[level-membership-for-neonatal-and-perinatal-medicine-category]
Immune system and host defense mechanisms
“Remarkable adaptations . . . allow a genetically and antigenically disparate conceptus to develop in direct contact with a fully competent maternal immune system.”100 Why doesn’t the mother reject the fetus and placenta? Knowledge of maternal, fetal, and neonatal immune physiology and understanding of the immune relationship between a mother and her fetus are still evolving. Current theories regarding this question—along with alterations in host defense mechanisms in the mother and neonate and implications for clinical practice—are examined in this chapter.
The immune system is made up of organs and specialized cells whose primary purpose is to defend the body from foreign substances (antigens) that may cause tissue injury or disease. These defense mechanisms consist of nonspecific and specific factors. Nonspecific factors include physical and biochemical barriers including skin and mucosal barriers, bone marrow, lymphoid tissue, digestive enzymes, pH, temperature, proteins, and enzymes such as lysozyme, transferrin, and interferon. Specific factors include cellular and humoral components that respond to foreign substances. In addition, individual genetic susceptibilities affect both nonspecific factors and specific factors. Nonspecific and specific factors make up two arms of the immune response—the innate (also known as natural or native immunity) and the adaptive (also known as specific or acquired immunity) (Table 13-1). Mechanisms of innate and adaptive immunity work cooperatively through complex interactions to prevent, control, and eradicate foreign antigens in the body without doing harm to the host. Host immune mechanisms and terminology are summarized in Table 13-2 and Figures 13-1 and 13-2. Innate and adaptive immunity are summarized in Boxes 13-1 and 13-2 on page 446. Roles of cytokines (interleukins and interferons) in innate and adaptive immunity are summarized in Table 13-3.
Table 13-1
Components of Innate and Adaptive Immunity
| CELLULAR COMPONENTS | HUMORAL COMPONENTS | MEDIATORS | |
| Innate | |||
| Adaptive | Antibodies | Cytokines |
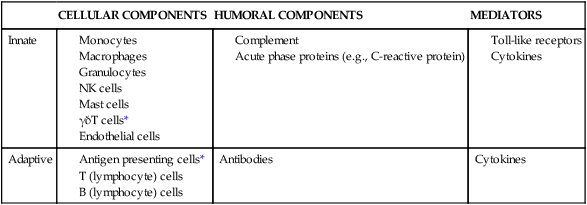
*These gamma delta T cells are found primarily in the gut mucosa. Antigen presenting cells are monocytes and/or macrophages that have engulfed a foreign object, processed it, and displayed it, within an MHC-I or MHC-II complex at the cell surface, to a T cell to initiate the adaptive immune response.
Table 13-2
Active immunity: The response produced by an immunocompetent individual following exposure to foreign antigens (bacteria, viruses, attenuated or inactivated/killed viruses).
Adaptive immunity: Also known as “acquired immunity.” The result of active immunity wherein specific antibody (humoral) and cell-mediated responses establish memory cells and antibodies that provide immunity during subsequent exposures to a specific antigen.
Allograft: Graft taken from an organism that is the same species as the recipient but not genetically identical. The fetus is a “semi-allograft.”
Antibody: Proteins (immunoglobulins) that react with specific antigens. The five classes are IgA, IgD, IgE, IgG, and IgM.
Antibody (humoral)-mediated immunity: Adaptive immune mechanism mediated by B cells, which produces antibodies and protects the body from extracellular antigens.
Antigen: Substances perceived by one’s host defense mechanisms as “foreign” (may include bacteria, viruses, pollutants, dust, certain foods, for example).
Antigen presenting cell (APC): A cell that displays MHC with antigen complexes on the surface of the cell. Any cell can be an APC in that all cells have class I MHC. The term APC is used to refer to cells that also express class II MHC and can activate T cells, dendritic cells, macrophages, and B cells.
Cell-mediated immunity: Adaptive immune mechanism provided by immune cells. TCD4+ (T-helper cells), TCD8+ (T-cytotoxic cells), and TCD4+/CD26+ (T-regulatory cells) provide protection against certain organisms, regulate B-cell function, defend against cancer, and mediate graft rejection.
Chemotaxis: Movement of neutrophils and other phagocytes in an organized fashion toward a site of antigenic invasion.
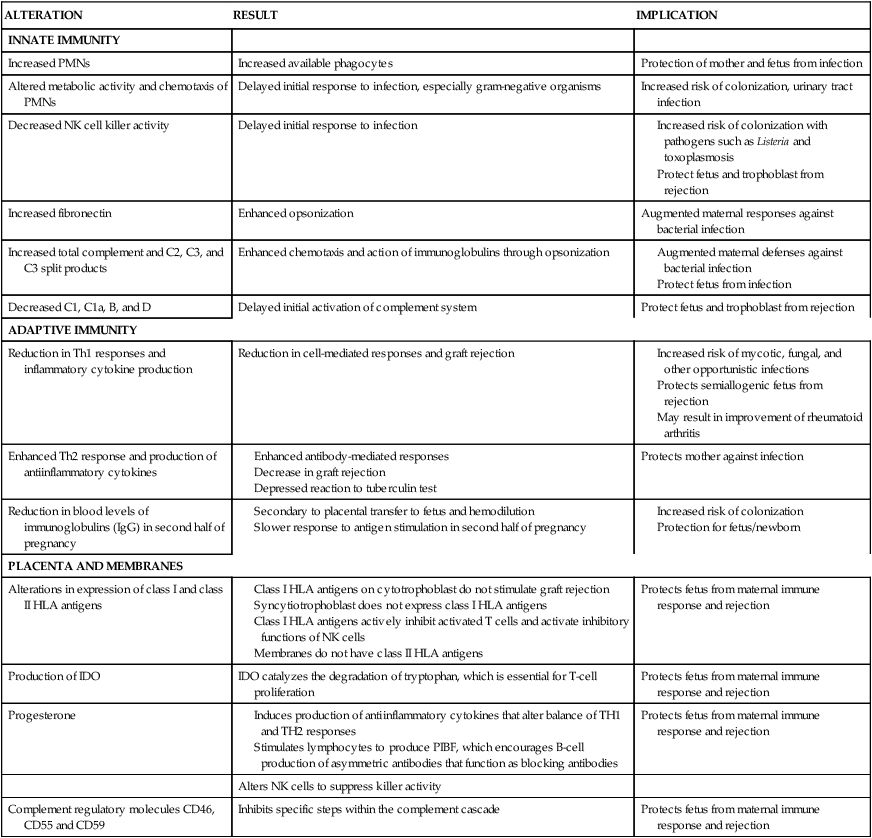
Complement: Thirty discrete plasma proteins that, as part of the innate immune response, function in a cascade of reactions to form a membrane attack complex that lyses cells. The effector response of the complement cascade also opsonizes antigen, increases vascular permeability, and stimulates chemotaxis to aid phagocytosis. The complement cascade can proceed via the classical pathway or alternate pathway.
Cytokines: Glycoproteins such as lymphokines, interleukins, interferon, and tissue necrosis factor that are produced by various components of the immune system, especially T lymphocytes and macrophages. Cytokines function as autocrine or paracrine agents to activate different arms of the immune response.
Cytotoxic/killer T cell (TCD81): A type of T lymphocyte that acts directly on specific antigens or target cells that exhibit a specific antigen and induces cell lysis.
Fibronectin: Nonspecific opsonin, inhibitor of bacterial adherence to epithelial cells, and clot-stabilizing protein found in plasma and endothelial tissue.
Human leukocyte antigen (HLA): The major forms of specific tissue antigens found on tissue surfaces that are unique to each person (includes HLA-A, HLA-B, HLA-C, HLA-D, and in the placenta HLA-G).
Histocompatibility: The ability of tissue to accept a transplant from another individual.
Immunoglobulin: Antibodies produced by B lymphocytes (IgG, IgM, IgA, IgD, and IgE).
Innate immunity: Initial physical and nonspecific responses. Includes skin, mucosa, digestive enzymes. Primary effectors are polymorphonuclear neutrophils (PMNs), macrophages, monocytes, mast cells, natural killer (NK) cells, and complement. Innate immune response results in elimination of foreign substance or antigen and stimulation of the adaptive immune response.
Interleukin: A polypeptide member of the cytokine family. Produced by T cells, APCs, NK cells, and macrophages. Interleukins regulate and facilitate immune responses.
Interferon: Glycoprotein in the cytokine family. Primary role is in fighting viral infections.
Lymphokine: Subclass of cytokines. Mediators released by activated T cells that facilitate or act as costimulatory for immune reaction.
Memory cell: Form of B lymphocyte sensitized to specific antigens that has the ability to produce specific antibodies with subsequent stimulation by the specific antigen.
Major histocompatibility complex (MHC): Specific antigens found on tissue surfaces divided into two groups: MHC I (HLA-A, HLA-B, and HLA-C, which are found on most of a person’s cells), and MHC II (found on surface of immune cells such as T and B lymphocytes).
Natural killer (NK) cells: A subpopulation of lymphocytes in the innate immune response that form a first line of defense against cells infected with a virus and cancerous cells. NK cells have receptors that recognize class I MHC antigens and can attack without prior sensitization. They secrete cytokines that provide costimulation for T and B cells.
Opsonization: Processing and marking or altering the cell surface of an antigen by actions of immunoglobulin or complement; substances acting in this manner are called opsonins. This process is critical in allowing phagocytosis of organisms with capsular polysaccharide coats such as group B streptococci.
Passive immunity: Transfer of antibodies from an actively immunized to a nonimmunized person.
Plasma cell: Form of B lymphocyte able to secrete immunoglobulins.
Regulatory T cell: TCD4+/CD25+ cells. The exact function of these cells in unclear. They may inhibit production of interleukins (IL-10) that are necessary products and facilitators of the adaptive immune response. Thus they may help to slow or terminate immune response once the antigens are destroyed.
T helper cell (TCD41): A type of T lymphocyte that proliferates when in contact with an APC that exhibits class II MHC/antigen complexes (B cells, dendritic cells). TCD4+ cells differentiate into Th1 or Th2 cells based on the cytokines produced. Th1 cells participate in cell-mediated immunity and Th2 cells participate in antibody-mediated immunity. These cells thus enhance the activity of B lymphocytes, other T cells, and macrophages via secretion of cytokines.
T suppressor cells: T lymphocytes that express CD8+ cells and function to suppress the cellular immune response.
Th1 cells: A subset of TCD4+ (T-helper cells) that facilitates cell-mediated immunity via secretion of interleukin-12 (IL-12), tumor-necrosis factor (TNF)-β, and interferon (IFN)-γ. The result is stimulation of macrophages and inflammation.
Toll-like receptors (TLRs): Molecules on the surface of phagocytes and other cells that recognize the patterns of microbial products and generate signals to activate an innate immune response.
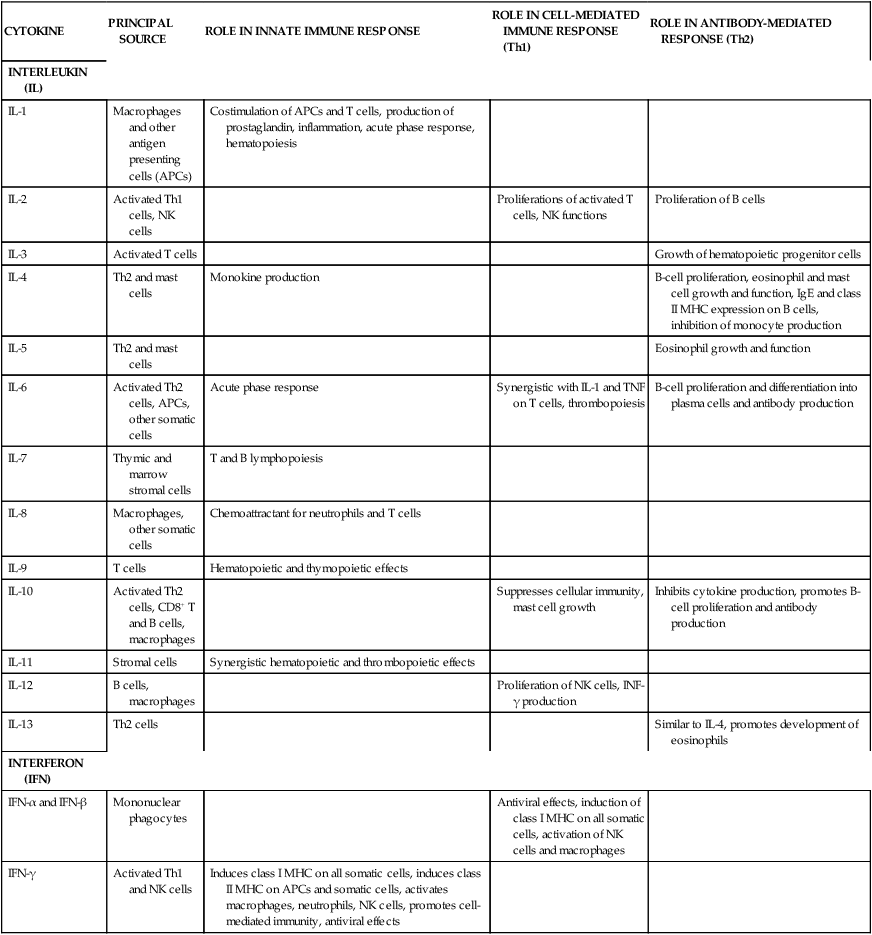
Table 13-3
Cytokines: Interleukins and Interferons
| CYTOKINE | PRINCIPAL SOURCE | ROLE IN INNATE IMMUNE RESPONSE | ROLE IN CELL-MEDIATED IMMUNE RESPONSE (Th1) | ROLE IN ANTIBODY-MEDIATED RESPONSE (Th2) |
| INTERLEUKIN (IL) | ||||
| IL-1 | Macrophages and other antigen presenting cells (APCs) | Costimulation of APCs and T cells, production of prostaglandin, inflammation, acute phase response, hematopoiesis | ||
| IL-2 | Activated Th1 cells, NK cells | Proliferations of activated T cells, NK functions | Proliferation of B cells | |
| IL-3 | Activated T cells | Growth of hematopoietic progenitor cells | ||
| IL-4 | Th2 and mast cells | Monokine production | B-cell proliferation, eosinophil and mast cell growth and function, IgE and class II MHC expression on B cells, inhibition of monocyte production | |
| IL-5 | Th2 and mast cells | Eosinophil growth and function | ||
| IL-6 | Activated Th2 cells, APCs, other somatic cells | Acute phase response | Synergistic with IL-1 and TNF on T cells, thrombopoiesis | B-cell proliferation and differentiation into plasma cells and antibody production |
| IL-7 | Thymic and marrow stromal cells | T and B lymphopoiesis | ||
| IL-8 | Macrophages, other somatic cells | Chemoattractant for neutrophils and T cells | ||
| IL-9 | T cells | Hematopoietic and thymopoietic effects | ||
| IL-10 | Activated Th2 cells, CD8+ T and B cells, macrophages | Suppresses cellular immunity, mast cell growth | Inhibits cytokine production, promotes B-cell proliferation and antibody production | |
| IL-11 | Stromal cells | Synergistic hematopoietic and thrombopoietic effects | ||
| IL-12 | B cells, macrophages | Proliferation of NK cells, INF-γ production | ||
| IL-13 | Th2 cells | Similar to IL-4, promotes development of eosinophils | ||
| INTERFERON (IFN) | ||||
| IFN-α and IFN-β | Mononuclear phagocytes | Antiviral effects, induction of class I MHC on all somatic cells, activation of NK cells and macrophages | ||
| IFN-γ | Activated Th1 and NK cells | Induces class I MHC on all somatic cells, induces class II MHC on APCs and somatic cells, activates macrophages, neutrophils, NK cells, promotes cell-mediated immunity, antiviral effects |
Modified from Silver, R.M., Peltier, M.R., & Branch, D.W. (2004). The immunology of pregnancy. In R.K. Creasy, R. Resnik, & J.D. Iams (Eds.), Maternal-fetal medicine: Principles and practice (5th ed.). Philadelphia: Saunders.
Maternal physiologic adaptations
Antepartum period
“Pregnancy is an immunological balancing act in which the mother’s immune system has to remain tolerant of potential major histocompatibility (MHC) antigens and yet maintain normal immune competence for defense against microorganisms.”167 The immune changes that occur systemically in the innate and adaptive systems and those that occur at the site of the fetal-maternal interface are described in this section.
Alterations in innate immunity
Mediators of the innate response are altered during pregnancy (Table 13-4). Chemotaxis (see the definition in Table 13-2) is decreased during pregnancy, which may delay initial maternal responses to infection.48,107 In addition, production of interleukin (IL)-4 and interferon-γ (IFN-γ) may also be decreased.107 Functionally, monocyte and granulocyte activity is enhanced, which results in faster and more efficient phagocytosis. This may help protect the mother so she does not mount a cell-mediated immune response to trophoblastic and fetal cells that appear in the maternal circulation. Natural killer (NK) cell activity is altered in a more complex manner. Systemic NK activity is down-regulated during pregnancy, secondary to the effects of progesterone, which appears to induce formation of a blocking factor that decreases lymphocyte proliferation and NK activity.39 The result is that systemic NK cytolytic activity is normal in the first trimester but lower during the second and third trimesters and immediately after delivery.
Table 13-4
Alterations in Host Defense Mechanisms during Pregnancy
| ALTERATION | RESULT | IMPLICATION |
| INNATE IMMUNITY | ||
| Increased PMNs | Increased available phagocytes | Protection of mother and fetus from infection |
| Altered metabolic activity and chemotaxis of PMNs | Delayed initial response to infection, especially gram-negative organisms | Increased risk of colonization, urinary tract infection |
| Decreased NK cell killer activity | Delayed initial response to infection |
Total white blood cell (WBC) volume increases slightly beginning in the second month and levels off during the second and third trimesters. The total WBC count in pregnancy varies among individual women, ranging from 5000 to 12,000 per mm3, with values as high as 15,000 per mm3 reported (see Chapter 8).129 The increase is primarily due to increased numbers of polymorphonuclear neutrophils (PMNs), monocytes, and granulocytes.129 A slight shift to the left may occur, with occasional myelocytes and metamyelocytes seen on the peripheral smear. Changes in other WBC forms are minimal (see Chapter 8).
Alterations in PMN function have been reported.48 PMN attachment, ingestion, and digestion of Candida albicans have been found to be increased, possibly secondary to the effects of human chorionic gonadotropin (hCG). Even so, pregnant women have higher rates of fungal infection, which may be secondary to the effects of estrogen on nutrient availability for fungal growth in the reproductive tract.129 Estrogens may alter local mucosal barrier function, allowing adherence of pathogenic organisms and increasing the risk of colonization. Conversely, improved PMN antibody expression during pregnancy may enhance phagocyte recognition and destruction of antigen-antibody complexes.
