[level-membership-for-critical-care-medicine-category]26
Hypovolemic Shock
Although hypovolemic shock has been recognized for more than 100 years, Wiggers1 in 1940 first offered a definition of hypovolemic shock that has remained significant until now: “Shock is a syndrome resulting from depression of many functions, but in which the reduction of the effective circulating blood volume is of basic importance, and in which impairment of the circulation steadily progresses until it eventuates in a state of irreversible circulatory failure.” Today, hypovolemic shock can be defined as an acute disturbance in the circulation leading to an imbalance between oxygen supply and demand in the tissues, caused by a decrease in circulating blood volume, mostly caused by trauma and hemorrhage.2 An oxygen debt develops when uptake no longer matches the demand for oxygen and leads to cellular ischemia and ultimately cell death. The condition is life threatening and, if left untreated, becomes irreversible after a certain period. Rapid and adequate resuscitation is mandatory to save lives. Conversely, hypovolemic shock carries a relatively favorable prognosis, if rapidly and adequately recognized and treated.
Hypovolemic shock can occur outside and inside the hospital, in trauma or surgery complicated by excessive loss of blood, but also in the course of burns, gastrointestinal hemorrhage, diarrhea, uncontrolled diabetes mellitus, addisonian crisis, and other conditions (Box 26.1). Some other types of shock, including septic, anaphylactoid, cardiogenic, and burn shock, may be accompanied by hypovolemia. The types of shock not primarily caused by hypovolemia are beyond the scope of this chapter.
Pathogenesis and Pathophysiology
Circulatory Changes
General Changes
Because hypovolemia results in a decrease in preload of the heart and low filling pressures or volumes, the cardiac output decreases.3–9 After unloading of the baroreceptor and activation of the sympathetic nervous system, tachycardia ensues, although some patients may respond with transient sympathetic inhibition and vagal nerve–mediated bradycardia during a sudden, severe loss of circulating blood volume.3,10–16 Tachycardia partially compensates for the decrease in stroke volume. A moderate decrease in cardiac output can be recognized from a decline in pulse pressure, orthostatic hypotension, and fall in regional perfusion indices.8,17,18 Hypovolemia results in wider than usual swings in central venous pressure (CVP) and arterial blood pressure during the respiratory cycle of spontaneous and mechanical ventilation because of increased sensitivity of the underfilled heart in the ascending part of the cardiac function curve to fluctuations in venous return associated with varying intrathoracic pressure.19,20 Although activation of the sympathetic nervous system and resulting arterial vasoconstriction during a moderate decrease in cardiac output prevent a severe reduction in arterial blood pressure, a further decrease in cardiac output leads to hypotension and shock.8,10 Systemic vascular resistance increases early after development of hypovolemic shock but may decrease in the later stages of shock, and this may herald irreversibility and death. The increase in resistance (and heart rate) may be transiently attenuated after an imbalance between sympathetic and vagal activity, possibly associated with release of opioids within the central nervous system and into the systemic circulation.*
Shock is characterized by an oxygen debt in the tissues.9,24–26 In the presence of sufficient oxygen, aerobic combustion of 1 mol of glucose yields 38 mol of energy-rich adenosine triphosphate (ATP), which can be hydrolyzed to provide energy for the vital and metabolic functions of the cell.27 In the absence of oxygen, glucose taken up by cells cannot be combusted because of insufficient uptake of pyruvate into the mitochondrial tricarboxylic acid cycle having a reduced turnover rate. Partly inactivated pyruvate dehydrogenase may play a role in the latter reductions. Pyruvate is converted into lactate, and the lactate-to-pyruvate ratio increases, concomitantly with a reduction in mitochondrial redox potential.24,27–29
Anaerobic glycolysis in the cytosol ultimately yields, per mol of glucose, 2 mol of ATP.27 Hydrolysis of ATP yields hydrogen ions (H+) that lead, when buffers are exhausted, to intracellular and ultimately to extracellular metabolic acidosis.30 These mechanisms form the basis of the so-called lactic acidosis during hypovolemic shock, whereby the lactate level in arterial blood is elevated above the normal 2 mmol/L associated with acidosis, and constitutes a useful measure of the oxygen debt in the tissues.26,27,31–34 Nevertheless, the energy deficit and lactate production in the cells in response to a lack of oxygen can be limited and organ function can be improved by supplying pyruvate and pyruvate dehydrogenase activators, such as dichloroacetate.27,29,35–37 Intracellular acidosis may otherwise protect ischemic cells from dying.36
The specificity of elevated lactate-to-pyruvate levels for an oxygen debt in the tissues has been doubted.27,38 Aerobic glycolysis is probably linked to the membrane Na+/K+-ATPase and stimulation of β2-receptors during sympathetic activation. Catecholamine (epinephrine) secretion may temporarily increase, rather than decrease, ATPase activity, and augment glycolysis and circulating lactate levels in tissues such as skeletal muscle, without a lack of oxygen and reduced ATP resources, during development and resuscitation from hypovolemic shock.38,39 Conversely, adrenergic antagonists may reduce lactic acidosis during hypovolemic shock.38 Epinephrine may increase glycogenolysis. Together, increased glycolytic fluxes independent of oxygen uptake may lead to equal elevations of pyruvate and lactate in the tissues, without the acidosis resulting from ATP hydrolysis with an oxygen debt.27 This situation may partly explain why the extent to which changes in the lactate level parallel changes in the anion gap or bicarbonate/base excess concentration during shock and resuscitation is controversial, and why elevated lactate levels sometimes may fail to predict an increase in oxygen uptake during an increase in oxygen delivery.40–42 This also may explain in part the discrepancies in the course of oxygen-related variables and lactate levels during catecholamine treatment of shock when attempting to boost oxygen delivery.42
The lactate level in blood is determined by production, distribution, and elimination.27 Produced lactic acid in the presence of oxygen may be converted via pyruvate to glucose or oxidized. Bicarbonate is then released. The liver plays a central role in this process, so that the elimination of lactate and clearance from plasma is impaired in case of liver ischemia or prior hepatic disease, even though renal uptake may increase.27,43,44 Nevertheless, changes in the lactate level in blood, rather than absolute values, mainly reflect changes in production and are a fair measure for the course of shock and the response to therapy, even in the presence of liver disease.27,45 Although not beyond doubt, the origin of lactate in hypovolemic shock can be skeletal muscle, lung, and gut, particularly if severe liver ischemia, hypoxia, and acidosis in shock attenuate the hepatic uptake of lactate delivered by the gut through the portal vein.27,43,44,46–48 The respiratory muscles also may contribute to lactic acidosis in a spontaneously breathing patient because, first, the respiratory muscles may demand a share of the cardiac output at the cost of other tissues, and, second, this share may be insufficient to meet oxygen demands of the diaphragm, which may be increased in view of hyperventilation.33,49–52
Notwithstanding the aforementioned limitations, an increase in the lactate level in blood and a decrease in the bicarbonate content/base excess or pH and an increase in the anion gap may be fair predictors of morbidity (multiple organ failure [MOF]) and mortality, whereas clearance of lactic acidosis usually indicates a better outcome. A decrease in the blood lactate level during resuscitation from hypovolemic shock is usually a favorable sign and associated with survival, whereas an increase in the lactate level and progressive acidosis usually are associated with morbidity and mortality, even though successful resuscitation may transiently increase the lactate level because of washout of lactate from ischemic tissues.* The mentioned variables may thus serve as guides for resuscitation.
Oxygen Balance
Because insufficient uptake of oxygen relative to demand in the tissues during shock is central, insight into the factors that determine oxygen uptake in shock is important.25,27 Oxygen delivery is determined by the cardiac output and the content of oxygen in arterial blood, that is, the arterial blood hemoglobin concentration and the saturation of hemoglobin with oxygen. The oxyhemoglobin dissociation curve determines the saturation of hemoglobin with oxygen for a given partial pressure of oxygen (PO2) in blood. During hypovolemic shock, a decrease in hemoglobin concentration, oxygen saturation, or both aggravates the effect of a decrease in cardiac output in compromising oxygen delivery to the tissues. Cardiac output is determined by preload, afterload, contractility, and heart rate.7
During a decrease in oxygen delivery with hypovolemic shock, the body maintains sufficient uptake of oxygen only if the extraction of oxygen increases, and the arteriovenous oxygen content gradient widens, resulting in a decrease in oxygen saturation of venous blood.† Associated with a decrease in oxygen delivery, tissue PO2 declines, and its heterogeneity increases, possibly indicating focal ischemia.‡ The decline in tissue PO2 may be even greater than the decrease in draining venous blood because of some increase in microvascular oxygen shunting at low blood flows.68,69 In animals, it has been shown that the increase in oxygen extraction to compensate for a decrease in oxygen delivery is maximum (but not 100%) if oxygen delivery decreases to less than 8 to 15 mL/kg per minute, that is, the critical oxygen delivery (Fig. 26.1).§ Although the critical oxygen delivery may vary widely among studies, following differences in species, basal oxygen needs, and methods to decrease oxygen delivery, data obtained in patients suggest that the critical oxygen delivery in humans may also amount to approximately 8 mL/kg per minute.58,64 During a decrease in oxygen delivery below this critical value in hypovolemic shock, oxygen uptake decreases to less than tissue demand, cellular ischemia ensues, and the body must rely on anaerobic metabolism to meet energy requirements.¶ Blood lactic acidosis, lacticacidemia, results. Conversely, oxygen uptake is supply-dependent if oxygen delivery is lower than the critical value and blood lactate levels are elevated, whereas oxygen uptake may not be supply-dependent if the lactate level in blood is normal. Treatment of hypovolemic shock, by infusing fluids and blood, is aimed at an increase in cardiac output and the oxygen content of blood and in oxygen delivery above the critical value so that oxygen uptake increases to meet body requirements and the lacticacidemia decreases.*
Treatment of hypovolemic shock, by infusing fluids and blood, is aimed at an increase in cardiac output and the oxygen content of blood and in oxygen delivery above the critical value so that oxygen uptake increases to meet body requirements and the lacticacidemia decreases.*
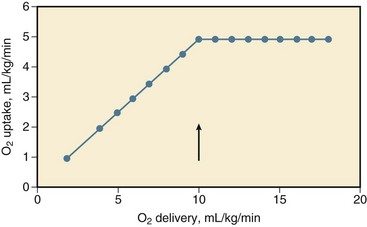
The critical oxygen delivery is a function of the body oxygen needs and the capability of the body to extract oxygen during a decline in delivery. The body oxygen needs may increase during hypovolemic shock, as a consequence of increased respiratory muscle activity and increased levels of catecholamines in the blood after activation of the sympathetic nervous system, but downregulation of the metabolic stimulant effect of catecholamines has been described.† The critical extraction of oxygen is a function of the adaptation of regional blood flow to tissue needs, the number of perfused capillaries and of diffusion distances, and the exchange surface area for oxygen.63,75 During a reduction in oxygen delivery, however, oxygen uptake is limited by convective transport of oxygen to the tissues, rather than by diffusion of oxygen to respirating mitochondria.76
In experimental animals, a change in hemoglobin affinity for oxygen, by altering the storage duration of reinfused blood, hardly changes the critical oxygen extraction, but changes in acid-base status that affect the position of the oxyhemoglobin dissociation curve may have some effect on the oxygen extraction capabilities of the body.60,76 Acid infusion may increase slightly, and base infusion may reduce oxygen extraction during supply-limited oxygen uptake.76 Nevertheless, hypercapnia may decrease critical oxygen extraction and increase critical oxygen delivery because of blood flow redistribution.77 A leftward shift of the oxyhemoglobin dissociation curve may impair maximum oxygen extraction during a reduction in oxygen delivery and may increase mortality rate in experimental animals with hypovolemic shock.60 Although the oxyhemoglobin dissociation curve may shift to the left in critically ill patients, for example, after transfusion of old, stored blood,78 the effect on oxygen uptake is unclear.
The effect of changes in body temperature is twofold: Changes are accompanied by changes in total body oxygen needs and by changes in critical oxygen extraction, probably by a vascular tone–associated altered distribution of blood flow.61 Hyperthermia increases critical oxygen delivery in hemorrhaged dogs, primarily through an increase in body oxygen needs and despite an increase in critical oxygen extraction, whereas hypothermia, which may be more common in traumatized or hemorrhaged patients, may decrease the critical oxygen delivery.61 Finally, blood viscosity may influence the extent to which a decrease in circulating blood volume affects oxygen uptake by the tissues. Experimental data suggest, however, that prior anemia does not ameliorate the decrease in oxygen uptake during a decrease in oxygen delivery with hypovolemic shock, indicating that the convective transport of oxygen is the major determinant of oxygen uptake when delivery is impaired, even though prior hemodilution may increase oxygen extraction capabilities and decrease critical oxygen delivery.6,70
Taken together, these factors may influence the extent to which oxygen uptake decreases during reduced delivery and how far oxygen delivery should be enhanced during resuscitation from hypovolemic shock. The critical oxygen delivery varies among tissues. The oxygen needs of the kidney may decline during a decrease in renal oxygen delivery because a decrease in renal perfusion may lead to a reduction in glomerular filtration and to a reduction in energy-consuming tubular resorption.63 In contrast, during progressive hypovolemia, the gut may experience supply dependency of oxygen uptake earlier than nongut tissue, partly because of a higher critical oxygen delivery (higher needs and less extraction of oxygen) and partly because of redistribution of blood flow away from the gut mucosa after more intense vasoconstriction in gut than in nongut tissue.* Clinically, this may result in nonocclusive bowel ischemia. Respiratory muscles may also have a higher critical oxygen delivery than the body as a whole during progressive hemorrhage.
Concomitant with an increased arteriovenous oxygen extraction during a decrease in oxygen delivery, the arteriovenous gradient of the carbon dioxide (CO2) content widens.8,52,81 The latter is associated with an increase in tissue and venous partial pressure of carbon dioxide (PCO2) relative to arterial PCO2 and a decrease in venous pH exceeding the decrease in pH in arterial blood.44,52,68 This widening of gradient is caused by the Fick principle and a greater decline in cardiac output than in oxygen uptake and CO2 production in the tissues because of inhibited oxidative metabolism. Nevertheless, the oxygen uptake usually decreases more than CO2 production, leading to an increase in respiratory quotient.49,65 This increase is likely to be caused by buffering of lactic acid by bicarbonate in the tissues and effluent blood, a shift toward glucose instead of fat use for residual oxidation in ischemic tissues, or a combination of both. The end-tidal expiratory CO2 fraction decreases in association with a reduction in oxygen uptake and CO2 production for a given ventilation.65 Conversely, a decrease in arterial PCO2 during a decline in CO2 production versus ventilation may be attenuated by an increase in dead-space ventilation resulting from a decrease in pulmonary blood flow/ventilation ratio.49 An increase in deadspace ventilation leads to widening of the gap between the arterial and expiratory PCO2.49
It has been suggested that the severity and duration of the oxygen debt accumulated during hypovolemic shock is a major determinant of survival in animals3,6 and in patients with trauma/hemorrhage and after major surgery.26,42,82 After trauma and hemorrhage, the defect in circulating blood volume and tissue oxygenation may be greater in patients who develop acute respiratory distress syndrome (ARDS) and MOF than in patients without these complications.* In patients undergoing major surgery, the oxygen debt during and after surgery may relate directly to the development of postoperative organ damage (i.e., MOF) and demise.73,82 Conversely, a high oxygen delivery and uptake during resuscitation may be associated with survival, whereas values that may be too low for elevated tissue demands are believed to contribute to ultimate demise, at least in animals with hypovolemic shock and critically ill patients after trauma or major surgery.† An increase of oxygen delivery and oxygen uptake to supranormal values has been suggested to improve survival further, although the latter debate has not been settled yet.‡ Extensive ischemic mitochondrial damage may limit an increase in oxygen consumption during resuscitation and reperfusion.
