CHAPTER 330 Imaging of Traumatic Brain Injury
TBI lesions can be classified into primary and secondary injuries (Table 330-1). Primary injuries are the direct result of trauma to the head. Secondary injuries arise as a complication of the primary lesions. This classification is important because secondary injuries are potentially preventable, whereas primary injuries, by definition, have already occurred by the time the patient first presents for medical attention. This classification also underscores the fact that TBI is not just a static event, but is rather a progressive injury with varying therapeutic windows. TBI can be further divided according to location (intra-axial or extra-axial) and mechanism (penetrating/open or blunt/closed). Clinical classification of the severity of TBI is currently based on the Glasgow Coma Scale (mild: 13 ≤ GCS ≤ 15; moderate: 9 ≤ GCS ≤ 12; severe: 3 ≤ GCS ≤ 8).1 The goal of neuroimaging is to identify treatable injuries, assist in the prevention of secondary damage, and provide useful prognostic information.
TABLE 330-1 Imaging Classification of TBI
| “Primary” Injury |
| Extra-axial Injury |
| Intra-axial Injury |
| Vascular Injury |
| “Secondary” Injury |
| Acute |
| Chronic |
Imaging Options
Conventional Radiography
Skull fracture, even without clinical signs, is an independent risk marker for a neurosurgically relevant intracranial lesion.2 However, skull films are mainly used for the identification of skull fractures in cases of suspected child abuse, and not for the evaluation of intracranial pathology in children or adults. In fact, conventional radiography is a poor predictor of intracranial pathology and should not be performed to evaluate TBI.3–5 In mild TBI, skull films rarely demonstrate significant findings. In severe TBI, the lack of abnormality on skull films does not exclude major intracranial injury. In one large autopsy series of patients with fatal head injuries, only 75% had skull fractures.6 Negative findings may even mislead medical management. Patients who are at high risk for acute intracranial injury, even without clinical evidence of skull fractures, must be imaged by computed tomography (CT).
Computed Tomography
In the setting of acute head trauma, CT is recommended for patients with moderate and severe TBI (GCS ≤ 12); who are older than 60 years; and have persistent neurologic deficit, headache or vomiting, amnesia, loss of consciousness of 5 minutes or more, depressed skull fracture, penetrating injury, bleeding diathesis, or had anticoagulation therapy.7–13 CT is the modality of choice because it is fast, widely accessible, and highly accurate in the detection of skull fractures and intracranial hemorrhage. Life support and monitoring equipment can be more easily accommodated in the CT scanner suite than in the magnetic resonance imaging (MRI) suite. In addition, CT is superior to MRI in revealing skull fractures and radiopaque foreign bodies. Indeed, MRI is contraindicated in the presence of certain foreign bodies. Noncontrast CT scans can provide rapid and accurate localization of space-occupying hematomas, associated mass effect, and impending complications that would require immediate medical and/or surgical intervention. Intravenous contrast administration should not be performed without a baseline noncontrast examination because the contrast can both mask and mimic underlying hemorrhage.
CT angiography (CTA) uses intravenous iodinated contrast to delineate the vascular structures at high (submillimeter) resolution. CTA is best performed with multidetector CT (MDCT) and rapid bolus contrast injection using a tracking technique. In suspected vascular injury, such as a fracture traversing the carotid canal or venous sinus, CTA can serve as a useful screening method.14
Dynamic perfusion CT is a technique for measuring brain hemodynamics by tracking transient attenuation changes in the blood vessels and brain parenchyma during the first-pass passage of an intravenously injected contrast bolus. Perfusion CT typically involves continuous cine scanning with a scan rate interval of 1 second and a total scanning duration of 40 to 45 seconds.15 Color maps of cerebral blood volume (CBV), mean transit time (MTT), and cerebral blood flow (CBF) can be generated from a voxel-by-voxel analysis of the change in attenuation over time. CT perfusion in patients with severe head trauma has been shown to provide independent prognostic information regarding functional outcome, with normal brain perfusion or hyperemia in patients with favorable outcome, and oligemia in patients with unfavorable outcome.16 One limitation of dynamic perfusion CT is limited anatomic coverage because only a few slices of the brain can be imaged during the 1-second window. Wider coverage can be achieved using a 40-mm wide detector and toggling table technique or a scanner with more multidectors.17,18 Another limitation of CT perfusion is the additional radiation exposure due to cine imaging. Radiation exposure is not an issue with MR perfusion imaging.
Magnetic Resonance Imaging (MRI)
On occasion, MRI may be indicated in patients with acute TBI if the neurologic findings are unexplained by the CT findings (e.g., hemiparesis). However, because of the risks to the patient imposed by the high-strength magnets in modern MRI units and the long scan times, routine MRI during the first week after severe TBI, especially with multiple injuries, is rarely performed. MRI is the preferred imaging modality for subacute and chronic TBI because of its superior sensitivity to both gray and white matter injury. MRI is comparable to CT in the detection of the majority of acute epidural and subdural hematomas.19,20 However, MRI is more sensitive to subtle extra-axial “smear” collections, nonhemorrhagic lesions, and brainstem injuries. It can also be more sensitive to subarachnoid hemorrhage.21,22
Fluid attenuated inversion recovery (FLAIR) imaging increases the conspicuity of focal cortical injuries (e.g., contusions), white matter shearing injuries, and subarachnoid hemorrhage by suppressing the adjacent bright cerebrospinal fluid (CSF) signal typically seen on conventional T2-weighted images. Note, however, that the abnormal high signal in the sulci and cisterns of ventilated patients receiving a high inspired oxygen fraction (>0.60) can be observed in uninjured patients and should not be mistaken for a hemorrhage.23 Sagittal and coronal FLAIR images are particularly helpful in the detection of diffuse axonal injury (DAI) involving the corpus callosum and the fornix, two areas that can be particularly difficult to evaluate on routine axial T2-weighted images.24
Gradient-recalled-echo (GRE) T2*-weighted imaging is highly sensitive to the presence of blood breakdown products, including deoxyhemoglobin, intracellular methemoglobin, ferritin, and hemosiderin. The presence of these molecules alters the local magnetic susceptibility of tissue, resulting in areas of signal loss on GRE T2*-weighted images. Because hemosiderin can persist indefinitely, its detection on GRE T2*-weighted images allows for improved evaluation of remote TBI. Unfortunately, small foci of hemosiderin can sometimes be resorbed; therefore, the lack of hemosiderin on GRE T2*-weighted images does not exclude old hemorrhage.25
Susceptibility-weighted imaging (SWI) can be likened to a supercharged GRE sequence. It amplifies the susceptibility changes among tissues and blood products by combining magnitude and phase information from a high-resolution, full velocity compensated 3-D T2-weighted gradient echo sequence.26 Conventional MRI relies only on the magnitude images and ignores the phase images, the latter of which contain valuable information regarding tissue susceptibility differences. In SWI, phase images are unwrapped and high-pass filtered to highlight phase changes. These are then converted into a mask that is multiplied with the corresponding magnitude images. SWI images are displayed using minimum intensity projection (minIP) reconstruction. The increase in tissue magnetic susceptibility contrast afforded by SWI improves the sensitivity to small TBI hemorrhages, as well as other diseases associated with hemorrhage (e.g., amyloid angiopathy). SWI is 3 to 6 times more sensitive than GRE T2*-weighted imaging in detecting hemorrhagic DAI.27–29
Diffusion-weighted imaging (DWI) measures the random motion of water molecules in brain tissue. Because of its sensitivity to acute shearing injury, DWI has been particularly useful for the detection of DAI.30–33 DWI reveals more DAI lesions than fast spin echo T2-weighted and/or GRE T2*-weighted images in patients imaged within 48 hours of injury. Acute DAI lesions typically show reduced apparent diffusion coefficient (ADC), which measures the magnitude of water diffusion averaged over a three-dimensional space. The fractional anisotropy (FA), which measures the preferential motion of water molecules along the white matter axons, is frequently reduced in chronic DAI. The integrity of the white matter tracts can be further assessed with diffusion tensor imaging (DTI) with 3-D tractography. Unfortunately, the white matter tracts generated with DTI tractography are quite variable and depend on the parameters used to create the tracts. Abnormalities within the tractogram, while visually very appealing, are not specific for TBI. More research is necessary before DTI tractography can be reliably incorporated into the imaging armamentarium.
