellular matrix-degrading enzymes, which act as spreading factors.
Antibody can also interfere with motility by binding to flagellae.
An important function on external and mucosal surfaces, often performed by secretory IgA (sIgA, see Chapter 3), is to stop bacteria binding to epithelial cells – for instance, antibody to the M proteins of group A streptococci gives type-specific immunity to streptococcal sore throats.
It is likely that some antibodies to the bacterial surface can block functional requirements of the organism such as binding of iron-chelating compounds or intake of nutrients (Fig. 14.5).
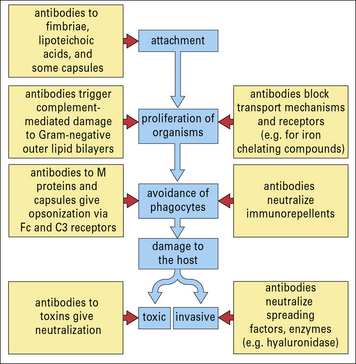
With the aid of antibodies, even organisms that resist the alternative (i.e. innate) complement pathway (see below) are damaged by complement or become coated with C3 products, which then enhance the binding and uptake by phagocytes (Figs 14.6 and 14.7).
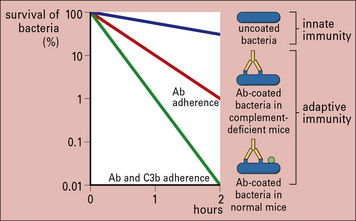
Fig. 14.6 Effect of antibody and complement on rate or clearance of virulent bacteria from the blood
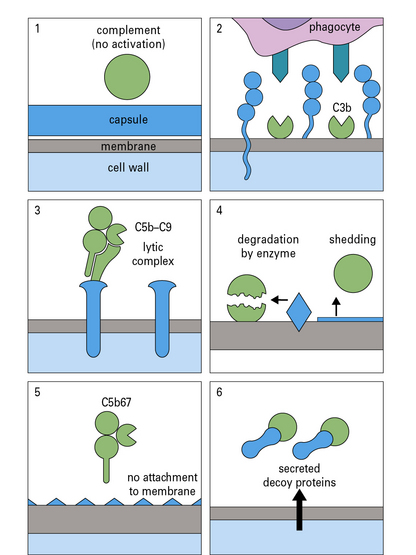
Pathogenic bacteria may avoid the effects of antibody
Neisseria gonorrhoeae is an example of a pathogenic bacterium that uses several immune evasion strategies (Fig. 14.8) and humans can be repeatedly infected with N. gonorrhoeae with no evidence of protective immunity.

Bactericial killing by phagocytes
Bacterial components attract phagocytes by chemotaxis
• bacterial components such as f-Met-Leu-Phe (which is chemotactic for leukocytes);
• complement products such as C5a; and
• locally released chemokines and cytokines derived from resident macrophages and epithelial cells (see Chapter 6).
• acute infection with encapsulated bacteria such as Streptococcus pyogenes give rise to tissue lesions rich in neutrophils (typical of so-called pyogenic or pus-forming infections);
• at the other extreme, chronic infections with M. tuberculosis result in granulomas rich in macrophages, macrophage-derived multinucleated giant cells, and T cells;
• other organisms, such as Listeria and Salmonella spp., result in lesions of more mixed composition.
The choice of receptors is critical
• whether killing mechanisms are triggered;
• whether the process favors the pathogen by subverting immunity.
• direct binding is mediated by pattern recognition molecules including Toll-like receptors and scavenger receptors (such as SRA, MARCO), mannose receptor, and dectin-1;
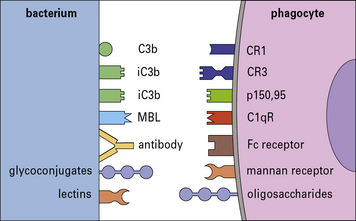
• opsonization is mediated through complement receptors such as CR1, CR3, and CR4, which recognize complement fragments deposited on the organism via the alternative or classic complement pathways.