Macrocirculation
During loss of blood volume, various mechanisms come into play that may counteract the resultant decrease in cardiac output and tissue oxygenation. First, a decrease in cardiac output during hypovolemic shock results in a redistribution of peripheral blood flow.§ This redistribution is partly the result of regional autoregulation to maintain blood flow, in which endothelial cells and production of endogenous partly gaseous vasodilators, including endothelial nitric oxide synthase–derived nitric oxide (NO), heme oxygenase–derived carbon monoxide, hydrogen sulfide, and metabolic by-products in the tissues including CO2, potassium, and adenosine, may play a central role.86–94 Endothelium-derived NO relaxes underlying smooth muscle in the vessel wall, via stimulation of guanylate cyclase and cyclic guanosine monophosphate (cGMP), which can be inhibited by methylene blue.87,88,95,96 Carbon monoxide also acts via cGMP.91 Some authors describe that inhibition of endothelial NO synthase ameliorates early hypotension and even the mortality risk during bleeding.92 When NO is released, the reactivity to endogenous and exogenous vasoconstrictors may be diminished, even early in hypovolemic shock.92,97 Other authors describe endothelial injury and dysfunction in various organs with diminished endothelium and NO-dependent vasorelaxation, which could be overcome by L-arginine and other NO donors, including ATP-MgCl2, pentoxifylline, or heparin, so that blockade of endothelial NO synthase–derived NO may be detrimental.87–89,97,98
The opposing vasoconstricting factors include catecholamines, liberated by the activated sympathetic nervous system and the adrenal medulla; direct sympathetic stimulation of the vessel wall; angiotensin II, liberated through an activated renin-angiotensin-aldosterone system; and vasopressin, released by the pituitary in hypovolemic shock.* Endothelin is an endothelium-derived potent vasoconstrictor, released on catecholamine stimulation or hypoxia, and its release may contribute to vasoconstriction, particularly in hepatic and renal vascular beds.100,101 Finally, a decrease in cardiac filling may reduce cardiac secretion of atrial natriuretic peptides, reducing the vasodilating and diuretic effect of these factors.102 Levels may also increase as a consequence of diminished renal clearance.103,104
Depending on the degree that the mechanisms are operative, the general result of the interplay is that blood flow to intestines, skeletal muscle, and skin is diverted toward vitally more important organs, such as heart and brain, so that the increase of overall peripheral resistance during hypovolemic shock is distributed differently among various organs, with greater increases in gut, skeletal muscle, and skin than in heart and brain.† The kidney also is a target for hypovolemic shock; renal perfusion may be maintained during mild hypotension after hypovolemia, but it rapidly decreases if severe hypotension supervenes, and the decrease may exceed that in other organs.‡ In hypovolemic human volunteers, this redistribution of blood flow accords with the patterns described.74
The redistribution of blood flow results in a greater share of oxygen delivery going to organs with high metabolic demand, such as heart and brain, than tissues with less metabolic demands, including skin, skeletal muscle, kidney, gut, and pancreas.§ The redistribution is probably necessary to optimize the uptake of delivered oxygen to the tissues and partly accounts for the increase in oxygen extraction during a decrease in oxygen delivery.63,75 In dogs, the ability of the body to extract oxygen diminishes with α-receptor blockade of sympathetic activity, suggesting that redistribution of blood flow aided by the sympathetic nervous system is a major determinant of critical oxygen extraction.63
Microcirculation
Vasoconstriction after activation of the sympathetic nervous system during hypovolemia (hemorrhage) occurs in the arteries and medium-sized arterioles but not in terminal arterioles, which may even dilate, as judged from vital microscopy studies in animals.¶ Relatively spared terminal arteriolar blood flow is presumably caused by vasodilating metabolic responses to a decline in nutrient blood flow. Nevertheless, capillary flow usually diminishes, and heterogeneity, both in space and time, increases, particularly in irreversible shock and independent of cardiac output. Traumatic/hypovolemic shock may induce expression of adhesion molecules on primed neutrophils and vascular endothelium and this, together with a reduced flow rate, may promote adherence of neutrophils to endothelium.95,111–119 This adherence may impair red blood cell flow, particularly in capillaries and postcapillary venules.
Traumatic/hypovolemic shock may induce expression of adhesion molecules on primed neutrophils and vascular endothelium and this, together with a reduced flow rate, may promote adherence of neutrophils to endothelium.95,111–119 This adherence may impair red blood cell flow, particularly in capillaries and postcapillary venules. Other authors suggest that capillary leukostasis is pressure-dependent and not receptor-dependent and reversible when perfusion pressure has been restored.121 Finally, endothelial cells may swell and may hamper capillary red and white blood cell flow.95,98,110,122 The microcirculation can be visualized, even in humans, by buccal or sublingual orthogonal polarization spectroscopy and side stream dark-field imaging.123
Other authors suggest that capillary leukostasis is pressure-dependent and not receptor-dependent and reversible when perfusion pressure has been restored.121 Finally, endothelial cells may swell and may hamper capillary red and white blood cell flow.95,98,110,122 The microcirculation can be visualized, even in humans, by buccal or sublingual orthogonal polarization spectroscopy and side stream dark-field imaging.123
Vasoconstriction is not confined to arteries, but also occurs in the venous vasculature, more in large than in small venules and particularly in the splanchnic area, and, again, this is largely mediated by increased activity of the sympathetic nervous system and vasopressin and angiotensin II release.5,12,68,108 Because most of the circulating blood volume is located in small venules, splanchnic venoconstriction results in a decrease in compliance and less volume for a given intravascular pressure in the venous system, increasing return of blood to the heart.5,7 Hence, partitioning circulating volume in stressed and unstressed portions now favors the former. During hypovolemic shock, the precapillary to postcapillary resistance increases, resulting in a decrease in capillary hydrostatic pressure and in fluid resorption from the interstitial space as opposed to normal filtration from capillary to interstitium, even though interstitial hydrostatic pressure decreases.4,5 This is accompanied by diminished transport of protein from blood to interstitium.124
Cellular water is mobilized, unless, at a later stage, the cell swells following Na+ overload.21,98,110,125–129 Studies on fluid volumes in hypovolemic shock are not equivocal, but generally suggest that the interstitial and cellular compartments are depleted in defense of the circulating blood volume to promote venous return to the heart.5,7,125,126 Mobilization of fluid from the interstitial and cellular compartment can be promoted by plasma hyperosmolarity, through an increase in the glucose concentration.130,131 Chronically starved rats with depleted glycogen stores more rapidly die of hypovolemic shock than fed ones, and this can be prevented by prior glucose infusion.130 In addition, the lymphatics may show increased pumping ability, increasing return of fluid into the systemic circulation independently of the reduced capillary fluid filtration rate.132 Lymphatic return of interstitial protein and fluid may contribute to repletion of circulating protein and fluid volume.132
Hemorrhage and hypovolemic shock lead to a decrease in hematocrit and a decrease in plasma proteins through transfer of fluid (and protein) from the interstitial to the intravascular space.4,5,124 Refilling of the intravascular space diminishes in time after a sudden decrease in circulating volume, when a decline in colloid osmotic pressure, associated with hypoproteinemia, and an increase in hydrostatic pressure accomplish a new steady state in capillary exchange through readjustment of the pericapillary hydrostatic and colloid osmotic pressures, which determine fluid and protein transport.5 Conversely, hypoproteinemia can promote transcapillary fluid transport and expansion of the interstitial space, if hydrostatic pressure returns toward normal (e.g., during crystalloid fluid resuscitation).126,133–136 During a sudden decrease in circulating blood volume by hemorrhage, some time is needed before the decrease in hematocrit and of proteins in blood is completed, and this decrease is aggravated by nonsanguineous fluid resuscitation.126,134,137 Finally, increased sympathetic discharge results in contraction of the spleen, releasing red blood cells into the circulation and defending a fall in hematocrit.14
Cells
During hypovolemic shock, the oxygen lack in the tissues causes a decline in the mitochondrial production and concentration of high-energy phosphates in the tissues because of greater breakdown than production of these compounds.24,29,46,138,139 This decline is a function of the severity and duration of regional hypoperfusion relative to oxygen demand. The decrease in the redox status and high-energy phosphates during experimental hypovolemic shock is more pronounced in some tissues (diaphragm, liver, kidney, and gut) than in others (heart and skeletal muscle), so regional lactate production may vary.*
A decrease in high-energy phosphates heralds irreversible cell injury during ischemia, whereas a less severe decline may result only in prolonged programmed cell death—apoptosis. In animals with hypovolemic shock and in critically ill patients, the circulating levels of ATP can be diminished, and ATP degradation products, including adenosine, inosine, hypoxanthine, and xanthine, can be elevated, suggesting breakdown of ATP following a lack of oxygen in the tissues.28,29,56,141,142 Conversely, reperfusion is associated with restoration of energy charge, depending on the effect of ischemia, the oxygen demand, and the level of reperfusion. The intravenous administration of energy in the form of ATP-MgCl2 may help tissues (kidney, liver, heart, gut) to recover from ischemia and resume function, independently of the vasodilating effects of the compound.24,128,143,144 Also, pretreatment with coenzyme Q10, involved in the respiratory chain reactions in mitochondria, has a beneficial effect during hypovolemic shock and resuscitation, at least in dogs.81 Nevertheless, part of the mitochondrial dysfunction after trauma and hypovolemic shock has been suggested to be independent of a lack of oxygen.53 Near-infrared spectroscopy, which can be applied in animals and patients, may indeed reveal normal absorption spectra for tissue oxyhemoglobin and low mitochondrial cytochrome aa3 redox status.53,123,145
About 60% of the energy produced by respirating mitochondria is needed to fuel the Na+/K+ pump of the cell, through which the gradient in electrolyte concentrations and electrical potential over the cell membrane are controlled.24 When ATP becomes insufficient because of a decline in production associated with lack of oxygen and production of protons increases, the Na+/K+ pump is inhibited and the Na+/H+ exchanger is activated, and this results, together with a possibly selective increase in cell membrane permeability for ions, in an influx of Na+ into and efflux of H+ and K+ out of the cell, leading to cellular uptake of fluid.† Measurement of membrane potentials of skeletal muscle and liver in experimental animals has shown that hypovolemic shock rapidly decreases the transmembrane potential (a less negative inner membrane potential), associated with electrolyte and fluid shifts across the cell membrane.‡ A decrease in activity of the Na+/K+ pump may contribute to hyperkalemia because of potassium exchange between cells, interstitial fluid, and vascular space.38,46,111,125 Finally, calcium (Ca2+) influx into cells and their mitochondria inhibits cellular respiration and ultimately contributes to cellular damage and swelling, particularly during resuscitation, and this can be prevented by administration of Ca2+ antagonists.* Because of cellular influx, the plasma-free Ca2+ levels may decrease in experimental and human hypovolemic shock.127,149,150 Intracellular lysosomes lose their integrity so that proteolytic enzymes are released and contribute to cell death.4,24,107,151 These enzymes eventually may reach the systemic circulation and may damage remote organs.4,24,107,151
As has become apparent in past years, the cellular response to stress, such as heat and tissue hypoxia, involves the expression of certain genes, coding for synthesis of the so-called heat-shock proteins, which play an important role in protecting the cells against stress.152–155 The clinical significance of these molecular cellular changes is unknown. The response may be partially responsible, however, for the decreased susceptibility to and tissue injury by hemorrhagic shock in animals with a prior challenge by endotoxin or other forms of preconditioning.113,156
Organ Perfusion and Function in Shock
Heart
According to Starling’s law of the heart, a change in preload, approximated by the end-diastolic volume and determined by the venous return of blood to the heart, directly results in a change in stroke volume, defining myocardial function.7 The relationship between end-diastolic filling pressure and volume reflects compliance. Apart from preload, cardiac output also depends on afterload, which is approximated by the end-systolic volume of the heart, and contractility, reflected by the peak systolic pressure-to-volume relationship (maximal elastance).7,55,157 A diminished response of the stroke work by the heart, that is, the product of stroke volume and arterial blood pressure, to an increase in preload during resuscitation from hypovolemic shock may indicate diminished cardiac contractility that is associated with a worse outcome (e.g., caused by preexisting cardiac disease, hypovolemic shock itself, myocardial contusion, or combinations).4,55,158,159 The effect of hypovolemic shock on myocardial function in animal models is controversial. Depending on models, methods, and definitions of cardiac dysfunction, some authors describe a decrease, but others describe an unchanged function of the left side of the heart.† The latter can be explained if a decrease in contractility of the heart is masked by the inotropic effect of catecholamines and other positive inotropic substances, such as endothelin, liberated during hypovolemic shock, even though receptor-mediated catecholamine responses may decline.4,23,100,160
Although coronary blood flow may be defended, and the oxygen demands of the heart may decrease associated with a decrease in filling (preload) and arterial blood pressure (afterload) during initial hypovolemic shock, hypotension may become so severe that coronary vasodilation to compensate for a decline in perfusion pressure becomes exhausted, so that myocardial oxygen delivery decreases to less than the oxygen needs of the heart and ischemia ensues, particularly if tachycardia is present.3,21,140,161–163 This sequence leading to ischemia may occur primarily in endocardium because of more rapidly exhausted vasodilation in endocardium than epicardium and redistribution of blood flow from the inner to the outer layer of the heart.140 The subendocardium may become ischemic, and patchy necrosis may ensue. Because of regional transmural and intramural differences in vasodilator reserve, myocardial ischemia may be heterogeneously distributed and associated with a diminished redox state, lactate production, and creatine phosphate breakdown.140,164 Ischemia ultimately may contribute to a decrease in myocardial contractility during hypovolemic shock. Smooth muscle–dependent and, particularly, endothelium-dependent coronary vasomotion may be impaired after hypovolemic shock.89,165 Myocardial edema and compression of capillaries with resultant impairment of diffusion and extraction of oxygen may also contribute to a decrease in regional coronary blood flow, regional myocardial ischemia, and decreased myocardial function in hemorrhaged animals.127,131,140,161
Hypovolemic shock may induce a decrease in left ventricular compliance and relaxation.160,161 The diastolic dysfunction may be particularly pronounced during resuscitation from hypovolemic shock.157,160,161 Postischemic failure (stunning) also may play a role during resuscitation, at least temporarily. Ischemia-reperfusion of the heart results in accumulation of intracellular Ca2+.127 This may impair mitochondrial and sarcoplasmic reticulum function and contribute to impaired cardiac function after hypovolemic shock.127,160 In dogs, the administration of Ca2+ blockers may prevent such deterioration during resuscitation from hypovolemic shock.127 Finally, systemic release or intramyocardial production of negative inotropic substances and inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL) 6 and platelet activating factor, oxidant damage, metabolic acidosis, diminished adrenoreceptor density, and resultant diminished sensitivity of the heart to circulating catecholamines may contribute to myocardial dysfunction during hypovolemic shock.* Pentoxifylline may improve endothelial and myocardial function.166 Reversibility of dysfunction is associated with survival.161
The clinical evidence for myocardial dysfunction during hypovolemic shock is scarce.46,55,163 Nevertheless, it is conceivable that severe hypotension reduces the balance between oxygen delivery and demand of the heart because many patients with hypovolemic shock may be elderly with coronary artery disease, compromising coronary vasodilation. Some may have preexisting impaired function while on beta blockers. For a patient with hypovolemic shock, a decrease in left ventricular compliance, contractility, or both may imply that a relatively high filling pressure would be needed to restore cardiac output during fluid resuscitation.55,71,161,163,171 The averaged optimal pulmonary capillary wedge pressure (PCWP), that is, the pressure above which cardiac output does not increase further, may not be elevated in patients with hypovolemic shock (i.e., 12 to 15 mm Hg), although in some patients, abnormally elevated filling pressures may be needed to increase cardiac output, or cardiac output does not increase at all during fluid resuscitation.55,71,171,172 A diminished function of the heart may hamper restoration of oxygen delivery to the tissues during resuscitation necessary for survival.9,45,55,159,160 Myocardial dysfunction may thus be greater in nonsurvivors than in survivors. There may be some electrocardiographic or enzymatic evidence for myocardial ischemia and injury, and some patients may experience a myocardial infarction as a complication of severe hypovolemic shock after hemorrhage.163,173
Lung
Hypovolemic shock often induces an increase in ventilatory minute volume, resulting in tachypnea or hyperventilation and a decrease in arterial PCO2.33,49,50,52,174 Unless complicated by pulmonary abnormalities, these changes are, at least initially, not the result of hypoxemia but an increase in dead-space ventilation following a decrease in pulmonary perfusion so that a higher minute ventilation is necessary for a given CO2 production to eliminate CO2 from the blood and to maintain a normal PCO2 in arterial blood.33,49,50 Minute ventilatory volume may increase further if a decrease in PCO2 is necessary to compensate for metabolic acidosis after accumulation of lactate in the blood.* The imbalance between increased demands of the diaphragm and reduced blood flow in shock may finally lead to respiratory muscle fatigue and a subsequent decline in ventilatory minute volume.50
Hypovolemic shock caused by trauma and hemorrhage and followed by extensive transfusion therapy of red blood cell concentrates can be complicated by pulmonary edema and impaired gas exchange.51,175–180 In some patients, fluid overloading, overtransfusion, and an elevated filtration pressure (PCWP) may be responsible: transfusion-associated circulatory overload (TACO). In others, pulmonary edema may be due to a pulmonary vascular injury, however, and increased vascular permeability at a relatively low PCWP, indicating noncardiogenic permeability edema or ARDS.51,174,177,178 The reaction to diuretics may help to differentiate between hydrostatic and permeability edema of the lungs. The latter seems relatively rare in polytransfused, polytraumatized patients unless associated with complications, but other studies suggest that about 30% of patients with severe trauma/hemorrhage, particularly if polytransfused, may develop ARDS.177,178,180,181
Experimental studies are at variance concerning alterations in capillary permeability of the lungs during hypovolemic shock and resuscitation.4,133,174,182,183 According to some investigators, hypovolemic shock following bleeding and transfusion mildly increases transvascular filtration of fluid and proteins and results in accumulation of interstitial fluid as a consequence of increased permeability,182 but other authors do not observe such changes.131,133,182,184 In other animal studies, however, traumatic/hypovolemic shock resulted in extensive morphologic changes of the lung, with endothelial and interstitial edema, accumulation of degranulated neutrophils, and scattered fat emboli, which may resemble the pulmonary changes after traumatic/hypovolemic shock in humans.156,174,185–187 As measured by the transvascular albumin flux in the lungs, almost 80% of patients with multiple trauma may show increased pulmonary vascular permeability in the disease course.51 This leak ultimately may contribute to pulmonary edema, impaired mechanics, and gas exchange.51 As suggested by animal experiments, among others, several factors may play a role, including release of proinflammatory mediators (TNF-α) and priming and activation of blood neutrophils after ischemia-reperfusion, contusion or ischemia-reperfusion of the lungs themselves, pulmonary microemboli of neutrophils, platelets and fat particles from the medulla of fractured long bones and pelvis, and neutrophilic antibodies or humoral or cellular breakdown products and released cytokines in long-stored and transfused blood products (transfusion-related acute lung injury, TRALI).* Translocated endotoxin may also play a role.185 Finally, aspiration of foreign material or gastric contents and posttraumatic pneumonia and sepsis may contribute to the development of ARDS in trauma patients. When pulmonary edema has developed, active resorption by alveolar cells becomes necessary for clearance. This process is cylic AMP–dependent and can be disturbed by inducible nitric oxide synthase (iNOS)–derived NO and peroxynitrite and enhanced by expression of heme oxygenase, which may mitigate lung injury in animal models.156 How this translates clinically is unclear.