MR spectroscopy (MRS) measures the relative amount of metabolites in brain tissue. Common neurochemicals that are measured with proton ([1H]) MRS include N-acetylaspartate (NAA), creatinine (Cr), choline (Cho), glutamate, lactate, and myo-inositol. NAA is a cellular amino acid and is a marker of neuronal health. Creatinine is a marker of energy metabolism and cellular density. Creatine is especially abundant in glial cells, and can serve as a marker for posttraumatic gliosis. A reduction in NAA:Cr ratio has been found in patients with history of TBI, and this finding correlated with a worse prognosis.34 An increase in Cho is observed in myelin injury. A reduction of NAA and an elevation of Cho correlate with the severity of injury as measured by the GCS and duration of posttraumatic amnesia.35 MRS can be useful since it can detect abnormalities that are invisible on conventional MRI.35,36
Magnetization transfer imaging (MTI) exploits the longitudinal (T1) relaxation coupling between bound (hydration) protons and free water (bulk) protons. When an off-resonance saturation (radiofrequency) pulse is applied, it selectively saturates protons that are bound in macromolecules. These protons subsequently exchange longitudinal magnetization with free water protons. The magnetization transfer ratio (MTR), a relative measure of the reduction in signal intensity because of the MT effect, provides a quantitative measure of the structural integrity of tissue. A reduction of the MTR correlates with a worse clinical outcome in patients with history of TBI.34
Magnetic Source Imaging (MSI)
Magnetic source imaging (MSI) uses magnetoencephalography (MEG) to localize a weak magnetic signal generated by neuronal electrical activity. Electrical currents flowing within dendrites give rise to a surrounding magnetic field that can be measured by superconducting quantum interfering devices (SQUID). MEG provides a selective reflection of activity in dendrites oriented parallel to the skull surface. MSI integrates anatomic data obtained with conventional MRI and electrophysiologic data obtained with MEG. In two recent studies, MSI showed excessive abnormal low-frequency magnetic activity in mild TBI patients with postconcussive syndromes.37,38 Application of MSI in the evaluation of TBI has been lacking because of the limited availability of MEG, mainly because of cost. Additional research is necessary before MSI is adopted in the clinical setting.
Single Photon Emission Tomography (SPECT)
Single photon emission tomography (SPECT), a nuclear medicine study, uses gamma-emitting isotopes (e.g., [133Xe] and technetium-99-m-hexamethyl-propylamine-oxime [99Tc-HMPAO]) to measure cerebral blood flow (CBF). It can potentially provide a better long-term prognostic predictor in comparison to CT or conventional MRI.39 For example, a worse prognosis has been associated with multiple CBF abnormalities, larger CBF defects, and defects that involve the basal ganglia, temporal and parietal lobes, and brainstem. However, due to its inherent low spatial resolution, SPECT is less sensitive in detecting smaller lesions that are visible on MRI. Therefore, SPECT imaging is complementary to, but not a replacement for, MRI in the evaluation of TBI.
Positron Emission Tomography (PET)
Positron emission tomography, as the name implies, uses positron-emitting isotopes, commonly 15-oxygen (15O) to measure cerebral perfusion and oxygen metabolism, and 2-fluoro-2-deoxy-D-glucose (18F-FDG) to measure cerebral glucose metabolism. PET is relatively expensive and not widely available, serving mainly as a research tool. 15O-PET can define a potential ischemic area after brain injury, which is associated with poor outcome.40–42 Acutely injured brain cells show increased glucose metabolism following severe TBI because of intracellular ionic perturbation.43 Following the initial hyperglycolysis state, injured brain cells show a prolonged period of regional hypometabolism, lasting up to 1 month. Early human studies in TBI have had limited success in demonstrating consistent results regarding regional glucose metabolism. Due to the heterogeneous nature of TBI, studies have found both hypermetabolism and hypometabolism in the same regions across different TBI patients.44 The “metabolic abnormalities” can also be found extending far beyond the lesions, especially with subdural and epidural hematomas.45 The cortical contusion, intracerebral hematoma, and encephalomalacia all tend to show more regional metabolic abnormalities confined to the specific lesions. A more recent study was able to show almost consistent hypermetabolism within the cerebellar vermis of the injured brain.46
Imaging Findings
Scalp and Skull Injury
Scalp injury is often the only reliable evidence of head impact, and thus can identify the coup site. When reviewing CT images, first look at the AP and lateral scout images (Fig. 330-1). Sometimes, the scout image can reveal unsuspected upper cervical spine injury or a horizontal skull fracture that will be missed if only the axial CT images are reviewed. When evaluating the CT and MR cross-sectional images, begin by examining the extracranial structures for evidence of scalp trauma (Fig. 330-2; see Figs. 330-6, and 330-15), such as a laceration, subgaleal hematoma, or radiopaque foreign body. The subgaleal hematoma is located beneath the subcutaneous fibrofatty tissue and superficial to the temporalis muscle (see Fig. 330-15).
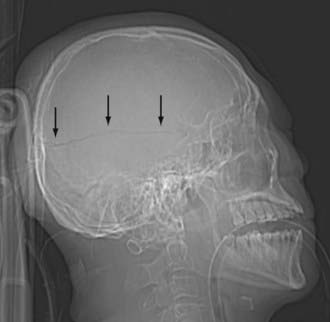
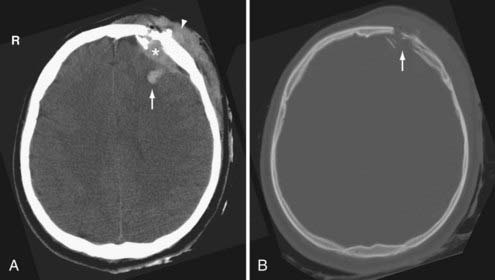
Skull fractures can be classified as linear, depressed, comminuted, compound (open), or diastatic. The linear fracture is the most common type of skull fracture. As mentioned earlier, the nondisplaced linear skull fractures may be difficult to detect, even on CT, if the fracture plane is parallel to the plane of section. Fortunately, isolated linear fractures without associated intracranial pathology are usually clinically insignificant. Depressed and comminuted (i.e., multiple bone fragments) skull fractures are easily detectable on CT (Fig. 330-2). Depressed fractures are often associated with an underlying cortical contusion. When a fracture involves the skull base, communicates with the overlying scalp, or involves the paranasal sinus, it is called a compound fracture. An external compound fracture is typically caused by a penetrating injury and is commonly associated with a depressed, comminuted skull fracture. An internal compound fracture is usually caused by blunt injury and includes those that extend from the mastoid, middle ear, and paranasal sinuses into the cranium. Compound fractures frequently result in pneumocephalus (intracranial air), and they have an increased risk of infectious complications.47 In a compound depressed fracture, the underlying dura is exposed under the fracture site. This is usually a neurosurgical emergency warranting removal of the contaminated bone fragments and dural repair. Diastatic fractures occur from separation of the cranial sutures and are more frequent in children than in adults.
Temporal bone fractures (TBF) are classified either according to their orientation (transverse versus longitudinal) relative to the long axis of the petrous bone (Ulrich’s original classification)48 or based on their involvement of the otic capsule (otic capsule sparing versus otic capsule violating).49,50 The older Ulrich’s classification is still more commonly used, while the newer classification is more predictive of clinical outcome. Longitudinal fractures are oriented parallel to the long axis of the petrous pyramid (Fig. 330-3). They frequently result from direct impact to the side of the head. They represent 70% to 90% of temporal bone fractures.51 Longitudinal fractures can lead to subluxation or fracture of the ossicles, conductive hearing loss, CSF otorhinorrhea, and facial nerve palsy (often partial and delayed). In contrast, transverse fractures are oriented perpendicular to the long axis of the petrous bone (Fig. 330-4). They typically result from impact to the frontal or occipital region. Sensorineural hearing loss, vertigo, nystagmus, perilymphatic fistula, and facial palsy are common complications of transverse fractures. Facial palsy (often complete) occurs in 30% to 50% of transverse fractures.51
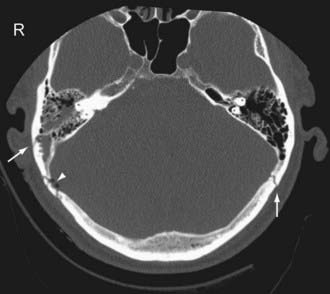
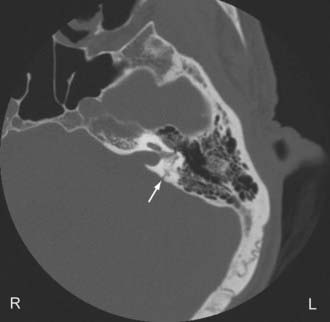
Most temporal bone fractures are oblique and mixed, and the simple classification of fractures as longitudinal or transverse may not be sufficient.52 Otic capsule sparing fractures run anterolateral to the otic capsule, and are usually caused by direct blows to the temporoparietal region. With otic capsule violating fractures, the cochlea and the semicircular canals are damaged. These fractures are the results of direct impacts to the occipital region. Compared with otic sparing fractures, patients with otic capsule violating fractures are 2 to 5 times more likely to develop facial nerve injury, 4 to 8 times more likely to develop CSF leak, and 7 to 25 times more likely to experience hearing loss, as well as more likely to sustain intracranial injuries such as epidural hematoma and subarchnoid hemorrhage.49,50
Primary Extra-Axial Injury
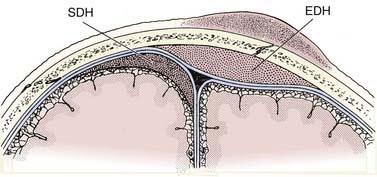
The epidural hematoma (EDH) develops within the potential space located between the dura and the inner table of the skull. The hematoma dissects the dura from the inner table of the skull, forming an ovoid collection that compresses the underlying brain. Since the EDH is subperiosteal, it rarely crosses cranial sutures, where the outer periosteal layer of the dura is firmly attached at sutural margins (Fig. 330-5). At the vertex, however, where the periosteum is not tightly attached to the sagittal suture, the EDH can cross the midline.