Toll-like receptors (TLR) are essential for the innate recognition of microorganisms and endogenous signals from tissue breakdown products, such as fetal fibronectin, hyaluronan, matrix proteins, and biglycans.31 Both TLRs and the recently identified Nod-like receptors (NLR) are thought to be involved in regulating innate responses of the trophoblast to infections and tissue damage signals.4 Changes in the cervix and fetal membranes with tissue remodeling near parturition may release these endogenous signals as part of the cascade leading to labor onset (Chapter 4).31
Alterations in the inflammatory response
Throughout pregnancy, the maternal circulation is seeded with small particles from the trophoblast. These cell clusters function as antigens of fetal origin and stimulate a systemic inflammatory response. The point of direct maternal contact with the placenta is at the junction with maternal endothelial cells in the uterine spiral arteries (see Chapter 3).132 This contact activates endothelial cells and initiates an inflammatory response. However, a glycoprotein known as pregnancy zone protein (PZP) is produced during pregnancy, which has an inhibitory effect on the inflammatory process.150 PZP levels increase 100- to 200-fold during pregnancy. PZP inhibits phagocytosis and suppresses inflammatory responses and IL-2 function near the decidua-trophoblast interface.150,157
Alterations in the complement system
The complement system has a role in both innate and adaptive responses. Although pregnancy stimulates an overall activation of the complement system, this activation does not result in rejection of the fetus.134 Alterations in the function of the complement system during pregnancy begin at 11 weeks’ gestation with an increase (due to greater hepatic synthesis) in both total serum complement and specific proteins of the complement system, including C2, C3, and C3 split products (Figure 13-3).92,134 These components enhance chemotaxis and actions of immunoglobulins through opsonization, thereby augmenting maternal defenses against bacterial infection. Other protein fragments of the complement system, such as C1, C1a, B, and D, are decreased.134 C1q (involved in activation of the classic complement pathway, immune cell modification, cell processes, and maintenance of immune tolerance) is synthesized and secreted by decidual epithelial cells during pregnancy and is thought to have a role in changes in decidual blood vessels and mediation of cell-to-cell interaction between decidual and trophoblast cells.115 Because complement fragments are involved in activation of the complement system, through either the classic or the alternative pathway, activity of the complement system early in the immune response may be delayed during pregnancy.
Alterations in adaptive immunity
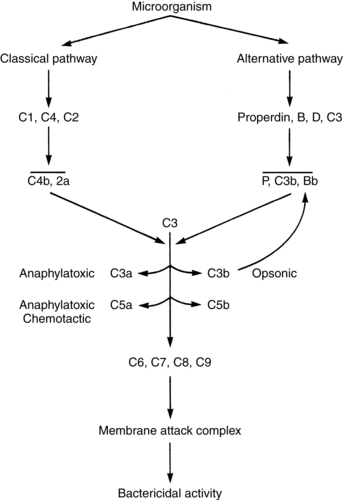
Pregnancy is characterized by a switch in the balance of Th1 and Th2 cell subsets that results in enhanced Th2 function. This change is probably mediated by progesterone, which stimulates production of Th2 cytokines (e.g., IL-3, IL-4, IL-6, IL-10) and reduces the number of Th1 cytokines (e.g., IL-2, IFN-γ, tumor necrotic factor-β [TNF-β]) (see Table 13-3).31,47 Because Th2 responses tend to enhance antibody-mediated responses and Th1 responses enhance cell-mediated immunity (Figure 13-4), this change alters the function of both antibody-mediated and cell-mediated responses. This has a potentially protective role in the maternal-fetal immune relationship.31 Decreases in the number of Th1 cytokines may decrease maternal resistance to the spread of bacterial and viral organisms.31,48 If there are alterations in this pattern so that Th1 predominates during pregnancy, inflammatory cytokine production increases, and is associated with spontaneous abortion, preeclampsia, preterm labor, and fetal growth restriction.31,138 Alterations in adaptive immunity during pregnancy are summarized in Table 13-4.
Alterations in cell-mediated immunity.
Cell-mediated immunity is somewhat suppressed during pregnancy. Lymphocyte and macrophage synthesis, activation, and function are altered slightly in pregnancy secondary to influences from estrogens, corticosteroids, progesterone, α-fetoprotein (AFP), hCG, human placental lactogen (hPL), prostaglandins, and serum proteins.47 Corticosteroids suppress activation of T-cell lymphokines, phagocytic activity, and lymphokine responsiveness of the macrophages.47 Prostaglandins (especially PGE1 and PGE2), hPL, and AFP also appear to have an immunosuppressive role during pregnancy. AFP may induce production of regulatory T lymphocytes (CD4+/CD25+).48
Most investigators report that the total number of lymphocytes does not change significantly during pregnancy.48,168 Some investigators have reported a decrease in the T-helper–to–T-suppressor (CD4+/CD8+) ratio. However, most reports indicate that the number of TCD4+ (T-helper cells) decrease progressively to term, while the number of T-suppressor cells (TCD8+) tend to remain relatively unchanged.48
Because TCD4+ cells normally augment the cytotoxic responses involved in graft rejection, a decreased number of these cells may help protect the fetus from rejection by the mother. T-suppressor cell function may increase in late pregnancy and suppress B-cell function.107 Decreased T-lymphocyte function and efficiency may increase the risk of viral and mycotic infections.
Alterations in antibody-mediated (humoral) immunity.
While a few studies have reported an increase in the number of B cells during pregnancy, most studies do not support this finding.48 Some investigators have found that levels of maternal IgG fall as gestation progresses, with decreases ranging from 30% to 40% after 28 weeks.48 However, others have found relatively little change. A decrease in IgG has been attributed to hemodilution of pregnancy, enhanced loss of IgG in the urine, and transfer of maternal IgG to the fetus in the last trimester. This relative decrease in IgG, along with alterations in the WBC population, may increase the risk of bacterial colonization with certain pathogens (e.g., streptococci).
Immunoglobulin A (IgA) decreases or remains stable during gestation, immunoglobulin M (IgM) remains stable or may decrease slightly, immunoglobulin E (IgE) undergoes minimal or no change in levels, and immunoglobulin D (IgD) increases until term. The slight decrease in serum IgA may reflect increased levels of IgA found in saliva and other mucosal fluids.48 The specific role of IgD in pregnancy is unknown.
Immune function at the fetal-maternal interface
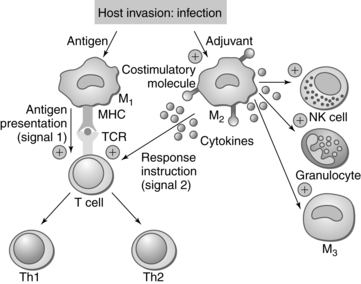
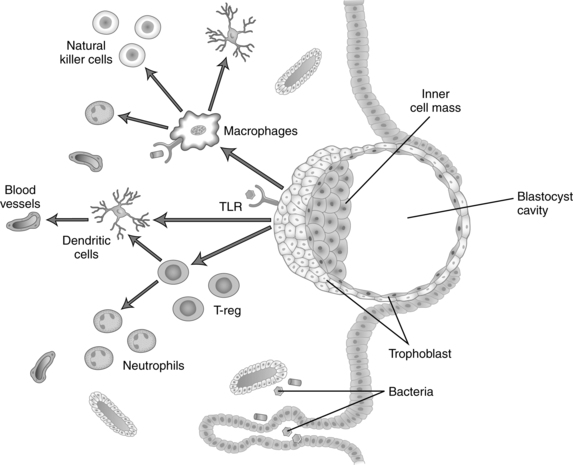
The fetus is a semiallograft, that is, foreign tissue from the same species but with a different antigenic makeup. Because the fetus has maternal, paternal, and embryonic antigens, major antigenic differences may exist between the fetus and mother, including blood group antigens and tissue antigens such as human leukocyte antigen (HLA). Protection of the fetus from rejection seems to be predominantly a localized uterine response, although there are also systemic responses mediated primarily by endocrine factors.158 Maternal tolerance of the fetal-placental unit has traditionally been believed to be secondary to elaboration of an immune “barrier” between the placenta and maternal tissue.158 However, it is now understood that maternal immune systems are “aware of fetal antigens, that they respond vigorously to the presence of the fetus and that under normal circumstances they are tolerant to these antigens.”124 Thus tolerance of the fetus by the maternal immune system is “an active mechanism whereby fetal tissues are prevented from being recognized as foreign and/or from being rejected by the cells of the maternal immune system.”158 During pregnancy the trophoblast and maternal immune system have a regulatory and supportive interaction that is critical for both pregnancy maintenance and protection against infectious microorganisms.107 Figure 13-5 illustrates interactions between the maternal immune system and the placental trophoblast.
Sperm are MHC class I– and II–negative, which usually protects them from recognition as foreign by the maternal immune system, although both sperm and seminal fluid carry other antigens. Seminal fluid also contains immunosuppressors. The woman responds to specific seminal fluid–induced cytokine and chemokine signals to increase immunosuppressive T-regulating (Treg) cells.137,138 Exposure to paternal antigens on sperm and seminal fluid before pregnancy may lead to decreased responsiveness and maternal tolerance to paternal MHC antigens.138,167
How does trophoblast eliminate or evade the immune responses of B-cell and T-cell activation given the general enhanced innate and humoral immune function that is seen in the systemic circulation during pregnancy? Factors that are currently thought to have a role in maternal tolerance of the fetus include trophoblast HLA-G; progesterone, progesterone induced-blocking factor, altered NK cell function, a Th1/Th2 balance that favors Th2 responses; indolamine 2,3 dehydrogenase (IDO) (catalyzes tryptophan in lymphocytes, an amino acid needed for T-cell proliferation and survival); Fas ligand (immune modulating protein that induces apoptosis of maternal lymphocytes that are fetal antigen reactive); suppressor macrophages; annexin II; lowered complement activity; toll-like receptor (TLR) expression and function; local immune suppression (mediated by Fas/Fas ligand system), and possibly other mechanisms that hide trophoblastic antigens from the maternal immune system.2,39,50,107,142,158
Implantation, the first point of interaction between maternal and fetal tissues, involves an inflammatory process with cross-talk (molecular dialogues) between the decidua and conceptus that increases activity of innate immune cells and is mediated by hormones, growth factors, cytokines, chemokines, adhesion molecules, extracellular matrix components and matrix metalloproteins.51 The inflammatory process at the maternal-placental interface is most marked in the first and third trimesters. In the first trimester these changes occur with the initial contact between the embryonic trophoblast and maternal tissues with implantation and the remodeling of maternal blood vessels.107 In the third trimester inflammation is a critical component of cervical changes in preparation for parturition and labor onset (see Chapter 4).107
After the placenta is established, the two points of contact between maternal and fetal tissues are the syncytiotrophoblast and the extravillous trophoblast. The syncytiotrophoblast is in contact with the endothelial cells of maternal spiral arteries and with the intervillous spaces, and is in direct contact with maternal blood. The extravillous trophoblast (primarily cytotrophoblast) forms the anchoring villi, which are in direct contact with maternal decidual tissue, and remodels maternal spiral arteries (see Chapter 3). Beginning in early pregnancy, small quantities of trophoblast cells detach and enter maternal blood through the uterine veins. These cells form minute emboli that eventually lodge in pulmonary capillaries and are cleared by proteolysis. This appears to be a normal process that does not lead to a maternal inflammatory response or other respiratory distress in women experiencing a normal pregnancy.
Trophoblast and major histocompatibility antigens.
The syncytiotrophoblast does not express either class I or class II MHC antigens, which are the main T cell targets in transplantation rejection.124 The cytotrophoblast has a unique nonclassical MHC-I surface antigen, HLA-G, which does not stimulate the classic cytotoxic T-cell response.85 HLA-G has a Fas/Fas ligand pathway for killing activated T cells and may help prevent maternal rejection of the fetus by inducing apoptosis of activated maternal T cells.32,85 HLA-G inhibits NK cell cytotoxicity and dendritic cell (an antigen presenting cell) maturation.50,176 HLA-G synthesis may be stimulated by IL-10 from the placenta. Extravillous trophoblast expresses HLA-G, HLA-C, and HLA-E (nonclassical MHC-I molecules that help these cells to evade maternal NK toxicity).111,136,176 HLA-G acts on cells of both the innate and adaptive immune systems and has a major role in reprogramming maternal immune responses at the maternal-fetal interface.136 Soluble HLA-G (sHLA-G) molecules are found in maternal plasma and increase from the first trimester on, peaking in the third trimester.136 Decreased expression of HLA-G on the extravillous trophoblast, seen in women with recurrent abortion and preeclampsia, alters conversion of the spiral arteries (see Chapter 3).136,176 Decreased sHLA-G is found with placental abruption, miscarriage, and preeclampsia.135
Trophoblastic cells (specifically the villous cytotrophoblast and extravillous trophoblast) express TLRs and may be able to initiate an innate immune response in the presence of microorganisms at the site of implantation.2,106 In addition, trophoblastic cells contain IDO, which enzymatically degrades tryptophan, a protein essential for T-cell proliferation, and TNF ligands that induce apoptosis. Together these molecules can kill activated immune cells.158
Natural killer cells.
Two subsets of uterine NK cells are seen: endometrial NK cells, seen during the menstrual cycle, and decidual NK cells, which are seen during pregnancy.124,182 Decidual NK cells are critical for pregnancy maintenance.72 The first direct contact between maternal and fetal tissue occurs 6 to 7 days after conception at the time of implantation. Subsequently, the extravillous trophoblast cells invade the maternal endometrial lining and erode the endothelial tissues that line the maternal spiral arteries (see Chapter 3). NK cells in the pregnant decidua are phenotypically distinct from the NK cells in systemic circulation. Peripheral NK cells express CD56dim, CD16+, and CD160+, a combination of characteristics that allow them to be highly cytotoxic. Conversely, decidual NK cells express CD56bright, CD16−, and CD160−, which allows them to produce cytokines such as IL-8 and IL-10 that are beneficial for trophoblastic invasion.69,72,95,111,131 These NK cells also secrete vascular epithelial growth factor (GF) and placental GF, which are angiogenic factors that promote trophoblast invasion and vascular remodling.111,124 NK cells accumulate at the site of implantation and in the decidua. They recognize HLA-G, HLA-C, and HLA-E on the invading trophoblast and influence production of cytokines and cytolytic factors that facilitate decidualization and control of trophoblast invasion, angiogenesis, and growth.86,95,107,119,176 Galectin-1 (Gal-1) is a carbohydrate binding protein with immunoregulatory functions that is found in high levels in uterine NK cells; Gal-1 is also found on trophoblast cells and may help reduce their cytotoxicity.72,130 Up-regulation of Gal-1 induces differentiation of dendritic cells into a phenotype that dampens Th1 responses and suppresses autoimmune inflammation.115 Trophoblast HLA-G can block receptors on the NK cell that initiate cell lysis. Thus the NK cell is altered to augment those functions that protect the conceptus while the general cytotoxic roles are down-regulated. Figure 13-6 illustrates how HLA-G on the trophoblast may inhibit maternal NK cells and block NK cytotoxicity toward fetal cells. Maternal stress, smoking, infection, and other factors can alter the NK phenotype and increase the risk of pregnancy complications.72
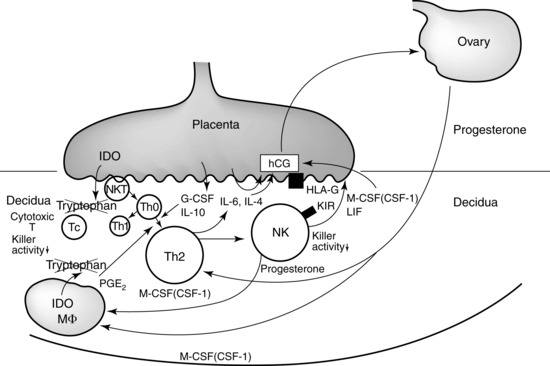
Decidual macrophages.
Macrophages cluster at the maternal-fetal interface and near the spiral arteries during trophoblast invasion (see Figure 13-5).3 These macrophages produce IL-10, IDO, PGE2, and other anti inflammatory molecules that trigger alloreactivity by the T cells to further protect the fetus.113,158 Placental growth hormone (see Chapter 19) may also alter the maternal immune system.158 Annexin II, a glycoprotein that binds to phospholipids, is produced by the placenta and may inhibit lymphocyte proliferation and IgG and IgM secretion at the placental site. Macrophages stimulated by Th1 cytokines or bacterial products develop a M1 phenotype and produce nitric oxide and free radicals to defend against microorganisms. Macrophages stimulated by Th2 cytokines develop a, M2 phenotype and have immunosuppressive properties. M2 is the common phenotype of decidual macrophages involved in costimulatory signaling.113
T cells and the TH1/TH2 switch.
Overall, there are more TCD8+ cells than CD4+ cells in the maternal decidua, which causes immunosuppressive activity to predominate.107 Changes in the balance of the different T-helper subsets (Th1 versus Th2) result in strengthening of Th2 (antibody-mediated) responses and cytokines (IL-3, IL-4, IL-5, IL-6, and IL-10) and a reduction in Th1 (cell-mediated and graft rejection) responses and cytokines (IL-2, TNF-α, and IFN-γ). This distribution of cytokines at the maternal-fetal tissue interface (see Figure 13-6) is important in implantation, pregnancy maintenance, and maternal tolerance of the fetus. The Th2-specific cytokines IL-4, IL-5, and IL-10 inhibit inflammation and some macrophage functions, and down-regulate IFN-γ production, whereas the Th1 cytokines are the primary effectors of phagocytosis.39,111,126,158 T-regulatory (Treg) cells (see Box 13-2 on page 446) normally account for 5% to 10% of peripheral CD4+ T cells. These increase two- to threefold during pregnancy beginning prior to implantation, peaking in the second trimester, and decreasing by term (this decrease correlates with the decrease in progesterone).111,127 Decreased Treg cells are seen with spontaneous abortion.111,127 The maternal T cells interact with trophoblast cells to support remodeling of the uterine vasculature by removing apoptotic cells, producing matrix proteases, mediating immunotolerance of the fetus, and protecting the fetus from pathogens.113
Nevertheless, the mother initially creates a weak immune response to the fetus/placenta, as evidenced by production of antibody to paternal MHC antigens, production of specific T cells, and weak cellular sensitivity to paternal and fetal antigens.75 This response usually results in an activation (facilitation) reaction rather than rejection response that is mediated by Th2 helper cells with release of Th2 cytokines, which down-regulate Th1 cytokines such as IL-2, IFN-γ, and TNF-α. As a result, maternal immune responses are down-regulated locally at the maternal-placental interface, but are not significantly affected at the systemic level.39 Failure of Th2 responses to increase or of Th1 responses to decrease is associated with an increased risk of recurrent abortion.25,101
Role of progesterone.