Brain
Classically, brain perfusion and microcirculation are considered to be relatively spared during progressive hypovolemia because of the extensive autoregulatory capacity of cerebral arteries.15,189 In case of autoregulation impairment after neurotrauma, however, brain perfusion may decrease, and subsequent reperfusion may contribute to secondary cerebral damage during hypovolemic shock and resuscitation. Hemorrhagic shock and resuscitation per se may also impair autoregulatory capacity of brain vessels, however, because of endothelial dysfunction and diminished NO-dependent vasodilator reactivity, so that the brain may experience an oxygen debt and subsequent metabolic and functional deterioration.29,88
Kidney
Hypovolemic hypotension is an important risk factor for acute kidney injury and failure after trauma.138 During a decrease in cardiac output following progressive hemorrhage, renal blood flow can be maintained because of renal vasodilation, so that the kidneys may not participate in the systemic vasoconstriction that characterizes hypovolemic shock.3 Vasodilating prostaglandins are released in the kidney through activation of the cyclooxygenase pathway of arachidonic acid metabolism in response to ischemia, increased sympathetic activity, and angiotensin II, so that renal vasodilation during the early phase of hemorrhage can be blocked by prostaglandin synthesis inhibition, resulting in a profound decrease in blood flow even if accompanied by an increase in arterial blood pressure.3 When blood pressure decreases during progressive hypovolemia, the renal vessels constrict, impairing blood flow to the kidneys more than to other organs.* This is partly caused by a baroreflex-mediated increase in sympathetic activity; activation of the renin-angiotensin-aldosterone system; and release of catecholamines, angiotensin II, endothelin, and vasopressin.13,14,74 During prolonged hypovolemic shock, sympathetic inhibition may protect against renal ischemia.14 This propensity for vasoconstriction is thus partly offset if NO and other factors with vasodilatory actions are released intrarenally.4,86 Inhibition of NO synthesis increases blood pressure, however, and increases renal perfusion and glomerular filtration during hypovolemic shock.86 In another study, endothelium-dependent renal vasodilation was impaired after hypovolemic shock.87
Renal ischemia results in a decrease in glomerular filtration (prerenal renal failure) that is less than the decline in blood flow so that the filtration fraction often increases.138 The latter is caused by greater constriction of efferent than of afferent arterioles in glomeruli, in which high levels of circulating angiotensin II are probably involved. The decrease in glomerular filtration together with an increase in tubular resorption of electrolytes and fluids, mediated by increased levels of antidiuretic hormone released by the pituitary and decreased levels of atrial natriuretic peptides through low atrial filling, results in oliguria or anuria (<0.3 mL/kg/hour) and a low sodium content of urine.138
The decrease in renal perfusion during hypovolemic shock is often accompanied by redistribution of blood flow from outer to inner cortex and medulla, which is already borderline hypoxic even in the normal state.127 If long-lasting and severe, the cortical kidney becomes ischemic, despite a decrease in oxygen needs associated with fewer energy needs for tubular resorption in the presence of less filtration, so that the levels of high-energy phosphates decline.138,139 Severe and prolonged renal ischemia and metabolic deterioration finally result in acute kidney injury and failure with morphologic changes, particularly in proximal tubules and medullary segments (acute tubular necrosis) when an increase in renal perfusion does not immediately restore filtration and diuresis, but rather injures renal structures (reperfusion injury), limiting a return of blood flow and glomerular filtration during resuscitation.129,138,185,186 This is often recognized by a persistent oliguria and a gradual increase in creatinine and urea levels in blood. In addition, the plasma levels and urinary excretion of biomarkers of injury and dysfunction may increase.104
Gut
During hypovolemic shock, blood flow from stomach to colon is redistributed to other organs, and this may be primarily mediated by elevated sympathetic activity and increased levels of vasopressin and angiotensin II even though vascular reactivity to the latter may diminish.† Vasoconstriction may overwhelm NO and other vasodilating mechanisms, and endothelium-dependent vasodilation may be impaired after oxidant endothelial injury.194 Gut ischemia is aggravated further by the countercurrent mechanism in mucosal (villous) blood flow, promoting diffusional shunting of oxygen from arteries to veins, bypassing tissues. Other studies reported that gut mucosal blood flow may be relatively spared during hypovolemia, however.75,105 Portal blood flow decreases, and portal blood levels of lactate increase after gut ischemia.27,48,195
Gastric mucosal ischemia may result in diminished energy-consuming acid production and may predispose to mucosal stress ulceration.192,196 Microscopic studies in experimental animals show damage of gastric mucosa, villous epithelium in small bowel, and mucosa of the large bowel after hypovolemic shock.114,192,197–199 Gastric mucosal ischemia-reperfusion injury after bleeding may be aggravated by gastric acid itself, neutrophils, inflammatory mediators, endothelin, reactive oxygen species (ROS), and proteases.198,200 Bowel ischemia and mucosal damage during hypovolemic shock in the dog may ultimately lead to leakage of fluid from the bloodstream to the bowel lumen, instead of normal resorption of luminal fluids.115,197 Diarrhea may contribute to intravascular volume depletion during severe and prolonged hypovolemic shock, at least in animals.
Gut mucosal ischemia, energy depletion, injury, and inflammation may compromise the barrier function of the mucosa, enhancing the likelihood that bacteria and endotoxins in intestinal lumen (large bowel) translocate through the damaged gut wall to lymph nodes, portal venous blood, or both.* The gut epithelial (lumen to plasma) permeability for small molecules also is increased. Indigenous flora, generated toxic ROS, cytokines, Ca2+ overload, iNOS, peroxynitrite, phospholipase A2 activation, and activated and adhering neutrophils during ischemia and reperfusion probably all play a role in the injury, promoting hyperpermeability and translocation.199,203 Mucosal injury and translocation can be inhibited by compounds targeted against these factors.199 Impaired detoxifying capacity of the Kupffer cells of the liver because of ischemia or preexistent liver disease may contribute further to bacteria and endotoxins reaching the systemic circulation and contributing to progression of shock by triggering an inflammation cascade, ultimately resulting in release of vasoactive substances.4,204,205 This translocation has been shown to contribute to the lethality of hypovolemic shock in experimental animals because clearance or blockade of translocated bacteria and endotoxins is associated with survival, and germ-free animals survive an episode of bleeding more often and longer than ones with normal intestinal flora.4,143,203,205 Finally, it has been shown that the absorptive capacity of the gut for carbohydrates, amino acids, and lipids decreases during hypovolemic shock.148,195 Although enteral feeding during hypovolemic shock and after resuscitation may increase metabolic demands of the gut, there is experimental evidence that luminal application of nutrients, particularly of enterocyte-fueling glutamine, induces an increase in small vessel blood flow, ameliorates damage, and diminishes the likelihood for translocation of endotoxins and bacteria during resuscitation from hemorrhage.206
In humans, hypovolemia leads to a decline in hepatosplanchnic perfusion.74 Stomach mucosal lesions may be common after prolonged hypovolemic shock, but overt bleeding is a relatively rare event, particularly in a rapidly, adequately resuscitated patient.207 Agents that decrease energy-demanding gastric acid production may protect against stress ulcers during mucosal ischemia.196 The gut is usually quiescent during hypovolemic shock in humans. Ileus is often present, and the patient is managed expectantly until bowel sounds return and enteral feeding is likely to be tolerated. Occasionally, a bowel infarction and perforation may complicate hypovolemic shock as a consequence of nonocclusive ischemia.148 Gut absorptive capacity may decrease,148 perhaps caused by gut ischemia. The adequacy of gastrointestinal blood flow can be monitored in humans with the help of a balloon catheter in the stomach (or gut), in which fluid or air is installed, or sublingually or buccally with help of a sensor (tonometry).* The mucosal PCO2 thus measured decreases, and the mucosal-to-blood PCO2 gradient increases, during a decrease in mucosal blood flow relative to demand. An increase of this gradient may occur at an earlier stage than an increase in heart rate or decrease in arterial blood pressure during progressive hypovolemia, constituting an early and sensitive sign of shock.208 Gastrointestinal tonometry can be used as a guide for resuscitation.† The clinical occurrence and significance of translocation of intestinal bacteria and endotoxins to mesenteric lymph nodes and the bloodstream are unclear, although the capacity of the human gut wall to resorb orally administered small molecules, including lactulose relative to mannitol, may increase, indicating epithelial barrier dysfunction.54,204,205,211–214
Liver
Liver microvascular and sinusoidal perfusion decline during hypovolemic shock because of diminished portal and hepatic arterial blood flow, roughly in proportion to the decrease in cardiac output so that in contrast to the gut there is no angiotensin II–mediated selective vasoconstriction in the hepatic arterial bed.‡ Endogenous mechanisms, including release of NO, carbon monoxide, and hydrogen sulfide in the absence of endothelial dysfunction, may counteract a decrease in perfusion, which is promoted by thromboxane A2 and endothelin.91,155 A decrease in blood flow may result in liver ischemia, a decrease in high-energy phosphate contents and clearance function as evidenced by insufficient capacity to clear indocyanine green from blood and a decrease in the bile excretion rate.§ The capacity to clear gut-derived endotoxin, cytokines and lactate also may decrease, and the ischemic liver produces lactate.43 Hepatic ischemia may result in a diminished capacity for metabolism of drugs such as lignocaine219 and for gluconeogenesis from lactate and amino acids, contributing to hypoglycemia in the late stage of hypovolemic shock.12,139 Inflammation of the liver causes cytokine expression; hepatic sinuses become filled with adherent neutrophilic aggregates, lining cells may swell, and microcirculatory failure and centrilobular necrosis/apoptosis may ensue with leakage of enzymes into the circulation.* ROS and NO-derived and toxic peroxynitrite and damage of endoplasmatic reticulum and mitochondria may be involved. Clinically, bilirubin and transaminases may be transiently elevated in blood, abnormalities attributed to ischemic hepatitis.222,223 A clinically useful measure of hepatic oxygen debt is an increase in the plasma ratio of β-hydroxybutyrate to acetoacetate (ketone body ratio), which occurs concurrently with a decrease in the hepatic mitochondrial redox state.28,91,95,223
Spleen
The spleen contracts during hypovolemic shock, probably caused by increased sympathetic activity, and this results in release of red blood cells into the circulation.3,14 Changes in hematocrit during the early phase of bleeding probably underestimate the severity of plasma losses. The spleen also releases stored platelets.