Additionally, Fc receptors on the phagocyte (FcγRI, FcγRII, and FcγRIII, see Chapter 3) bind antibody that has coated bacteria (see Fig. 14.7), whereas various integrins can bind fibronectin and vitronectin opsonized particles.
Phagocytic cells have many killing methods
The killing pathways of phagocytic cells can be oxygen dependent, with the generation of reactive oxygen intermediates, or oxygen independent (see Chapter 7). In neutrophils, the oxidative burst may also act indirectly, by promoting the flux of K+ ions into the phagosome and activating microbicidal proteases.
Some cationic proteins have antibiotic-like properties
The defensins (Fig. 14.10) are cysteine- and arginine-rich cationic peptides of 30–33 amino acids found in phagocytes such as neutrophils, where they comprise 30–50% of the granule proteins.
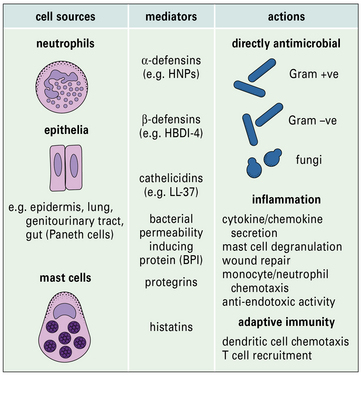
Defensins also have important immunostimulatory properties including:
Macrophage killing can be enhanced on activation
Macrophage activation occurs most effectively by the combination of exposure to:
• microbial products (through the receptors described above); and
• cytokines (particularly IFNγ) derived from cells of the innate and adaptive immune system.
Optimal activation of macrophages is dependent on TH1 CD4 T cells
The most important source of IFNγ during the adaptive immune response to intracellular bacteria is from TH1 CD4+ T cells (Fig. 14.11).
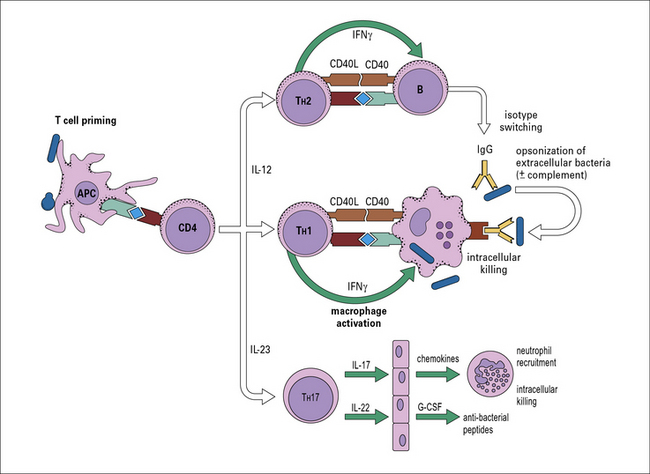
Fig. 14.11 Overview of CD4+ T cell-mediated immunity to bacteria and fungi
Naive CD4+ T cells are stimulated by class II MHC positive antigen-bearing dendritic cells (DCs) via the TCR, in conjunction with co-stimulatory molecules such as CD80/86 and CD28, which induce T cell activation and proliferation. Differentiation into TH1, TH17, or TH2 effector cells is strongly influenced by the cytokine environment during this interaction – microbial pattern recognition events that favor production of IL-12 promote TH1 development, low IL-12 favors TH2 responses, whereas combinations of IL-6, IL-1 TGFβ, IL-21, and IL-23 are required for development and maintenance of TH17 responses. TH17 cells mediate their biological activities via secretion of IL-17 which induces G-CSF and CXC family chemokines to enhance neutrophil differentiation and recruitment, and of IL-22 which induces antimicrobial peptides such as defensins and mucins. Although not shown here, conditions with high levels of IL-10 or TGFβ can induce regulatory T cells, rather than effector (i.e. TH1, TH2, or TH17) subsets. Optimal T cell help for either B cells or macrophage responses involves T cell-derived cytokines and direct cell contact. TH1 cells also promote opsonizing antibody production of high affinity (Fig. 9.8), which complements their activation of phagocytes by IFNγ, but the main activators of antibody synthesis are THz cells, themselves induced by IL-4, producing IL-4, 5, 6, 10 and 13 (Fig. 9.7).