Progesterone promotes production of IL-4 and IL-6, the Th1-to-Th2 switch, and IL-4.31,50,126 Progesterone also stimulates activated lymphocytes and decidual cells to synthesize progesterone-induced blocking factor (PIBF).39,155 PIBF stimulates B-cell production of asymmetric antibodies, which are antibodies that have a mannose-rich oligosaccharide group present on one of the Fab regions. Asymmetric antibodies cannot initiate complement or phagocytosis but they can combine with antigen. The result of this combination is an antigen-antibody complex that is univalent and functions as a “blocking antibody” that prevents further antigen-antibody interaction. PIBF also inhibits the cytotoxicity of NK cells.39,59 Intrauterine inflammation and infection lead to decreased progesterone function and increased proinflammatory cytokines.31 Progesterone may up-regulate TLR-4 and suppress TLR-2 to protect against preterm delivery.31
Transplacental passage of maternal antibodies
Both protective and potentially damaging maternal antibodies cross the placenta. Maternal IgG antibodies are the only ones to cross in significant amounts. IgG has a specific carrier that facilitates active placental transport. Maternal antibody has multiple functions in the fetus and neonate including passive immunity against pathogens, epigenetic inheritance of immunologic memory, immunological imprinting, suppression of IgE responsiveness and suppression of tumor development.87
Fetal levels of IgG are low until 20 to 22 weeks’ gestation, when passive and active transfer of IgG across the placenta increases.164 All four IgG subclasses cross, although the IgG1 and IgG3 subclasses predominate and are transferred more efficiently.164 A carrier attached to a trophoblast surface receptor that is specific for the Fc fragments of IgG (and does not bind to other immunoglobulins) mediates active transfer of IgG across the placenta.70,164 This receptor has the greatest affinity for IgG1 and IgG3, followed by IgG4 with low affinity for IgG2; the receptor has no affinity for IgM or IgA.81 Active transfer allows for movement of IgG to the fetus even when maternal levels are low.70 IgG1 crosses earliest in pregnancy and is the primary immunoglobulin transferred before 28 weeks. IgG3 crosses later and does not reach maternal levels until after 32 to 33 weeks.81 In most infants, IgG levels near term are higher than maternal levels.57,82 Depending on maternal antibody complement, the newborn may have passive immunity against tetanus, diphtheria, polio, measles, mumps, group B streptococcus (GBS), Escherichia coli, hepatitis B virus (HBV), Salmonella enterica, and other pathogens.
Protection of the fetus from infection
Most viruses and many bacteria are capable of being transferred across the placenta, although relatively few are.70 Immune factors that prevent maternal rejection of the fetus also protect the placenta from infectious agents.
All 10 TLRs found in humans are expressed in the placenta, primarily in the trophoblast.107 The TLRs expressed vary with gestational age.107,135 Each TLR is specific for the molecular pattern of certain organisms. For example TLR-4 has a receptor for gram-negative bacterial lipopolysaccharide; TLR-3 binds double-stranded viral DNA. TLRs help the trophoblast to recognize microorganisms. TLR simulation initiates an inflammatory response with up-regulation of cytokines and interferon, and up-regulation of antimicrobial proteins and peptides.180 The placental trophoblast also secretes various antimicrobial factors to inhibit infectivity, recruit monocytes and macrophages, and prevent transmission to the fetus.107 Aggressive microorganism invasion may overcome these defenses and lead to fetal infection and preterm labor. Ascending infection can result in a maternal immune response, production of PMNs in the amniotic fluid, and a fetal inflammatory response.56,77,180 Altered TLR signal transduction is seen with preterm labor, preeclampsia, and intrauterine growth restriction.77,107,135
Occasionally organisms may reach the fetus by directly infecting placental tissue. Once the organism avoids cytokines at the maternal-placental interface, it must interact with trophoblast receptors to pass through the placental stroma and enter fetal blood. The primary placental defense is the Hofbauer (macrophage) cells. The placenta also contains phagocytes and lymphocytes and produces cytokines such as interferon.39 The organisms most commonly transferred across the placenta are Listeria monocytogenes, Treponema pallidum, human immunodeficiency virus (HIV), parvovirus B19, rubella, Toxoplasma gondii, and cytomegalovirus (CMV).114
Amniotic fluid contains antibacterial and other protective substances similar to many of those found in human milk. These substances include transferrin, beta-lysin, peroxidase, fatty acids, IgG and IgA immunoglobulins, and lysozyme.73,107 The antibacterial capacity of amniotic fluid improves with advancing gestation. Additionally, the fetal membranes provide barrier protection against ascending infection, although some organisms can penetrate intact membranes. Often pathogenic organisms enter the fetus via the respiratory tract, where surfactant protein (SP)-A and SP-D, alveolar macrophages, and PMNs provide additional defenses.173,180,181
Intrapartum period
Initiation of labor, cervical ripening, membrane rupture, and myometrial contractility involve an inflammatory process (see Chapter 4).31 Innate immune cells such as macrophages, dendritic cells, NK cells, and mast cells have roles in the onset of labor.78 During labor and in the early postpartum period, the WBC count increases to values of up to 25,000 to 30,000 per mm3. This increase is primarily due to an increase in neutrophils and may represent a normal response to physiologic stress.35 Impairment in the functional activity of peripheral blood lymphocytes and a decrease in the absolute number of total CD4+ and CD8+ lymphocytes may be observed immediately after birth. Cytokines may also have a role in the initiation of labor (see Chapter 4) and in reversing the pregnancy-induced reduction of maternal immune responsiveness at the decidual-trophoblast barrier during labor and birth.47 For example, IL-1 and IL-6 also increase with the onset of labor. TNF-α may assist in the initiation of contractions. IFN-γ may play a role in placental separation via activation of NK cells.47
Labor also has an effect on the fetal immune system. Labor alters neonatal neutrophil responses (see Neonatal Physiology). For example, newborns born vaginally exhibit leukocytosis, elevation of neutrophil count, and delayed apoptosis compared to infants born by cesarean section. These changes may be immunologically beneficial to the fetus.104
Postpartum period
It is unclear how quickly the immune system returns to prepregnant function after delivery, with various studies reporting timelines from a few weeks to 3 to 9 months due to differences in measurement techniques.153 The WBC count, which increases in labor and immediately after birth, gradually returns to normal values by 4 to 7 days. Progenitor cells from the fetus can be found in maternal tissue for many years postpartum.111
Immunologic properties of human milk
The newborn is immunologically immature and therefore vulnerable to infection. Human milk contains many immunologic components, including leukocytes, immunoglobulins, and other proteins (see Table 5-4). Immunologic benefits for the breastfeeding child include lowered risk of developing asthma, cow’s milk allergy, food allergy, gastrointestinal (GI) and respiratory infections, necrotizing enterocolitis, diabetes mellitus, and some immune disorders such as Crohn’s disease, celiac disease, and multiple sclerosis.45,87,116
The leukocytes in human milk are primarily monocytic macrophages (85% to 90%) and lymphocytes (10% to 15%), with some neutrophils and epithelial cells.45,116 Monocytic macrophages in human milk synthesize complement, lysozyme, and lactoferrin; transport immunoglobulin; and protect against necrotizing enterocolitis. These cells also have phagocytic activity against Staphylococcus aureus, E. coli, and C. albicans and may help regulate T-cell function.116,175
IgM levels are highest in colostrum, decrease after 5 days, and then remain constant for at least 180 days.70 Levels of IgG in human milk are also consistent for at least the first 180 days. The immunoglobulin found in highest concentrations (90%) in colostrum and human milk is secretory IgA (sIgA). Levels of sIgA are highest in colostrum, fall gradually until 12 weeks, and then remain stable for the next 2 years of lactation.90 The secretory component attached to the IgA monomer protects the IgA molecule from proteolytic digestion in the GI tract. sIgA does not enter the circulation from the gut but provides localized barrier protection by attaching to the mucosal epithelium and preventing attachment and invasion by specific infectious agents.116 sIgA also neutralizes certain viruses and bacterial enterotoxins and inhibits intestinal absorption of proteins and other macromolecules found in foods.116 The latter function may provide protection against the development of allergies. sIgA actions are enhanced by complement.
Human milk contains C3 and C4 and produces complement by the alternative pathway, which can be activated by factors such as bacterial products and circulating proteins. Most of the immunoglobulins in human milk are produced by sensitized antibody cells in the breast that have been transported to that site from gut-associated (i.e., maternal intestinal) lymphatic tissue and bronchotracheal-associated lymphatic tissue. Thus sIgA that is specific for a wide variety of respiratory and enteric bacterial and viral organisms can be found in human milk.70 Depending on the mother’s immunologic experience, the immunoglobulins in human milk may provide the infant with protection against pathogens such as diphtheria, pertussis, Shigella, Salmonella, poliovirus, and echoviruses.
Human milk contains other nonspecific protective factors that act synergistically with each other and sIgA to provide immunity to the newborn.70,83,169,180 Lactoferrin, α-lactalbumin, and sIgA form the major whey proteins in human milk and constitute 60% to 80% of total human milk protein. Lactoferrin is an iron-binding protein that restricts the availability of iron needed for growth by certain fungi and bacteria such as S. aureus and E. coli. Folic acid and vitamin B12-binding protein restrict available folate and vitamin B12 for bacterial and fungal growth.
Other immune components of human milk include. MIF; antiviral and antistaphylococcal factors; interferon (which prevents viral replication); lipase (which increases levels of free fatty acids and monoglycerides that may act against certain viruses); and oligosaccharides (which are complex carbohydrates that prevent attachment of bacteria and other antigens to gut epithelial receptors). Human milk acetic and lactic acid are two by-products of breast milk digestion in the gut. These acids decrease stool pH and inhibit growth of Shigella and E. coli. In addition, human milk can induce the production of immune factors such as sIgA by the infant. Maternal T cells in breast milk stimulate development of immune cells in the newborn thymus; thus the thymus gland of a breastfed infant is larger than the thymus in an infant receiving formula.45,116
In preterm infants, human milk may have a critical role in providing protection against infection and the development of allergies. The preterm infant’s gastrointestinal tract lacks many intrinsic local host defense factors including sIgA, lysozyme, and gastric acid. In addition, the intestinal mucosal barrier function is more immature (see Gut Host Defense Mechanisms).175 As a result, the preterm infant is at higher risk for bacterial penetration through the intestines and development of sepsis and necrotizing enterocolitis compared to term infants. The lack of sIgA and immaturity of the intestinal mucosa increase the likelihood of foreign macromolecules entering the circulation, which may increase the risk of allergies in genetically susceptible infants.175 Human milk may enhance maturation of the intestinal mucosal barrier, thereby reducing the risk of both infections and allergies. Increased levels of sIgA, lactoferrin, and lysozyme have been found in milk from mothers of preterm infants.175 Thus mothers of preterm infants may have an important immune adaptation that provides additional protection from infection for their infants.
Clinical implications for the pregnant woman and her fetus
Spontaneous abortion
There are multiple etiologies for spontaneous abortion. Immunologic factors include activation of an immune response secondary to microbial infection (e.g., chlamydia, listeriosis, parvovirus B19, or toxoplasmosis), an exaggerated maternal immune response to trophoblastic invasion (e.g., predominance of Th1 reactivity), a cytokine-induced failure of the ovary to produce sufficient progesterone, and the presence of autoimmune antibodies such as antiphospholipid antibodies that interfere with placentation.138,142,176 Decreased expression of HLA-G on the extravillous trophoblast is seen in some women with recurrent abortion and is associated with altered conversion of the spiral arteries.136,176 Decreased sHLA-G is also seen with spontaneous abortion.135 Spontaneous abortion is also associated with decreased Treg cells in the decidua and increased peripheral T-cell activation.30,111,127 Voisin suggests that some spontaneous abortions before 3 months in primiparous women may be secondary to a defect in active maternal tolerance of the fetus, whereas rejection after the third month or in women who have had a prior pregnancy may be due to excess rejection-type responses with increased levels of Th1 inflammatory cytokines such as IFN-γ, TNF-α, and TNF-β.168
Women who experience recurrent pregnancy loss, that is, three or more consecutive spontaneous abortions, exhibit decidual NK cell activity that is similar to the activity of NK cells in peripheral circulation, and a concomitant shift toward exaggerated Th1 immune responses instead of the usual pregnancy-exaggerated Th2 responses.38 It is unclear whether the altered NK cells are the result of the Th1 predominance or whether the Th1 response is the result of a different population of decidual NK cells that cause spontaneous abortion instead of trophoblast expansion.
Recent evidence suggests that inadequate progesterone production occurs when the hypothalamic-pituitary-gonadal axis is inhibited by systemic immune activation that triggers a noninflammatory pathway for pregnancy failure.43 This cytokine-induced failure of the ovary to produce progesterone results in resorption of the embryo. Both NK cells and TNF-α are effectors that can block this process by inhibiting the suppressor of cytokine signaling (SOCS) proteins, which are involved in the inhibition of progesterone production by the ovary.43,142
Risk of maternal infection
Even with alterations in host defense mechanisms, most women are not significantly immunocompromised during pregnancy nor do they experience a significant increase in the severity of bacterial infections during pregnancy.107 However, suppression of maternal Th1 cell-mediated immunity may increase maternal susceptibility to, but not necessarily severity of, certain infections, especially viruses and certain opportunistic pathogens, including C. albicans, Pneumocystis jiroveci (formerly Pneumocystis carinii), T. gondii, L. monocytogenes, Streptococcus pneumoniae, Neisseria gonorrhoeae, Mycobacterium tuberculosis, polio, rubella, influenza, varicella, CMV, and herpes simplex virus (HSV).40,47,107 Genetic polymorphisms in the genes that encode modulators of innate mucosal immunity, such as IL-6, IL-8, and TNF-α, may increase the risk of altered vaginal microflora.52 Alterations in neutrophil chemotaxis and function also contribute to the persistence of infections during pregnancy.48,129 Changes in the frequency of infections during pregnancy are also due to other reasons. For example, the increased incidence of urinary tract infections and pyelonephritis during pregnancy is primarily due to anatomic alterations in the urinary tract that result in urinary stasis (see Chapter 11).
Viral infections, however, are seen more frequently during pregnancy, especially during the second and third trimesters, and tend to be more severe. For example, in pregnant women, primary varicella is more likely to result in pneumonia and increased morbidity and the risk of complications with influenza is increased (see Chapter 10).40
Although methodological limitations are prevalent, most studies of HIV infection find that pregnancy does not appear to significantly increase the risk of death, disease progression, or CD4+ counts below 200, although there are suggestive trends for these events to occur in some studies.8 Antiviral therapies during pregnancy have significantly reduced perinatal transmission and improved outcomes for HIV-infected women and their infants.8,40,104 Infected women experience a greater reduction in TCD4+ cells during pregnancy and a longer time to return to normal after pregnancy than noninfected women, but the effects are equivocal.8,80
Fungal infections are also seen more frequently during pregnancy with an increase in symptomatic vulvovaginitis during the third trimester. The increased incidence of fungal infections is related to increased adherence of Candida to vaginal mucous membranes, increased glycogen in the vagina, enhanced proliferation of Candida under the influence of estrogen, and alterations in cell-mediated immunity.40 The incidence of infection with protozoa (e.g., malaria, amebiasis) and helminthic (intestinal parasites) organisms is increased in countries where these organisms are prevalent. During pregnancy, malaria is more frequent and more severe, with an increased risk of sequelae. The malaria parasite has an affinity for placental tissue, leading to stillbirth and preterm birth. In addition, malaria is a major cause of low birth weight in endemic areas.47 Latent malaria can be reactivated by iron overload during pregnancy.
Inflammation, infection, and preterm labor
Inflammation (decidual, chorioamnionic, or systemic) is a significant etiologic factor in preterm labor (see Chapter 4 and Figure 4-7). “Preterm labor is a syndrome initiated by multiple mechanisms including intrauterine infection, uteroplacental ischemia or hemorrhage, uterine overdistention, cervical disease, stress, endocrine disorders, and other immunologically mediated processes.”31,p. 209 Many of these processes lead to inflammation at the maternal-fetal interface. Onset of labor involves inflammatory mediators, which these events can trigger.
Both acute and chronic maternal infections are associated with preterm labor. Intrauterine infection leads to activation of the innate immune system. Recognition of microorganisms by TLRs results in release of inflammatory chemokines and cytokines. Proinflammatory cytokines (IL-1β, TNF-α, IL-8, Il-6) in combination with microbial endotoxins increase prostaglandin production and release of matrix metalloproteases and other substances that can lead to cervical ripening, fetal membrane rupture, and increased myometrial contractility.31
Urinary tract infections have also been associated with preterm labor (see Chapter 11). Maternal fever can lead to maternal dehydration, resulting in increased uterine activity, which, along with elaboration of prostaglandins, may result in initiation of labor. In women with acute infections, high temperature alone may lead to the release of catecholamines and corticotropin-releasing hormone and increase uterine irritability. Preterm labor is discussed further in Chapter 4.
Although high levels of IgG and sIgA in maternal cervical mucus may provide protection against infection from vaginal pathogens, vaginal and cervical organisms have been linked with the initiation of preterm labor and premature rupture of membranes.63,139 Both these events are probably mediated via inflammatory cytokines that stimulate prostaglandin E2α production.139
Chorioamnionitis and cerebral palsy
Chorioamnionitis or inflammation of the placental membranes increases the risk that the fetus will develop cerebral palsy.53,178 Although the exact mechanism has not been completely determined, several processes may contribute to the underlying pathophysiology. Elevated levels of fetal cytokines produced in response to maternal infection may directly injure the fetal brain or cause damage indirectly by increasing the permeability of the blood-brain barrier and facilitating passage of cytokines and microbial products into the brain. The presence of microbial products may induce an inflammatory process in the brain that damages the fetal brain and induces periventricular leukomalacia. Inflammation of the placental membranes may contribute to cerebral palsy by interfering with gas exchange and inducing a hypoxic-ischemic injury in the fetus (see Figure 6-4). Maternal fever may raise the core temperature of the fetus, which could be harmful to the fetal brain. Finally, maternal infection could cause direct infection of the fetal brain. Although the fetus is more susceptible to adverse effects of maternal infection early in gestation, the placenta is a poorer barrier against some pathogens late in gestation. Consequently, while adverse fetal effects of maternal infection are more likely at early gestational ages, the incidence of fetal infection is more likely late in pregnancy.178 The attributable risk of cerebral palsy secondary to chorioamnionitis is estimated as approximately 12% in term infants and approximately 28% in preterm infants.178
Immunization and the pregnant woman
Alterations in the immune system during pregnancy do not seem to significantly alter the woman’s responses to immunization.42,47,49 Potential risks associated with immunization during pregnancy include fetal viremia, teratogenesis, interference with development of the infant’s response to immunizations during childhood, and maternal side effects that might compromise uteroplacental function.143
Ideally, immunization of women of childbearing age should occur at least 3 months before conception (or immediately after delivery) to reduce the risk of infection and adverse fetal effects.13,42 Immunity can be achieved by either passive or active means. At times, passive immunizations may be recommended during pregnancy after accidental or potential exposure to an organism associated with significant maternal or fetal risks.13 Passive immunity involves transfer of antibodies from an actively immunized to a nonimmunized person and confers short-term protection until the antibodies are catabolized by the nonimmunized person’s system. The physiologic transfer of antibodies from mother to fetus and injection of γ-globulin are both examples of passive immunity.