Pancreas
The pancreas is severely ischemic during hypovolemic shock.107 Ischemic pancreatitis may lead to autodigestion of acinar cells and liberation of pancreatic lysosomal enzymes into the systemic circulation, including proteases and factors with negative inotropic properties on the heart, although the latter factors may also come from ischemic gut.4,107,166 Ligation of the pancreatic duct may be beneficial in experiments by preventing gut injury and barrier failure, among others.224
Hormones and Metabolism
As mentioned before, a severe decrease in cardiac output resulting in a decrease in arterial blood pressure during hypovolemic shock results in activation of the sympathetic nervous system through the baroreceptor reflex and liberation of norepinephrine from nerve endings and epinephrine from adrenal medulla so that circulating levels of these catecholamines increase.11–13,74,81,99 The insulin secretion by the pancreas is inhibited, and glucagon secretion is enhanced by high circulating norepinephrine levels.99 The renin-angiotensin-aldosterone system is activated, and the pituitary secretion of vasopressin/antidiuretic hormone and opioids increases.11–13,23,99 The pituitary response to stress further includes an increase in adrenocorticotropic hormone (ACTH) with resultant corticosteroid release by the adrenal cortex, unless limited by the so-called relative adrenal insufficiency following hypoperfusion-induced adrenal damage.12,99,152 These factors may be essential for survival because prior adrenalectomy decreases survival of animals subjected to hypovolemic shock, and steroid repletion is protective in this respect.12 This protective effect can be attributed to, among others, less overactivation of the sympathetic nervous system and increased sensitivity of the heart and vasculature to circulating levels of catecholamines.12
Finally, the secretion of atrial natriuretic peptides by the myocardium declines in response to hypovolemia and diminished wall stress of the atria. These factors, among others, result in tachycardia and a diminished renal excretion of water and salt to restore circulating blood volume. Endogenous opioids could play a role in maintaining shock by their vasodilating and myocardial depressant properties, however.11,13,23 Administration of the opioid antagonist naloxone and its derivatives augment arterial blood pressure in hypovolemic shock.13,23,143,225 Similarly, thyrotropin-releasing hormone depresses the opioid system and increases arterial blood pressure, cardiac function, and survival during hypovolemic shock in animals.23 Thyroid hormone may have a similar effect.226
During trauma, hypovolemic shock, and cellular ischemia, intermediary metabolism undergoes profound changes, partly caused by an altered hormonal milieu.99 The early hyperglycemic response to traumatic/hypovolemic shock is the combined result of enhanced glycogenolysis, caused by the hormonal response to stress and elevated epinephrine, cortisol, and glucagon levels; increased gluconeogenesis in the liver, partly mediated by glucagon; and peripheral resistance to the action of insulin, the secretion of which may be diminished shortly after onset of shock but may be enhanced later after shock.24,47,99,129 This resistance is most likely the result of an altered hormonal milieu—the increase in circulating epinephrine and cortisol levels. During the late, irreversible stage of hypovolemic shock, however, hypoglycemia supervenes, at least in animal models, because glycogen stores may be depleted and the capacity for gluconeogenesis by the liver may decrease because of ischemia.12,24,47,111,130
Increased gluconeogenesis in the liver, and to a lesser extent in the kidneys, follows increased efflux of amino acids such as alanine and glutamine from the muscle to the liver because of breakdown of muscle protein.99,222 The latter is evidenced by increased urinary losses of nitrogen and a negative nitrogen balance.99,222 Amino acid metabolic changes may contribute to the immunodepression of trauma. Lactate produced in muscle also can be converted to glucose in the liver.99 Finally, fatty acid metabolism undergoes profound changes, with depressed lipolysis, ketogenesis, and combustion of fatty acids during shock and an increase in the resuscitation phase.99,222 Some investigators regard a deranged intermediary metabolism of primary importance for the eventual outcome of shock, whereas others merely consider these changes a result of the shock process itself.222
Inflammatory and Immunologic Changes
Activation of the xanthine-oxidase system and formation of uric acid from the ATP breakdown products hypoxanthine and xanthine during reperfusion could liberate ROS, which damage vascular endothelium and parenchymal cell membranes through peroxidation of lipids.* The release of ROS during ischemia-reperfusion may activate macrophages and attract neutrophils, partly mediated by release of cytokines via activated nuclear factor-κB (NF-κB).194,228 The interaction of ROS fueled by oxygen and NO may further play a role in inflammation and vascular tone after perfusion.90,228 ROS scavengers may inhibit formation of toxic peroxynitrite via NO, and ROS and may inhibit breakage of DNA single strands and activation of poly(ADP-ribose) polymerase, which contributes to cellular injury.90,92,221 Some time after hypovolemic shock and resuscitation, iNOS may become active particularly in the gut and liver; circulating NO breakdown products may increase and inhibition of the excessive NO release may ameliorate hemodynamic changes, organ inflammation, and neutrophil accumulation and function, partly via less peroxynitrite formation, unless inhibition leads to a decrease in cardiac output.90,92,155,220 Increased iNOS-derived NO also may be prevented and treated by corticosteroids or ACTH fragments.93,229
Proinflammatory mediators may be expressed locally in a variety of organs in response to hemorrhagic shock, including heart and lungs, and this is partly under control of α-sympathoadrenergic and neuroimmune stimuli, toll-like receptor 4, NF-κB, hypoxia-inducible factor, glycogen synthase kinase-3β, and other factors involved in cell signaling.228,230–233 During and after hemorrhage, hypovolemic shock, and resuscitation, macrophages, including lung macrophages and Kupffer cells in the liver, may release cytokines, including TNF-α, IL-1, IL-6, and IL-8. This inflammatory response is attenuated when reperfusion takes place in hypoxic, rather than normoxic, conditions.234 The response can be ameliorated by blockade of NF-κB, administration of the macrophage-inhibitor pentoxifylline, or ATP-MgCl2 increasing hepatic blood flow.*
Ischemia per se and the immune consequences of gut barrier injury may play a role in Kupffer cell responses. The reperfused gut, together with the liver, may be a source of systemically released cytokines, as suggested by animal experiments and observations in humans after trauma, and translocated endotoxin may play a role.† During reperfusion after resuscitation, cytokines may induce and amplify the inflammatory response to ischemia and may induce further local and remote organ damage with circulatory changes.166,187,236,240,241 Spillover of mediators into the mesenteric lymph or portal and systemic circulations during reperfusion of prior ischemic gut may have deleterious effects on remote organs by inducing neutrophil activation and adherence, which may contribute to a lung vascular injury with increased permeability.185,186,188,240,241 Circulating levels of proinflammatory cytokines may be of predictive value for remote organ damage, including ARDS, after trauma in patients.230,237,238 Endotoxin binding or antibodies and cytokine antibodies may ameliorate remote tissue damage after bleeding, hypovolemic shock, and resuscitation.185,186
Trauma and shock/resuscitation have also been shown to activate the complement and the arachidonic acid systems.151,242–245 Complement activation may yield potent vasodilating and leukoattractant substances and contribute to remote inflammatory organ damage (ARDS). Ischemia may generate phospholipase A2, catalyzing arachidonic acid metabolism into prostaglandins via the cyclooxygenase pathway, releasing thromboxane A2 and prostacyclin, and into leukotrienes via the lipoxygenase pathway.10,151,243 Thromboxane A2, released from platelets, neutrophils, and cell membranes, has potent vasoconstricting properties and promotes aggregation of platelets and neutrophils, whereas prostacyclin has vasodilating properties and inhibits platelet and neutrophil aggregation.243 Leukotrienes have vasoconstricting properties, increase capillary permeability, and attract neutrophils.151 Vasoconstricting prostaglandins may be involved in tissue damage during ischemia-reperfusion, and vasodilating prostaglandins may be involved in the vasodilated state of terminal hypovolemic shock.10,242 Another lipid mediator that may be released is platelet-activating factor, but the precise action of this mediator is unclear.149,170
The interplay of these factors may result in endothelial activation throughout the body and an inflammatory reaction, ultimately involving attraction, activation, and endothelial adherence of neutrophils, as shown in animal models of hypovolemic shock after bleeding and ischemia-reperfusion.* Neutrophils release vasoconstricting, platelet-aggregating, and damaging thromboxane A2 and may inhibit vasodilating prostacyclin, via secreted ROS and proteases such as elastase.117,228,238,244,246 Neutrophil aggregation and secreted activation products also may also play a role in the reperfusion injury by impairing resumption of small vessel blood flow, even in the presence of a seemingly adequate cardiac output and arterial blood pressure.† In humans, the activation of neutrophils after trauma, with increased adhesion molecule expression and propensity for degranulation, is associated with morbidity after trauma, such as development of MOF and predisposition to sepsis.115–117,233,237,247 After initial leukopenia (neutropenia) following trapping of leukocytes in the microcirculation, activation of the pituitary-adrenal axis and release of corticosteroids and catecholamines during hypovolemic shock result in an increase of circulating neutrophils following demargination and release from bone marrow, together with eosinopenia and lymphocytopenia.46,112–115,216 A tertiary decrease of circulating neutrophils in patients with a downhill course may be explained by microcirculatory sequestration.115 The hemodynamics, organ function, and survival of rats with hypovolemic shock/resuscitation are improved if rats are made neutropenic before the challenge, and this may relate to improved regional and capillary blood flow.112,243 A monoclonal antibody against or antagonists of neutrophil-endothelial adhesion molecules decrease reperfusion injury in lungs, liver, stomach, and intestines after hypovolemic shock or ruptured aortic aneurysm and may improve survival, at least in animal models.114,117,118,187,228 This does not impair host defense against subsequent bacterial infections.114
However, neutrophils may later become downregulated after initial stimulation by circulating proinflammatory and anti-inflammatory mediators.111,229,248 Neutrophil dysfunction is evidenced by a diminished potential to migrate and to digest and kill bacteria, perhaps in the presence of an inhibited respiratory burst.111,188,247,248 In hemorrhaged mice, the infusion of granulocyte colony-stimulating factor or IL-6 after hemorrhage may partly prevent neutrophil defects and protect against death from subsequent pulmonary sepsis.248 Also, the opsonization function of macrophages, that is, the reticuloendothelial system, is depressed so that removal from the circulation of fibrin, cell aggregates, and bacteria by the liver is at least transiently impaired.* This may relate to the appearance after hypovolemic/traumatic shock of substances in blood that depress reticuloendothelial system function or to a decrease of the α2-glycoprotein fibronectin in plasma, a substance that aids the reticuloendothelial system in opsonization.4,167,236,249,251 This deficiency may contribute to development of MOF and might be reversed by infusion of plasma cryoprecipitate.251 Hypovolemic shock and gut-derived factors may blunt the increase in bone marrow cytopoiesis after soft tissue trauma and endotoxin and contribute to susceptibility to sepsis.252,253
Hemorrhagic/hypovolemic shock and subsequent resuscitation depress the immune system by suppressing the function of not only neutrophils but also lymphocytes and macrophages; this depresses humoral and cellular immune responses, decreasing antigen presentation and delayed hypersensitivity to skin test antigens and increasing susceptibility to sepsis.† Part of this may be mediated via neuroimmune modulation and resulting efferent sympathetic, adrenergic, and vagal stimulation.37,93 In patients, the immune defect correlates with the extent and severity of trauma and the degree of blood resuscitation required, but animal experiments document that hemorrhage/resuscitation per se depresses immune function, although trauma and blood transfusions may only be synergistic in this respect.236,250,254,255,258 Priming of immune cells may explain in part the increased sensitivity to endotoxin and sepsis after hypovolemic shock, although other authors have described that prior hypovolemic shock and priming decreased the immune response and increased the tolerance to endotoxin or sepsis.114,184,187,259 Hemorrhage decreases the capability of lymphocytes to proliferate and to produce lymphokines (IL-2) in response to mitogens, an effect that seems dependent on an energy or NO deficit or on Ca2+ influx in these cells after ischemia because the defect can be overcome by administration of Ca2+ influx blockers.236,250,254,255,258 Increased macrophage production of cytokines during hypovolemic shock and resuscitation may be followed by decreased ability of the cells to release mediators such as TNF-α and to express HLA-DR, upon challenges, and to process and present antigens to lymphocytes. This may relate to a cellular energy deficit, accumulation of Ca2+, and enhanced prostaglandin E2 synthesis.‡ The immunodepression after hypovolemic shock and predisposition to sepsis may finally include the release by Kupffer cells, among others, of anti-inflammatory mediators, such as IL-10 and soluble receptors (receptor antagonists) for previously released proinflammatory cytokines, and this may relate to sepsis-induced MOF and increased risks of morbidity and mortality in trauma patients.250,260 Otherwise, the immunologic consequences of trauma, hemorrhage, and hypovolemic shock depend on numerous additional factors, including gender and other genetic influences.230,247,250 Men may exhibit more immunodepression after trauma/hemorrhage than women. The clinical implication may be that men are more susceptible than women to microbial infections after trauma.261
Circulating coagulation factors and platelet counts may decrease after hypovolemic shock and resuscitation, whereas fibrin products may increase. This is the consequence of coagulation activation and fibrinolysis inhibition by endothelial activation, tissue injury, and inflammatory responses, even though dilution after fluid resuscitation may heavily contribute.181,237,262 Disseminated intravascular coagulation (DIC) and fibrin deposits, if insufficiently removed by the fibrinolytic system, are believed to contribute to a decrease in plasma coagulation factors and to widespread microvascular organ dysfunction.181,237,263 Proinflammatory responses, some resuscitation fluids, hypothermia, and acidosis may contribute to DIC and the coagulation defect of severe hemorrhagic/traumatic shock.237,264
Reperfusion and Irreversible Shock
Reperfusion of various organs, including the heart, gut, skeletal muscle, brain, kidneys, and liver, after a transient episode of ischemia, as occurs during hypovolemic shock, results in the so-called reperfusion injury, which limits the possibility for resumption of microvascular tissue blood flow and function of organs, particularly of the liver, even if cardiac output and arterial blood pressure have been restored to normal value.* Redistribution of blood flow during hypovolemia may be only partly attenuated by reperfusion.