• IFNγ upregulates induced NO• synthetase expression; whereas
• IL-4 and -13 promote the expression of arginase, which inhibits NO• production, reducing the macrophage killing potential and diverting it to a profibrotic phenotype. In some cases these cells are called alternatively activated macrophages.
Other cytokines such as GM-CSF and TNF can also contribute to macrophage activation.
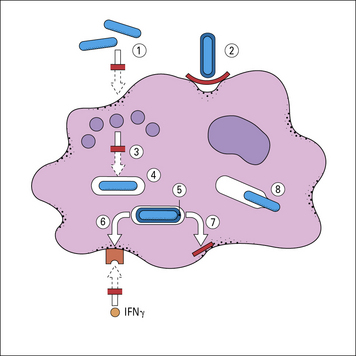
Successful pathogens have evolved mechanisms to avoid phagocyte-mediated killing
Because most organisms are ultimately killed by phagocytes, it is not surprising that successful pathogens have evolved an array of mechanisms to counteract this risk (Fig. 14.12).
Direct anti-bacterial actions of T cells
Infected cells can be killed by CTLs
Although antigen processing and presentation via the class I MHC pathway (see Chapter 8) is most efficient for microbial antigens derived from the cytosol, nevertheless, CTLs are also clearly induced by bacteria that never escape the phagosome such as M. tuberculosis, salmonellae, and chlamydiae. This occurs either by cross-presentation of antigens within the same cell or by cross-priming where antigens are released from infected cells undergoing apoptosis and then transferred to nearby DCs for efficient presentation via the MHC I pathway. In some cases, lysis of infected host cells by CTLs can result in killing of the organism inside. This can be due to the action of granulysin – an antibacterial peptide stored in the cytotoxic granules and released during the cytotoxic process.
CTLs can also secrete IFNγ when they recognize infected targets, providing an additional pathway of macrophage activation and protective immunity (Fig. 14.13).
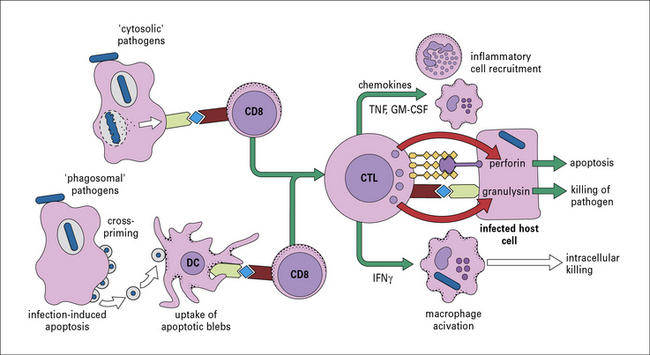
Other T cell populations can contribute to antibacterial immunity
T cells bearing γδ (rather than αβ) receptors (see Chapter 5) proliferate in response to bacterial infection.
NK T cells are a diverse group of T cells, some of which have an invariant T cell antigen receptor. They recognize not proteins, but hydrophobic antigens, particularly microbial glycolipids such as the lipoarabinomannan from M. tuberculosis, presented via CD1 molecules (see Fig. 5.w3 ).
).
Examples to illustrate the relationship between the nature of an organism, the disease, and immunopathology caused, and the mechanism of immune response that leads to protection, are given in Figure 14.14.