Active immunity involves exposure to some form of the antigen (attenuated or live organisms rendered noninfectious, inactivated or killed organisms, inactivated exotoxins or toxoids) that stimulates the production of antibodies and memory cells against that antigen by the nonimmunized person. Active immunization induces long-lasting or permanent immune responses.42,143
Immunization of pregnant women with live attenuated viruses or bacteria is not recommended because there is a small risk of transfer of the antigen to the fetus.42,154 Immunization with measles (rubeola), mumps, and rubella vaccines is specifically contraindicated because of potential adverse fetal consequences. However, if a woman is accidentally vaccinated for rubella early in pregnancy, the risk of adverse fetal effects appears to be low, and therapeutic abortion is not recommended.42 Inactive (i.e., dead) viruses (e.g., pneumococcal, influenza, diphtheria, tetanus, hepatitis B) are considered safe for use in pregnancy.13,14,42,154 Since the risk of influenza-related morbidity is increased during pregnancy, influenza vaccinations are recommended for all pregnant women during flu season, regardless of trimester.15,42 Administration of immunization for rubella during the first few days after birth is often recommended. The woman who receives rubella vaccine should be counseled to use methods to prevent conception for at least 4 weeks.13,143
Blood and blood products given within 14 days of active immunization may interfere with the effectiveness of the vaccine, because blood may contain antibodies against the antigens in the vaccine and therefore interfere with the woman’s own immune response.172 Although there is a small theoretic risk of transfer of antigens through breast milk or nasopharyngeal secretions from a recently immunized woman to her infant, this is a rare event without significant reported morbidity.172 Current recommendations regarding specific immunizations during pregnancy can be found in several reviews.13,49,143,154
Malignancy and pregnancy
Theoretically, alterations in the maternal immune system could increase the risk of malignancy because cell-mediated immunity, which is suppressed in the pregnant woman, normally protects against virally induced tumors. Some investigators have suggested that there is an increase in rate of progression and decreased survival times in women with breast and cervical cancer during pregnancy, but this has generally not been supported.33 However, a delay in diagnosis (due to breast changes during pregnancy with increased engorgement and nodularity) may result in a more advanced stage at time of diagnosis.33 Gestational breast cancer (diagnosis during pregnancy, lactation, or the first year postpartum) tends to involve “larger tumors, more frequent nodal involvement, metastasis and vascular invasion.”33, p. 893 The effect of pregnancy on malignant melanoma is also variable.33 Melanoma may intensify during pregnancy and, then recede or disappear after delivery.171 The basis for this change has been attributed to increased levels of melanocyte-stimulating hormone during pregnancy (see Chapter 14) and hormonal stimulation of estrogen receptors found on melanoma cells.
Immunologic aspects of preeclampsia
Preeclampsia is a complex, multifaceted disorder involving alterations in many major body systems (see Chapter 9 and Table 9-5). There is increasing evidence that vascular endothelial cell injury is an early event in the pathogenesis of preeclampsia.132,133,148 The exact cause of this damage is unclear but an immunologic role has been suggested.9,132,133,148 Factors that support an immunologic basis for preeclampsia include an increased incidence of the disorder in primigravidas or, in the case of multiparas, a pregnancy with a new partner or different sperm donor; an increase in pregnancies with a large placental mass (e.g., multiple pregnancies) or hydatidiform mole; a decreased incidence in repeat pregnancies (even if the previous pregnancy ended in a miscarriage) with the same father and consanguineous marriages; a decrease with longer cohabitation with the father before pregnancy (and thus exposure to paternal antigens in semen); and the presence of pathologic changes in the uterine vessels near the placental site that are similar to those with allograft rejection.132,133,148
Specific alterations in the immune system have been observed in women with preeclampsia. Many of these changes are exaggerations of the inflammatory changes normally found during pregnancy. However, women with preeclampsia have a further reduction of NK cell activity with increased peripheral and reduced decidual NK cells (which alters placental development and spiral artery remodeling), altered macrophage activation, more TCD8+ cytotoxic cells, decreased TCD4+-to-TCD8+ ratio, more immune (antibody-antigen) complexes, increased levels of fibronectin, decreased IDO activity, and alterations in complement.9,25,132,133,148 In women with preeclampsia the usual Th1/Th2 pattern during pregnancy is altered so that Th1 predominates with increased levels of inflammatory cytokines, including TNF-α, IL-2, IL-6, and IL-8, but reductions in IL-10.9,31,133,138,148 In addition, increased expression of TLRs by the interstitial trophoblast or alterations in TLR signal transduction may contribute to local abnormal cytokine levels that lead to the development of preeclampsia.9,77,107,135 Decreased placental HLA-G expression and sHLA-G are also reported.100,133,136,148,176 These changes alters conversion of the spiral arteries and increase the risk of vascular endothelial injury, which appears to be the underlying mechanism that results in preeclampsia.
The pregnant woman with an autoimmune disease
The effect of pregnancy on autoimmune disorders is varied; individual women experience improvement, exacerbation, or no change depending on the disorder. These differences may be related to the immune mechanism involved in the specific disorder; that is, whether the disorder involves alteration in Th1 responses (which decrease during pregnancy) or Th2 responses (which increase). Autoimmune disorders are thought to impair T-suppressor cell activity, resulting in hyperactive B-lymphocyte response and production of autoantibodies that form immune (antibody-antigen) complexes with their target antigens.122 Autoantibodies damage tissue either by directly reacting with antigens on the surface of cells or by combining with antigens to form immune complexes.121,122 The immune complexes activate the complement cascade, which in turn stimulates phagocytosis, inflammatory responses, and tissue damage.
Because the usual changes in the immune system during pregnancy are the exact opposite of these events, women with autoimmune disorders may experience an improvement in their symptoms.121,122 For example, 75% of women with rheumatoid arthritis experience improvement during pregnancy (which is associated with increased Th1 mediators, which are decreased in pregnancy), followed by a relapse within the first 3 months postpartum.92,111,184 Other changes during pregnancy that might lead to this amelioration include changes in plasma levels of maternal humoral factors, depression of cell-mediated immunity, and suppression of inflammatory reactions.92,97,111,122,184
Pregnancy is unlikely to alter the natural history of SLE unless the woman has nephritis or is experiencing a flare at the time of conception.97 However, SLE can adversely impact the course of pregnancy.30 Women with mild disease that is under control for 6 to 12 months before and during pregnancy usually experience no exacerbation of SLE during pregnancy, and some may improve. Exacerbation, when it occurs, has been reported to be more frequent in early pregnancy and during the first 6 to 8 weeks after birth.184 Postpartum exacerbation may represent a rebound phenomenon as suppression of cell-mediated activity is terminated.122
SLE involves production of autoantibodies of the IgG class and increases in Th2 mediators, which are increased in pregnancy normally.122 Women with SLE who have anticardiolipin antibodies or lupus anticoagulant antibodies have an increased risk of thrombosis, spontaneous abortion, thrombocytopenia, preeclampsia, fetal growth restriction, and preterm birth.92,97,159,170 Lupus anticoagulant factor is an immunoglobulin that binds to prothrombin-activating complexes and predisposes the woman to recurrent thrombosis in the spiral arteries and placental infarction. Some women with SLE who have anti-Ro/SS-A or anti-LA/SS-B will have a fetus with neonatal lupus erythematosus.92,103 These infants have congenital heart block, cutaneous lupus rashes, cytopenia, and possibly other systemic manifestations. This syndrome is usually transient and resolves after birth, although complete heart block may not be transient.92 Fetal heart block occurs because the anti-Ro/SS-A or anti-LA/SS-B antibodies lodge in the fetal cardiac conducting system and interfere with conduction pathways.24
Fetal and neonatal complications associated with transplacental passage of maternal antibodies
Potentially damaging IgM class antibodies, such as the ABO antigens, and allergy-producing IgE antibodies generally do not cross the placenta. Smaller, potentially damaging maternal IgG antibodies may cross the placenta as a result of Rh incompatibility (see next section) or one of several maternal chronic diseases, leading to transient disorders in some neonates.64 Neonatal effects from the passage of these IgG antibodies range from mild to severe. In general, if the disorder is not fatal during the perinatal period, manifestations are transient and regress as maternal antibody is catabolized.
Maternal Graves’ disease may involve transplacental passage of a thyroid-stimulating immunoglobulin resulting in transient neonatal hyperthyroidism (in approximately 1% of offspring) that can persist for up to 2 to 6 months.84,128 Myasthenia gravis is associated with passage of maternal IgG against acetylcholine receptors, resulting in transient myasthenia gravis in 10% to 20% of offspring. Symptoms can last from a few hours to up to 3 months.152 Because symptoms usually develop after 12 hours of age, infants who appear healthy at birth may later develop respiratory failure.18 The delay in symptom onset may be due to transfer of anticholinesterase drugs from the mother or elevated AFP.152 Maternal antiplatelet antibodies induce fetal thrombocytopenia in up to 50% of the offspring of women with immune thrombocytopenia. The greatest neonatal risk is for severe hemorrhage in the perinatal period. In most infants, platelet levels reach lowest levels at 4 to 6 days after birth and return to normal by 1 to 2 months.18 Risk to infants from antibody passage in women with SLE is described in the previous section.
Rho(D) isoimmunization and ABO incompatibility
The Rh system involves a group of at least 45 different RBC surface antigens controlled by genes encoding Rh proteins (DCE). The Rh locus is on the short arm of chromosome 1.102 A related protein, Rh glycoprotein, is essential for expression of the Rh antigen. Antigens of the D group are the ones usually involved in incompatibility between mother and fetus. Rho(D) isoimmunization occurs when an Rho(D)-negative mother carrying an Rho(D)-positive fetus produces antibody against the D antigen on the fetal RBC. Isoimmunization can also occur with other RBC antigens such as Kell, Duffy, Kidd, MNS, and the ABO system.
Rho(D) isoimmunization is of particular clinical importance because the D antigen is a potent antigen, is present in large amounts on the fetal RBC, appears as early as 6 weeks’ gestational age, and stimulates formation of IgG-type antibody and memory cells in the mother.62 The amount of antigen necessary to trigger an immune response in the mother varies for each person, as does the intensity of the response by the mother’s immune system. The incidence of Rho(D) isoimmunization is relatively low, ranging from 10% to 14% if Rho(D) immune globulin (RhIG) is not given after birth to less than 2% if RhIG is given.23,61 The R2 phenotype (cDE) expresses the most D antigen and increases the risk of maternal sensitization.162 Rho(D)-negative women are unlikely to have antibodies against the D antigen unless they have been immunized during a previous pregnancy or from a mismatched blood transfusion. As a result, isoimmunization is rare in first pregnancies. Normally during pregnancy the small amounts of fetal blood (less than 0.05 mL) that cross the placenta and enter the maternal circulation are too small to trigger production of antibodies by the mother’s immune system. In a few women, however, as little as 0.01 mL of fetal blood has been reported to cause maternal immunization (sensitization).23 Detectable fetal blood cells can be found in approximately 3% of pregnant women in the first trimester, 12% in the second trimester, and 45% in the third trimester.62,162 Approximately 1% to 2% of Rho(D)-negative women develop anti-D antibodies during their first pregnancy.23 This number can be reduced by prophylactic administration of RhIG during pregnancy.
At birth, larger quantities of fetal blood (0.5 mL or more) may enter the maternal circulation. This amount of fetal blood (if the fetus is Rho[D] positive) is sufficient to stimulate formation of both anti-D antibody and memory cells in many women (Figure 13-7, A). Formation of memory cells results in permanent immunization. During subsequent pregnancies, even a very small amount of blood from an Rho(D)-positive fetus entering the mother’s system may be enough to trigger memory cells to produce antibodies against the D antigen on the fetal RBC. The antibodies that are produced in this secondary response are predominantly of the IgG1 and IgG3 subclasses and are thus actively transported across the placenta (see Transplacental Passage of Maternal Antibodies) to the fetal circulation where they hemolyze fetal RBCs (see Figure 13-7, B). With development of RhIG (a human γ-globulin concentrate of anti-D), initial active immunization of most Rh-negative women can be prevented. RhIG is given prophylactically at 28 weeks to prevent immunization during pregnancy; in the first 72 hours after birth if the newborn is Rh-positive; or after any potentially immunizing events such as an abortion, ectopic pregnancy, amniocentesis, or significant antepartal bleeding. A dose of RhIG at 28 weeks provides protection for approximately 12 weeks (i.e., until term).62,102 The incidence of immunization during pregnancy is 1.6% to 2%, decreasing to 0.1% to 0.18% with administration of both antenatal and postpartum RhIG.62,102,162 RhIG does not cause hemolysis in the fetus even though the majority of the antibodies are transported across the placenta.61,93,162
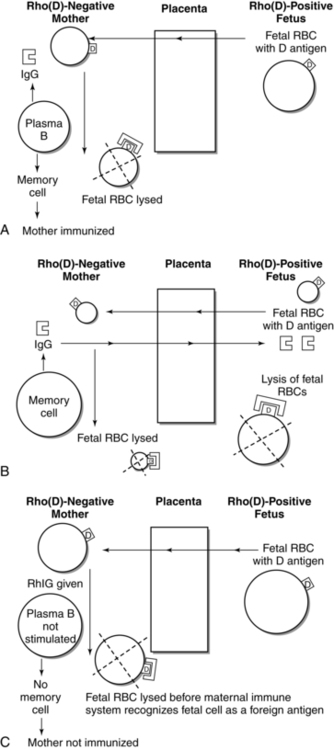
RhIG destroys fetal RBCs in the mother’s system before the foreign D antigen on these cells can be recognized by her immune system and trigger formation of antibodies and, more importantly, memory cells (see Figure 13-7, C). However, the exact mechanism by which RhIG acts to prevent alloimmunization is unclear.102 Three potential mechanisms have been suggested for the action of RhIG: (1) clearance of antigen from the mother’s system; (2) antigen-blocking by attaching to antigenic sites on the fetal cells, thereby preventing interaction with maternal lymphocytes; or (3) central inhibition of antibody production.23,26,62,162 The last theory suggests that RhIG forms immune complexes with the Rho(D) antigen. Immune complexes suppress the stimulating effect of helper T lymphocytes, which in turn suppresses antibody formation by B lymphocytes.62,162
The standard dose (300 mcg) provides protection for up to 15 mL of fetal RBCs.62 Because development of an adequate antibody response to a specific antigen can take days or weeks in persons not previously sensitized, RhIG can probably be given up to at least 2 weeks after birth (possibly longer) if omitted earlier for some reason.62 After antenatal administration of RhIG, some women may develop a low (1:4 or less) anti-D serum antibody titer. This positive titer reflects a passive immunity from the RhIG. The infant of a mother who received RhIG prophylaxis within 12 weeks may have a weakly positive direct Coombs test due to placental transfer of RhIG antibodies. Neither of these responses indicates maternal immunization, and postpartum RhIG administration is indicated.23,62
ABO incompatibility
The potential for isoimmunization also exists with ABO incompatibility, although severe hemolytic disease in the fetus and newborn is rare even though ABO incompatibility is three times as common as Rho(D) incompatibility. Previous exposure and immunization are not necessary with ABO incompatibility because the mother already has naturally occurring antibodies against fetal RBC antigens. The most common situation in which ABO incompatibility occurs is with a type O mother and a type A (Figure 13-8, A) or, less frequently, type B infant. The A antigen seems to be more antigenic than the B antigen. The type O mother has naturally occurring anti-A and anti-B antibodies in her serum that can react against the A or B antigens on the fetal RBCs. ABO incompatibility could also occur with a type AB infant but never with a type O infant (type O RBCs have neither A nor B antigens for maternal anti-A or anti-B antibodies to identify and react against).
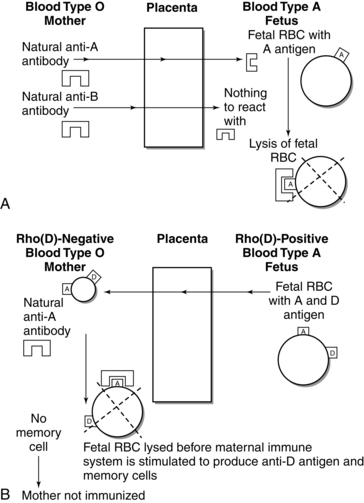
The simultaneous occurrence of Rho(D) and ABO isoimmunization has a protective effect that reduces the likelihood of maternal Rho(D) sensitization. This is illustrated in Figure 13-8, B, with an Rho(D)-negative, type O (naturally occurring anti-A and anti-B serum antibodies) woman who is carrying a fetus who is Rho(D)-positive and type A (with A and B RBC antigens). The naturally occurring maternal anti-A antibody destroys fetal cells entering maternal circulation (during pregnancy or at delivery) before they can trigger the mother’s immune system to produce anti-D antibodies and memory cells. If anti-D antibodies and memory cells are not formed, the woman remains unimmunized.23,162
Summary
Alterations in maternal host defense mechanisms are critical for maintenance of the pregnancy, ensuring that the mother does not reject the fetus, and protecting the fetus against infection. Yet these same mechanisms may increase the risk of infection and other immune system disorders in the pregnant woman. Transplacental passage of maternal immunoglobulins provides protection against specific pathogens but can also lead to fetal disease. Concerns related to organisms that colonize the maternal genital tract (e.g., GBS) and sexually transmitted diseases such as HSV, HBV, and HIV have increased interest in host defense mechanisms and maternal-fetal interrelationships, and altered clinical practice in recent years. Recommendations for clinical practice based on changes in host defense mechanisms are summarized in Table 13-5. By recognizing the changes in maternal host defense mechanisms, health care providers can identify women and infants at risk and initiate appropriate interventions and counseling.
Table 13-5
Recommendations for Clinical Practice Related to Changes in Host Defense Mechanisms: Pregnant Woman
Recognize normal parameters for immune system components and patterns of change during pregnancy and the postpartum period (pp. 443, 445, 449-451, Table 13-4).
Obtain and evaluate complete history for possible exposure to infectious organisms, current illness, or presence of autoimmune disorders (pp. 456-459).
Recognize risk factors for development of specific infections (p. 457, Table 13-4).
Monitor and counsel women regarding early signs and symptoms of infection (p. 457).
Recognize clinical and laboratory changes during labor and delivery and postpartum that may mask signs of infection (p. 455 and Chapter 8).
Assess women with infections during the second and third trimesters for signs of preterm labor (p. 457 and Chapter 4).
Teach women methods to prevent or reduce the risk of infection while pregnant (pp. 456-459).
Counsel woman regarding the immunologic advantages of human milk and its potential role in preventing gastrointestinal infections and allergies (pp. 455-456, Table 5-4, and Chapter 12).
Monitor iron intake and serum ferritin levels in women with a history of malaria (p. 457).
Counsel women regarding risks of immunizations with live attenuated or killed vaccine during pregnancy and to avoid pregnancy for 3 months after immunization (p. 458).
Avoid giving immunizations within 14 days of administration of blood or blood products (except RhIG) (p. 458).
Give RhIG to unsensitized Rho(D)-negative women prophylactically at 24 to 30 weeks’ gestation and after potentially immunizing events (pp. 460-461).
Recognize that women who have received antenatal RhIG may have low antibody titers for approximately 13 weeks yet may still need RhIG postpartum if the newborn is RH positive (pp. 460-461).