Reperfusion after a certain period of shock and diminished oxygen uptake results in an increase in oxygen uptake above baseline levels, provided that oxygen delivery and cellular function are adequate.42,62,73,99 This repayment of the oxygen debt is largely determined by the increased demands for oxygen to resynthesize ATP from adenosine and phosphates and to rebuild the lost energy stores. This repayment is determined by the extent to which mitochondria are damaged during ischemia and the availability of substrates to resynthesize high-energy phosphates and restore cellular contents of these compounds because the substrates needed for synthesis may have been washed out, necessitating de novo synthesis.24,142,143 Resuscitation may not completely restore energy levels, the activity of the Na+/K+ pump, and the membrane potential of skeletal muscle and liver necessary to remove accumulated fluid and Na+ in the cell.125,127,142
Reperfusion not only results in resumption of oxygen delivery but also of Ca2+ to the tissues. This Ca2+ may be taken up by cells and may contribute to the reperfusion injury by damaging cell organelles, inhibiting mitochondrial respiration, and activating proteases and prostaglandin synthesis.127,129,148,149 Reperfusion injury of heart, gut, kidneys, and liver after resuscitation from hypovolemic shock in animals may be prevented in part by administration of Ca2+ influx blockers independently of their vasodilating effects, suggesting that Ca2+ overload is partly responsible for the reperfusion injury.127,129,148,149 Finally, endothelial damage and swelling and cellular aggregation may hamper the regional regulation of blood flow during resuscitation from hypovolemic shock.79,95,110,122 Neutrophil-mediated endothelial injury may increase capillary permeability and contribute to fluid losses during resuscitation.112,178,179,228,267 Conversely, the intravenous administration of energy in the form of ATP-MgCl2 or adenosine-regulating compounds may help tissues to recover from ischemia and resume function, independently of the vasodilating effects of the compounds, by providing energy, improving the microcirculation, and reducing cell swelling to promote survival.* Nevertheless, the ability of organs or the whole body to increase oxygen uptake during reperfusion above normal may be associated with survival in experimental animals with hypovolemic shock and in hypovolemic patients after trauma or major surgery, whereas inability may be associated with ultimate demise.9,73,82 Also, ischemic preconditioning may protect against hemorrhagic shock and reperfusion-induced tissue injury.156
If shock syndrome with hypotension and subnormal oxygen uptake persists after optimal fluid repletion and attempts at reperfusion with inotropic and vasoactive drugs, the condition can be regarded as irreversible and terminal.4,9 The term irreversible shock has been mainly used in animal experiments, however, in which reinfusion of the shed blood after a certain period is unable to reverse the shock syndrome.4,268 Various factors may play a role.4 First, vascular decompensation may contribute to a further decrease in blood pressures and may include diminished constrictive reactivity, dilation of arterioles, and insensitivity to circulating or exogenous catecholamines.97,108 The decline in vascular resistance may be partly caused by metabolic vasodilation in ischemic and acidotic tissues, overcoming vasoconstrictive influences.4,10,108 Other factors that may be involved include dysfunction of vascular smooth muscle after induction of iNOS and resultant increased production of vasodilating NO in the vessel wall, acidosis and activation of low ATP-activated K+ channels, histamine release, and prostaglandin-induced neurotransmission failure.† Circulating levels of NO breakdown products, nitrate and nitrite, may be elevated already early after hemorrhage in animals and trauma in humans, although other authors described low levels in humans.269 iNOS upregulation and NO production may be prevented by NO blockers, corticosteroids, or ACTH fragments.92,93,229 Finally, central cerebral or humoral mechanisms may contribute to the irreversible hemorrhagic shock, and this may relate to endogenous opioids, thyrotropin-releasing hormone, or macrophage-derived cannabinoids.13,23,270
The decrease in arterial vascular resistance may be particularly pronounced in the tissues, showing most intense vasoconstriction during hypovolemic shock, including gut and skeletal muscle, offsetting the redistribution of blood flow during hypovolemic shock and increasing blood flow to these organs at the expense of blood flow to vital tissues.10,22 In contrast, venous compliance and resistance increase, leading to peripheral pooling of blood and a decrease in venous return to the heart.4,166 The latter changes may be particularly pronounced in the splanchnic region.166 During prolonged or irreversible hypovolemic shock, capillary hydrostatic pressure may increase after arteriolar vasodilation and venular constriction, resulting in a decrease in the precapillary-to-postcapillary resistance ratio and promoting fluid filtration into the interstitium.4,5 Capillary permeability also may increase, resulting in a high capillary hydraulic conductance and a decrease in the reflection coefficient for plasma proteins. Increased permeability for proteins increases capillary filtration for a given intravascular hydrostatic pressure and promotes the formation of edema.178,267 The increase in permeability may be the consequence of endothelial damage and loss of protective glycocalyx by ischemia-reperfusion, possibly involving ROS and proinflammatory mediators.271 It may contribute further to a decline in circulating blood volume.5,267 Cells may swell, and this may diminish circulating blood volume further.4,125,127–129 Expansion of the cellular and interstitial fluid volume at the expense of the intravascular volume is manifested by a preterminal increase of the hematocrit.4,5,126
Irreversible hypovolemic shock may contribute to MOF and death of patients.2,207 An inflammatory response to ischemic tissue and patchy necrosis/apoptosis may contribute to organ damage and dysfunction and thereby to the irreversibility of hypovolemic shock.4,114,228 Reperfusion injury may aggravate organ damage and contribute to irreversible shock.114,228,266 The pump function of the heart may diminish after a decrease in systolic contractility and compliance, and this may contribute to irreversibility of shock during resuscitation.158 Myocardial dysfunction may contribute to the development of pulmonary alveolar edema if aggressive fluid infusion in attempts to increase cardiac output results in an elevated PCWP.4 Diminished function of the heart may hamper restoration of oxygen delivery and uptake to the tissues during resuscitation.9,45,157,158 Damage of the gut mucosa may cause translocation of luminal bacteria and endotoxins from gut lumen to systemic circulation, at least in experimental animals, and the resultant sepsis may contribute to the irreversibility of hypovolemic shock.*
Clinical Features
Causes
One of the most frequent causes of hypovolemic shock is blood loss after trauma (see Box 26.1), including blood loss during or after major surgery.2 Ruptured aortic aneurysm and gastrointestinal hemorrhage are other frequent causes of hypovolemic shock. Upper gastrointestinal bleeding can be caused by peptic ulcer disease, reflux esophagitis, variceal bleeding, erosive gastritis (stress ulcer), or aortoduodenal fistula after vascular surgery. Lower gastrointestinal bleeding can result from diverticular disease, carcinomas, or polyps in the colon. Sometimes, massive hemoptysis resulting from a tumor, tuberculosis, fungal infection, or bronchiectasis can be the cause of hypovolemic shock. Hematuria as a result of a tumor or trauma is a rare cause of hypovolemic shock. During multiple trauma, blood loss is essential in causing hypovolemic shock, but trauma itself can activate various mediator systems, with resultant release of vasoactive substances that contribute to the development of shock. In contrast to pure hypovolemic shock, cardiac output can be elevated, and peripheral vascular resistance is often decreased in cases of multiple trauma.25
Signs and Symptoms
For a clinical diagnosis of shock, hypotension and clinical signs of organ ischemia should be present. Arterial blood pressure and clinical signs are relatively insensitive for small blood losses (Table 26.1).208 This sensitivity can be improved by using the shock index, calculated from heart rate divided by systolic blood pressure.18 The clinician can recognize shock from a decrease in systolic blood pressure to less than 90 mm Hg or a decrease of more than 40 mm Hg below preshock levels, with a reduction in pulse pressure. Hypotension may be so severe that blood pressure is unrecordable noninvasively. There can be a large gradient between invasively and noninvasively measured arterial blood pressure and between central and radial artery pressure during shock and drug-induced vasoconstriction.272 Hypotension may become particularly marked when the patient sits or stands versus when the patient is supine (orthostatic hypotension).8,17,18 Postural dizziness, tachycardia, and hypotension are reliable and early signs of hypovolemia, whereas dryness of mucous membranes and axillae, decreased turgor, supine hypotension, and other signs have less diagnostic value.17,18
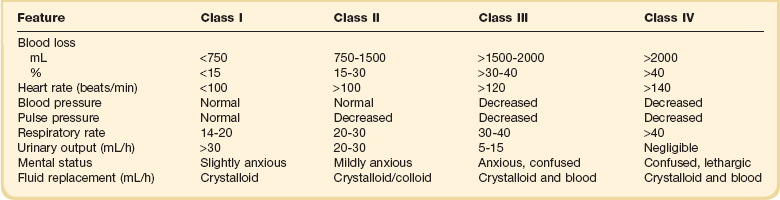
Tachycardia may be absent in case of prior use of beta blockers. Elderly patients may have atrial fibrillation and a high ventricular response. Occasionally, bradycardia is present, particularly when vagally mediated fainting supervenes.16 The peripheral veins are collapsed, and the jugular venous pressure is low. Conversely, an elevated jugular venous pressure should warn the clinician of associated obstruction of the circulation, following pneumothorax, pericardial tamponade, and others, or of pump failure following myocardial contusion or infarction. The respiratory cycle–induced changes in stroke volume and in systolic arterial blood pressure and CVP are exaggerated.20 Although dependent on tidal volume and respiratory compliance, these variations in a mechanically ventilated patient may constitute fair indices of hypovolemia, so high variations may predict an increase in cardiac output upon fluid loading.19 Noninvasive, pulse contour–based techniques are suitable for these purposes. Fluid responsiveness also can be predicted by an increase in blood pressure and decrease in stroke volume and pressure variations during leg raising or similar maneuvers. The body temperature may decrease, particularly in elderly patients. The gradient between the ambient and toe temperature may be a fair index of peripheral blood flow and a measure for the severity of hypovolemic shock because a reduction in skin blood flow (cold, clammy skin) is an early and ominous sign of shock in view of selective cutaneous vasoconstriction.123 Other signs of hypovolemic shock include tachypnea, oliguria/anuria, diaphoresis, cold and clammy skin with diminished capillary refill, and peripheral cyanosis.
Diagnostic Approach
General
If trauma and external blood loss are the cause of shock, control of external bleeding, crossmatching of blood, and infusion of fluids and blood components have a higher priority than further diagnostic procedures. Treat first what kills first. Blunt chest trauma can be complicated by aortic rupture, tension pneumothorax, hemothorax or hemopericardium, and tamponade. A chest radiograph can be useful to diagnose these conditions. After blunt abdominal trauma, splenic or hepatic ruptures are possible, and an abdominal tap and analysis of the fluid can be performed to exclude or establish intra-abdominal bleeding or hollow-organ perforation.273,274 This diagnostic procedure has been largely replaced, however, by imaging, if time permits, with help of so-called focused assessment sonography for trauma (FAST) or computed tomography of the abdomen in the emergency department. This helps in selecting patients for explorative laparotomy in order to avoid negative surgery or for percutaneous coiling.273,274 The abdominal viscera show characteristic lesions in hemorrhagic shock with low filling of large veins, decreased perfusion of some organs, wall thickening, submucosal edema, and enhancement of the gut.275 A ruptured abdominal aortic aneurysm can be diagnosed via ultrasonography or angiography if the patient’s condition allows the use of such an invasive, time-consuming procedure. The usefulness of emergency aortic clamping or balloon tamponade for massive abdominal hemorrhage is controversial.276 In the case of gastrointestinal hemorrhage, diagnostic procedures also are performed after initial resuscitation, including gastroscopy for upper gastrointestinal bleeding, sigmoidoscopy for lower gastrointestinal bleeding, and angiography. Introduction of a nasogastric tube (and early intubation) can be useful to aspirate blood, diagnose bleeding, prevent aspiration during vomiting, and follow the course of bleeding.
Laboratory Investigations
At admission of a patient with suspected hypovolemic shock, blood samples should be taken to determine the hemoglobin/hematocrit and leukocyte and platelet counts; electrolyte, creatinine, and lactate concentrations; arterial blood gases and pH; and blood typing (crossmatching). Immediately after hemorrhage, the hemoglobin content and hematocrit of blood are normal, but they decrease in time with refilling of the plasma compartment, as does the protein content.* A high hemoglobin content and hematocrit can be encountered during pure loss of plasma (water), as occurs during burn wounds or severe diarrhea. Acute hypovolemic shock may be accompanied by slight leukopenia followed by leukocytosis.46,111,112,248,253 If coagulation disorders are suspected (therapy with anticoagulants, liver disease, bleeding tendency), platelet counts and coagulation tests should be performed. Transient thrombocytopenia may ensue if shock is severe and massive amounts of whole blood are lost and rapidly replaced by erythrocyte concentrates or nonsanguineous fluids (i.e., through dilution). Isolated thrombocytopenia without DIC may thus occur.
The concentrations of electrolytes (sodium, potassium, chloride) in blood are essentially normal unless the concentrations in the fluid lost deviate from those in plasma (hypertonic and hypotonic dehydration) and resuscitation fluids and shock is accompanied by severe metabolic acidosis. In the latter example, potassium leaves the cell, potentially leading to hyperkalemia.111,125 More often, however, less severe forms of shock are accompanied by hypokalemia because of adrenergic receptor–stimulated Na+/K+-ATPase. Saline fluid loading or overloading can result in hyperchloremic metabolic acidosis. Adrenocortical insufficiency may result in hyponatremia, hyperkalemia, and hyperchloremic acidosis, caused by changes in urinary excretion induced by mineralocorticoid deficiency. In a patient with liver disease, the corresponding abnormalities can be found in laboratory studies. In the case of uncontrolled diabetes mellitus, hyperglycemia and glucosuria are observed. As previously mentioned, the glucose concentrations in blood can be elevated in early shock and, occasionally, depressed in late shock. Finally, the concentration of unbound Ca2+ in blood may diminish during hypovolemic shock after cellular uptake and polytransfusion of red blood cell concentrates if they contain calcium-binding citrate as an anticoagulant.127,150
During hypovolemic shock, metabolic acidosis, often associated with an elevated lactate level in blood, is common and of prognostic significance, although the decrease in bicarbonate and base excess may not parallel the increase in lactate.27,31–33,38,41 The pH can be subnormal after lactic acidosis and a decrease in the bicarbonate content, even if ameliorated by hyperventilation and a decrease in PCO2.27,32,33,49,50 The lactate level in blood can be determined rapidly and followed frequently (every 2 hours). The lactate level and its course during treatment also is of prognostic significance during shock because during successful treatment the lactate level decreases and the bicarbonate concentration and pH increase, whereas an unchanged or even increased lactate level during resuscitation is usually associated with morbidity, including sepsis, MOF, and death.27,31,32,56 An elevated anion gap, the difference between the sodium on the one hand and the sum of the bicarbonate and chloride concentrations in blood on the other hand, can be a first sign of lactic acidosis, although, as previously mentioned, elevated lactate levels may not be associated with acidosis in the absence of an oxygen debt.27 The serum creatinine concentration is initially normal. The urea content increases following prerenal renal insufficiency, catabolism, or breakdown of blood in the gut during gastrointestinal hemorrhage. In the urine, the osmolarity is increased.
The sodium content is low, together with a low fractional excretion of sodium (FENa),138 calculated as the quotient of urinary (U) and plasma (P) sodium (Na) and creatinine (creat) concentrations:
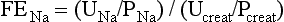
In case of acute renal injury (acute tubular necrosis), the urinary sodium content and fractional excretion are increased.138 This increased sodium also occurs during adrenocortical insufficiency. Prior diuretic therapy may invalidate this diagnostic tool, however, whereas the fractional excretion of urea may not be affected by diuretics.104 Tubular injury may be tracked from increased urinary excretion of biomarkers, which may help predict the need for renal replacement therapy.104
Miscellaneous abnormalities may include elevated levels of nitrate and nitrite, the stable breakdown products of NO.269 Transient elevations of bilirubin, alkaline phosphatase, γ-glutamyltransferase, and transaminases in blood may be severe and denote ischemic liver damage.222,223 Elevations of creatinine kinase may be caused by skeletal muscle, cardiac, gut, or, less likely, brain damage. Elevated troponin concentrations may specifically indicate cardiac injury.163,173
Monitoring
Noninvasive monitoring of arterial blood pressure to judge the course of shock and its response to treatment suffices for some patients with hypovolemic shock. Nevertheless, there may be substantial differences between the invasive and noninvasive readings of arterial blood pressure, favoring arterial catheterization and invasive monitoring. Urinary output should be measured hourly in patients with shock to judge the adequacy of treatment because transition of oliguria to a diuresis exceeding 40 mL/hour is an indicator of adequate renal perfusion. The gradient between toe and body temperature and capillary refill time can be used as noninvasive indices of peripheral perfusion.123
Unless hypovolemic shock is rapidly reversed by initial infusion of fluids, there is often a need for hemodynamic and respiratory monitoring in the intensive care unit for a patient with hypovolemic shock. The goal of monitoring is to document the course of shock and its reaction to treatment. Complications can be diagnosed in an early phase so that action can be rapidly undertaken, if necessary. Respiratory monitoring is meant to detect, at an early stage, respiratory insufficiency and muscle fatigue, which are caused by an imbalance in oxygen supply to demand and which may necessitate intubation and mechanical ventilatory support.50
Arterial blood pressure can be monitored invasively via a catheter in the radial, axillary, or femoral artery, introduced percutaneously using the Seldinger technique, under aseptic conditions. Percutaneous insertion of a double-lumen or triple-lumen central venous catheter may be useful but does not allow for more rapid fluid infusion than through two peripheral cannulas. The internal jugular, subclavian, or femoral vein may be used for that purpose. This also permits monitoring of CVP and oxygen saturation and a measure of total body oxygen supply-to-demand ratio and predictor of fluid responsiveness. Pressures in the lesser circulation (pulmonary arterial pressure and PCWP) can be measured with the help of a balloon-tipped pulmonary artery catheter inserted percutaneously and advanced under pressure monitoring until the inflated balloon wedges in a pulmonary artery side branch. This catheter also allows for thermodilution measurement of cardiac output and obtaining mixed venous blood for blood gas analysis.73,279 Together with arterial blood measurement of oxygen variables, this allows the calculation of oxygen delivery, extraction, and uptake.9,25,73 These calculations may contribute to judging the severity of shock and its response to treatment.6,9,25,73
The CVP reflects the filling pressure of the right ventricle, and the PCWP reflects the left atrial pressure and, in the absence of mitral valve disease, the filling pressure of the left ventricle.19 Under certain circumstances, however, including ventilation with positive end-expiratory pressure or measurement above the level of the left atrium, when the measured pressure is more influenced by alveolar than by venous pressure, the CVP and PCWP may overestimate true (i.e., transmural) right and left atrial pressures. The response of filling pressure and cardiac output to fluid loading, as measured with the central venous or pulmonary artery catheter, is an index of myocardial function and can be useful to assess fluid responsiveness, particularly in case of preexistent and prognostically unfavorable cardiac disease.* Measurement of PCWP is important if function or compliance of the left ventricle is altered (e.g., in case of preexistent heart disease), when the CVP may underestimate PCWP.55,71 Conversely, the CVP may overestimate PCWP in cases of severe pulmonary hypertension and right ventricular failure. It has been suggested that changes in CVP during fluid loading do not predict changes in PCWP.71 The intensity and speed of therapy can be guided by the response of filling pressures and cardiac output, as measured with the use of the central venous or pulmonary artery catheter.9,19,31,42,55
These measurements also can help to time, choose, and dose concomitant therapy with inotropic or vasopressor agents. Together with the plasma colloid osmotic pressure, the PCWP determines filtration of fluid across pulmonary capillaries according to the Starling equation. Monitoring of the PCWP during infusion of fluids may prevent pulmonary edema because infusion can be guided by the filling pressures of the heart. Taken together, data obtained with the pulmonary artery catheter are useful if the hypovolemic origin of shock is not immediately apparent in complicated cases, as in patients with preexistent cardiac disease.9,25,73 Data obtained with the catheter are of diagnostic value in complicated forms of shock because hypovolemic shock is characterized by low filling pressures and cardiac output and a high peripheral vascular resistance. These characteristics may serve to differentiate from other types of shock. The indications for insertion may also include a high risk for shock in patients undergoing major surgery and shock of unknown origin when clinical judgment fails to recognize severe hypovolemia.