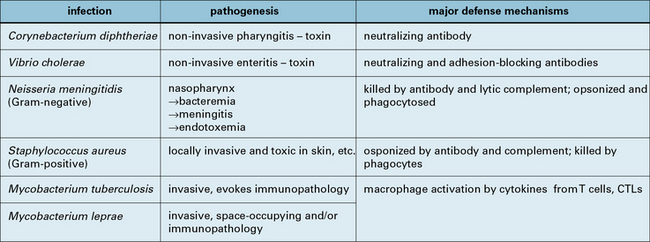
Immunopathological reactions induced by bacteria
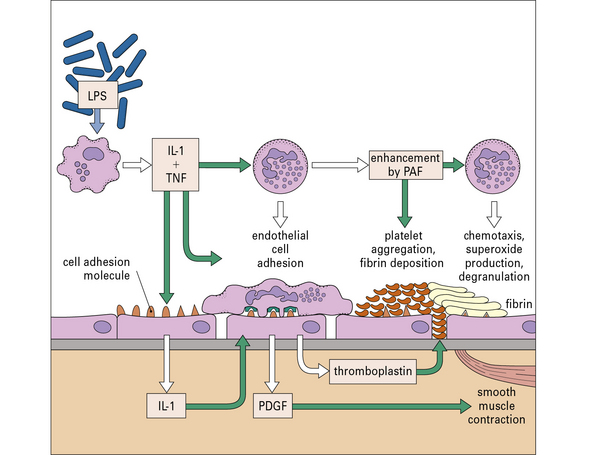
Excessive cytokine release can lead to endotoxin shock
One of the most severe examples of this is endotoxin (septicemic) shock, when there is massive production of cytokines, usually caused by bacterial products released during septicemic episodes. Endotoxin (LPS) from Gram-negative bacteria is usually responsible, though Gram-positive septicemia can cause a similar syndrome. There can be life-threatening fever, circulatory collapse, diffuse intravascular coagulation, and hemorrhagic necrosis, leading eventually to multiple organ failure (Fig. 14.15).

The Schwartzman reaction is a form of cytokine-dependent tissue damage
Schwartzman observed that if Gram-negative organisms were injected into the skin of rabbits, followed by a second dose given intravenously 24 hours later, hemorrhagic necrosis occurred at the prepared skin site. This is known as the Schwartzman reaction (Fig. 14.w1).
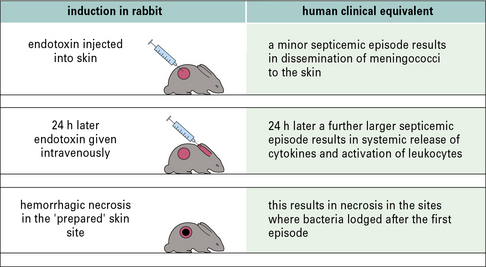
Endotoxin (LPS) is the active component of the intravenous ‘triggering’ injection.
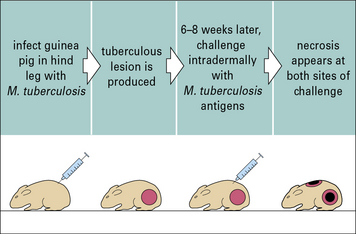
The Koch phenomenon is necrosis in T cell-mediated mycobacterial lesions and skin test sites
The Koch phenomenon is a necrotic response to antigens of M. tuberculosis, originally demonstrated by Robert Koch in tuberculous guinea pigs (Fig. 14.w2). It may be related to the necrosis that also occurs in the lesions in tuberculosis. It is at least partly due to the release of cytokines into a T cell-mediated inflammatory site (delayed hypersensitivity site). Such sites can be extremely sensitive to the tissue-damaging effects of cytokines, as seen in the Schwartzman reaction, particularly when there is mixed TH1 and TH2 activity.
Some individuals suffer from excessive immune responses
Heat-shock proteins are prominent targets of immune responses
Immune responses are generated against both mammalian and pathogen-derived HSPs during infection.
HSPs can enhance the presentation of antigens via the class I MHC/CD8 pathway (see Chapter 5) and are themselves immunogenic for both CD4 and CD8 T cells.
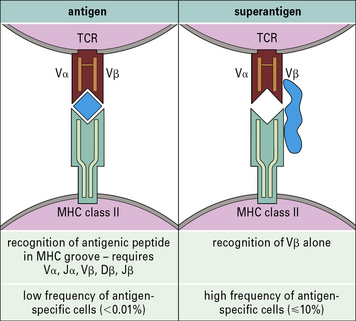
The toxicity of superantigens results from massive cytokine release
Certain bacterial components called superantigens bind directly to the variable regions of β chains (Vβ) of antigen receptors on subsets of T cells, and cross-link them to the MHC molecules of APCs, usually outside the normal antigen-binding groove (Fig. 14.16). Between them staphylococci and streptococci have some 21 different superantigens and these molecules can also be found in other bacteria such as mycoplasmas. The full biological significance of this bacterial adaptation is not yet clear – it could be to the organism’s advantage to exhaust or deplete T cells that would otherwise be protective.