EDHs are usually arterial in origin. The vast majority of EDHs occur at the site of impact (i.e., the coup site) and are usually associated with a skull fracture, commonly involving the temporal squamosa region where the fracture disrupts the partially embedded middle meningeal artery.13 In children, EDHs may occur from stretching or tearing of meningeal arteries without an associated fracture. EDHs are less common in children and in the elderly for several reasons. First, the overall incidence of severe head trauma is less in children and the elderly. Second, the skull is more compliant and the meningeal groove is more shallow in children. Third, the dura is more adherent to the inner table of the skull and is therefore not easily displaced in the elderly.
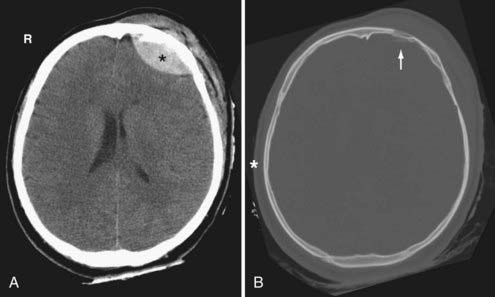
On CT, an acute EDH appears as a well-defined biconvex hyperdense collection within the epidural space that causes focal brain compression (Fig. 330-6). Mass effect, sulcal effacement, and midline shift are frequently seen with large EDHs. The attenuation value of an acute EDH usually ranges between 50 to 70 Hounsfield units (HU).
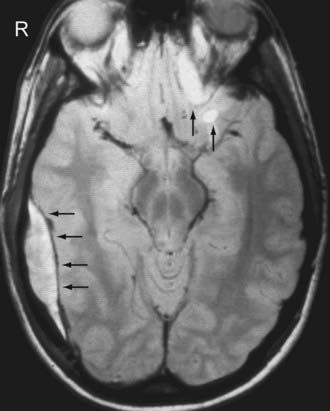
On MRI, a thin dark line is observed at the inner margin of the EDH (Fig. 330-7). This line represents the two layers of displaced dura and confirms the epidural location of the hematoma. Inward displacement of the venous sinuses also serves as a clue that the hematoma is located within the epidural space. As is the case with hematomas elsewhere, the signal characteristics of the EDH are dependent on the age of the blood products.
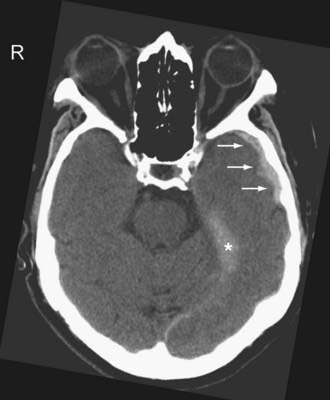
Venous EDHs are less common than arterial EDHs and they tend to occur in three locations: (1) posterior fossa (from rupture of the torcula or transverse sinus), (2) middle cranial fossa (from disruption of the sphenoparietal sinus) (Fig. 330-8A), and (3) vertex (due to injury to the superior sagittal sinus or cortical veins).53 Unlike the arterial EDH, the venous EDH rarely expands beyond its initial size because of the lower pressure imposed by venous extravasation. The venous EDH is less frequently associated with a skull fracture than is the arterial EDH.
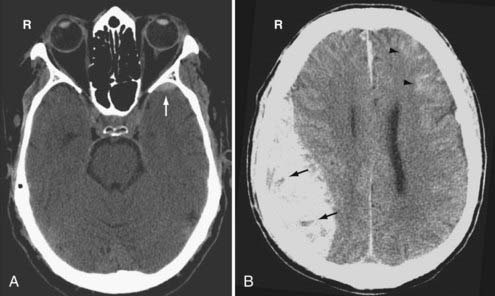
Certain imaging characteristics of the EDH are correlated with a worse prognosis and require more aggressive management. One important imaging finding is the presence of low-density areas within the hyperdense hematoma (the so-called swirl sign), thought to represent active bleeding (Fig. 330-8B).54,55 It is an ominous sign that forewarns expansion of an arterial EDH. Patients with this type of EDH tend to present early, with a poorer GCS and a higher mortality rate.56 Contrast extravasation within the low-density areas of the EDH due to active hemorrhage from an underlying dural vessel laceration has also been reported.57 Thus, active extravasation on CT may be another potential biomarker for EDH expansion, and when used in conjunction with other imaging characteristics such as mixed density, it may warrant more aggressive clinical management. Midline shift greater than 1 cm and brainstem distortion are additional imaging findings that often require aggressive management.
Subdural hematomas (SDH) are located between the arachnoid and the inner meningeal layer of the dura. Because the dura and arachnoid are not firmly attached, the SDH is frequently seen layering along the entire hemispheric convexity from the anterior falx to the posterior falx (see Fig. 330-5). The SDH usually develops from laceration of bridging cortical veins during sudden head deceleration. It can also arise from injury to pial vessels, pacchionian granulations, or penetrating branches of superficial cerebral arteries. Another cause of the SDH is rapid decompression of obstructive hydrocephalus. In this setting, the brain surface recedes from the dura quicker than the brain parenchyma can reexpand after being compressed by the distended ventricles, causing disruption of the bridging cortical veins. The incidence of the SDH is higher in the elderly due to cerebral atrophy because the increase in the extra-axial space allows for increased motion between the brain parenchyma and the calvarium.
On CT, the typical acute SDH is hyperdense, homogeneous, and crescent-shaped (Fig. 330-9). The majority of SDHs are supratentorial and located over the convexity. They are frequently seen along the falx and tentorium (Fig. 330-10). Unlike the EDH, they tend to occur at the contrecoup site. Because the SDH is often associated with parenchymal injury, the degree of mass effect may appear more extensive than the size of the collection.
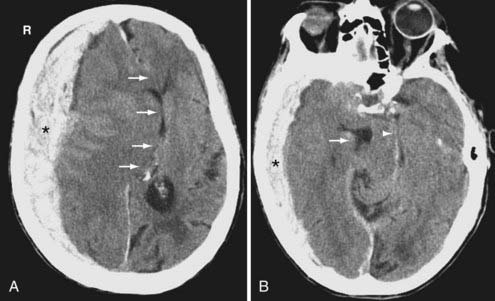
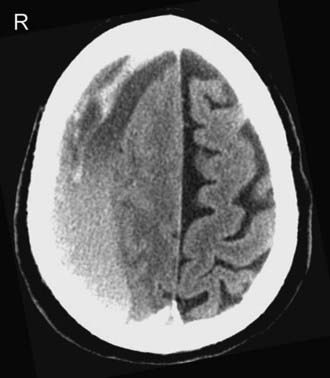
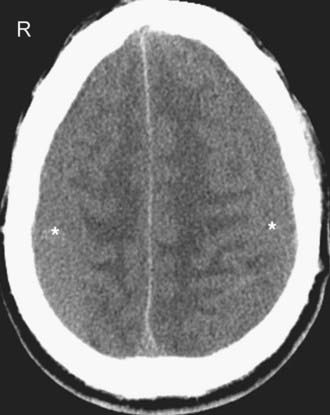
Compared with the normal brain (20 to 30 HU), the density (attenuation) of an acute SDH (50 to 60 HU) is higher because of clot retraction. The density of the SDH will progressively decrease as protein degradation occurs. Rebleeding during evolution of an SDH is not uncommon, and appears as a heterogeneous mixture of fresh blood and a partially liquefied hematoma (Fig. 330-11). A sediment level or “hematocrit effect” may be seen from either rebleeding or in patients with clotting disorders. The chronic SDH has density similar to, but slightly higher than, cerebrospinal fluid (Fig. 330-12). It may be difficult to distinguish from prominent subarachnoid space in patients with cerebral atrophy. In these patients, a contrast-enhanced CT can improve detection of the chronic SDH by demonstrating an enhancing capsule or displaced cortical veins. Over time, activated fibroblasts and blood vessels from the dura organize within the SDH. The fragile penetrating vessels are prone to bleeding, however, which can lead to the dreaded “chronic recurrent” SDH. This collection may not be crescentic in shape because of dural adhesions; it is typically heterogeneous with multiple internal septations, loculations, and fluid levels.
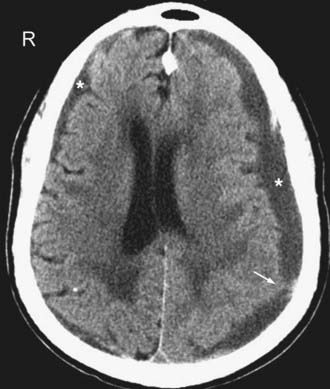
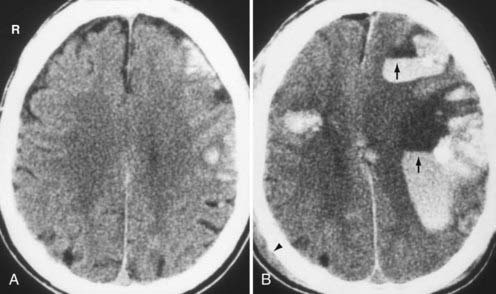
In the subacute setting—usually between 1 to 3 weeks following injury depending on the patient’s hematocrit level, clotting capability, and presence or absence of rebleeding—an isodense phase occurs. During this transition from the acute to the chronic phase, a small thin convexity isodense SDH can be difficult to identify (Fig. 330-13). Recognition of indirect imaging findings such as sulcal effacement, displacement of the gray-white junction away from the inner calvarium (white matter “buckling,”), distortion of the ventricles, and midline shift can improve detection of such isodense SDH.