Difficulties during treatment also may constitute indications for pulmonary artery catheterization, including hypovolemic shock unresponsive to liberal fluid repletion in the absence of a low jugular venous pressure or CVP and hypovolemic shock together with preexistent cardiac disease unresponsive to fluid repletion if a large discrepancy between CVP and PCWP is suspected and if vasoactive drugs are considered. Monitoring the PCWP may help to lessen the risk for pulmonary edema during fluid loading. Contraindications for pulmonary artery catheterization include those for central venous catheterization. The complications of the technique are discussed elsewhere. Although the use of pulmonary artery catheters is hotly debated because of lack of direct evidence that they help to increase survival, there are some indications that therapy guided by variables collected with the catheter improves the outcome of selected critically ill patients after trauma or surgery.9,25,73,280 Nevertheless, the exact hemodynamic and metabolic resuscitation goals are difficult to define, so the usefulness of the pulmonary artery catheter is difficult to prove.9,26,42,67,280
As an alternative to invasively inserted catheters, various less invasive or even noninvasive (pulse contour–based) systems have been developed that circumvent some of the problems associated with filling pressures as preload indicators and predictors of responsiveness of cardiac output to fluid loading.19,20,281 Among others, the transpulmonary thermodilution technique with detection of thermal changes after central venous injection of cold dextrose 5% in water in the iliac artery allows for calculation of cardiac output, global end-diastolic volume, and extravascular lung water—measures of cardiac preload, pulmonary fluid filtration, and edema.281 Assessment of cardiac volumes can be helpful to judge function, similar to echocardiography.157,281 The latter technique also evaluates filling or injury of large vessels, suspected cardiac contusion, and pericardial tamponade.282 The diameter (changes) of the large veins can be used as an indicator of filling status. The use of pulse-contour techniques for beat-to-beat evaluation of arterial pressure curve–derived stroke volume, as well as pulse pressure and stroke volume variations invoked by the respiratory cycle to guide fluid treatment in mechanically ventilated patients, remains controversial.20,281 The esophageal Doppler flow probe with which flow time, stroke volume, and cardiac output can be estimated is somewhat operator dependent.
Developments in monitoring the circulation of a patient in hypovolemic shock further include continuous monitoring of central venous or mixed venous oxygen saturation with the help of the fiber-optic technique introduced via catheters, allowing for the continuous evaluation of the oxygen supply-to-demand ratio; right ventricular end-diastolic volume monitoring as an index of filling status; and measurement of tissue blood flow, PO2, PCO2, and oxygenation by electrodes and optic techniques.* Venous O2 saturations may indeed help to guide fluid resuscitation in clinical practice because a low saturation increases upon adequate fluid challenges in fluid-responsive patients. Tissue PO2 decreases and PCO2 increases during regional perfusion failure, and these events (in skin, conjunctiva, muscle, or bladder) are probably early signs of hypovolemia following redistribution of blood flow before hypotension ensues and have be used as guides to treatment.* The adequacy of gastrointestinal blood flow can be judged noninvasively with the help of a tonometer balloon catheter in the stomach (or gut) or with help of a sensor sublingually or buccally, in which fluid or air is instilled, and the PCO2 is measured to calculate the mucosal-to-blood PCO2 gradient, as explained previously.† Fluid treatment guided by the adequacy of gastrointestinal mucosal perfusion as judged by tonometry could improve the outcome of hemorrhaged trauma patients compared with resuscitation based on standard hemodynamic variables alone.40,123,209,211
Monitoring the end-tidal CO2 fraction, determined by and directly related to the blood flow–dependent tissue CO2 production, and the gradient to arterial PCO2, determined by blood flow–dependent dead-space ventilation, can help to judge the response to resuscitation.49,65 Hydrostatic pressure measurements in the urinary bladder may reflect the measurements in the abdominal compartment and may help to identify intra-abdominal hypertension and abdominal compartment syndrome developing during extensive fluid resuscitation.285 This may impair gut and renal perfusion with subsequent dysfunction and warrant decompression laparotomy.285
Approach to Management
General
Treatment of shock cannot be delayed, so in practice diagnosis and treatment are done simultaneously. Treatment of hypovolemic shock is aimed at the restoration of the circulation and treatment of the underlying cause. Box 26.2 describes some general guidelines, but we do not specifically address burn wound shock requiring a special approach. The main therapeutic goal in hypovolemic shock is to restore circulating blood volume and to optimize oxygen delivery so that oxygen uptake plateaus (see Fig. 26.1) and meets tissue needs.72,286 Optimization of cardiac output, stroke work, and tissue oxygenation and maintenance of arterial blood pressure are physiologically reasonable resuscitation targets for patients.42 Optimization does not imply maximization above levels adequate for tissue needs.26,67 Studies by some investigators have suggested that supranormal rather than normal oxygen delivery and consumption may be associated with survival from severe trauma or hemorrhage, including a ruptured aortic aneurysm, and that therapeutic targeting at these values (with oxygen delivery >600 mL/minute/m2 and oxygen consumption >170 mL/minute/m2) improves the outcome of severe trauma in humans.9,25,40,42,83 These concepts are highly controversial, however. In any case, resuscitation based on blood pressure alone probably does not fully restore tissue oxygenation, particularly in the case of myocardial dysfunction after shock and resuscitation.42,55
Adequate resuscitation from hypovolemic shock should result in an increase in oxygen delivery and uptake, a decrease in lactate levels, and amelioration of metabolic acidosis.* Concomitantly with the increase in cardiac output, arterial blood pressure increases, but normal levels may not be necessary to aim at during resuscitation.71,72,172 Indeed, successful resuscitation in terms of a restored cardiac output and arterial blood pressure may poorly reflect effective recovery of tissue perfusion, even though reversal of oliguria is considered a favorable return of renal perfusion.† Animal experiments have suggested that early circulatory optimization also ameliorates inflammatory changes after trauma/hemorrhage. In attempts to restore oxygen delivery, hypoxemia should be prevented or corrected, although recent evidence suggests that hypoxic resuscitation ameliorates proinflammatory responses to reperfusion as compared to normoxic resuscitation.234 The arterial oxygen saturation, important for the oxygen content of delivered blood, usually exceeds 90% if the PO2 is greater than 60 mm Hg. If arterial PO2 is less than 60 mm Hg, supplemental oxygen can be given through a nasal cannula or mask, but hyperoxemia should be avoided. The patient should be intubated and artificially ventilated in case of impending respiratory insufficiency from whatever cause. Prophylactic positive end-expiratory pressure in attempts to prevent the development of ARDS in at-risk patients, including patients with traumatic/hemorrhagic shock, is useless.
For the initial stabilization of trauma patients in shock with severe abdominal trauma or bleeding from lower extremities, passive leg raising or Trendelenburg positioning has been used, but a large benefit on preload-augmented tissue oxygen delivery has been doubted.8,19,20,287 The pneumatic antishock garment can be applied as a temporary, immediately lifesaving procedure to (1) stop the hemorrhage and splint pelvic and lower extremity fractures for transportation of the patient after inflation, (2) mobilize blood volume by exerting external pressure on the leg and abdomen, and (3) redirect flow toward vital organs such as the heart and brain.288–290 Use of the garment and hypertonic saline may act synergistically.288 The disadvantages of the procedure include aggravation of pressure-dependent and uncontrolled bleeding and ischemia of intra-abdominal organs. Deflation should be gradual to prevent lethal hypotension after return into the circulation of vasoactive substances and lactic acid from ischemic tissue.288,289 The use of the pneumatic antishock garment has greatly declined in recent decades. Spontaneous breathing against an inspiratory threshold or decreasing expiratory pressure during mechanical ventilation may increase venous return and has been studied experimentally.291 Measures to establish a way to the bloodstream for infusing fluids include insertion of a large-bore catheter in a peripheral vein or, if cannulation of peripheral veins seems impossible because of collapse, a catheter in a central (i.e., jugular/subclavian) vein. The latter catheter also allows monitoring of CVP. The intraosseous route for administration of hypertonic fluids is particularly useful in traumatized children with hypovolemic shock.292 To this end, a marrow screw is inserted in the sternum or tibia. Fluids can be given by gravity or pressure.
Resuscitation Strategies
The speed with which shock can be reversed depends on the delay from onset to treatment and the severity of shock, as estimated from the clinical condition and hemodynamic status. The speed of fluid infusion can be guided by the clinical condition of the patient, heart rate and blood pressure, diuresis, and determinations every 2 hours of the arterial blood lactate level and acid-base balance.31 Repeated assessment of jugular venous pressure, auscultation of the lungs, and arterial blood gases are indicated to prevent overhydration and pulmonary edema. If a central venous or pulmonary artery catheter is in place, a fluid challenge protocol can be used (Table 26.2).31 Monitoring of central venous or PCWP or preload volumes allows for rapid infusion of solutions and evaluation of the response of oxygen delivery and uptake to resuscitation.9,25,31,73 When in doubt, predicting fluid responsiveness in the patient, for instance with cardiac failure on mechanical ventilation, by passive leg raising and other tests that reversibly challenge the circulation can be helpful to further guide fluid therapy and avoid harmful fluid overloading.19,20
Table 26.2
CVP, central venous pressure; PCWP, pulmonary capillary wedge pressure.
Adapted from Weil MH, Henning RJ: New concepts in the diagnosis and fluid treatment of circulatory shock. Anesth Analg 1979;58:124.
The usefulness of immediate and vigorous resuscitation in the course of uncontrolled bleeding (e.g., after penetrating vascular trauma) has been challenged in recent years.279,293–295 Studies have shown that infusion of substantial amounts of nonsanguineous fluids and increasing pressure-dependent bleeding lead to dilution of blood components, coagulation disturbances, and increased mortality risk unless the bleeding is controlled before resuscitation.279,290,294,295 The clinical implication is that resuscitation perhaps should not take place, at least not vigorously, at the scene of the accident when bleeding cannot be controlled and transport to the hospital where the bleeding can be controlled is practicable. This may apply not only to trauma patients but also to patients with a ruptured abdominal aneurysm. Authors have proposed slow (low volume) rather than rapid (early and large volume) infusion rates and controlled hypotensive resuscitation (e.g., with help of vasodilators) at least as long as bleeding is uncontrolled.290,293,294,296–298 The value of this type of resuscitation in humans with hyperoncotic/hypertonic solutions, including acetate with vasodilating and buffering properties, is debatable.210,279,298 Also, the levels of hypotension that are safe remain unclear.299 Conversely, early vasopressor therapy by vasopressin, for instance, may benefit patient outcomes more than large-volume infusions.300 The treatment of massive bleeding by blood products requires an institutional protocol. Closed-loop control of fluid therapy may become clinically applicable in the near future.301
Fluids
During hypovolemic shock, the repletion of intravascular volume is of primary importance to restore cardiac output and oxygen transport to the tissues before repletion of the interstitial and intracellular fluids.* Available fluids are described in Tables 26.3 to 26.5. Electrolyte solutions (the crystalloid fluids) are shown in Table 26.3, and high-molecular-weight solutions (the colloid fluids) are shown in Tables 26.4 and 26.5. In the first group, the hypertonic fluids (i.e., fluids with a higher osmolarity than plasma) are presented. Among the colloid solutions are solutions with a higher colloid osmotic pressure than plasma—the hyperoncotic solutions. Hypertonic and hyperoncotic solutions are plasma expanders because they are able to mobilize cellular and interstitial fluid during resuscitation and to expand plasma volume rapidly.

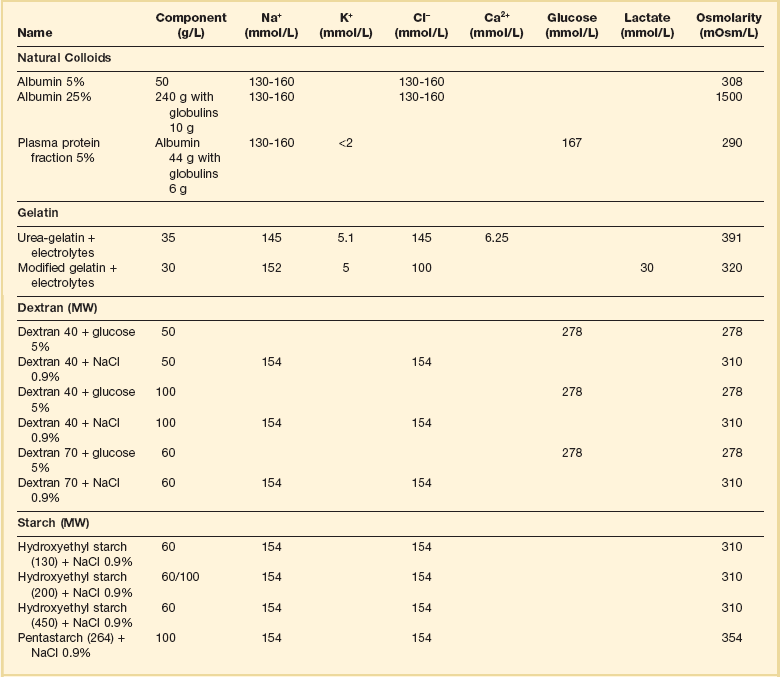
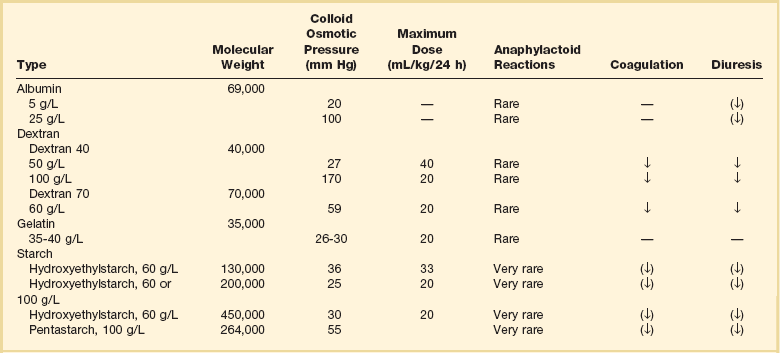
Table 26.6 shows how the volume of the various compartments is replenished during fluid resuscitation. Because infusion of glucose 5% hardly increases intravascular volume, this solution has no place in resuscitation from hypovolemic shock. Lactated Ringer’s solution containing K+ should not be used during renal insufficiency because of the danger of inducing hyperkalemia. Infusion of lactate-containing solutions during lactic acidosis may be controversial because the capacity of the liver to regenerate bicarbonate from lactate may be impaired.33 Nevertheless, infusion of lactate-containing solutions such as lactated Ringer’s for resuscitation from hypovolemic shock generally does not substantially increase lactate levels, worsen acidosis, or adversely affect outcome.305 However, racemic Ringer’s lactate may be proinflammatory and proapoptotic as compared to normal saline and only L-lactate containing solutions are therefore currently recommended.191 Adverse effects of overzealous fluid administration of any type include promotion of pulmonary edema and ascites formation with subsequent aggravation of abdominal compartment syndrome.285,286
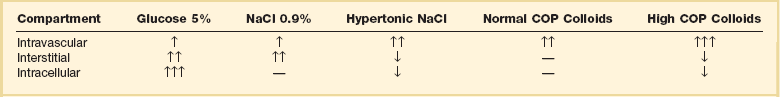
The natural colloids consist of albumin and plasma solutions. Albumin is an effective colloidal solution for intravascular volume repletion, and its intravascular half-life is approximately 16 hours.* For the artificial colloids, the volume-expanding effect and the duration of action generally increase with increasing in vivo molecular weight (Table 26.7). About one third of the hyperoncotic fluids with high molecular weight (dextran 70 and hydroxyethylstarch) may still be in the circulation after 24 hours, whereas dextran 40 is retained for approximately 3 hours. Dextrans and gelatins are perhaps equally effective in restoring circulating volume.309,310 The latter substances might increase tissue blood flow in the microcirculation. Dextrans are currently less often used than gelatins and starches and institutional and geographic habits largely determine the choice among fluids.