Understand the implications of a weakly positive direct Coombs’ test in some infants of women who have received RhIG during the prenatal period and the need for RhIG postpartum (p. 461).
Give RhIG postpartum to unsensitized Rho(D)-negative women with a Rho(D)-positive newborn in the first 72 hours following birth (pp. 460-461).
Counsel women with autoimmune disorders about potential effects of pregnancy on course of disease (p. 459).
Use Standard Precautions for blood and body fluids during the perinatal period (pp. 457, 475).
Counsel women with HBV, HSV, HIV, and other viral infections regarding long-term effects on infant (including shedding of virus) and use of precautions to prevent spread of the infection (p. 457).
Counsel women with chronic disorders associated with transplacental passage of antibodies regarding the potential impact on their fetus and neonate (pp. 459-461).
Development of host defense mechanisms in the fetus
Cellular components of the immune system develop from precursor cells within blood islands of the yolk sac (see Chapter 8). Multipotential stem cells arise in these islands and migrate into the liver and spleen and later to the bone marrow and thymus. The mucosal immune system is developed by 28 weeks’ gestation, but not activated until after birth, unless the infant is exposed to an intrauterine infection.54
B and T lymphocytes arise from common lymphoid stem cells. Pre–B cells are seen in the liver by 7 to 8 weeks; immature B lymphocytes with surface IgM receptors and complement are found in the fetal liver by 10 weeks.70,82 By 12 weeks, B lymphocytes that have IgG and IgA cell surface receptors are seen in peripheral blood, bone marrow, liver, and spleen; numbers reach adult values by 15 weeks.114,174 Fetal synthesis of immunoglobulins begins by 10 to 14 weeks; however, levels of fetal immunoglobulins produced by the fetus normally remain low throughout gestation.66,174 Fetal serum IgG levels rise in the second and third trimesters primarily secondary to increased transplacental passage from the mother (see Transplacental Passage of Maternal Antibodies). Secretory IgA is not produced by the fetus/neonate until after birth. Elevated IgM levels in cord blood (greater than 20 mg/dL [greater than 0.20 g/L]) suggest intrauterine infection and may be seen in newborns with congenital cytomegalovirus (CMV), rubella, or toxoplasmosis. This IgM is of fetal origin as IgM does not cross from the mother.
Differentiation of the thymus begins at 6 to 7 weeks.105 The thymus develops as an outgrowth of the third and fourth pharyngeal pouches. The rudimentary thymus on each side of the body grows toward midline. These structures fuse by 8 weeks and then gradually descend into the chest.99 Stem cells from the liver and spleen migrate to the thymus to form thymocytes. T lymphocytes appear in the thymus by 7 weeks and in the blood by 12 weeks.174
Differentiation of CD4+ and CD8+ surface antigens begins around 10 weeks.70 Mature CD4+ and CD8+ T lymphocytes are detected in the liver and spleen by 14 weeks, and in the fetal circulation by 15 to 16 weeks’ gestation.66,114 By midgestation, T cells are capable of secreting interleukin (IL)-2; tumor necrosis factor (TNF)-α; TNF-β; and, to a lesser extent, interferon (IFN)-γ, IL-3, IL-4, IL-5, and IL-6.114 TNF, IL-1B, and IL-6 are found in amniotic fluid and increase to term; elevation of IL-6 is seen with intrauterine infection.74,180 Surfactant protein (SP)-A and SP-D are elevated in amniotic fluid and may also have an immunoregulatory function.173,181 Th1 responses tend to be inhibited by IL-10 from the placenta.96 Innate responses are characterized by IL-23 directed immunity, which promotes development of Th17 CD4+ and enhances defenses against extracellular pathogens.176 Production of IL-12, which induces IFN-γ production by CD4+ T cells and NK cells, and differentiation of Th1 cells, is decreased.176 Transplantation responses develop relatively early. Antigen recognition can be demonstrated by 12 weeks, and graft-versus-host reactions by 13 weeks’ gestation. The fetus can increase production of proinflammatory cytokines with intrauterine infections and mount an inflammatory response. However, this may increase the risk of brain injury, especially white matter damage, and cerebral palsy (see section on Chorioamnionitis and Cerebral Palsy and Chapter 15).74
The fetal immune system is dominated by Th2 to “avoid proinflammatory Th1 type alloimmune responses to maternal tissue that may trigger preterm birth or spontaneous abortion.”125 IL-6, IL-10, and IL-23, and Th17 mediated immune responses are all up-regulated with increased production in response to bacterial lipopolysaccharide-binding protein.176 TNF-α, IL-1B, and Th1 mediated immune responses are down-regulated in the fetus and amniotic fluid. This decreases the extent and duration of proinflammatory immune responses that can lead to preterm birth.176 An exaggerated fetal immune response has been linked to periventricular leukomalacia and cerebral palsy in addition to preterm labor onset.176
White blood cells arise initially from yolk-sac stem cells (see Chapter 8). Neutrophils and macrophages arise from colony-forming unit–granulocyte-macrophage progenitor stem cells.70 Myelopoiesis is 10 times more active in the fetus than in the adult and increases fourfold by the second trimester.76 Granulocytic cells from which neutrophils arise can be found in fetal blood by 6 to 8 weeks and mature neutrophils by 14 to 30 weeks.82,114 After 5 months, neutrophils are produced primarily in the bone marrow. Few granulocytes are found in peripheral blood during the first half of gestation (e.g., at 22 to 23 weeks’ gestation, the fetus has approximately 2% of the neutrophils found in cord blood), but by term the number of granulocytes is similar to those observed in adults.70
Embryonic macrophages are found by 3 to 4 weeks in the yolk sac. By 4.5 weeks, approximately 70% of the blood cells in the liver are macrophages, decreasing to 1% to 2% over the next 6 weeks.145 IL-1 produced by fetal macrophages up-regulates granulocyte colony stimulating factor production by fetal monocytes in the fetal liver and bone marrow.82 True monocytes appear in the blood at 5 months, with levels increasing after 6 months when bone marrow hematopoiesis begins to predominate.145
Natural killer cells (which have a major role in surveillance against viral infection) are found in the liver and blood by 5 to 6 weeks; however, basal activity of these cells is almost undetectable until after 20 weeks’ gestation and does not increase significantly until after 32 weeks.108 “The fetus develops the capacity to recognize and respond to invading microorganisms through the action of toll-like receptor–initiated pathways and the production of proteins, such as antimicrobial peptides and lipopolysaccharide (LPS)-binding protein.”176, p. 141
Complement protein synthesis by the fetal liver begins as early as 6 weeks, but individual components develop gradually and are not seen until 8 to 14 weeks; serum complement is seen after 20 to 22 weeks.19 Complement increases rapidly after 26 to 28 weeks, but still may be only 50% to 75% of that of an adult by term.70,174 Complement is not transferred across the placenta.
In summary, an innate immune response can be identified in the fetus early in gestation, while adaptive immune functions remain inactive until term. However, adaptive immune functions may be activated in some fetuses in the presence of fetal infection. In terms of the functional development of the immune system, 32 to 33 weeks’ gestation seems to be a critical time. Before this point, the immune responses of the fetus or preterm infant are compromised in comparison with the term neonate.108 However, after this time the preterm infant’s immune system rapidly approaches that of term infants, although the immune system of these infants is still immature compared to that of adults.
Neonatal physiology
Newborns are vulnerable to infection because of (1) altered host defense mechanisms, particularly in the preterm infant, and (2) a lack of experience and exposure to many common organisms, resulting in delayed or diminished adaptive immune responses to foreign antigens. The newborn’s immune system is adapted for fetal and early newborn life with decreased Th1 cytokine production and increased Th2 cytokines.180 These adaptations are an advantage for the fetus, because the lower levels of Th1 cytokines reduce both the risk of alloimmune rejection between the mother and fetus and the risk of excessive inflammation that can lead to preterm birth, and during the early newborn period to allow for skin and gut colonization and induction of host defense functions.125,177,180 However, after birth these adaptations increase the neonate’s vulnerability to infection. Alterations in neonatal host defense mechanisms are found in innate immune system responses, inflammatory responses, antibody and cell-mediated immunity (i.e., adaptive immunity), and the complement system. (See Boxes 13-1 and 13-2 on page 446 for a review of innate and adaptive immunity.) Table 13-2 includes definitions of immune terminology. The newborn is also susceptible to infections as a result of other factors, including (1) entry of pathogenic organisms due to the breaks in mucosal or cutaneous barriers that may occur during birth; (2) gastrointestinal (GI) infections and potentially later development of allergies due to immaturity of the gut host defense mechanisms; and (3) disruption of skin barriers from invasive procedures, the use of tape and monitor leads, and intravenous catheters in infants who are born prematurely or who are ill.
Transitional events
Skin colonization
Newborns are initially colonized with organisms from the maternal genital tract acquired during the birth process. This is followed by colonization with maternal skin flora and other organisms present in the newborn’s environment. The maternal genital tract flora at delivery normally includes Lactobacillus, Escherichia coli, and protective anaerobes, but it may also contain potentially dangerous organisms such as group B streptococcus (GBS) and Chlamydia trachomatis. Colonization occurs initially on the skin, umbilical cord, and genitalia, followed by mucous membranes of the eyes, ears, throat, and nares.107 Skin flora is more extensive in infants with little or no vernix caseosa, which normally acts as a protective mechanical barrier.107
Intestinal colonization
The normal intestinal microorganisms have both nutritional and immune system functions.112,163 Immune functions include providing a barrier against pathogens, stimulating gut host defenses, and modulating inflammatory responses and intestinal permeability.112,163 Nutritional functions include synthesis of vitamin K, short chain fatty acids, and biotin.112
Colonization of the GI system occurs in two stages. During the first stage (birth to 1 week), the infant is inoculated with organisms that he or she comes into contact with during and after birth. In the second stage (1 to 4 weeks), the infant’s diet significantly influences the pattern of bacterial flora. The normal gut flora provides an important protective mechanism against GI infections by occupying potential pathogen-binding sites on intestinal mucosa. The upper small intestine is usually sterile or sparsely colonized, probably due to the gastric pH, or possibly due to the antibacterial properties of bile, secreted immunoglobulins, and normal intestinal motility (see Chapter 12). Coliform colonization of the upper small intestine can occur in very low–birth weight (VLBW) infants and infants who are intubated or fed by transpyloric tube. The decreased intestinal motility of preterm infants may predispose them to intestinal bacteria overgrowth.27 Gut colonization occurs by attachment of organisms to glycoconjugate receptors in the intestinal mucosal surface. Oligosaccharides in human milk help prevent colonization with pathogenic organisms by competing for mucosal bacterial receptors.46
Meconium is usually sterile at birth, except perhaps after prolonged ruptured membranes. Bacteria can be found in meconium within a few hours of birth and increase rapidly over the next few days. Use of antimicrobial drugs during labor partially suppresses but does not prevent colonization and may result in a shift in the dominant gut flora and an increase in resistant flora.160
In the first few days after birth, the neonate may be further protected from potentially pathogenic gut organisms by the acidity of gastric secretions (which inhibits growth of gram-positive and gram-negative bacteria) and by immaturity of the gut epithelium (which alters attachment of pathogenic organisms). Infants for whom oral feedings are delayed are more likely to have no bacterial growth in fecal samples. The intestines of these infants are similar to the germ-free state. This state is associated with a slower mucosal cell turnover (which allows toxins to have a more profound effect), fewer lymphoid cells in the gut, and alterations in gut immune responses that may increase the risk of infection.94
Breastfed infants develop different colonization patterns from formula-fed infants. These different patterns may reflect physicochemical differences and the influence of antimicrobial factors in human milk.46,110,139 The lower buffering capacity of human milk (due to lower concentrations of proteins and minerals) allows acids from bacterial metabolic end products to increase. The resulting acidic environment is one in which Lactobacillus and Bifidobacterium thrive, preventing growth of acid-sensitive organisms such as bacteroides and enterobacteria. The low protein and phosphate and high lactose contents of human milk also promote growth of these flora. Protective substances in human milk (see Table 5-4 and Immunologic Properties of Human Milk) may stimulate secretory IgA (sIgA), strengthen epithelial tight junctions, and possibly modify intestinal inflammatory and allergic responses.116 Antimicrobial factors found in human milk may also alter gut colonization patterns. For example, lactoferrin is an iron-binding protein that is only 9% saturated in human milk. As a result, lactoferrin can bind iron entering the gut that is not absorbed and reduce the availability of iron for bacteria metabolism.116
Formula feedings buffer the acid produced by gut bacteria. This leads to an alkaline environment in which the bifidobacteria cannot compete with gut enterobacteria. Gram-negative enterococci become the dominant gut organism. As formulas become more similar to human milk, differences in colonization, although present, are less apparent.34,46,139 Current research is examining the use of prebiotics such as galacto-oligosaccharides and fructo-oligosaccharides in formula-fed infants to stimulate intestinal flora similar to that of breastfed infants in order to enhance intestinal mucosal development.118
Alterations in innate immunity
The inflammatory response and phagocytosis (see Figures 13-1, 13-2, and Box 13-1 on page 446) in the newborn are different when compared with adult function because functional limitations of the infant’s polymorphonuclear monocytes (PMNs) affect leukocyte metabolic activities, mobilization, chemotaxis, adhesion, opsonization, phagocytic activity, and intracellular killing. The neonate’s PMNs are more rigid (due to an altered cytoskeleton), less deformable (due to generation of less actin), and have a poorer response to chemotactic stimulators and impaired receptor mobilization.76,144 These limitations alter movement kinetics and orientation of the PMNs. The neonate’s PMNs are less able to leave the blood vessel to reach the site of pathogen invasion because of the decreased deformability. The newborn’s PMNs have less ability to adhere to the vascular epithelium, leading to poorer aggregation of PMNs along the vessel wall near the site of injury.22,66,76 L-selectin expression, which is critical for initial neutrophil adhesion to vascular endothelium and migration, is low in neonates and decreases in the first 24 to 72 hours; levels are even lower in preterm infants.22,76,144,180 Neonatal neutrophils have a three- to fourfold decrease in bactericidal permeability-increasing protein, which may increase the risk of gram-negative sepsis.22
Neonatal PMNs do not respond as readily as those of older individuals to chemotactic factors.28,70 Chemotaxis (see the definition in Table 13-2) in neonates is about half as efficient as chemotaxis in adults, resulting in slower, more random movement of neutrophils and monocytes to the site of antigenic invasion.28,76,180 This is secondary to the structural and functional limitations of neonatal PMNs and due to lower levels of chemotaxis-stimulating substances in the neonate’s blood. These limitations are exacerbated in septic infants.180 The increased IL-6 production reduces PMN migration; however, IL-8 priming during labor improves PMN chemotaxis.180,183 Other factors that alter chemotaxis include impaired PMN calcium metabolism, altered actin polymerization and adhesion defects.76 In the fetus, the chemotaxis response is consistent from 24 weeks to term, decreases after birth, and then increases to reach adult levels in term infants by about 2 weeks of age.28,22,140 In preterm infants, chemotaxis does not begin to respond efficiently until 2 to 3 weeks after birth and develops slowly.28 The reason for this is unclear. Achievement of chemotaxis function at adult levels is delayed in more immature preterm infants, with less than two thirds of them achieving adult values by 42 weeks.140 Therefore these infants remained at higher risk for infection even after reaching their due dates.
Newborns, particularly preterm infants, have decreased serum opsonic activity, resulting from low levels of immunoglobulins and complement components. In addition, neonates have difficulty recognizing and destroying encapsulated bacteria unless the pathogen is first opsonized by attaching complement fragments and immunoglobulins (see Table 13-2).144
Most studies report that phagocytic activity in the healthy term newborn is similar to phagocytic activity in adults, although newborn phagocytic cells are less responsive to stimulation by chemotaxic factors.66,70 Thus phagocytosis is diminished to some extent by the decreased availability of neutrophils (the primary circulating phagocyte) at the invasion site due to altered chemotaxis. Macrophage (the primary tissue phagocyte) activity in response to cytokines (see Tables 13-2 and 13-3) from T cells is also decreased. Phagocytic activity and intracellular bactericidal activity of PMNs are probably not significantly different from the same function in adults in most healthy infants and are similar to adult levels by about 2 weeks.18,28,70 However, some authors have reported that even healthy preterm infants born at less than 33 weeks’ gestation have a poorer phagocytosis response for the first few months of life, possibly due to poor opsonization.22
Activated neutrophils normally undergo apoptosis within 24 to 48 hours. Neonatal neutrophils have a delay in apoptosis, prolonging their survival, possibly related to decreased expression of caspase-3, a component of several apoptosis pathways. The increased survival may be an advantage in fighting infection, but the elongated activation may increase the risk of chronic inflammation and disorders such as chronic lung disease.76
Phagocytosis has been found to be normal in most stressed neonates, although decreased phagocytosis toward some gram-negative bacteria has been reported. Infants who experience health complications either in utero or after birth (e.g., hypoxic-ischemic events, respiratory distress, meconium aspiration, sepsis, or hyperbilirubinemia) demonstrate significantly decreased bactericidal activity in response to both gram-positive and gram-negative organisms. Bactericidal activity may be reduced even further in VLBW infants who weigh less than 600 g.18 The cause is unclear but may relate to developmental immaturity with intrinsic alterations or defects in leukocyte metabolic activity, or it may result from peroxidative damage to the cell.66 There are several changes in cellular metabolism associated with phagocytosis including the respiratory “burst” with a rapid increase in oxygen consumption, the hexose monophosphate (HMP) shunt that is involved with glycolysis activity, and the production of toxic oxygen metabolites necessary for bactericidal activity. These activities are reduced in term infants with sepsis and in preterm infants of less than 34 weeks’ gestation.22,28,180 In preterm infants these metabolic processes remain low for the first 1 to 2 months of life.22,28
The number of neutrophils varies during the first few days after birth, with an increase after birth that peaks at 12 to 24 hours, declines to 72 hours, then remains stable during the neonatal period.70,76 This increase is blunted and lower levels are seen in preterm infants.76 The increase after birth is probably due to movement of marginated neutrophils into the main circulation.76,144 PMN levels below 3000 m3 in the first 3 days or below 1500 m3 after the first 3 days are unusual in term infants; neutrophil levels may normally be as low as 1100 m3 in preterm infants.70 Although low PMN levels are normally associated with neonatal infection, they are also seen in infants born prematurely or after hypoxic-ischemic events, and in infants with intraventricular hemorrhage or Rh hemolytic disease.70
Bone marrow reserves, which are neutrophil storage pools (NSPs), are lower in the neonate, especially preterm infants. This is due to a reduction in granulocyte macrophage–colony-stimulating factor (GM-CSF).28,76,82 Approximately one third of preterm infants have a functional neutropenia.122 In neonates, especially preterm infants, the marrow is probably functioning at or near capacity for neutrophil proliferation. Therefore these infants are less able to increase production much further with infection and the NSP becomes exhausted more rapidly.89,122,180 This may account for the neutropenia and shift to the left (release of immature forms of neutrophils) often seen with sepsis, especially in VLBW infants.28
Toll-like receptors (TLRs) and antigen presenting cells (APC) (see Box 13-1 on page 446) are different in neonates than adults. Neonatal TLR-mediated cytokine response is characterized by decreased Th1 proinflammatory cytokines (TNF-α, IL-12, IL-1B, IFN-γ) due in part to progesterone, PGE2, and plasma adenosine levels.91,125,180 Plasma adenosine is increased with hypoxia and stress and inhibits TNF-α production by the monocytes and production of Th1 cytokines.180 Labor up-regulates TLR-4 on monocytes and PMNs.147,180 SP-A, SP-D, and alveolar macrophages act as pattern recognition and signaling molecules via interaction with TLRs, CD-14+, and other components of the innate system.173 TLRs may play a role in the pathogenesis of necrotizing enterocolitis, in particular, abnormal TLR-4 signaling in the intestines.7,179 TLR-4 is a receptor for lipopolysaccharides (a component of the outer membrane of gram-negative bacteria). The altered signaling by TLR-4 limits innate responses to these organisms.7
APC function is decreased in the neonate resulting in increased reliance of the innate system for pathogen removal.180 Cord blood dendritic cells have an immature phenotype and low chemokine levels, decreased endocytoxic activity, decreased antigen presenting and costimulatory capacity, and produce low levels of IL-12.91 Neonatal dendritic cells require greater stimulation for activation and are poorer stimulants of T-cell proliferation with impaired IL-12 production.180 Neonatal APCs express low levels of costimulatory molecules such as CD40, CD80, and CD86.70 This characteristic, along with low IL-12 production by APCs and IFN-γ by neonatal T-helper cells reduces the infant’s ability to induce Th1 responses and may lead to immune tolerance rather than response.91,180
The number of monocytes in infants is highest in the first 24 hours after birth and then decreases over the next month.145 Neonatal monocyte chemotaxis and phagocytosis are altered during sepsis with a decreased delivery of these macrophages to infection sites.70,82,89,125,180 Neonatal monocytes and dendritic cells have impaired expression of MHC II and costimulatory molecules, reducing antigen presentation to CH4+ T cells.180 Synthesis of interferon (IFN)-γ, interleukin (IL)-8, IL-10, extracellular matrix proteins, fibronectin, and leukotrienes by neonatal monocytic macrophages is reduced.82 Monocyte antibody-dependent cellular cytotoxicity is about 50% of adult values.82 Monocyte adhesion is similar to that of adults.57 Monocytic bactericidal activity and processing of antigens are generally considered to be adequate.