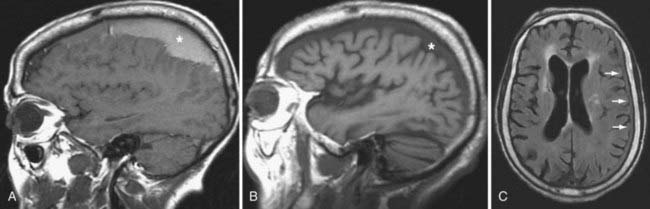
On MRI, the acute SDH is isointense to the brain on T1-weighted images and hypointense on T2-weighted images. During the subacute phase, when the SDH is isodense on noncontrast CT images, the SDH is a high signal intensity on T1-weighted images because of the presence of methemoglobin (Fig. 330-14A). The chronic liquefied SDH appears hypointense on T1-weighted and hyperintense on T2-weighted images relative to the normal brain. The signal intensity of the chronic SDH is slightly higher than CSF signal intensity on T1-weighted, FLAIR, and proton-density T2-weighted images (Fig. 330-14B and C). Because of beam-hardening artifact and the lack of multiplanar capability that limits CT, MRI is particularly useful in identifying small convexity and vertex hematomas that may not be readily detected on axial CT.
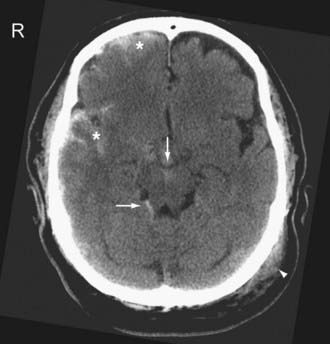
On CT, acute SAH appears as linear or serpentine areas of high density that conform to the morphology of the cerebral sulci and cisterns (Fig. 330-15; see Fig. 330-8B). SAH along the convexity or tentorium can be difficult to differentiate from an SDH. A useful clue is the extension of the SAH into adjacent sulci. Occasionally, “effacement” of the sulci because of the presence of intrasulcal SAH is the only imaging clue of the presence of SAH.
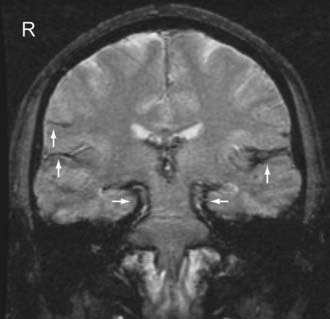
Acute SAH is sometimes more difficult to detect on MR imaging than on CT because intracellular oxyhemoglobin and/or deoxyhemoglobin is isointense to brain parenchyma. However, FLAIR has been shown to be more sensitive than CT in detecting acute SAH in an animal model, especially when a high volume (1 to 2 mL) is present.22 Subacute SAH, when the blood is isointense to the CSF on CT, is better appreciated on MRI because of the high signal intensity of extracellular methemoglobin. Chronic SAH can also be seen on MRI and is invisible on CT. Old blood products such as ferritin and hemosiderin in the subarachnoid space appear as linear areas of decreased signal intensity on T1- and T2-weighted images (“superficial hemosiderosis”). Old blood is best detected on SWI and GRE T2*-weighted images (Fig. 330-16).
On CT, IVH usually appears as a CSF-blood fluid level layering dependently within the ventricular system (Fig. 330-17). Sometimes, a tiny collection of increased density layering dependently in one occipital horn may be the only clue to IVH. Occasionally, the IVH may appear “tumefactive” as a cast within the ventricle.
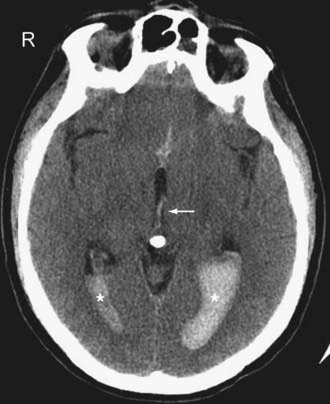
Primary Intra-Axial Injury
Axonal injury refers to white matter damage arising from shear-strain deformation of brain tissue following rotational acceleration and deceleration injury. When the skull is rapidly rotated, axial stretching, separation, and disruption of the white matter fibers occur because the nonrigid brain and brainstem lag behind. Diffuse axonal injury (DAI) indicates extensive injury to the white matter and occurs in about half of all severe head trauma cases.58 DAI is of special interest because it is thought to be responsible for the majority of TBI cognitive deficits, yet it is underdiagnosed by conventional imaging techniques.59–61
With respect to the imaging findings, DAI is somewhat of a misnomer because the injury does not always appear diffuse. Nevertheless, the location of DAI tends to correlate with the severity of the trauma.62 Specifically, mild DAI (grade I) involves only the peripheral gray-white junction of the lobar white matter, commonly the parasagittal regions of the frontal lobes and temporal stem (Fig. 330-18). Patients with moderate DAI (grade II) have injury to the corpus callosum, particularly the posterior body and splenium, in addition to the lobar white matter (Fig. 330-19). In severe DAI (grade III), the dorsolateral midbrain, in addition to the lobar white matter and corpus callosum, is affected.
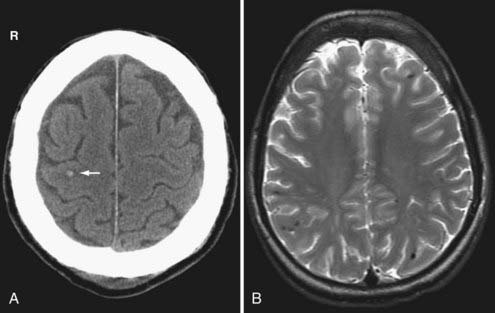
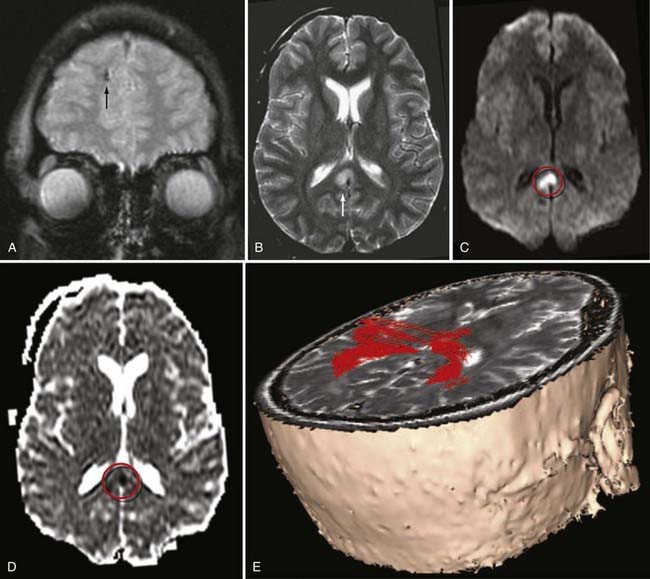
On CT, DAI lesions appear as small petechial hemorrhages seen at the gray-white junction of the cerebral hemispheres (see Fig. 330-18A), corpus callosum, and the dorsolateral midbrain, depending on the severity of the trauma. Because of its higher sensitivity to blood products, GRE T2*-weighted MRI reveals more hemorrhagic DAI lesions than CT.60 However, only a minority of DAI lesions is associated with hemorrhage.63 FLAIR MR imaging can identify additional nonhemorrhagic foci of DAI, yet it still underestimates the true extent of traumatic white matter damage.24 In addition, nonhemorrhagic lesions on FLAIR images are nonspecific for TBI, especially in elderly patients. Nonhemorrhagic acute DAI lesions appear as multiple small foci of increased signal on T2-weighted images and decreased signal on T1-weighted images. Acute DAI can also show reduced apparent diffusion coefficient (Fig. 330-19D) and reduced fractional anisotropy (FA) on DWI. In subacute DAI, intracellular methemoglobin from petechial hemorrhage appears as central hypointensity on T2-weighted images and hyperintensity on T1-weighted images. The conspicuity of DAI on MRI eventually diminishes as the damaged axons degenerate and the edema resolves. Chronic DAI imaging findings include nonspecific atrophy, gliosis, wallerian degeneration and hemosiderin staining, which can persist indefinitely on gradient-echo T2*-weighted images. The FA is frequently reduced in chronic DAI.
MRI is clearly superior to CT in detecting axonal injuries, especially when susceptibility-weighted sequences and higher field strength magnets (3T) are used.64 Yet even with MRI, the incidence of DAI is still underestimated. As mentioned earlier, newer imaging methods, such as diffusion tensor imaging (DTI) with 3-D tractography (Fig. 330-19E), have shown potential in improving the detection of white matter injury in both acute and chronic DAI.30,31,65 MRS and MTI can also provide additional prognostic value in DAI.34
The cortical contusion is a hemorrhagic parenchymal brain “bruise” involving the superficial gray matter with occasional extension into the underlying white matter. Regions of the brain that are in close contact with the rough inner surfaces of the skull are typically affected. These regions include the temporal lobes above the petrous bone and posterior to the greater sphenoid wing, and the frontal lobes above the cribriform plate, orbital roof, and lesser sphenoid wing. Contusions along the parasagittal convexity are less common, and the cerebellum is rarely involved.66 Contusions are common beneath depressed skull fractures. Contusions are usually associated with a better prognosis than DAI unless they are accompanied by brainstem injury or significant mass effect.