Fungal infections
• single cells (yeasts) small enough to be ingested by host phagocytes; or
• long slender, branching hyphae, which may require extracellular killing processes.
There are four categories of fungal infection
• patients with untreated AIDS;
• patients with cancer and undergoing chemotherapy;
• patients with transplants on immunosuppressive agents; and
Human fungal infections fall into the following four major categories:
• superficial mycoses caused by fungi known as dermatophytes, usually restricted to the non-living keratinized components of skin, hair, and nails, and including infection by Trichophyton and Microsporum spp. (which cause ringworm and athlete’s foot) and Malassezia spp. (which causes pityriasis);
• subcutaneous mycoses in which saprophytic fungi cause chronic nodules or ulcers in subcutaneous tissues following trauma (e.g. chromomycosis, sporotrichosis, and mycetoma);
• systemic mycoses caused by soil saprophytes, which are inhaled from the environment and produce subclinical or acute lung infections that can disseminate to almost any tissue in the immunocompromised host – Histoplasma, Blastomyces, Coccidioides, and Paracoccidioides spp. can all cause primary disease in otherwise immunocompetent individuals, whereas Aspergillus spp., Pneumocystis jiroveci, and C. neoformans act more as opportunists;
• candidiasis caused by Candida albicans, a ubiquitous commensal and the most common opportunistic fungal pathogen – disturbance of normal physiology by immunosuppressive drugs, of normal flora by antibiotics, or T cell function (as in severe combined immune deficiency, thymic aplasia, and AIDS), results in superficial infections of the skin and mucous membranes, and systemic disease can occur in intravenous drug users and patients with lymphoma or leukemia.
Innate immune responses to fungi include defensins and phagocytes
Phagocytes, particularly neutrophils (Fig. 14.17) and macrophages, are essential for killing fungi, either by:
Fig. 14.17 Evidence for neutrophil-mediated immunity to mucormycosis
(Courtesy of Professor RJ Hay.)
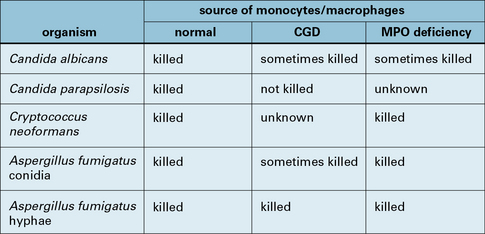
The oxidative burst plays a crucial role in some antifungal responses, as seen in the susceptibility to severe aspergillosis by patients with CGD who have defects in the NADPH oxidase system. However, phagocytes from such patients with defective oxygen reduction pathways nevertheless kill other yeast and hyphae with near normal efficiency, so demonstrating the role of other killing mechanisms (Fig. 14.18). For instance, NO• and its derivatives are important for resistance to C. neoformans.
T cell-mediated immunity is critical for resistance to fungi
Most fungi are highly immunogenic and induce strong antibody and T cell-mediated immune responses, which can be detected by serology and delayed-type (type IV) hypersensitivity skin reactions (see Chapter 26).
Resistance to most pathogenic fungi (including dermatophytes and most systemic mycoses including C. neoformans, Histoplasma capsulatum, etc., but not Aspergillus spp.) is clearly dependent upon T cell-mediated immunity, particularly CD4+ TH1 cells secreting IFNγ and to a lesser extent CD8 T cells (Fig. 14.19). As in the case of bacteria, dendritic cells are necessary for this response and produce IL-12 after engulfing fungi.