Fibronectin, a glycoprotein found in plasma and tissues, inhibits bacterial adherence to epithelial cells; enhances antibody binding of bacteria such as staphylococci and GBS; and is involved in opsonization and clearance of fibrin, immune complexes, and platelets. Neonatal levels of plasma fibronectin average 50% of adult values, with the lowest levels seen in preterm infants and those with hypoxic-ischemic events or respiratory distress.18,70,89 C-reactive protein (CRP) synthesis is similar in uninfected neonates and adults. A diminished CRP response has been observed in the first 12 to 24 hours after birth in infants infected with group B streptococcus.67
Numbers of natural killer (NK) cells, which provide protection against tumors or virus-infected cells, are similar to or greater than those in adults, but NK activity is reportedly decreased to 15% to 60% of adult activity.58,70,89 Neonatal NK cells are phenotypically different from those of adults with a lower cytolytic capacity.58 Most adult NK cells are CD56high, whereas most neonatal NK cells are CD56low, a more immature form.57 Neonatal NK cells are less able to bind to target cells and have limited lytic ability.89 The altered activity is probably mediated by the decreased levels of interferon and other cytokines that normally augment NK cell function. NK cells in preterm infants have decreased cytotoxic function.57
In general, the total number of circulating lymphocytes is similar to adult levels at birth but falls during the first 3 days and plateaus by 10 days of age. The ratio of T lymphocytes to B lymphocytes is lower in cord blood than in adult blood.70 Neonatal mast cells secrete more histamine than adult cells, which alters dendritic cell cytokine production. Mast cells may play a role in development of erythema toxicum.180
APCs (including monocytes, macrophages, and dendritic cells) in the neonate “exhibit deficits in pathogen recognition, activation after stimulation, phagocytic function, bactericidal function and amplification of the immune response that may increase the risk for development and progression of EONS [early-onset neonatal sepsis].”180, p. 314 The ability of neonates to localize infection is limited. As a result, septicemia is more likely to develop in neonates from spread of the pathogenic organism to the blood and cerebrospinal fluid. A major factor in this inability to localize infection is impaired chemotaxis. Adequate phagocytosis and killing of many pathogenic organisms such as GBS depend on complement (especially C3) and opsonic activity. Serum concentrations of C3 and thus opsonic activity are proportional to gestational age, further increasing the risk of sepsis in preterm infants.70
The inability to localize infection results in clinical manifestations of infection that are different in the newborn compared to the adult, and this can make the diagnosis of sepsis in the neonate difficult. Specifically, signs of sepsis in the neonate are often subtle and nonspecific, involving changes in activity, tone, color, or feeding. In addition, neonates have a poor hypothalamic response to pyrogens; therefore fever is not a reliable indicator of infection in the neonate. Alterations in innate immunity are summarized in Table 13-6.
Table 13-6
Alterations in Host Defense Mechanisms in the Neonate
| ALTERATION | RESULT | IMPLICATION |
| INNATE IMMUNITY | ||
| Structural alterations of PMNs | Altered movement kinetics and orientation of PMNs | Delayed initial response to invasion by pathogenic organisms |
| Altered PMN chemotaxis, adherence, and L-selectin expression |
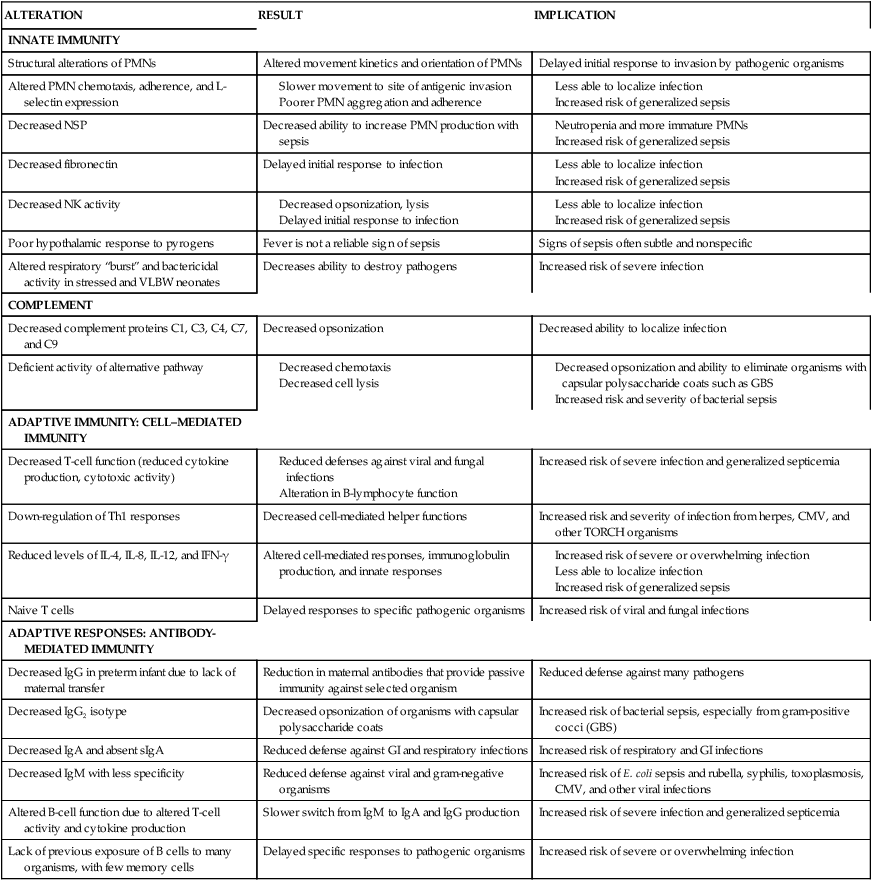
Alterations in the complement system
Newborns have decreased serum levels of both classical and alternative pathway components (see Figure 13-3), defective complement activation, and a decrease in complement receptors.18,19,174,180 Components of the classical system in the term neonate are slightly reduced or comparable to adult values, whereas levels of these factors are significantly reduced in preterm infants and correlate with gestational age (see Table 13-7).70 For example, levels of several key complement proteins (C1, C3, C4, C7, C9) are 55% to 75% of adult values in term infants, and 10% to 30% of adult levels in 28-week preterm infants. The lower levels of complement proteins result in decreased opsonization, decreased chemotaxic activity, and decreased cell lysis. Limited alternative pathway activity and marked reductions in properdin are seen in two thirds of newborns.18,70 The classical and alternative pathways are further depressed in infants with documented sepsis.
Table 13-7
Published Ranges of Complement Levels in Neonates
| Mean % of Adult Levels (Number of Studies) | ||
| COMPLEMENT COMPONENT | TERM NEONATE | PRETERM NEONATE |
| CH50 | 56-90 (5) | 45-71 (4) |
| AP50 | 49-65 (4) | 40-55 (3) |
| Clq | 61-90 (4) | 27-58 (3) |
| C4 | 60-100 (5) | 42-91 (4) |
| C2 | 76-100 (3) | 67-96 (2) |
| C3 | 60-100 (5) | 39-78 (4) |
| C5 | 73-75 (2) | 67 (1) |
| C6 | 47-56 (2) | 36 (1) |
| C7 | 67-92 (2) | 72 (1) |
| C8 | 20-36 (2) | 29 (1) |
| C9 | <20-52 (3) | <20-41 (2) |
| B | 35-64 (4) | 36-50 (4) |
| 9 | 33-71 (6) | 16-65 (3) |
| H | 61 (1) | — |
| C3bi | 55 (1) | — |
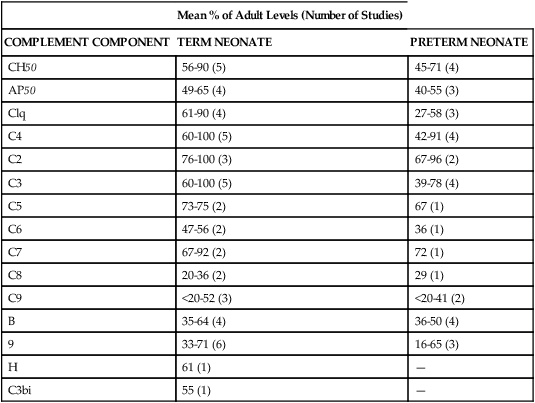
From Lewis, D.B. & Wilson, C.B. (2001). Developmental immunology and role of host defenses in fetal and neonatal susceptibility to infection. In J.S. Remington & J.O. Klein (Eds.), Infectious diseases of the fetus and newborn infant (5th ed.). Philadelphia: Saunders.
Alterations in adaptive immunity
Cell-mediated and antibody-mediated (humoral) immunity is developmentally immature in both preterm and term neonates (see Table 13-6). Response of the adaptive system to APCs is less efficient than in adults.180Box 13-2 on page 446 and Figure 13-2 provide a review of adaptive immunity.
Alterations in cell-mediated immunity
Absolute numbers of T lymphocytes in the newborn are higher than in adults, but the functional ability of newborn T cells is decreased, as is the diversity of specific T-cell receptors, which results in a delay in the response to specific organisms.5,89 Cytotoxic activity of neonatal T cells is 30% to 60% less than in adults, and cytokine production is reduced.101 Examples of cytokines and their roles are found in Table 13-3; cytokine production in the newborn is summarized in Table 13-8.
Table 13-8
Production of Selected Cytokines in the Newborn
| CYTOKINE | SOURCE | ACTIVITY | NEWBORN PRODUCTION |
| IL-1 | Macrophage | T and B proliferation; Ig synthesis | Normal |
| IL-2 | T cell | T and B proliferation; induction of IL-2 receptors | Normal to low |
| IL-4 | T cell | B-cell proliferation; Ig switch | Low |
| IL-6 | T and B cells, macrophages, endothelial cells | T and B activation; Ig production | Normal to low |
| IL-10 | T cell | Inhibits T help; Ig production | Low |
| IL-12 | T and B cells; macrophages | Promotes NK killing and antigen-dependent cytolysis | Low |
| IFN-γ | T cell | Inhibits Ig production; increases MHC expression; promotes antigen presentation | Low |

From Schelonka, R.L. & Infante, A.J. (1998). Neonatal immunology. Semin Perinatol, 22, 7.
T-suppressor activity tends to dominate in the newborn.108 Although Th2 (antibody-mediated) helper responses are the dominant response in the neonate, the T-helper cells provide relatively poor support of B cells.144 For example, neonatal T cells do not induce neonatal B cells to produce some of the immunoglobulins that adult B cells would produce.37 CD4+/CH25high Treg cells are found in high levels in cord blood.44,125 These cells suppress T-cell proliferation in response to cytokines and Th1 cytokine (especial IFN-γ) production thus limiting adaptive responses. This is an advantage in limiting autoimmunity but also limits responses to infection and vaccines.125
At birth most of the T lymphocytes are primary cells (i.e., naive cells) with few secondary memory T cells. Neonatal naive T cells differ from adult naive T cells in that the neonatal cells have a higher turnover rate and limited Th1 responses.96 Primary T cells release less cytokine and have a diminished response to antigens.37,114,139,144 The most prevalent T-cell phenotype in the neonate is the CD45RA+ (naive) cell. These cells account for 90% of the neonate’s T cells, decreasing to 50% (similar to adults) by 10 years of age. CD45RA+ cells are “naive” because of the fetus’s lack of exposure to foreign antigens. When exposed to a foreign antigen presented by a B lymphocyte or dendritic cell, the neonatal CD45RA+ cells differentiate into CD45RO+ (memory) cells.99 The neonatal CD45RO+ cell is capable of producing significant amounts of cytokines (e.g., IFN-γ, TNF-α, GM-CSF) and acting as a helper cell for B-cell function.37,114,139,144 However, because the newborn’s T cells have no previous experience with most antigens until exposed and sensitized after birth, it probably takes 4 to 6 weeks for the newborn to achieve minimal adaptive immune protection.18
Impairment of cytokine production alters cell-mediated immunity, immunoglobulin production, and innate responses.94,98 Although some cytokine production by neonatal T cells is altered, IL-1, IL-2, and IL-6 levels are normal or slightly reduced. However, levels of IL-4, IL-8, IL-12, and IFN-γ are reduced.82,88,98 The decrease in IL-4 and IFN-γ is consistent with the “naive” state of CD45RA+ cells.55,96 IL-8 levels are markedly lower in preterm infants, limiting neutrophil mobilization and chemotaxis.55 Development of necrotizing enterocolitis and bronchopulmonary dysplasia in preterm infants has been postulated to be related in part to unregulated cytokine production.70 Genomic polymorphisms in gene encoding for production of cytokines may help explain individual differences in neonatal susceptibility to sepsis.
Neonates have down-regulation of Th1 responses with a decreased capacity to produce cytokines thus promoting Th1 (cell-mediated) responses with a blunted response to activation by IFN-γ, possibly due to decreased signal transduction.6,17,88,96,99 This may increase susceptibility to oropharyngeal candidiasis.98 Neonatal T cells are able to increase cytokine production when stimulated. For example, CD8+ cytotoxic T cells can be stimulated and reach near adult levels of function in some cases with cytomegalovirus (CMV) exposure.65,96 However, neonatal T cells require greater stimulation as well as stimulation from additional factors (e.g., accessory cells and signaling molecules) than adult cells to achieve full activity.5,70,144 Cell-mediated immunity is further depressed in small-for-gestational-age (SGA) infants. These infants have a smaller proportion of T lymphocytes and altered lymphocyte function than appropriate-for-gestational-age (AGA) infants, perhaps secondary to alterations in thymic activity. The thymus of SGA infants is smaller in weight and volume and demonstrates histologic alterations and decreased activity of thymic inductive factors, which may contribute to immune dysfunction in these neonates.
Alterations in antibody-mediated (humoral) immunity
The fetal and neonatal differences in antibody-mediated immunity are primarily due to the alterations in T-cell activity, immature cytokine function, and the effects of passive immunization from immunoglobulins acquired transplacentally and/or through breastfeeding. Activity of B cells requires signaling from the interaction of surface antigen and T-helper cells to amplify the immune response.70 Because T-cell activity and cytokines are needed to enhance B-cell function, there is a reduction in fetal and neonatal production of immunoglobulins.66,82 T-cell–dependent B responses have a delayed onset, peak at lower levels, are of shorter duration, and show different IgG isotypes than in adults.70,96 T-cell–independent B responses are also deficient, especially in response to antigens with capsular polysaccharide coats such as group B streptococcus.96,149 Neonatal B cells have fewer IL-5 receptors, decreased activation by IL-10, and poorer responses to Th2 antigens.79,149 Many neonatal B cells have CD5+ surface antigens (B1a cells). B1a cells are not dependent on T-cell help for activation as are conventional B cells (B2).79,114 However, neonatal T cells do not stimulate B cells to switch production of immunoglobulins from IgM to IgG or IgA. As a result, neonates produce primarily IgM and little IgG and IgA, even when exposed to bacteria with polysaccharide capsules.79,144 Neonatal B cells in response to vaccine antigens tend to differentiate into memory B cells rather than antibody-producing plasma B cells.
The total amount of immunoglobulin at birth is 55% to 80% of adult values, and primarily consists of IgG from the mother.82 Levels of serum immunoglobulins after birth are illustrated in Figure 13-9. Neonatal IgG values reflect the IgG acquired from the mother either transplacentally or via breast milk.89 The newborn can produce IgG1 in a manner similar to that of an adult; however, IgG2 production is reduced to about 2 years. IgG2 is important for defense against polysaccharide antigens such as are seen with GBS, K1 forms of E. coli, Haemophilus influenzae, pneumococcus, and meningococcus.82 Development of antibody responses is primarily dependent on postbirth age rather than Post-menstrual age.57
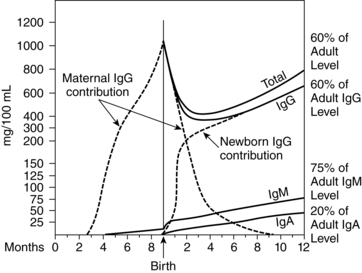
IgG is important for immunity to bacteria, especially gram-negative organisms, bacterial toxins, and viruses.70,174 Neonatal values depend on gestational age because placental transfer of IgG increases during the third trimester. Thus preterm infants may have inadequate protection. For example, cord blood values of IgG are minimal in infants born at 24 to 25 weeks’ gestation and average 400 mg/dL (4.0 g/L) at 32 weeks and 1500 mg/dL (15 g/L) at term.114,174,180 Term IgG levels are 90% to 95% of adult values and 5% to 10% higher than maternal values.70,140 Levels of IgG are lower in SGA infants, possibly due to impaired placental transport of maternal IgG.89 After birth, levels fall gradually as maternal IgG is catabolized. Because significant production of IgG by the infant does not occur until after 6 months, all infants experience a transient “physiologic hypogammaglobulinemia” (see Physiologic Hypogammaglobulinemia) in the first 6 months. However, as a result of IgG transfer in breast milk, the neonate continues to get antibodies against infectious agents for which the mother has circulating antibodies because of previous exposure or immunization (see Table 5-4).