On CT, hemorrhagic contusions appear as mottled areas of high density within the superficial gray matter (Fig. 330-20A). They may be surrounded by larger areas of low density from associated vasogenic edema. As the contusion evolves, the characteristic “salt and pepper” pattern of mixed areas of hypodensity and hyperdensity becomes more apparent. Nonhemorrhagic contusions appear as low attenuation areas and can be difficult to detect initially until the development of sufficient edema. Due to its superficial location, the cortical contusion can be difficult to detect on CT, especially in the presence of patient motion and beam hardening streak artifacts.
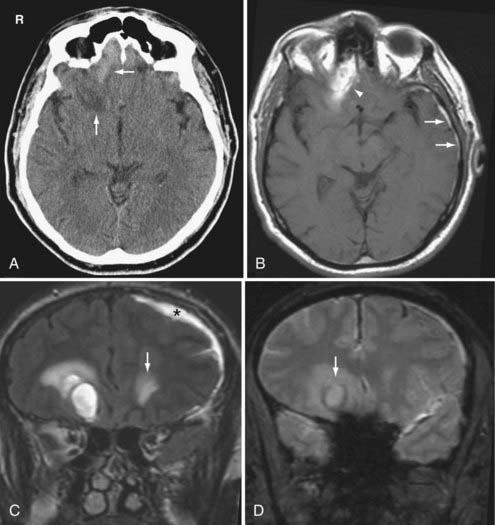
MRI provides better detection of contusions than CT because the skull does not distort the MR images. In addition, different MR techniques allow for emphasis on blood products at different ages.67 On MRI, contusions appear as ill-defined areas of variable signal intensity on both T1-and T2-weighted images, depending on the age of the lesions (Fig. 330-20B–D). Again, because contusions involve the surface of the brain, they may have a “gyral” morphology. Hemosiderin from an old contusion can persist indefinitely and serves as an important marker of prior TBI. An old contusion commonly evolves into a wedge-shaped area of peripheral encephalomalacia with the broad base facing the skull. Therefore, it can mimic an old ischemic infarction.
The intracerebral hematoma may result from expansion and coalescence of adjacent cortical contusions or it may result from microcavitation or shear-induced hemorrhage of small intraparenchymal blood vessels. The bleeding is more well-defined and tends to have less surrounding edema than the cortical contusion. Intracerebral hematomas are located deeper in the brain than cortical contusions and frequently involve the frontotemporal white matter. Involvement of the basal ganglia has been described; however, hemorrhage in the basal ganglia should alert the clinician that an underlying hypertensive bleed may be the culprit. Intracerebral hematomas are often associated with skull fractures and other primary intracranial injuries, including contusions and DAI, especially in patients who are unconscious at the time of injury. The intracerebral hematoma is the most common cause of clinical deterioration in patients who have experienced a lucid interval after the initial injury. Delayed hemorrhage is the most common cause of clinical deterioration during the first several days after head trauma.68
On CT, the acute intracerebral hematoma is a rounded hyperdense mass lesion. As the hematoma evolves, a low-density rim, due to edema and pressure necrosis, can be observed. Ring enhancement can be seen within a subacute hematoma because of the proliferation of new capillaries lacking a complete blood-brain barrier (Fig. 330-21). Indeed, the enhancing subacute hematoma can be difficult, if not impossible, to differentiate from an abscess, infarct, or neoplasm without accurate clinical history or additional novel imaging methods such as MRS or DWI. The imaging characteristics of the chronic intracerebral hematoma are nonspecific for TBI, but involvement of the orbitofrontal and anteroinferior temporal lobes is characteristic.
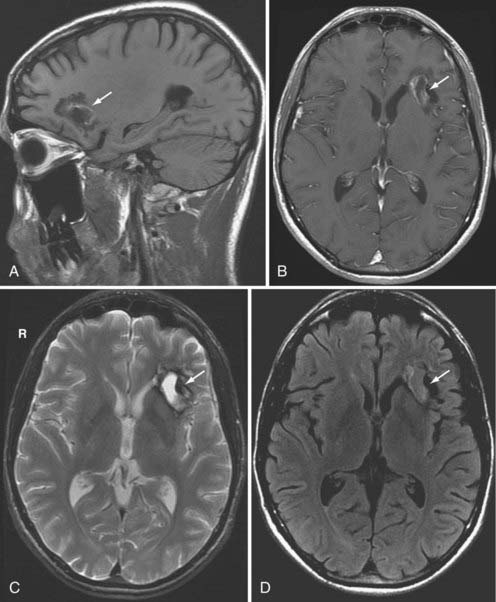
On occasion, approximately 1 to 4 days following the onset of the initial trauma, delayed intracerebral hematomas can occur in areas that previously demonstrated focal contusions on CT or MRI.69,70 These delayed hematomas tend to occur in multiple lobar locations and are associated with a poor prognosis (Fig. 330-22). The proposed pathogenesis is due to reperfusion hemorrhage secondary to vasospasm with subsequent vasodilation and hypotension, and subsequent hypertension exacerbated by an underlying coagulopathy.
Vascular Injury
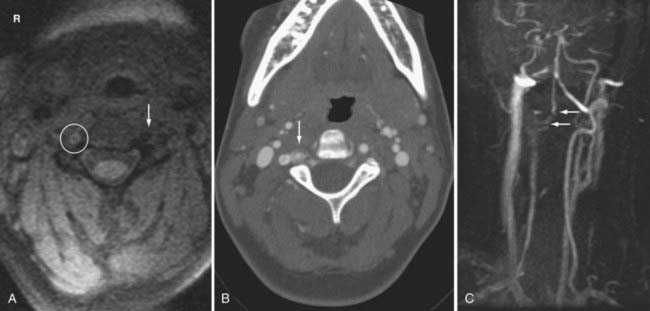
An arterial dissection occurs when there is incomplete disruption of the vessel wall with formation of a subintimal or intramural hematoma. The identification of the dissection is often best with T1-weighted MR images with fat suppression where it appears as a bright “crescent sign” (Fig. 330-23). Other imaging findings include an irregular and small caliber of the affected vessel. Absence of the normal vascular flow void and abnormal flow-related enhancement secondary to slow flow, intraluminal thrombus, or vessel occlusion may be identified on MR and MRA, respectively. An intimal flap is rarely seen. A watershed and/or embolic parenchymal infarction supplied by the injured vessel may also be seen.
Conventional catheter angiograms have long been thought to be the “gold standard” for confirmation and delineation of the vascular dissection, and they can also show vasospasm and pseudoaneurysm formation. However, MR angiography and MDCT angiography serve as important screening tools in the evaluation of patients with suspected vascular injury. In addition, because catheter angiography only demonstrates the caliber of the patent lumen, MR and CTA can identify a dissected vessel that is normal on catheter angiography. MR and CT are equally sensitive for the detection of intramural hematoma and subintimal flap, but a recent study shows that CTA is more sensitive in depicting vertebral artery pseudoaneurysms.14
Traumatic pseudoaneurysms are rare in adults but they account for 11% of all pediatric aneurysms.71 The wall of the pseudoaneurysm is actually an encapsulated hematoma in communication with the artery. On occasion, the adventitia can still be intact. Nevertheless, the wall of the pseudoaneurysm provides little support, and hence it has a propensity to hemorrhage. On imaging, the pseudoaneurysm frequently has an irregular contour (Fig. 330-24) and a wide neck. The size of a partially thrombosed pseudoaneurysm is underestimated on conventional angiography because the angiogram only depicts the patent portion of the lesion. MRI and CT can better reveal the true extent of a partially thrombosed pseudoaneurysm than angiography. In the absence of thrombosis or turbulent flow, the pseudoaneurysm appears as a round area of signal void on both T1- and T2-weighted images. Pulsation within the pseudoaneurysm shows phase artifacts on MRI, and this can be a helpful imaging clue to the presence of a vascular lesion. Thrombosis within the pseudoaneurysm manifests as a rounded mass with concentric laminated rings of heterogeneous signal intensity, consistent with thrombus in various stages of evolution.