Table 26.7
Distribution of Artificial Colloids 24 Hours After Intravenous Infusion in Normal Volunteers
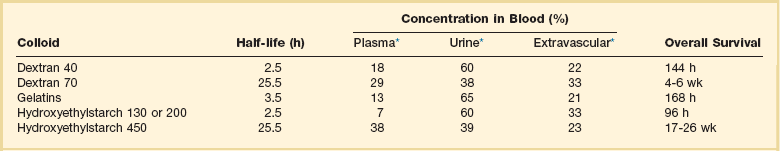
*Percent total dose administered.
Adapted from Mishler JM: Systemic plasma volume expanders: Their pharmacology, safety, and clinical efficacy. Clin Haematol 1984;13:75.
The starch compounds have gained wide interest for the resuscitation of hypovolemic shock.* These colloids are at least as effective as albumin but less expensive.135,136,172,308,312 Because the molecular range of the starch compounds varies enormously, the pharmacokinetics are complex.311 Nevertheless, about 40% of the infused hetastarch (molecular weight >200) still remains in the circulation after 24 hours because 30% of the infused substance may have a half-life of 67 hours. Ninety percent of smaller starch is cleared in 24 hours. The duration of the volume-expanding effect of the starches depends not only on molecular range and concentration but also on the so-called substitution grade, which is the number of hydroxyethyl groups per glucose unit, and the substitution type, the ratio of C2 to C6 hydroxyethylation.311 A high molecular weight, high substitution grade, and high C2/C6 ratio retard breakdown by plasma amylase and prolong intravascular retention. The residual starch compounds are partly excreted by urine and partly taken up by the reticuloendothelial system. Accumulation may also occur in dendritic cells of the skin and the liver and in the renal tubules, with subsequent adverse effects. Starch compounds may increase the amylase level in blood and may confound the diagnosis of acute pancreatitis. Experimental evidence shows that some starch compounds, particularly in the 100,000 to 300,000 D molecular weight range, have the advantage in sealing the capillary endothelium in case of increased permeability after ischemia or trauma, diminishing fluid and protein filtration and preventing edema.136,179,311,312
In the resuscitation of hypovolemic shock (e.g., after trauma or burns), the use of hypertonic solutions with sodium concentrations greater than 0.9% also has gained wide interest.* The solutions essentially consist of hypertonic sodium chloride, to which colloids have often been added. The combinations include NaCl 7.5% with dextran 70 (6%/10%), NaCl 7.2% with dextran 60 (10%), and NaCl 7.5% with hydroxyethylstarch 6%.33,62,122,314–317 Hypertonic solutions usually result, at a much lower infusion volume than isotonic solutions (small volume resuscitation), in a rapid hemodynamic improvement, that is, an increase in cardiac output and in oxygen delivery and uptake and arterial blood pressure in experimental animals and patients with traumatic/hypovolemic shock.† Infusion of rapidly acting hypertonic saline, particularly if combined with hyperoncotic colloids, increases survival in bleeding animals compared with infusion of either component or other isotonic or hypertonic (nonelectrolyte) solutions.316 Clinical trials also have shown some value of hypertonic solutions in the initial treatment of hypovolemic shock after burns and trauma with uncontrolled bleeding.119,286,314,316,317 The use of hypertonic saline solutions, however, warrants close monitoring of plasma sodium levels to prevent excessive hypernatremia and hyperosmolarity.313–316
The hypertonic fluids primarily act through resorption of interstitial and cellular fluid volume and expansion of the plasma volume.7,131,314 It has been calculated that only 4 mL/kg 7.5% saline solution can increase circulating plasma volume by 8 to 12 mL/kg body weight. A hyperosmolarity-induced increase in cardiac contractility may also contribute to the increase in cardiac output, although this effect has been doubted.318 Other potential mechanisms include activation of pituitary and pulmonary osmoreceptors, leading to release of vasopressin and vagal afferent-mediated venoconstriction, and hyperosmolarity-induced arterial vasodilation.7,62,313,314 Infusion of hypertonic sodium combined with hyperoncotic colloid solutions more rapidly and completely increases cardiac output and arterial blood pressure, and the effects last longer than those produced with infusion of hypertonic or colloid solutions alone.313–317 During infusion of hypertonic saline, particularly if combined with hyperoncotic colloid solutions, the distribution of peripheral oxygen delivery is reversed to a more favorable pattern with preferential perfusion of vital organs, including gut and kidney.62,313 The increase in oxygen uptake in bled dogs was less rapid and complete during resuscitation with hypertonic saline plus hydroxyethylstarch, however, than during infusion of relatively large volumes of the latter.33,62
The hypertonic solutions may also ameliorate immunodepression, the translocation of bacteria, and susceptibility to sepsis after hypovolemic shock in rodents.201,319 Hypertonic solutions (plus dextrans) restore capillary blood flow and organ function during resuscitation better than iso-osmotic fluids because of their ability, among others, to reduce endothelial cell swelling, adhesion molecule expression, neutrophil activation and adherence, and cellular apoptosis compared with normotonic crystalloids such as racemic Ringer’s lactate.191,320 Hypertonic solutions may also prevent lung injury after hemorrhage/resuscitation, probably via these mechanisms.95,119,122,319
Fluid Controversies
The choice between available fluids should be guided by the estimated extent and type of fluid losses; their composition and localization; and the properties of infusion fluids, their distribution over body compartments, and, perhaps, the associated costs, which are high for albumin, intermediate for artificial colloids, and low for crystalloid solutions.131,302–304 Nevertheless, the use of various solutions for resuscitation from hypovolemic shock is hotly debated, partly because the importance of the colloid osmotic pressure for resuscitation and prevention of pulmonary edema is uncertain.302 Also, the relative merits and detriments (i.e., safety) of natural and artificial colloids remain unclear.302,321
Capillary filtration depends on the pericapillary hydrostatic and the colloid osmotic pressure gradient, according to the Starling equation. If at a given permeability an imbalance in pressures augments capillary filtration of fluids, a decrease in interstitial colloid osmotic pressure, an increase in interstitial hydrostatic pressure (which also depends on the compliance of the interstitium), and increased lymph flow can either alone or in combination partially prevent gross accumulation of interstitial fluid (edema). The colloid osmotic pressure of plasma is primarily determined by the plasma albumin content and normally measures about 24 mm Hg.31,126,137,322 The pressure can be estimated from albumin and protein concentrations in plasma, but infusion of artificial colloid solutions invalidates this calculation, so proper assessment of plasma colloid osmotic pressure necessitates direct measurement.137,323 Because of a decrease in circulating plasma protein levels, hypovolemic shock results in a decrease in plasma colloid osmotic pressure.* During hypoproteinemia and a reduced plasma colloid osmotic pressure, fluid filtration for a given hydrostatic pressure increases until the pericapillary colloid osmotic pressure gradient decreases and a new steady state, often at increased lymph flow, has been achieved.133,135,136 Evoking safety mechanisms such as a reduced interstitial colloid osmotic pressure and increased lymph flow may keep the interstitium relatively dry, and these mechanisms may be more effective in the lung than in the systemic circulation.133 During hypoproteinemia, the hydrostatic pressure needed to invoke pulmonary edema decreases, however, because of more rapid exhaustion of safety mechanisms.135,324 Conversely, increased lung water caused by an elevated hydrostatic pressure can be ameliorated by colloid infusion.324
For a given increase in hydrostatic pressure, the infusion of crystalloids decreases the plasma colloid osmotic pressure and tends to enhance, if insufficiently compensated by a decrease in the pericapillary colloid osmotic pressure gradient, pulmonary and systemic fluid filtration and interstitial fluid expansion more than infusion of albumin/colloids, which maintain plasma colloid osmotic pressure.* Crystalloid solutions replenish not only the intravascular but also the interstitial space by increased filtration, whereas colloid fluids tend to primarily fill the former compartment, at least initially.131,278 Widening of the intravascular-to-interstitial colloid osmotic pressure gradient may prevent increased fluid filtration, but the effect may be transient when some colloids have been filtered along with fluids into the interstitium and a new steady state of perimicrovascular pressure and draining lymph flow has been established.136 The mechanism may form the basis for the well-known observation that colloid solutions yield a twofold to threefold greater expansion of the intravascular space than crystalloids and that the latter have greater tendency for edema formation for a given amount of fluid infused, so less colloid than crystalloid is probably needed for resuscitation to similar hemodynamic end points in hypovolemic shock.† In some clinical trials, colloids proved to be superior to crystalloids in resuscitation from hypovolemic shock,137,172,309 in terms of both the speed and the extent of correction of the hemodynamic abnormalities. Conversely, this may also explain the observations of some investigators that during resuscitation from hypovolemic shock pulmonary edema can be prevented in part if the intravascular filtration pressure (i.e., the gradient between plasma colloid osmotic and PCWP) is kept greater than approximately 6 mm Hg, with an elevated risk for pulmonary edema, particularly in case of increased permeability, if the gradient is less than approximately 3 mm Hg, and that resuscitation with colloids less often induces evidence for pulmonary edema than infusion of crystalloids during hypovolemic shock.‡
If the permeability for proteins increases and the reflection coefficient decreases, the hydraulic conductance of the capillary membrane also increases.133 Increased permeability for proteins increases capillary fluid filtration for a given intravascular hydrostatic pressure and promotes the formation of edema.308 During increased permeability, the filtration of fluids and expansion of the interstitial fluid space depend more than normally on hydrostatic pressures and less on colloid osmotic pressures because the colloid osmotic pressure gradient is decreased.133 The differences between the types of solutions in fluid filtration and formation of edema in the lung and peripheral tissues diminish.326 This may explain in part why some clinical studies did not find a predictive value of the colloid osmotic pressure-PCWP gradient for pulmonary edema and lack of a difference between fluid types for formation of pulmonary edema and impaired gas exchange during resuscitation from hypovolemic shock.302,307 Moreover, an increase in CVP increases the back-pressure for lymph flow. Careful animal studies on hypovolemic shock combined with a lung vascular injury showed, however, that colloids are more effective than crystalloids in restoring the circulation and that the former increased lung water less than the latter unless permeability was severely increased.308,326 This can be explained by the fact that even in case of increased permeability the reflection coefficient is not zero, and that the pericapillary colloid pressure gradient still exerts some influence on the transcapillary movement of fluids.