Fungi possess many evasion strategies to promote their survival
Evasion strategies used by fungi to promote their survival include the following:
• Cryptococcus neoformans produces a polysaccharide capsule, which inhibits phagocytosis (similar in principle to that of encapsulated bacteria), though this can be overcome by the opsonic effects of complement and antibodies;
• similarly, Candida conceal the β glucans of their cell wall which would otherwise be efficiently recognized by host dectin-1, underneath an external coat of mannan, a molecule which is considerably less immune-reactive. β glucans are also differentially expressed in the yeast versus filamentous forms of some fungi, contributing to the differences in immune response to these two distinct phases of infection;
• Histoplasma capsulatum is an obligate intracellular pathogen that evades macrophage killing by entering the cell via CR3 and then altering the normal pathways of phagosome maturation, in parallel to the strategies of intracellular bacteria such as M. tuberculosis;
• dermatophytes suppress host T cell responses to delay cell-mediated destruction.
Anas A., van der Poll T., de Vos A.F. Role of CD14 in lung inflammation and infection. Crit Care. 2010;14:209.
Andrea M. Cooper cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422.
Borghetti P., Saleri R., Mocchegiani E., et al. Infection, immunity and the neuroendocrine response. Vet Immunol Immunopathol. 2009;130:141–162.
Cerf-Bensussan N., Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10:735–744.
Cunha C., Romani L., Carvalho A. Cracking the Toll-like receptor code in fungal infections. Expert Rev Anti Infect Ther. 2010;8:112.
Curtis M.M., Way S.S. Interleukin 17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009;126:177–185.
Davis M., Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 2009;136:37–49.
Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010;22:252–262.
Dietrrich J., Doherty T.M. Interaction of mycobacterium tuberculosis with the host: consequence for vaccine development. APMIS. 2010;117:440–457.
Flannagan R.S., Cosio G., Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366.
van de Veerdonk F.L., Mihai G. Netea T-cell subsets and antifungal host defenses. Curr Fungal Infect Rep. 2010;4:238–243.
Harty J.T., Tvinnereim A.R., White D.W. CD8 + T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308.
Hazlett L., Wu M. Defensins in innate immunity. Cell Tissue Res. 2011;343:175–188.
Hooper L.V., Macpherson A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169.
Hohl T.M., Pamer E.G. Cracking the fungal armor. Nat Med. 2006;12:730–732.
Hohl T.M., Rivera A., Pamer E.G. Immunity to fungi. Curr Opin Immunol. 2006;18:465–472.
Holt P.G., Strickland D.H. Soothing signals: transplacental transmission of resistance to asthma and allergy. J Exp Med. 2010;206:2861–2864.
Jo E.K. Innate immunity to mycobacteria: vitamin D and autophagy. Cell Microbiol. 2010;12:1026–1035.
Kaufmann S.H., Hussey G., Lambert P.H. New vaccines for tuberculosis. Lancet. 2010;375:2110–2119.
Kaufmann S.H., Schaible U.E. Antigen presentation and recognition in bacterial infections. Curr Opin Immunol. 2005;17:79–87.
Kronenberg M., Kinjo Y. Innate-like recognition of microbes by invariant natural killer T cells. Curr Opin Immunol. 2009;21:391–396.
Kumar V., Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10:1325–1334.
Lambris J.D., Ricklin D., Geisbrecht B.V. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142.
Mackenzie C.R., Heselar K., Muller A., Daubener W. Role of indole 2,3-dioxygenase in antimicrobial defence and immune regulation: tryptophan depletion versus production of toxic kynurenes. Curr Drug Metab. 2007;8:237–244.
MacLennan C., Fieschi C., Lammas D.A., et al. Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans. J Infect Dis. 2004;190:1755–1757.
Monack D.M., Mueller A., Falkow S. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol. 2004;2:747–765.
Papayannopoulos V., Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521.
Park S.J., Mehrad B. Innate immunity to Aspergillus species. Clin Microbiol Rev. 2009;22:535–551.
Philpott D.J., Girardin S.E. NOD like receptors: sentinels at host membranes. Curr Opin Immunol. 2010;22:428–434.
Puel A., Picard C., Cypowyj S., et al. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr Opin Immunol. 2010;22:467–474.