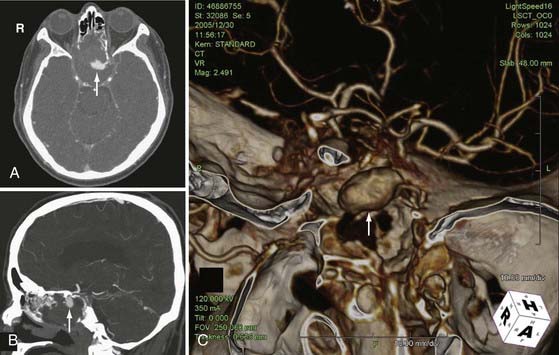
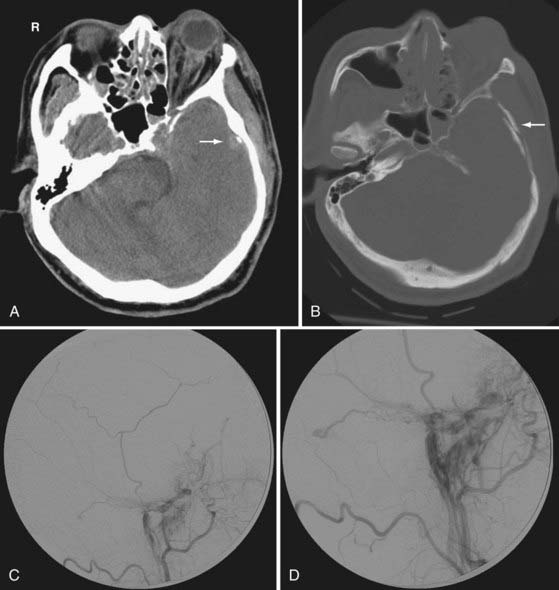
The traumatic arteriovenous fistula is a direct communication between an artery and a vein. The traumatic carotid-cavernous fistula (CCF) results from a defect in the cavernous portion of the internal carotid artery that allows direct communication with the adjacent cavernous sinus venous plexus. It typically results from a full-thickness arterial injury, and may be seen following both blunt and penetrating TBI. A CCF is thought to be preceded by a traumatic pseudoaneurysm of the internal carotid artery. Typical imaging features of the CCF include engorgement of the cavernous and petrosal sinuses, and a dilated tortuous ipsilateral superior ophthalmic vein (Fig. 330-25). When the superior ophthalmic vein exceeds 4 mm in diameter, a CCF should be suspected. Other imaging findings include enlarged extraocular muscles, proptosis, retrobulbar fat stranding, preseptal soft tissue swelling, and an ipsilateral convex cavernous sinus. However, the findings may be bilateral and symmetric because venous channels connect the cavernous sinuses. In severe cases, intracranial venous hypertension can lead to brain edema and hemorrhagic venous infarction. Skull base fractures, especially those involving the sphenoid bone, should alert the clinicians to search for associated cavernous carotid injury. Patients can present weeks or even months after the initial trauma. Therefore, a CCF can be overlooked if a detailed clinical history and ophthalmic examination is not performed.
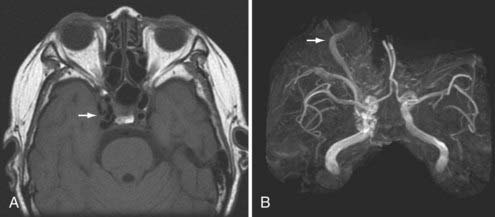
Another rare traumatic vascular injury is the arteriovenous dural fistula. It is most often caused by laceration of the middle meningeal artery with resultant meningeal artery to meningeal vein fistulous communication (Fig. 330-26). Because the fistula generally drains via the meningeal veins, the injured middle meningeal artery rarely leads to the formation of an EDH. Patients are often asymptomatic or have nonspecific complaints such as tinnitus.
Acute Secondary Injury
Effacement of the cerebral sulci (Figs. 330-27 and 330-28) and cisterns, as well as compression of the ventricles on CT, are typical imaging findings. In cytotoxic edema, the gray-white differentiation is typically lost, which is in contrast to hyperemia where the gray-white differentiation is preserved. With cytotoxic edema associated with circulatory arrest, the cerebellum and brainstem are usually spared and may appear hyperintense relative to the affected cerebral hemispheres.
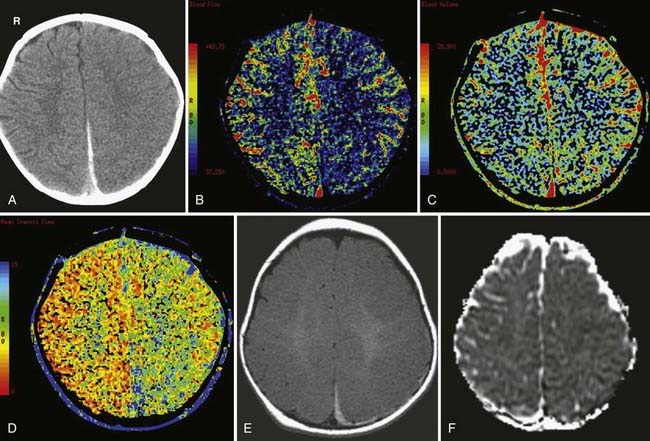
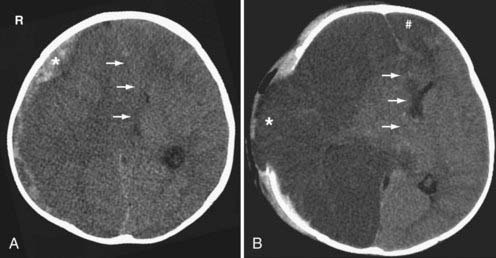
Traumatic brain herniation refers to displacement of brain tissue from one compartment to another secondary to the mass effect produced either by primary or secondary injuries. The compartmentalization is based on the dural partitions and skull openings. In subfalcine herniation, the most common form of herniation, the cingulate gyrus is displaced across the midline under the falx cerebri and above the corpus callosum (see Fig. 330-9A). Compression of the ipsilateral ventricle because of mass effect and enlargement of the contralateral ventricle because of obstruction of the foramen of Monro can be seen on imaging. In uncal (medial transtentorial) herniation, the medial temporal lobe is displaced over the free margin of the tentorium (Fig. 330-9B). Effacement of the lateral aspect of the suprasellar cisterns is an important early clue of the presence of uncal herniation. In transtentorial herniation, the brain herniates either upward or downward because of lesions within the posterior fossa or supratentorium, respectively. Upward herniation occurs when portions of the cerebellum and vermis displace through the tentorial incisura. In posterior fossa downward herniation, the cerebellar tonsils displace through the foramen magnum. Downward herniation of the cerebrum manifests as effacement of the suprasellar and perimesencephalic cisterns. Inferior displacement of the calcified pineal gland is another clue for the presence of downward herniation. External (transcalvarial) herniation occurs when elevated intracranial pressure (ICP) is combined with a skull defect (see Fig. 330-28). This type of herniation is being seen more often because of an increased use of decompressive craniectomies. With all types of brain herniation, the underlying culprit must be timely corrected to prevent additional secondary injury.
Chronic Secondary Injury
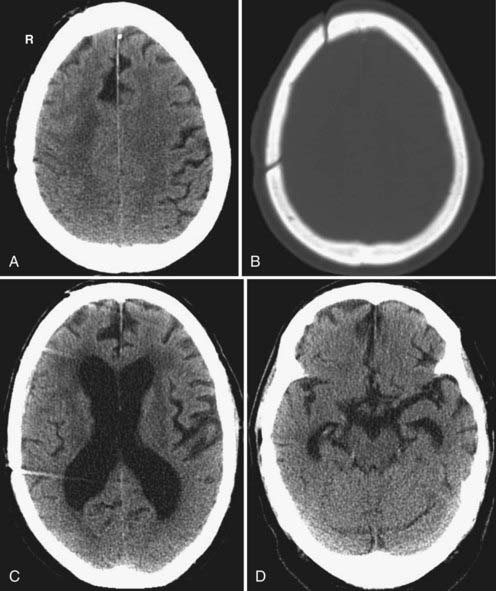
Traumatic hydrocephalus occurs secondary to impaired CSF reabsorption at the level of the arachnoid villi (communicating hydrocephalus) or, less commonly, secondary to intraventricular obstruction, typically at the level of the cerebral aqueduct (noncommunicating hydrocephalus). Mass effect from brain herniation or a hematoma can also cause noncommunicating hydrocephalus via compression of the aqueduct, foramen of Monro, or ventricular outflow foramina. As mentioned earlier, hydrocephalus can be a complication of prior SAH or IVH. On imaging, the ventricles are dilated, the sulci may be effaced, and there may be periventricular transependymal edema (Fig. 330-29).
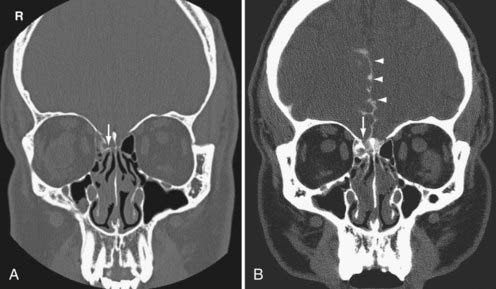
The cerebrospinal fluid (CSF) leak occurs from a dural tear associated with a skull base fracture. CSF otorrhea occurs when communication between the subarachnoid space and middle ear develops in association with a ruptured tympanic membrane. CSF rhinorrhea arises when there is communication between the subarachnoid space and the paranasal sinuses. In patients with recurrent meningeal infections and a prior history of trauma, a CSF leak should be suspected. They are often difficult to localize. Radionuclide cisternography is highly sensitive for the detection of CSF leak. However, CT scanning with intrathecal contrast is required for detailed anatomic localization of the defect (Fig. 330-30).