IgA is important for localized immunity in the GI and respiratory tracts. As stated above, IgA does not cross the placenta in significant amounts. Neonatal (term and preterm) values are low (0.1 to 5 mg/dL [0.001 to 0.05 g/L], which is less than 2% of adult values). Elevated IgA is found in cord blood after maternal-fetal transfusion and occasionally with intrauterine infection, although IgM is seen more commonly after fetal infection.114 sIgA is not found in neonates at birth but can be detected in saliva, tears, and intestinal mucosa by 2 weeks to 2 months.82 sIgA occurs as a monomer or is attached to a polypeptide chain (secretory component). The polypeptide chain provides resistance to pH changes and protects the IgA molecule from proteolytic digestion in the GI tract.
IgM, which also does not cross the placenta, is important for protection against blood-borne infections and can trigger multiple effector functions of the classical complement cascade.174 IgM is the major immunoglobulin synthesized in the first month of life.5,66,122 IgM levels increase rapidly by 2 to 4 days after birth, probably secondary to stimulation from environmental antigens, although neonatal values are low (5 to 15 mg/dL [0.05 to 0.15 g/L]), with means of 6 mg/dL (0.06 g/L) at 28 weeks’ gestation and 11 mg/dL (0.11 g/L) at term, which is approximately 10% of adult values.70,89 The fetus is capable of producing significant IgM in response to exposure to certain antigens, such as the TORCH organisms, after 19 to 20 weeks’ gestation, which may be due to activation of the fetal B1a cells.114 However, neonatal IgM has less specificity than adult IgM in responding to specific antigens, which may hinder the initial recognition of pathogenic organisms. IgE and IgD do not cross the placenta in significant amounts, and newborn values are less than 10% of adult values.66
Few B cells that have differentiated into immunoglobulin-secreting cells (IgSCs) are seen in the first 5 days after birth, and those that are found are mostly IgM-secreting cells. By a month of age, two thirds of neonates have IgSCs, most of which are IgA-secreting cells. Increased IgSCs are more common in both term and preterm infants with intrauterine infection. Low serum concentrations of IgG in preterm infants may also be due to a decreased ability of their B cell to switch immunoglobulin isotype forms.174
Gut host defense mechanisms
Maturation of the mucosal immune system occurs rapidly after birth in response to antigenic exposure and is well developed by 1 year of age in both preterm and term infants.54 Host defense mechanisms in the gut involve both nonimmune and immune factors (Table 13-9). Many of these factors are initially immature in the neonate, which reduces the effectiveness of the gut mucosal barrier and increases the risk of GI disorders. Neonatal GI mucosal immaturity includes (1) increased permeability to macromolecules (all potential antigens), (2) altered immune tolerance, (3) decreased sIgA, and (4) decreased cytokine production. This allows entry of pathogenic organisms into systemic circulation and may increase the risk of development of allergic disorders. As defense mechanisms mature, GI barriers become more impermeable, offering greater protection against uptake of antigenic substances.29,94 The development of gut defense mechanisms is sometimes referred to as gut closure.
Table 13-9
Alterations in Gut Host Defense Mechanisms in the Newborn
| DEFENSE MECHANISM | MODE OF ACTION | ALTERATIONS IN NEWBORN |
| NONIMMUNE FACTORS | ||
| Gastric acid | Decreased number of organisms entering intestines | Decreased in term and preterm infant until 4 weeks’ postnatal age |
| Intestinal motility and peristalsis | Remove organisms and antigen | Decreased to 29 to 32 weeks’ postconceptual age |
| Intraluminal proteolysis | Determines amount of macromolecular transport across intestinal epithelium | Decreased pancreatic enzyme function in preterm infant; some decrease in response in both term and preterm infants to 2 years; result is an increased absorption of intact proteins across small intestine |
| Mucosal surface | ||
| Mucous coat |
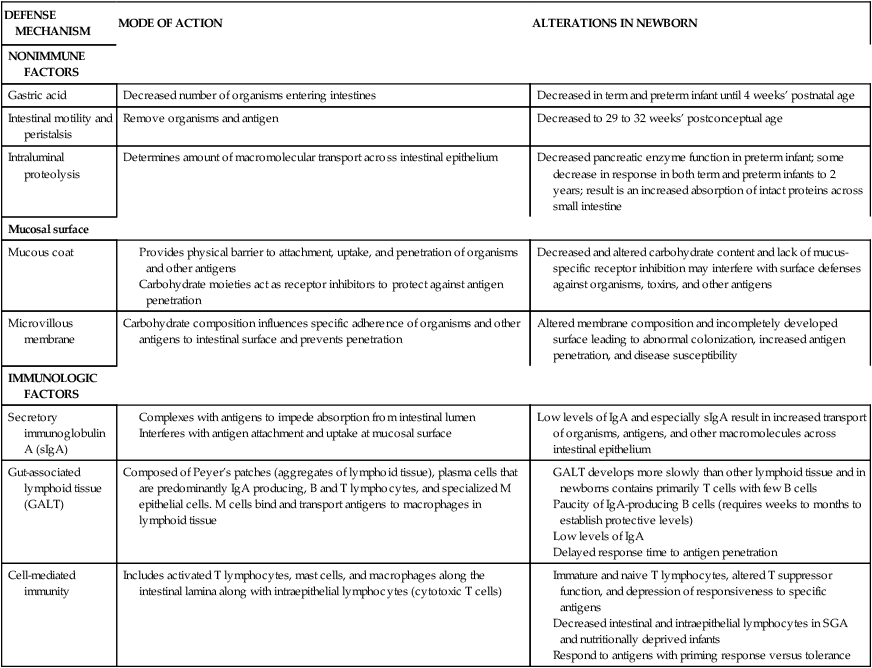
SGA, Small for gestational age.
Compiled from references 46, 54, 94, 179.
Gut closure is influenced by ingestion of food, especially colostrum, which enhances maturation of the intestinal lining via introduction of factors such as thyroxine, transforming growth factor, insulin-like growth factors, neurotensin, cortisol, lactoferrin, bombesin, and epidermal growth factor.94 Gut closure is delayed in preterm and SGA infants, who can absorb macromolecules across the intestinal epithelium for up to 8 to 12 months. Nonimmune factors that stimulate gut closure include maturation of gastric acid production, intestinal motility and peristalsis, intraluminal proteolytic activity, and the mechanical barrier properties of the gut mucosal surface.
The major specific immune factors that affect gut closure are sIgA and cell-mediated immunity. Fermentation of the prebiotic oligosaccharides contained in breast milk results in production of probiotic (“good”) bacteria such as bifidobacteria and lactobacilli. These probiotics stimulate synthesis and secretion of sIgA and help produce a balanced Th1/Th2 response. sIgA also coats the mucosa to protect against bacterial invasion.46
Neonatal cell-mediated immunity, especially a localized depression of T-lymphocyte suppressor activity, and the decreased IgA hinder an effective response to antigens that can cross the intestinal barrier. The mature immune response to the presence of antigens in the gut is immune tolerance. With immune tolerance, the absorbed antigen elicits a localized IgA response that destroys most of the antigen. As a result, there are fewer antigens available to enter the systemic system. Immune tolerance appears to be enhanced by the presence of partially digested polypeptide fragments. These fragments may not be formed in the neonate secondary to immature proteolysis. With decreased IgA, greater amounts of antigen are absorbed. The presence of antigens also triggers T-suppressor cell activity, which interferes with and inhibits systemic responses to these antigens.94 Decreased T-suppressor response can alter systemic immune responses and lead to inflammatory or allergic responses.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
Immune system and host defense mechanisms
“Remarkable adaptations . . . allow a genetically and antigenically disparate conceptus to develop in direct contact with a fully competent maternal immune system.”100 Why doesn’t the mother reject the fetus and placenta? Knowledge of maternal, fetal, and neonatal immune physiology and understanding of the immune relationship between a mother and her fetus are still evolving. Current theories regarding this question—along with alterations in host defense mechanisms in the mother and neonate and implications for clinical practice—are examined in this chapter.
The immune system is made up of organs and specialized cells whose primary purpose is to defend the body from foreign substances (antigens) that may cause tissue injury or disease. These defense mechanisms consist of nonspecific and specific factors. Nonspecific factors include physical and biochemical barriers including skin and mucosal barriers, bone marrow, lymphoid tissue, digestive enzymes, pH, temperature, proteins, and enzymes such as lysozyme, transferrin, and interferon. Specific factors include cellular and humoral components that respond to foreign substances. In addition, individual genetic susceptibilities affect both nonspecific factors and specific factors. Nonspecific and specific factors make up two arms of the immune response—the innate (also known as natural or native immunity) and the adaptive (also known as specific or acquired immunity) (Table 13-1). Mechanisms of innate and adaptive immunity work cooperatively through complex interactions to prevent, control, and eradicate foreign antigens in the body without doing harm to the host. Host immune mechanisms and terminology are summarized in Table 13-2 and Figures 13-1 and 13-2. Innate and adaptive immunity are summarized in Boxes 13-1 and 13-2 on page 446. Roles of cytokines (interleukins and interferons) in innate and adaptive immunity are summarized in Table 13-3.
Table 13-1
Components of Innate and Adaptive Immunity
| CELLULAR COMPONENTS | HUMORAL COMPONENTS | MEDIATORS | |
| Innate | |||
| Adaptive | Antibodies | Cytokines |
*These gamma delta T cells are found primarily in the gut mucosa. Antigen presenting cells are monocytes and/or macrophages that have engulfed a foreign object, processed it, and displayed it, within an MHC-I or MHC-II complex at the cell surface, to a T cell to initiate the adaptive immune response.
Table 13-2
Active immunity: The response produced by an immunocompetent individual following exposure to foreign antigens (bacteria, viruses, attenuated or inactivated/killed viruses).
Adaptive immunity: Also known as “acquired immunity.” The result of active immunity wherein specific antibody (humoral) and cell-mediated responses establish memory cells and antibodies that provide immunity during subsequent exposures to a specific antigen.
Allograft: Graft taken from an organism that is the same species as the recipient but not genetically identical. The fetus is a “semi-allograft.”
Antibody: Proteins (immunoglobulins) that react with specific antigens. The five classes are IgA, IgD, IgE, IgG, and IgM.
Antibody (humoral)-mediated immunity: Adaptive immune mechanism mediated by B cells, which produces antibodies and protects the body from extracellular antigens.
Antigen: Substances perceived by one’s host defense mechanisms as “foreign” (may include bacteria, viruses, pollutants, dust, certain foods, for example).
Antigen presenting cell (APC): A cell that displays MHC with antigen complexes on the surface of the cell. Any cell can be an APC in that all cells have class I MHC. The term APC is used to refer to cells that also express class II MHC and can activate T cells, dendritic cells, macrophages, and B cells.
Cell-mediated immunity: Adaptive immune mechanism provided by immune cells. TCD4+ (T-helper cells), TCD8+ (T-cytotoxic cells), and TCD4+/CD26+ (T-regulatory cells) provide protection against certain organisms, regulate B-cell function, defend against cancer, and mediate graft rejection.
Chemotaxis: Movement of neutrophils and other phagocytes in an organized fashion toward a site of antigenic invasion.
Complement: Thirty discrete plasma proteins that, as part of the innate immune response, function in a cascade of reactions to form a membrane attack complex that lyses cells. The effector response of the complement cascade also opsonizes antigen, increases vascular permeability, and stimulates chemotaxis to aid phagocytosis. The complement cascade can proceed via the classical pathway or alternate pathway.
Cytokines: Glycoproteins such as lymphokines, interleukins, interferon, and tissue necrosis factor that are produced by various components of the immune system, especially T lymphocytes and macrophages. Cytokines function as autocrine or paracrine agents to activate different arms of the immune response.
Cytotoxic/killer T cell (TCD81): A type of T lymphocyte that acts directly on specific antigens or target cells that exhibit a specific antigen and induces cell lysis.
Fibronectin: Nonspecific opsonin, inhibitor of bacterial adherence to epithelial cells, and clot-stabilizing protein found in plasma and endothelial tissue.
Human leukocyte antigen (HLA): The major forms of specific tissue antigens found on tissue surfaces that are unique to each person (includes HLA-A, HLA-B, HLA-C, HLA-D, and in the placenta HLA-G).
Histocompatibility: The ability of tissue to accept a transplant from another individual.
Immunoglobulin: Antibodies produced by B lymphocytes (IgG, IgM, IgA, IgD, and IgE).
Innate immunity: Initial physical and nonspecific responses. Includes skin, mucosa, digestive enzymes. Primary effectors are polymorphonuclear neutrophils (PMNs), macrophages, monocytes, mast cells, natural killer (NK) cells, and complement. Innate immune response results in elimination of foreign substance or antigen and stimulation of the adaptive immune response.
Interleukin: A polypeptide member of the cytokine family. Produced by T cells, APCs, NK cells, and macrophages. Interleukins regulate and facilitate immune responses.
Interferon: Glycoprotein in the cytokine family. Primary role is in fighting viral infections.
Lymphokine: Subclass of cytokines. Mediators released by activated T cells that facilitate or act as costimulatory for immune reaction.
Memory cell: Form of B lymphocyte sensitized to specific antigens that has the ability to produce specific antibodies with subsequent stimulation by the specific antigen.
Major histocompatibility complex (MHC): Specific antigens found on tissue surfaces divided into two groups: MHC I (HLA-A, HLA-B, and HLA-C, which are found on most of a person’s cells), and MHC II (found on surface of immune cells such as T and B lymphocytes).
Natural killer (NK) cells: A subpopulation of lymphocytes in the innate immune response that form a first line of defense against cells infected with a virus and cancerous cells. NK cells have receptors that recognize class I MHC antigens and can attack without prior sensitization. They secrete cytokines that provide costimulation for T and B cells.
Opsonization: Processing and marking or altering the cell surface of an antigen by actions of immunoglobulin or complement; substances acting in this manner are called opsonins. This process is critical in allowing phagocytosis of organisms with capsular polysaccharide coats such as group B streptococci.
Passive immunity: Transfer of antibodies from an actively immunized to a nonimmunized person.
Plasma cell: Form of B lymphocyte able to secrete immunoglobulins.
Regulatory T cell: TCD4+/CD25+ cells. The exact function of these cells in unclear. They may inhibit production of interleukins (IL-10) that are necessary products and facilitators of the adaptive immune response. Thus they may help to slow or terminate immune response once the antigens are destroyed.
T helper cell (TCD41): A type of T lymphocyte that proliferates when in contact with an APC that exhibits class II MHC/antigen complexes (B cells, dendritic cells). TCD4+ cells differentiate into Th1 or Th2 cells based on the cytokines produced. Th1 cells participate in cell-mediated immunity and Th2 cells participate in antibody-mediated immunity. These cells thus enhance the activity of B lymphocytes, other T cells, and macrophages via secretion of cytokines.
T suppressor cells: T lymphocytes that express CD8+ cells and function to suppress the cellular immune response.
Th1 cells: A subset of TCD4+ (T-helper cells) that facilitates cell-mediated immunity via secretion of interleukin-12 (IL-12), tumor-necrosis factor (TNF)-β, and interferon (IFN)-γ. The result is stimulation of macrophages and inflammation.
Toll-like receptors (TLRs): Molecules on the surface of phagocytes and other cells that recognize the patterns of microbial products and generate signals to activate an innate immune response.
Table 13-3
Cytokines: Interleukins and Interferons
| CYTOKINE | PRINCIPAL SOURCE | ROLE IN INNATE IMMUNE RESPONSE | ROLE IN CELL-MEDIATED IMMUNE RESPONSE (Th1) | ROLE IN ANTIBODY-MEDIATED RESPONSE (Th2) |
| INTERLEUKIN (IL) | ||||
| IL-1 | Macrophages and other antigen presenting cells (APCs) | Costimulation of APCs and T cells, production of prostaglandin, inflammation, acute phase response, hematopoiesis | ||
| IL-2 | Activated Th1 cells, NK cells | Proliferations of activated T cells, NK functions | Proliferation of B cells | |
| IL-3 | Activated T cells | Growth of hematopoietic progenitor cells | ||
| IL-4 | Th2 and mast cells | Monokine production | B-cell proliferation, eosinophil and mast cell growth and function, IgE and class II MHC expression on B cells, inhibition of monocyte production | |
| IL-5 | Th2 and mast cells | Eosinophil growth and function | ||
| IL-6 | Activated Th2 cells, APCs, other somatic cells | Acute phase response | Synergistic with IL-1 and TNF on T cells, thrombopoiesis | B-cell proliferation and differentiation into plasma cells and antibody production |
| IL-7 | Thymic and marrow stromal cells | T and B lymphopoiesis | ||
| IL-8 | Macrophages, other somatic cells | Chemoattractant for neutrophils and T cells | ||
| IL-9 | T cells | Hematopoietic and thymopoietic effects | ||
| IL-10 | Activated Th2 cells, CD8+ T and B cells, macrophages | Suppresses cellular immunity, mast cell growth | Inhibits cytokine production, promotes B-cell proliferation and antibody production | |
| IL-11 | Stromal cells | Synergistic hematopoietic and thrombopoietic effects | ||
| IL-12 | B cells, macrophages | Proliferation of NK cells, INF-γ production | ||
| IL-13 | Th2 cells | Similar to IL-4, promotes development of eosinophils | ||
| INTERFERON (IFN) | ||||
| IFN-α and IFN-β | Mononuclear phagocytes | Antiviral effects, induction of class I MHC on all somatic cells, activation of NK cells and macrophages | ||
| IFN-γ | Activated Th1 and NK cells | Induces class I MHC on all somatic cells, induces class II MHC on APCs and somatic cells, activates macrophages, neutrophils, NK cells, promotes cell-mediated immunity, antiviral effects |
Modified from Silver, R.M., Peltier, M.R., & Branch, D.W. (2004). The immunology of pregnancy. In R.K. Creasy, R. Resnik, & J.D. Iams (Eds.), Maternal-fetal medicine: Principles and practice (5th ed.). Philadelphia: Saunders.
Maternal physiologic adaptations
Antepartum period
“Pregnancy is an immunological balancing act in which the mother’s immune system has to remain tolerant of potential major histocompatibility (MHC) antigens and yet maintain normal immune competence for defense against microorganisms.”167 The immune changes that occur systemically in the innate and adaptive systems and those that occur at the site of the fetal-maternal interface are described in this section.