Sansonetti P.J. To be or not to be a pathogen: that is the mucosally relevant question. Mucosal Immunol. 2011;4:8–14.
Schaible U.E., Kaufmann S.H. Iron and microbial infection. Nat Rev Microbiol. 2004;2:946–953.
Stewart G.R., Young D.B. Heat-shock proteins and the host–pathogen interaction during bacterial infection. Curr Opin Immunol. 2004;16:506–510.
Thornton C.A., Macfarlane T.V., Holt P.G. The hygiene hypothesis revisited: role of maternal–fetal interactions. Curr Allergy Asthma Rep. 2010;10:444–452.
Umetsu D.T. Revising the immunological theories of asthma and allergy. Lancet. 2005;365:98–100.
van de Vosse E., Hoeve M.A., Ottenhoff T.H. Human genetics of intracellular infectious diseases: molecular and cellular immunity against mycobacteria and salmonellae. Lancet Infect Dis. 2004;4:739–749.
Voyich J.M., Musser J.M., DeLeo F.R. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 2004;6:1117–1123.
Wills-Karp M., Santeliz J., Karp C.L. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat Rev Immunol. 2001;1:69–75.
Zelante T., DeLuca A., D’Angelo C., et al. IL-17/TH17 in antifungal immunity: what’s new. Eur J Immunol. 2009;39:645.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 14 Immunity to Bacteria and Fungi
• Mechanisms of protection from bacteria can be deduced from their structure and pathogenicity. There are four main types of bacterial cell wall and pathogenicity varies between two extreme patterns. Non-specific, phylogenetically ancient recognition pathways for conserved bacterial structures trigger protective innate immune responses and guide the development of adaptive immunity.
• Lymphocyte-independent bacterial recognition pathways have several consequences. Complement is activated via the alternative pathway. Release of proinflammatory cytokines and chemokines increases the adhesive properties of the vascular endothelium and promotes neutrophil and macrophage recruitment. Pathogen recognition generates signals then regulate the lymphocyte-mediated response.
• Antibody provides an antigen-specific protective mechanism. Neutralizing antibody may be all that is needed for protection if the organism is pathogenic only because of a single toxin or adhesion molecule. Opsonizing antibody responses are particularly important for resistance to extracellular bacterial pathogens. Complement can kill some bacteria, particularly those with an exposed outer lipid bilayer, such as Gram-negative bacteria.
• Ultimately most bacteria are killed by phagocytes following a multistage process of chemotaxis, attachment, uptake, and killing. Macrophage killing can be enhanced on activation. Optimal activation of macrophages is dependent on TH1 CD4 T cells, whereas neutrophil responses are promoted by TH17 CD4 T cells. Persistent macrophage recruitment and activation can result in granuloma formation, which is a hallmark of cell-mediated immunity to intracellular bacteria.
• Successful pathogens have evolved mechanisms to avoid phagocyte-mediated killing and have evolved a startling diversity of mechanisms for avoiding other aspects of innate and adaptive immunity.
• Infected cells can be killed by CTLs. Other T cell populations and some tissue cells can contribute to antibacterial immunity.
• The response to bacteria can result in immunological tissue damage. Excessive release of cytokines caused by microorganisms can result in immunopathological syndromes, such as endotoxin shock and the Schwartzman reaction.
• Fungi can cause life-threatening infections. Immunity to fungi is predominantly cell mediated and shares many similarities with immunity to bacteria.
Innate recognition of bacterial components
The immune defense mechanisms elicited against pathogenic bacteria are determined by their:
• mechanism(s) of pathogenicity; and
• whether they are predominantly extracellular or also have the ability to survive inside mammalian cells.
There are four main types of bacterial cell wall
The four main types of bacterial cell wall (Fig. 14.1) belong to the following groups.