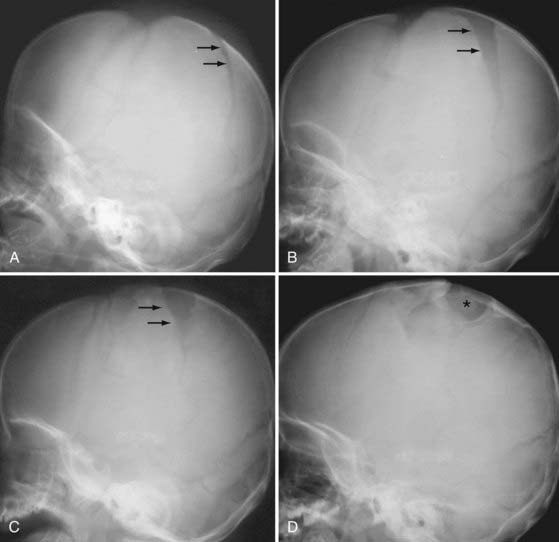
The leptomeningeal cyst, an almost exclusively pediatric lytic calvarial lesion commonly referred to as a “growing fracture,” is also caused by a tear in the dura. The dural defect allows expansion of the arachnoid at the site of the bony defect, presumably as a result of CSF pulsation. Such expansion leads to progressive, slow widening of the skull defect or suture. The leptomeningeal cyst appears as a lytic skull defect on CT or plain skull radiographs (Fig. 330-31).
Acknowledgements
We thank the residents, fellows, and attendings from the Neuroradiology Section of the University of California, San Francisco for their continuing effort in submitting interesting cases to the teaching file server (http://tfserver.ucsf.edu). Some of the cases presented in this article were the products of their effort.
Adams J. Pathology of nonmissile head injury. Neuroimaging Clin N Am. 1991;1:397-410.
Adams JH, Graham DI, Gennarelli TA, et al. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry. 1991;54:481-483.
Al-Nakshabandi NA. The swirl sign. Radiology. 2001;218:433.
Bell RS, Loop JW. The utility and futility of radiographic skull examination for trauma. N Engl J Med. 1971;284:236-239.
Bergsneider M, Hovda DA, McArthur DL, et al. Metabolic recovery following human traumatic brain injury based on FDG-PET: time course and relationship to neurological disability. J Head Trauma Rehabil. 2001;16:135-148.
Dahiya R, Keller JD, Litofsky NS, et al. Temporal bone fractures: otic capsule sparing versus otic capsule violating clinical and radiographic considerations. J Trauma. 1999;47:1079-1083.
Garnett MR, Blamire AM, Rajagopalan B, et al. Evidence for cellular damage in normal-appearing white matter correlates with injury severity in patients following traumatic brain injury: a magnetic resonance spectroscopy study. Brain. 2000;123(pt 7):1403-1409.
Gean AD. Imaging of Head Trauma. Philadelphia: Williams & Wilkins-Lippincott; 1994.
Gennarelli TA, Thibault LE, Adams JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-574.
Haacke EM, Xu Y, Cheng YC, et al. Susceptibility weighted imaging (SWI). Magn Reson Med. 2004;52:612-618.
Hardwood-Nash DC, Fritz CR. Neuroradiology in Infants and Children. St Louis: CV Mosby; 1976.
Hesselink JR, Dowd CF, Healy ME, et al. MR imaging of brain contusions: a comparative study with CT. AJR Am J Roentgenol. 1988;150:1133-1142.
Lewine JD, Davis JT, Bigler ED, et al. Objective documentation of traumatic brain injury subsequent to mild head trauma: multimodal brain imaging with MEG, SPECT, and MRI. J Head Trauma Rehabil. 2007;22:141-155.
Liu AY, Maldjian JA, Bagley LJ, et al. Traumatic brain injury: diffusion-weighted MR imaging findings. AJNR Am J Neuroradiol. 1999;20:1636-1641.
Mittl RL, Grossman RI, Hiehle JF, et al. Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. AJNR Am J Neuroradiol. 1994;15:1583-1589.
Munoz-Sanchez MA, Murillo-Cabezas F, Cayuela-Dominguez A, et al. Skull fracture, with or without clinical signs, in mTBI is an independent risk marker for neurosurgically relevant intracranial lesion: a cohort study. Brain Inj. 2009;23:39-44.
Newton MR, Greenwood RJ, Britton KE, et al. A study comparing SPECT with CT and MRI after closed head injury. J Neurol Neurosurg Psychiatry. 1992;55:92-94.
Orrison WW, Gentry LR, Stimac GK, et al. Blinded comparison of cranial CT and MR in closed head injury evaluation. AJNR Am J Neuroradiol. 1994;15:351-356.
Sinson G, Bagley LJ, Cecil KM, et al. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: correlation with clinical outcome after traumatic brain injury. AJNR Am J Neuroradiol. 2001;22:143-151.
Stiell IG, Lesiuk H, Wells GA, et al. Canadian CT head rule study for patients with minor head injury: methodology for phase II (validation and economic analysis). Ann Emerg Med. 2001;38:317-322.
Stiell IG, Lesiuk H, Wells GA, et al. The Canadian CT head rule study for patients with minor head injury: rationale, objectives, and methodology for phase I (derivation). Ann Emerg Med. 2001;38:160-169.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
Wintermark M, Sesay M, Barbier E, et al. Comparative overview of brain perfusion imaging techniques. Stroke. 2005;36:e83-e99.
Wintermark M, van Melle G, Schnyder P, et al. Admission perfusion CT: prognostic value in patients with severe head trauma. Radiology. 2004;232:211-220.
Woodcock RJJr, Short J, Do HM, et al. Imaging of acute subarachnoid hemorrhage with a fluid-attenuated inversion recovery sequence in an animal model: comparison with non-contrast-enhanced CT. AJNR Am J Neuroradiol. 2001;22:1698-1703.
1 Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
2 Munoz-Sanchez MA, Murillo-Cabezas F, Cayuela-Dominguez A, et al. Skull fracture, with or without clinical signs, in mTBI is an independent risk marker for neurosurgically relevant intracranial lesion: a cohort study. Brain Inj. 2009;23:39-44.
3 Bell RS, Loop JW. The utility and futility of radiographic skull examination for trauma. N Engl J Med. 1971;284:236-239.
4 Hackney DB. Skull radiography in the evaluation of acute head trauma: a survey of current practice. Radiology. 1991;181:711-714.
5 Masters SJ. Evaluation of head trauma: efficacy of skull films. AJR Am J Roentgenol. 1980;135:539-547.
6 Adams J. Pathology of nonmissile head injury. Neuroimaging Clin N Am. 1991;1:397-410.
7 Haydel MJ, Preston CA, Mills TJ, et al. Indications for computed tomography in patients with minor head injury. N Engl J Med. 2000;343:100-105.
8 Jagoda AS, Cantrill SV, Wears RL, et al. Clinical policy: neuroimaging and decision making in adult mild traumatic brain injury in the acute setting. Ann Emerg Med. 2002;40:231-249.
9 Lorberboym M, Lampl Y, Gerzon I, et al. Brain SPECT evaluation of amnestic ED patients after mild head trauma. Am J Emerg Med. 2002;20:310-313.
10 Stiell IG, Lesiuk H, Wells GA, et al. Canadian CT head rule study for patients with minor head injury: methodology for phase II (validation and economic analysis). Ann Emerg Med. 2001;38:317-322.
11 Stiell IG, Lesiuk H, Wells GA, et al. The Canadian CT head rule study for patients with minor head injury: rationale, objectives, and methodology for phase I (derivation). Ann Emerg Med. 2001;38:160-169.
12 Stiell IG, Wells GA, Vandemheen K, et al. The Canadian CT head rule for patients with minor head injury. Lancet. 2001;357:1391-1396.
13 Zee CS, Go JL. CT of head trauma. Neuroimaging Clin N Am. 1998;8:525-539.
14 Vertinsky AT, Schwartz NE, Fischbein NJ, et al. Comparison of multidetector CT angiography and MR imaging of cervical artery dissection. AJNR Am J Neuroradiol. 2008;29:1753-1760.
15 Wintermark M, Sesay M, Barbier E, et al. Comparative overview of brain perfusion imaging techniques. Stroke. 2005;36:e83-e99.
16 Wintermark M, van Melle G, Schnyder P, et al. Admission perfusion CT: prognostic value in patients with severe head trauma. Radiology. 2004;232:211-220.
17 Siebert E, Bohner G, Dewey M, et al. 320-slice CT neuroimaging: initial clinical experience and image quality evaluation. Br J Radiol. 2009.
18 Youn SW, Kim JH, Weon YC, et al. Perfusion CT of the brain using 40-mm-wide detector and toggling table technique for initial imaging of acute stroke. AJR Am J Roentgenol. 2008;191:W120-W126.
19 Gentry LR, Godersky JC, Thompson B, et al. Prospective comparative study of intermediate-field MR and CT in the evaluation of closed head trauma. AJR Am J Roentgenol. 1988;150:673-682.
20 Orrison WW, Gentry LR, Stimac GK, et al. Blinded comparison of cranial CT and MR in closed head injury evaluation. AJNR Am J Neuroradiol. 1994;15:351-356.
21 Noguchi K, Ogawa T, Seto H, et al. Subacute and chronic subarachnoid hemorrhage: diagnosis with fluid-attenuated inversion-recovery MR imaging. Radiology. 1997;203:257-262.