Alterations in innate immunity
Mediators of the innate response are altered during pregnancy (Table 13-4). Chemotaxis (see the definition in Table 13-2) is decreased during pregnancy, which may delay initial maternal responses to infection.48,107 In addition, production of interleukin (IL)-4 and interferon-γ (IFN-γ) may also be decreased.107 Functionally, monocyte and granulocyte activity is enhanced, which results in faster and more efficient phagocytosis. This may help protect the mother so she does not mount a cell-mediated immune response to trophoblastic and fetal cells that appear in the maternal circulation. Natural killer (NK) cell activity is altered in a more complex manner. Systemic NK activity is down-regulated during pregnancy, secondary to the effects of progesterone, which appears to induce formation of a blocking factor that decreases lymphocyte proliferation and NK activity.39 The result is that systemic NK cytolytic activity is normal in the first trimester but lower during the second and third trimesters and immediately after delivery.
Table 13-4
Alterations in Host Defense Mechanisms during Pregnancy
| ALTERATION | RESULT | IMPLICATION |
| INNATE IMMUNITY | ||
| Increased PMNs | Increased available phagocytes | Protection of mother and fetus from infection |
| Altered metabolic activity and chemotaxis of PMNs | Delayed initial response to infection, especially gram-negative organisms | Increased risk of colonization, urinary tract infection |
| Decreased NK cell killer activity | Delayed initial response to infection |
Total white blood cell (WBC) volume increases slightly beginning in the second month and levels off during the second and third trimesters. The total WBC count in pregnancy varies among individual women, ranging from 5000 to 12,000 per mm3, with values as high as 15,000 per mm3 reported (see Chapter 8).129 The increase is primarily due to increased numbers of polymorphonuclear neutrophils (PMNs), monocytes, and granulocytes.129 A slight shift to the left may occur, with occasional myelocytes and metamyelocytes seen on the peripheral smear. Changes in other WBC forms are minimal (see Chapter 8).
Alterations in PMN function have been reported.48 PMN attachment, ingestion, and digestion of Candida albicans have been found to be increased, possibly secondary to the effects of human chorionic gonadotropin (hCG). Even so, pregnant women have higher rates of fungal infection, which may be secondary to the effects of estrogen on nutrient availability for fungal growth in the reproductive tract.129 Estrogens may alter local mucosal barrier function, allowing adherence of pathogenic organisms and increasing the risk of colonization. Conversely, improved PMN antibody expression during pregnancy may enhance phagocyte recognition and destruction of antigen-antibody complexes.
Toll-like receptors (TLR) are essential for the innate recognition of microorganisms and endogenous signals from tissue breakdown products, such as fetal fibronectin, hyaluronan, matrix proteins, and biglycans.31 Both TLRs and the recently identified Nod-like receptors (NLR) are thought to be involved in regulating innate responses of the trophoblast to infections and tissue damage signals.4 Changes in the cervix and fetal membranes with tissue remodeling near parturition may release these endogenous signals as part of the cascade leading to labor onset (Chapter 4).31
Alterations in the inflammatory response
Throughout pregnancy, the maternal circulation is seeded with small particles from the trophoblast. These cell clusters function as antigens of fetal origin and stimulate a systemic inflammatory response. The point of direct maternal contact with the placenta is at the junction with maternal endothelial cells in the uterine spiral arteries (see Chapter 3).132 This contact activates endothelial cells and initiates an inflammatory response. However, a glycoprotein known as pregnancy zone protein (PZP) is produced during pregnancy, which has an inhibitory effect on the inflammatory process.150 PZP levels increase 100- to 200-fold during pregnancy. PZP inhibits phagocytosis and suppresses inflammatory responses and IL-2 function near the decidua-trophoblast interface.150,157
Alterations in the complement system
The complement system has a role in both innate and adaptive responses. Although pregnancy stimulates an overall activation of the complement system, this activation does not result in rejection of the fetus.134 Alterations in the function of the complement system during pregnancy begin at 11 weeks’ gestation with an increase (due to greater hepatic synthesis) in both total serum complement and specific proteins of the complement system, including C2, C3, and C3 split products (Figure 13-3).92,134 These components enhance chemotaxis and actions of immunoglobulins through opsonization, thereby augmenting maternal defenses against bacterial infection. Other protein fragments of the complement system, such as C1, C1a, B, and D, are decreased.134 C1q (involved in activation of the classic complement pathway, immune cell modification, cell processes, and maintenance of immune tolerance) is synthesized and secreted by decidual epithelial cells during pregnancy and is thought to have a role in changes in decidual blood vessels and mediation of cell-to-cell interaction between decidual and trophoblast cells.115 Because complement fragments are involved in activation of the complement system, through either the classic or the alternative pathway, activity of the complement system early in the immune response may be delayed during pregnancy.
Alterations in adaptive immunity
Pregnancy is characterized by a switch in the balance of Th1 and Th2 cell subsets that results in enhanced Th2 function. This change is probably mediated by progesterone, which stimulates production of Th2 cytokines (e.g., IL-3, IL-4, IL-6, IL-10) and reduces the number of Th1 cytokines (e.g., IL-2, IFN-γ, tumor necrotic factor-β [TNF-β]) (see Table 13-3).31,47 Because Th2 responses tend to enhance antibody-mediated responses and Th1 responses enhance cell-mediated immunity (Figure 13-4), this change alters the function of both antibody-mediated and cell-mediated responses. This has a potentially protective role in the maternal-fetal immune relationship.31 Decreases in the number of Th1 cytokines may decrease maternal resistance to the spread of bacterial and viral organisms.31,48 If there are alterations in this pattern so that Th1 predominates during pregnancy, inflammatory cytokine production increases, and is associated with spontaneous abortion, preeclampsia, preterm labor, and fetal growth restriction.31,138 Alterations in adaptive immunity during pregnancy are summarized in Table 13-4.
Alterations in cell-mediated immunity.
Cell-mediated immunity is somewhat suppressed during pregnancy. Lymphocyte and macrophage synthesis, activation, and function are altered slightly in pregnancy secondary to influences from estrogens, corticosteroids, progesterone, α-fetoprotein (AFP), hCG, human placental lactogen (hPL), prostaglandins, and serum proteins.47 Corticosteroids suppress activation of T-cell lymphokines, phagocytic activity, and lymphokine responsiveness of the macrophages.47 Prostaglandins (especially PGE1 and PGE2), hPL, and AFP also appear to have an immunosuppressive role during pregnancy. AFP may induce production of regulatory T lymphocytes (CD4+/CD25+).48
Most investigators report that the total number of lymphocytes does not change significantly during pregnancy.48,168 Some investigators have reported a decrease in the T-helper–to–T-suppressor (CD4+/CD8+) ratio. However, most reports indicate that the number of TCD4+ (T-helper cells) decrease progressively to term, while the number of T-suppressor cells (TCD8+) tend to remain relatively unchanged.48
Because TCD4+ cells normally augment the cytotoxic responses involved in graft rejection, a decreased number of these cells may help protect the fetus from rejection by the mother. T-suppressor cell function may increase in late pregnancy and suppress B-cell function.107 Decreased T-lymphocyte function and efficiency may increase the risk of viral and mycotic infections.
Alterations in antibody-mediated (humoral) immunity.
While a few studies have reported an increase in the number of B cells during pregnancy, most studies do not support this finding.48 Some investigators have found that levels of maternal IgG fall as gestation progresses, with decreases ranging from 30% to 40% after 28 weeks.48 However, others have found relatively little change. A decrease in IgG has been attributed to hemodilution of pregnancy, enhanced loss of IgG in the urine, and transfer of maternal IgG to the fetus in the last trimester. This relative decrease in IgG, along with alterations in the WBC population, may increase the risk of bacterial colonization with certain pathogens (e.g., streptococci).
Immunoglobulin A (IgA) decreases or remains stable during gestation, immunoglobulin M (IgM) remains stable or may decrease slightly, immunoglobulin E (IgE) undergoes minimal or no change in levels, and immunoglobulin D (IgD) increases until term. The slight decrease in serum IgA may reflect increased levels of IgA found in saliva and other mucosal fluids.48 The specific role of IgD in pregnancy is unknown.
Immune function at the fetal-maternal interface
The fetus is a semiallograft, that is, foreign tissue from the same species but with a different antigenic makeup. Because the fetus has maternal, paternal, and embryonic antigens, major antigenic differences may exist between the fetus and mother, including blood group antigens and tissue antigens such as human leukocyte antigen (HLA). Protection of the fetus from rejection seems to be predominantly a localized uterine response, although there are also systemic responses mediated primarily by endocrine factors.158 Maternal tolerance of the fetal-placental unit has traditionally been believed to be secondary to elaboration of an immune “barrier” between the placenta and maternal tissue.158 However, it is now understood that maternal immune systems are “aware of fetal antigens, that they respond vigorously to the presence of the fetus and that under normal circumstances they are tolerant to these antigens.”124 Thus tolerance of the fetus by the maternal immune system is “an active mechanism whereby fetal tissues are prevented from being recognized as foreign and/or from being rejected by the cells of the maternal immune system.”158 During pregnancy the trophoblast and maternal immune system have a regulatory and supportive interaction that is critical for both pregnancy maintenance and protection against infectious microorganisms.107 Figure 13-5 illustrates interactions between the maternal immune system and the placental trophoblast.
Sperm are MHC class I– and II–negative, which usually protects them from recognition as foreign by the maternal immune system, although both sperm and seminal fluid carry other antigens. Seminal fluid also contains immunosuppressors. The woman responds to specific seminal fluid–induced cytokine and chemokine signals to increase immunosuppressive T-regulating (Treg) cells.137,138 Exposure to paternal antigens on sperm and seminal fluid before pregnancy may lead to decreased responsiveness and maternal tolerance to paternal MHC antigens.138,167
How does trophoblast eliminate or evade the immune responses of B-cell and T-cell activation given the general enhanced innate and humoral immune function that is seen in the systemic circulation during pregnancy? Factors that are currently thought to have a role in maternal tolerance of the fetus include trophoblast HLA-G; progesterone, progesterone induced-blocking factor, altered NK cell function, a Th1/Th2 balance that favors Th2 responses; indolamine 2,3 dehydrogenase (IDO) (catalyzes tryptophan in lymphocytes, an amino acid needed for T-cell proliferation and survival); Fas ligand (immune modulating protein that induces apoptosis of maternal lymphocytes that are fetal antigen reactive); suppressor macrophages; annexin II; lowered complement activity; toll-like receptor (TLR) expression and function; local immune suppression (mediated by Fas/Fas ligand system), and possibly other mechanisms that hide trophoblastic antigens from the maternal immune system.2,39,50,107,142,158
Implantation, the first point of interaction between maternal and fetal tissues, involves an inflammatory process with cross-talk (molecular dialogues) between the decidua and conceptus that increases activity of innate immune cells and is mediated by hormones, growth factors, cytokines, chemokines, adhesion molecules, extracellular matrix components and matrix metalloproteins.51 The inflammatory process at the maternal-placental interface is most marked in the first and third trimesters. In the first trimester these changes occur with the initial contact between the embryonic trophoblast and maternal tissues with implantation and the remodeling of maternal blood vessels.107 In the third trimester inflammation is a critical component of cervical changes in preparation for parturition and labor onset (see Chapter 4).107
After the placenta is established, the two points of contact between maternal and fetal tissues are the syncytiotrophoblast and the extravillous trophoblast. The syncytiotrophoblast is in contact with the endothelial cells of maternal spiral arteries and with the intervillous spaces, and is in direct contact with maternal blood. The extravillous trophoblast (primarily cytotrophoblast) forms the anchoring villi, which are in direct contact with maternal decidual tissue, and remodels maternal spiral arteries (see Chapter 3). Beginning in early pregnancy, small quantities of trophoblast cells detach and enter maternal blood through the uterine veins. These cells form minute emboli that eventually lodge in pulmonary capillaries and are cleared by proteolysis. This appears to be a normal process that does not lead to a maternal inflammatory response or other respiratory distress in women experiencing a normal pregnancy.
Trophoblast and major histocompatibility antigens.
The syncytiotrophoblast does not express either class I or class II MHC antigens, which are the main T cell targets in transplantation rejection.124 The cytotrophoblast has a unique nonclassical MHC-I surface antigen, HLA-G, which does not stimulate the classic cytotoxic T-cell response.85 HLA-G has a Fas/Fas ligand pathway for killing activated T cells and may help prevent maternal rejection of the fetus by inducing apoptosis of activated maternal T cells.32,85 HLA-G inhibits NK cell cytotoxicity and dendritic cell (an antigen presenting cell) maturation.50,176 HLA-G synthesis may be stimulated by IL-10 from the placenta. Extravillous trophoblast expresses HLA-G, HLA-C, and HLA-E (nonclassical MHC-I molecules that help these cells to evade maternal NK toxicity).111,136,176 HLA-G acts on cells of both the innate and adaptive immune systems and has a major role in reprogramming maternal immune responses at the maternal-fetal interface.136 Soluble HLA-G (sHLA-G) molecules are found in maternal plasma and increase from the first trimester on, peaking in the third trimester.136 Decreased expression of HLA-G on the extravillous trophoblast, seen in women with recurrent abortion and preeclampsia, alters conversion of the spiral arteries (see Chapter 3).136,176 Decreased sHLA-G is found with placental abruption, miscarriage, and preeclampsia.135
Trophoblastic cells (specifically the villous cytotrophoblast and extravillous trophoblast) express TLRs and may be able to initiate an innate immune response in the presence of microorganisms at the site of implantation.2,106 In addition, trophoblastic cells contain IDO, which enzymatically degrades tryptophan, a protein essential for T-cell proliferation, and TNF ligands that induce apoptosis. Together these molecules can kill activated immune cells.158
Natural killer cells.
Two subsets of uterine NK cells are seen: endometrial NK cells, seen during the menstrual cycle, and decidual NK cells, which are seen during pregnancy.124,182 Decidual NK cells are critical for pregnancy maintenance.72 The first direct contact between maternal and fetal tissue occurs 6 to 7 days after conception at the time of implantation. Subsequently, the extravillous trophoblast cells invade the maternal endometrial lining and erode the endothelial tissues that line the maternal spiral arteries (see Chapter 3). NK cells in the pregnant decidua are phenotypically distinct from the NK cells in systemic circulation. Peripheral NK cells express CD56dim, CD16+, and CD160+, a combination of characteristics that allow them to be highly cytotoxic. Conversely, decidual NK cells express CD56bright, CD16−, and CD160−, which allows them to produce cytokines such as IL-8 and IL-10 that are beneficial for trophoblastic invasion.69,72,95,111,131 These NK cells also secrete vascular epithelial growth factor (GF) and placental GF, which are angiogenic factors that promote trophoblast invasion and vascular remodling.111,124 NK cells accumulate at the site of implantation and in the decidua. They recognize HLA-G, HLA-C, and HLA-E on the invading trophoblast and influence production of cytokines and cytolytic factors that facilitate decidualization and control of trophoblast invasion, angiogenesis, and growth.86,95,107,119,176 Galectin-1 (Gal-1) is a carbohydrate binding protein with immunoregulatory functions that is found in high levels in uterine NK cells; Gal-1 is also found on trophoblast cells and may help reduce their cytotoxicity.72,130 Up-regulation of Gal-1 induces differentiation of dendritic cells into a phenotype that dampens Th1 responses and suppresses autoimmune inflammation.115 Trophoblast HLA-G can block receptors on the NK cell that initiate cell lysis. Thus the NK cell is altered to augment those functions that protect the conceptus while the general cytotoxic roles are down-regulated. Figure 13-6 illustrates how HLA-G on the trophoblast may inhibit maternal NK cells and block NK cytotoxicity toward fetal cells. Maternal stress, smoking, infection, and other factors can alter the NK phenotype and increase the risk of pregnancy complications.72
Decidual macrophages.
Macrophages cluster at the maternal-fetal interface and near the spiral arteries during trophoblast invasion (see Figure 13-5).3 These macrophages produce IL-10, IDO, PGE2, and other anti inflammatory molecules that trigger alloreactivity by the T cells to further protect the fetus.113,158 Placental growth hormone (see Chapter 19) may also alter the maternal immune system.158 Annexin II, a glycoprotein that binds to phospholipids, is produced by the placenta and may inhibit lymphocyte proliferation and IgG and IgM secretion at the placental site. Macrophages stimulated by Th1 cytokines or bacterial products develop a M1 phenotype and produce nitric oxide and free radicals to defend against microorganisms. Macrophages stimulated by Th2 cytokines develop a, M2 phenotype and have immunosuppressive properties. M2 is the common phenotype of decidual macrophages involved in costimulatory signaling.113
T cells and the TH1/TH2 switch.
Overall, there are more TCD8+ cells than CD4+ cells in the maternal decidua, which causes immunosuppressive activity to predominate.107 Changes in the balance of the different T-helper subsets (Th1 versus Th2) result in strengthening of Th2 (antibody-mediated) responses and cytokines (IL-3, IL-4, IL-5, IL-6, and IL-10) and a reduction in Th1 (cell-mediated and graft rejection) responses and cytokines (IL-2, TNF-α, and IFN-γ). This distribution of cytokines at the maternal-fetal tissue interface (see Figure 13-6) is important in implantation, pregnancy maintenance, and maternal tolerance of the fetus. The Th2-specific cytokines IL-4, IL-5, and IL-10 inhibit inflammation and some macrophage functions, and down-regulate IFN-γ production, whereas the Th1 cytokines are the primary effectors of phagocytosis.39,111,126,158 T-regulatory (Treg) cells (see Box 13-2 on page 446) normally account for 5% to 10% of peripheral CD4+ T cells. These increase two- to threefold during pregnancy beginning prior to implantation, peaking in the second trimester, and decreasing by term (this decrease correlates with the decrease in progesterone).111,127 Decreased Treg cells are seen with spontaneous abortion.111,127 The maternal T cells interact with trophoblast cells to support remodeling of the uterine vasculature by removing apoptotic cells, producing matrix proteases, mediating immunotolerance of the fetus, and protecting the fetus from pathogens.113
Nevertheless, the mother initially creates a weak immune response to the fetus/placenta, as evidenced by production of antibody to paternal MHC antigens, production of specific T cells, and weak cellular sensitivity to paternal and fetal antigens.75 This response usually results in an activation (facilitation) reaction rather than rejection response that is mediated by Th2 helper cells with release of Th2 cytokines, which down-regulate Th1 cytokines such as IL-2, IFN-γ, and TNF-α. As a result, maternal immune responses are down-regulated locally at the maternal-placental interface, but are not significantly affected at the systemic level.39 Failure of Th2 responses to increase or of Th1 responses to decrease is associated with an increased risk of recurrent abortion.25,101
Role of progesterone.
Progesterone promotes production of IL-4 and IL-6, the Th1-to-Th2 switch, and IL-4.31,50,126 Progesterone also stimulates activated lymphocytes and decidual cells to synthesize progesterone-induced blocking factor (PIBF).39,155 PIBF stimulates B-cell production of asymmetric antibodies, which are antibodies that have a mannose-rich oligosaccharide group present on one of the Fab regions. Asymmetric antibodies cannot initiate complement or phagocytosis but they can combine with antigen. The result of this combination is an antigen-antibody complex that is univalent and functions as a “blocking antibody” that prevents further antigen-antibody interaction. PIBF also inhibits the cytotoxicity of NK cells.39,
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