Pathogenicity varies between two extreme patterns
The first lines of defense do not depend on antigen recognition
The second line of defense is mediated by recognition of bacterial components
If organisms do enter the tissues, they can be combated initially by further elements of the innate immune system. Numerous bacterial components are recognized in ways that do not rely on the antigen-specific receptors of either B cells or T cells. These types of recognition are phylogenetically ancient ‘broad-spectrum’ mechanisms that evolved before antigen-specific T cells and immunoglobulins, allowing protective responses to be triggered by common microbial components bearing so-called ‘pathogen-associated molecular patterns’ (PAMPs), recognized by ‘pattern recognition molecules’ of the innate immune system (see Chapter 6).
Q. List some examples of soluble molecules, cell surface receptors, and intracellular molecules that recognize PAMPs
A. Collectins and ficolins (see Fig. 6.w3), the Toll-like receptors (see Fig. 6.21), the mannose receptor (see Fig. 7.11), and the NOD-like receptor proteins (see Fig. 7.13) all recognize PAMPs.
Many organisms, such as non-pathogenic cocci, are probably removed from the tissues as a consequence of these pathways, without the need for a specific adaptive immune reaction. Figure 14.3 shows some of the microbial components involved and the host responses that are triggered.
LPS is the dominant activator of innate immunity in Gram-negative bacterial infection
Recognition of LPS is a complex process involving molecules that bind LPS and pass it on to cell membrane-associated receptors on leukocytes, and endothelial and other cells, which initiate this proinflammatory cascade (Fig. 14.4).
Lymphocyte-independent effector systems
Complement is activated via the alternative pathway
Perhaps more importantly, complement activation releases C5a, which attracts and activates neutrophils and causes degranulation of mast cells (see Chapter 3). The consequent release of histamine and leukotriene (LTB4) contributes to further increases in vascular permeability (see Fig. 14.3).
Release of proinflammatory cytokines increases the adhesive properties of the vascular endothelium
The rapid release of cytokines such as TNF and IL-1 (see Fig. 14.4) from macrophages increases the adhesive properties of the vascular endothelium and facilitates the passage of more phagocytes into inflamed tissue. Combined with the release of chemokines such as CCL2, CCL3, and CXCL8 (see Chapter 6), this directs the recruitment of different leukocyte populations.
Pathogen recognition generates signals that regulate the lymphocyte-mediated response
• display of MHC molecule–peptide complexes;
• expression of co-stimulatory molecules (such as CD40, CD80, and CD86); and
Some of this DC activation occurs secondary to their production of cytokines such as type I IFN.
Immunologists have made use of these effects for many decades (even without knowing their true molecular basis) in the use of adjuvants in vaccination. ‘Adjuvant’ is derived from the Latin adjuvare, to help. When given experimentally, soluble antigens evoke stronger T and B cell-mediated responses if they are mixed with bacterial components that act as adjuvants. Components with this property are indicated in Figure 14.1. This effect probably reflects that the antigen-specific immune response evolved in a tissue environment that already contained these pharmacologically active bacterial components.
Antibody dependent anti-bacterial defenses
• it neutralizes diphtheria toxin by blocking the attachment of the binding portion of the molecule to its target cells;
• similarly it may block locally acting toxins or extracellular matrix-degrading enzymes, which act as spreading factors.
Antibody can also interfere with motility by binding to flagellae.
An important function on external and mucosal surfaces, often performed by secretory IgA (sIgA, see Chapter 3), is to stop bacteria binding to epithelial cells – for instance, antibody to the M proteins of group A streptococci gives type-specific immunity to streptococcal sore throats.
It is likely that some antibodies to the bacterial surface can block functional requirements of the organism such as binding of iron-chelating compounds or intake of nutrients (Fig. 14.5).
With the aid of antibodies, even organisms that resist the alternative (i.e. innate) complement pathway (see below) are damaged by complement or become coated with C3 products, which then enhance the binding and uptake by phagocytes (Figs 14.6 and 14.7).
Fig. 14.6 Effect of antibody and complement on rate or clearance of virulent bacteria from the blood
Pathogenic bacteria may avoid the effects of antibody
Neisseria gonorrhoeae is an example of a pathogenic bacterium that uses several immune evasion strategies (Fig. 14.8) and humans can be repeatedly infected with N. gonorrhoeae with no evidence of protective immunity.