22 Woodcock RJJr, Short J, Do HM, et al. Imaging of acute subarachnoid hemorrhage with a fluid-attenuated inversion recovery sequence in an animal model: comparison with non-contrast-enhanced CT. AJNR Am J Neuroradiol. 2001;22:1698-1703.
23 Frigon C, Jardine DS, Weinberger E, et al. Fraction of inspired oxygen in relation to cerebrospinal fluid hyperintensity on FLAIR MR imaging of the brain in children and young adults undergoing anesthesia. AJR Am J Roentgenol. 2002;179:791-796.
24 Ashikaga R, Araki Y, Ishida O. MRI of head injury using FLAIR. Neuroradiology. 1997;39:239-242.
25 Messori A, Polonara G, Mabiglia C, et al. Is haemosiderin visible indefinitely on gradient-echo MRI following traumatic intracerebral haemorrhage? Neuroradiology. 2003;45:881-886.
26 Haacke EM, Xu Y, Cheng YC, et al. Susceptibility weighted imaging (SWI). Magn Reson Med. 2004;52:612-618.
27 Babikian T, Freier MC, Tong KA, et al. Susceptibility weighted imaging: neuropsychologic outcome and pediatric head injury. Pediatr Neurol. 2005;33:184-194.
28 Tong KA, Ashwal S, Holshouser BA, et al. Diffuse axonal injury in children: clinical correlation with hemorrhagic lesions. Ann Neurol. 2004;56:36-50.
29 Tong KA, Ashwal S, Holshouser BA, et al. Hemorrhagic shearing lesions in children and adolescents with posttraumatic diffuse axonal injury: improved detection and initial results. Radiology. 2003;227:332-339.
30 Huisman TA, Sorensen AG, Hergan K, et al. Diffusion-weighted imaging for the evaluation of diffuse axonal injury in closed head injury. J Comput Assist Tomogr. 2003;27:5-11.
31 Le TH, Mukherjee P, Henry RG, et al. Diffusion tensor imaging with three-dimensional fiber tractography of traumatic axonal shearing injury: an imaging correlate for the posterior callosal “disconnection” syndrome: case report. Neurosurgery. 2005;56:189.
32 Liu AY, Maldjian JA, Bagley LJ, et al. Traumatic brain injury: diffusion-weighted MR imaging findings. AJNR Am J Neuroradiol. 1999;20:1636-1641.
33 Niogi SN, Mukherjee P, Ghajar J, et al. Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. AJNR Am J Neuroradiol. 2008;29:967-973.
34 Sinson G, Bagley LJ, Cecil KM, et al. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: correlation with clinical outcome after traumatic brain injury. AJNR Am J Neuroradiol. 2001;22:143-151.
35 Garnett MR, Blamire AM, Corkill RG, et al. Early proton magnetic resonance spectroscopy in normal-appearing brain correlates with outcome in patients following traumatic brain injury. Brain. 2000;123(pt 10):2046-2054.
36 Garnett MR, Blamire AM, Rajagopalan B, et al. Evidence for cellular damage in normal-appearing white matter correlates with injury severity in patients following traumatic brain injury: a magnetic resonance spectroscopy study. Brain. 2000;123(pt 7):1403-1409.
37 Lewine JD, Davis JT, Bigler ED, et al. Objective documentation of traumatic brain injury subsequent to mild head trauma: multimodal brain imaging with MEG, SPECT, and MRI. J Head Trauma Rehabil. 2007;22:141-155.
38 Lewine JD, Davis JT, Sloan JH, et al. Neuromagnetic assessment of pathophysiologic brain activity induced by minor head trauma. AJNR Am J Neuroradiol. 1999;20:857-866.
39 Newton MR, Greenwood RJ, Britton KE, et al. A study comparing SPECT with CT and MRI after closed head injury. J Neurol Neurosurg Psychiatry. 1992;55:92-94.
40 Coles JP, Fryer TD, Smielewski P, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab. 2004;24:202-211.
41 Coles JP, Fryer TD, Smielewski P, et al. Defining ischemic burden after traumatic brain injury using 15O PET imaging of cerebral physiology. J Cereb Blood Flow Metab. 2004;24:191-201.
42 Menon DK. Brain ischaemia after traumatic brain injury: lessons from 15O2 positron emission tomography. Curr Opin Crit Care. 2006;12:85-89.
43 Bergsneider M, Hovda DA, McArthur DL, et al. Metabolic recovery following human traumatic brain injury based on FDG-PET: time course and relationship to neurological disability. J Head Trauma Rehabil. 2001;16:135-148.
44 Gross H, Kling A, Henry G, et al. Local cerebral glucose metabolism in patients with long-term behavioral and cognitive deficits following mild traumatic brain injury. J Neuropsychiatry Clin Neurosci. 1996;8:324-334.
45 Alavi A. Functional and anatomic studies of head injury. J Neuropsychiatry Clin Neurosci. 1989;1:S45-S50.
46 Lupi A, Bertagnoni G, Salgarello M, et al. Cerebellar vermis relative hypermetabolism: an almost constant PET finding in an injured brain. Clin Nucl Med. 2007;32:445-451.
47 Wilberger J, Chen DA. The skull and meninges. In: Eisenberg HM, EF A, eds. Management of Head Injury. Neurosurg Clin North Am. 1991:341-350.
48 Ulrich K. Verletzungen des Gehorlorgans bel Schadelbasisfrakturen(Ein Histologisch und Klinissche Studie). Acta Otolaryngol Suppl. 1926;6:1-150.
49 Dahiya R, Keller JD, Litofsky NS, et al. Temporal bone fractures: otic capsule sparing versus otic capsule violating clinical and radiographic considerations. J Trauma. 1999;47:1079-1083.
50 Little SC, Kesser BW. Radiographic classification of temporal bone fractures: clinical predictability using a new system. Arch Otolaryngol Head Neck Surg. 2006;132:1300-1304.
51 Gentry LR. Temporal bone trauma. Neuroimaging Clin N Am. 1991;1:319-340.
52 Ghorayeb BY, Yeakley JW. Temporal bone fractures: longitudinal or oblique? The case for oblique temporal bone fractures. Laryngoscope. 1992;102:129-134.
53 Gean AD, Kates RS, Lee S. Neuroimaging in head injury. New Horiz. 1995;3:549-561.
54 Al-Nakshabandi NA. The swirl sign. Radiology. 2001;218:433.
55 Greenberg J, Cohen WA, Cooper PR. The “hyperacute” extraaxial intracranial hematoma: computed tomographic findings and clinical significance. Neurosurgery. 1985;17:48-56.
56 Pruthi N, Balasubramaniam A, Chandramouli BA, et al. Mixed-density extradural hematomas on computed tomography-prognostic significance. Surg Neurol. 2009;71:202-206.
57 Kumbhani SR, Purcell DD, Gean AD. Contrast extravasation within a traumatic epidural hematoma: have we been missing something? Vancouver, Canada: American Society of Neuroradiology 47th Annual Meeting & NER Foundation Symposium; 2009.
58 Jennett B, Adams JH, Murray LS, et al. Neuropathology in vegetative and severely disabled patients after head injury. Neurology. 2001;56:486-490.
59 Inglese M, Makani S, Johnson G, et al. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298-303.
60 Mittl RL, Grossman RI, Hiehle JF, et al. Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. AJNR Am J Neuroradiol. 1994;15:1583-1589.
61 Scheid R, Preul C, Gruber O, et al. Diffuse axonal injury associated with chronic traumatic brain injury: evidence from T2*-weighted gradient-echo imaging at 3 T. AJNR Am J Neuroradiol. 2003;24:1049-1056.
62 Gennarelli TA, Thibault LE, Adams JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564-574.
63 Adams JH, Graham DI, Gennarelli TA, et al. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry. 1991;54:481-483.
64 Lee H, Wintermark M, Gean AD, et al. Focal lesions in acute mild traumatic brain injury and neurocognitive outcome: CT versus 3T MRI. J Neurotrauma. 2008;25:1049-1056.
65 Arfanakis K, Haughton VM, Carew JD, et al. Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol. 2002;23:794-802.
66 Gentry LR. Head trauma. In: Atlas SW, editor. Magnetic Resonance Imaging of the Brain and Spine. 2nd ed. Philadelphia: Lippincott-Raven; 1996:611-647.
67 Hesselink JR, Dowd CF, Healy ME, et al. MR imaging of brain contusions: a comparative study with CT. AJR Am J Roentgenol. 1988;150:1133-1142.
68 Soloniuk D, Pitts LH, Lovely M, et al. Traumatic intracerebral hematomas: timing of appearance and indications for operative removal. J Trauma. 1986;26:787-794.
69 Lipper MH, Kishore PR, Girevendulis AK, et al. Delayed intracranial hematoma in patients with severe head injury. Radiology. 1979;133:645-649.
70 Nanassis K, Frowein RA, Karimi A, et al. Delayed post-traumatic intracerebral bleeding. Delayed post-traumatic apoplexy: “Spatapoplexie.”. Neurosurg Rev. 1989;12(suppl 1):243-251.
71 Hardwood-Nash DC, Fritz CR. Neuroradiology in Infants and Children. St Louis: CV Mosby; 1976.