Clinical studies on the colloid/crystalloid controversy may be difficult to interpret because of differences in patient populations and end points between fluid types.302,321 Lack of similar end points used for resuscitation may partly explain why infusion of colloid solutions increased the risk for pulmonary failure compared with infusion of crystalloids because colloids, owing to their greater intravascular volume-repleting effect, tend to increase hydrostatic filtration pressure in the lung more rapidly than crystalloid fluids even though colloid osmotic pressure is maintained or increases during infusion of the former and decreases with the latter.306 The importance of a difference in hydrostatic pressure for the risk of pulmonary edema would be accentuated in case of increased permeability.306 Finally, pulmonary mechanics, gas exchange, and radiographic changes used to evaluate the effects of fluid infusions in many studies may not accurately reflect changes in lung water.52,172,306–308
There are safety concerns with artificial colloids, particularly in the presence of other risk factors for organ damage.304,321 Potential disadvantages of (artificial) colloid over crystalloid solutions include inhibition of the coagulation system; the risk for anaphylactoid reactions; inhibition of renal salt and water excretion; renal injury; and perhaps, at least for albumin, depression of myocardial function, possibly owing to binding of Ca2+, although this has not been seen in all studies.171,172,321,327 Of all artificial colloids, dextrans affect coagulation most adversely, independently of hemodilution, by interfering with coagulation factors and diminishing thrombocyte and red blood cell aggregation.327 The gelatins may also have some intrinsic effects on coagulation, whereas the anticoagulant effects of hydroxyethyl starches probably relate, in addition to hemodilution, to less endothelial release of von Willebrand factor.290,311,327 Anaphylactoid reactions to artificial colloids are extremely rare and vary from slight fever and skin reactions to life-threatening anaphylactic shock. Starch compounds elicit these reactions less often than dextrans and gelatins.311 Large-molecular-weight starches may accumulate in subcutaneous tissues and may cause pruritus, even for weeks after administration.311 However, resuscitation of trauma patients with hydroxyxethyl starch may result in less endothelial damage, renal injury, and pulmonary dysfunction than resuscitation with gelatins.179 Nevertheless, the renal damaging effect of starch is probably greater than that of gelatins in patients with prior risk factors for acute kidney injury.321 Hence, colloids may contribute to the development of acute kidney injury and failure, particularly in the case of overadministration, which may otherwise be more frequent with artificial colloids than with albumin/plasma solutions, which can be monitored by measurements of plasma albumin concentrations. As opposed to albumin/plasma, infusion of colloid solutions is often bound to a maximum (see Table 26.5). Side effects may be more frequent with artificial colloids than with albumin/plasma infusion even though the latter carries a very low risk of anaphylactoid reactions and disease transmission. Crystalloid and artificial colloid solutions may activate neutrophil-endothelial interactions and depress macrophage and immune functions more than albumin does.191,321,328,329 In contrast to lactated Ringer’s solution, Ringer’s ethyl pyruvate solution has favorable anti-inflammatory and cell-protecting actions.29,37
If used in the resuscitation from hypovolemic shock, artificial colloids may still be preferred over natural colloids because the latter are more expensive and less available, even though albumin and plasma solutions are effective volume expanders. There is some evidence that the type of fluids infused during hypovolemia may influence the extent and speed with which oxygen uptake is restored: infusion of colloid (plasma/albumin) solutions in hypovolemic postoperative and trauma patients may increase uptake of oxygen for a given increment in plasma volume and oxygen delivery more rapidly than infusion of crystalloids and may thereby improve outcomes.9,25,330 This is thought to result in part from increased diffusion distances for oxygen in the tissues subsequent to tissue edema, evoked by massive crystalloid infusion.9,25,307 Administration of nonbuffered (unbalanced) crystalloid solutions such as normal saline carries the risk of hyperchloremic metabolic acidosis, which can be avoided in part by infusion of buffered (balanced) solutions such as Ringer’s lactate.325 Nevertheless, meta-analyses suggest, at least in some groups, a slightly increased mortality risk after resuscitation with artificial colloids.302,321,331,332 In the SAFE study comparing albumin and saline for resuscitation in the intensive care unit, a slight but nonsignificant increase in mortality rate was observed in the (neuro)trauma subgroup treated by albumin infusions, although animal studies suggest some beneficial and anti-inflammatory effects of albumin resuscitation from hemorrhagic shock.333
Blood Products and Substitutes
Resuscitation with blood components may restore tissue oxygen delivery and energy metabolism more rapidly, completely, and persistently than resuscitation with crystalloid during hemorrhage and hypovolemic shock, although this is controversial.162,305 Nevertheless, it has been suggested that infusion of sodium salts may be essential, and that addition of saline to blood improves survival from hypovolemic shock after hemorrhage because of correction of both the intravascular and the interstitial volume deficits.313
In the treatment of hypovolemic shock following ongoing hemorrhage, infusion of red blood cells in the form of erythrocyte concentrates, or packed red blood cells, remains crucial.277,286,334 This is achieved by autotransfusion from uncontaminated areas during surgery, if possible; by infusion of blood group O Rh-negative donor blood in emergency situations; or by infusion of typed and stored/anticoagulated donor blood. The position of the oxyhemoglobin dissociation curve of old, stored blood is shifted to the left.60,78 Although this theoretically may impair the delivery and uptake of oxygen in the tissues, the effects of these changes in animal experiments are usually limited and clinical repercussions are unclear.60,76 Transfusion of substantial amounts of erythrocyte concentrates is preferentially accomplished through a microfilter to avoid alloimmunization and infusion of neutrophils and other cellular aggregates, which develop in time during storage of blood and which may lodge in the lung, promote pulmonary injury, and impair gas exchange, leading to TRALI.175–177 Today, prior leukocyte-reduced red blood cell concentrates are often used, but it is controversial whether this is associated with less risk. Excluding multiparous women who may have become sensitized to allogenic leukocytes and may carry leukocyte antibodies contributing to TRALI is another strategy applied in some countries. Also, humoral mediators released in stored blood or during infusion might be responsible in part for pulmonary vascular injury after massive transfusion of blood.180,190 Nevertheless, massive transfusion may remain a risk factor for bacterial sepsis, ARDS, and MOF, independently from bleeding and severity of hypovolemic shock.* Finally, transfusion of blood components and plasma carries a small risk of transmitting infectious diseases and depressing immune function.305,329
Because loss of blood also leads to loss of coagulation factors and platelets, and blood concentrations are diluted further during nonsanguineous fluid resuscitation, replenishing plasma levels by infusion of fresh frozen plasma and platelets is usually required to help stop ongoing bleeding.262 Fresh frozen plasma should not be used solely for the treatment of hypovolemia, even though plasma may diminish endothelial hyperpermeability through restoration of glycocalyx.262,271 The strategy of blood products infusion has undergone some changes in the last decade, in which studies suggest optimal hemostasis and outcome when fresh frozen plasma units are infused at a 1 : 1 ratio with packed red blood cell concentrates and random donor platelet units at a 1 : 3 to 1 : 5 ratio.334,337,338 If, during resuscitation, the clotting times are prolonged by a factor of 1.5 or more, more fresh frozen plasma can be given, and if platelet counts decrease to less than 50 to 100 ×109/L, platelets can be transfused, particularly in case of intracranial or life-threatening bleeding. The value of prophylactic administration of coagulation factors in a polytransfused, traumatized patient to prevent further bleeding after initial hemostasis is unclear, however. Supplementation of Ca2+ may be necessary only if more than 12 to 20 units of packed red blood cells, anticoagulated with Ca2+-binding citrate, have been given if rapidly transfused and particularly if liver function is impaired.150 Further treatment is guided by ionized Ca2+ determinations in plasma. Fibrinogen concentrates are increasingly used with increasing evidence that fibrinogen plays an important role in coagulation and that primary hyperfibrinolysis is common in trauma patients, as revealed by thrombelastometry.334,337 Antifibrinolytic drugs, such as tranexaminic acid, given prior to fibrinogen concentrates are useful adjuncts.339 The exact place of recombinant factor VIIa, a potent procoagulant, in the treatment of refractory bleeding has not been settled yet.286,340 The factor stops bleeding and saves blood transfusion, but cost-effectiveness is unclear. Adverse effects include a tendency for thromboembolic events.340 The bleeding tendency of trauma is also aggravated by hypothermia and acidosis, but it is unclear whether aggressive treatment of hypothermia or acidosis substantially ameliorates coagulation disturbances and to what extent DIC contributes.181,264,337 The importance of the latter also remains somewhat unclear, and treatment may consist of infusion of antithrombin III concentrates or, in case of severe bleeding, of fresh frozen plasma and platelets.181
To overcome some of the problems associated with donor or autologous red blood cell transfusions, investigators have intensively searched for safe and effective hemoglobin substitutes applicable in humans.230,286,341–343 These substitutes include chemical oxygen carriers, hemoglobin modifications, and liposome/vesicle-encapsulated hemoglobin.344 The chemical hemoglobin modifications have been designed to prevent or limit the renal toxicity of free hemoglobin. They include polymerized, modified, cross-linked, and recombinant hemoglobins.34,345 The use of hemoglobin substitutes has been under clinical investigation. Some nonrecombinant substitutes seemed to increase arterial and, particularly, pulmonary arterial blood pressure more than accounted for by fluid loading, but some compounds have more adverse effects than others.346 This may relate to the property of hemoglobin to scavenge NO or release endothelin and platelet-activating factor, or combinations, and the use of these solutions is therefore still not without hazard.346 Diaspirin cross-linked hemoglobin may beneficially influence intracranial hemodynamics during resuscitation from hypovolemic shock.296 A clinical trial on diaspirin-cross-linked hemoglobin in trauma failed to improve survival over resuscitation with saline, however.341 The use of hemoglobin substitutes such as perfluorocarbons has not yet reached the stage of widespread, routine clinical practice, although they may effectively carry oxygen in humans and may improve resuscitability from hypovolemic shock following bleeding in animals compared with nonhemoglobin-based solutions.341–343,347 Further research is ongoing.
Acidosis and Optimal Hematocrit
The underlying idea for partial correction of metabolic acidosis is that acidosis is detrimental for, among others, myocardial function by increasing pulmonary artery pressure and right ventricular afterload, impairing catecholamine sensitivity, and diminishing adrenergic receptors and intracellular Ca2+ transport necessary for contraction, even if masked by increased sympathetic activity.36,168 Metabolic acidosis may increase the tendency for life-threatening ventricular arrhythmias and may lessen defibrillation thresholds and vascular tone.27 The need for treatment of metabolic (lactic) acidosis (e.g., by intravenous administration of buffer solutions) remains unclear.* Administration of sodium bicarbonate may carry the risk for aggravation of intracellular acidosis in the tissues because bicarbonate releases CO2 during buffering and CO2 more rapidly traverses the cell membrane than the bicarbonate ion.* Alkali therapy with sodium bicarbonate carries the risks of shifting the oxyhemoglobin dissociation curve to the left and impairing tissue oxygenation, a decrease in ionized Ca2+, and causing hypernatremia and osmolarity, although the consequences of these theoretical drawbacks are unclear.27,36,44 Albeit not beyond doubt, experimental and clinical studies suggest that the administration of buffers such as sodium bicarbonate is not harmful, even though the hemodynamic and metabolic effects of the solution may not surpass those obtained by saline infusion.† In many institutions, small doses of alkali buffers such as sodium bicarbonate (50 to 100 mL of a 4.2%, 0.5 mmol/mL solution) are still given to treat metabolic (lactic) acidosis if arterial pH is less than 7.2 and acidosis persists despite optimal cardiovascular resuscitation.27,36 During sodium bicarbonate infusion, the patient should be hyperventilated to prevent hypercapnia in arterial blood and bicarbonate doses should be guided by the arterial blood acid-base status to prevent alkalosis and diminished oxygen release after overadministration.27,44,348 Prevention of hypercapnia may obviate increased CO2 diffusion and aggravation of intracellular acidosis.27,36 The value of buffers, including bicarbonate/carbonate, that do not generate CO2 and prevent aggravation of intracellular acidosis is still controversial.27,35,348,349 Dichloroacetate is a stimulator of pyruvate dehydrogenase, and the drug may ameliorate lactate accumulation and postresuscitation organ dysfunction, but there is probably no benefit for patient outcome.27,35,36
The hematocrit is the main determinant of blood viscosity, and the latter determines, together with the geometric features of the vascular bed, the blood flow in the microvasculature.120 Experimental studies suggest that during normovolemic hemodilution normal oxygen delivery is achieved at a range of hematocrit values from 12% to 65% for the heart; 30% to 65% for the brain; 30% to 55% for liver, intestine, and kidney; and 30% to 60% for the whole body because adaptations in vessel diameter and changes in blood flow in this hematocrit range are able to compensate for changes in oxygen content, maintaining a normal oxygen delivery.85,120,265,350 The optimal hematocrit for the whole body may not conform to the regional optimal hematocrit.
Because blood is a non-newtonian fluid, so that blood viscosity depends not only on hematocrit but also on blood flow velocity (shear stress), there may be differences along the vascular profile in blood viscosity, with propensity for red blood cell aggregation in postcapillary venules, where flow velocity is lower than in arterioles, particularly during hypovolemic shock.4,120 Increased blood viscosity in postcapillary venules may contribute to impaired tissue perfusion during hypovolemic shock.4 Conversely, the volume status, myocardial function, and vascular tone contribute to blood viscosity so that, for example, the optimal hematocrit for oxygen delivery is lower during hypovolemia than hypervolemia.21,120,350 Finally, red blood cell deformability is decreased in hemorrhagic shock, contributing to increased viscosity.224
Taken together, it is hard to generally define the optimal hematocrit for oxygen delivery to the body during hypovolemic shock, even though hematocrit-induced changes in the rheologic properties of blood may contribute to hemodynamic changes in critically ill patients.21,120,350,351 Most, but not all, authors believe, however, that mild hemodilution (hematocrit approximately 0.30) may benefit delivery and uptake of oxygen in the tissues and promote survival of critically ill patients with hypovolemia, whereas severe hemodilution or hemoconcentration may be detrimental.* A low hematocrit in the course of hypovolemic shock after major surgery may warrant red blood cell replacement, whereas a high hematocrit may necessitate infusion of nonsanguineous fluids.9,352 Mild hemodilution may benefit resumption of red blood cell flow and oxygen uptake after prior ischemia.
Vasoactive Drugs
Generally, catecholamines do not have a place in the treatment of hypovolemic shock unless they are used to bridge a period in which infusion fluids are not yet available, or if adequate fluid resuscitation has proved insufficient to reverse hypotension (irreversible shock) and to increase oxygen delivery to the point that tissue needs are met.† Persistent hypotension despite normovolemia can be caused by a low cardiac output following myocardial dysfunction or by peripheral vasodilation. Data obtained with advanced hemodynamic monitoring may help to identify these abnormalities, which can be important for choosing among the available vasopressor and inotropic drugs, which have widely differing receptor affinities and hemodynamic effects.9,45
Treatment with the drugs is best guided by the prevailing hemodynamic profile and aims at optimization of the circulation toward values associated with survival.9,42,45 Drugs are given as a continuous intravenous infusion, preferably via a central vein. The initial dose is low, and often combinations of drugs are used. The use of catecholamines should be judicious and carefully guided by hemodynamic parameters to reach predefined hemodynamic goals.9 β-Adrenergic drugs increase cardiac output by inotropic (β1) or vasodilating (β2) properties.9,353 Dopaminergic compounds may preferentially increase splanchnic and renal perfusion, glomerular filtration, and diuresis.66,284 Dobutamine, having vasodilating β2 properties, may exert greater effects on delivery and uptake of oxygen than dopamine at a lower PCWP.9,353 A decrease in the arterial blood pressure concomitantly with a decreased wedge pressure after dobutamine infusion may warrant additional fluid repletion.9 Drugs with α-adrenergic activity, such as norepinephrine, increase arterial blood pressure, but this increase may not lead to a decrease in cardiac output because they may increase venous return to the heart by decreasing venous compliance.7 The vascular reactivity to vasoconstrictors may diminish in the late phase of shock, but norepinephrine remains the agent of first choice in the (bridging to definitive) treatment of hypovolemic shock after fluid loading.354 The use of adrenergic drugs is not without hazards. They may enhance the metabolic demands of the body so that the oxygen supply-to-demand ratio is not favorably influenced even if oxygen delivery is enhanced.355 Particularly, epinephrine may increase lactic acid levels independently of oxygen balance.38 Low-dose dopamine has been shown, at least in bled dogs, to impede oxygen extraction by the gut during a decrease in oxygen supply, probably associated with transmural distribution of blood flow.66 Finally, vasoconstricting vasopressin and methylene blue, a guanylate cyclase inhibitor, have been tried to overcome intractable hypotension in this phase.96,300 Vasopressin has also been used in the initial management of uncontrolled hemorrhagic shock to safeguard arterial pressure for vital organ (e.g., cerebral) perfusion without overzealous fluid administration that may dilute coagulation factors and promote further bleeding.300
Brain Injury and Resuscitation
Hypovolemia and a decreased mean arterial blood pressure are considered as major threats for cerebral perfusion in brain injury. The latter may create intracranial hypertension following edema, bleeding, and contusion so that perfusion is more dependent on pressure than normal. Small volume resuscitation from hypovolemia with hypertonic (and hyperoncotic) solutions could increase mean arterial blood pressure at a small increase in plasma volume and could, by virtue of hypertonicity, decrease cerebral edema and intracranial pressure. The solutions are highly suitable for treatment of multiple trauma that includes the brain.314,315 Conversely, too much normotonic, certainly hypotonic, and perhaps albumin solutions may aggravate cerebral edema, but too little fluids and under-resuscitation with resulting hypotension and hypoperfusion may do the same. Vasopressor drugs such as vasopressin may be useful adjuncts in the initial treatment of hemorrhagic hypotension plus brain injury.
In Practice
In practice, the different types of fluids, including isotonic and hypertonic crystalloid and iso-oncotic or hyperoncotic colloid solutions, are often combined in the resuscitation from hypovolemic shock (see Box 26.2). For resuscitation of hypovolemic shock following hemorrhage, typed blood is often not immediately available, even though a blood sample for crossmatching has been sent to the blood bank as soon as possible after admission. If shock is severe and warrants immediate infusion of blood, type O Rh-negative erythrocyte concentrates can be safely used. In the absence of blood, resuscitation should begin with nonsanguineous fluids. During hypovolemic shock, initial resuscitation is often begun with hypertonic or isotonic (balanced) crystalloids, supplemented with colloid solutions, and finally accomplished through infusion of erythrocyte concentrates and plasma (Fig. 26.2).
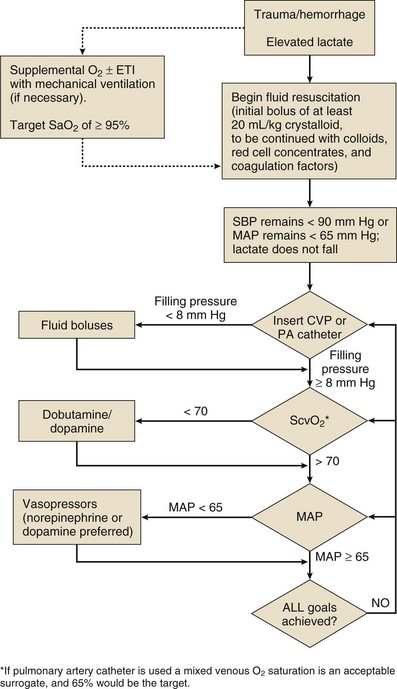
Supportive Care
A vomiting patient in hypovolemic shock should be protected against aspiration of gastric contents by early intubation. The value of specific measures for prevention of hemorrhagic gastric mucosal stress ulceration remains controversial.207 After resuscitation from shock, attention also should be paid to the nutritional status of the patient.222 It should be judged whether enteral or parenteral nutrition is necessary to improve nitrogen balance and energy intake.356 Although enteral feeding during hypovolemic shock and after resuscitation may increase metabolic demands of the gut, luminal application of nutrients such as glutamine may induce an increase in mucosal blood flow, ameliorate damage, and diminish the likelihood for translocation of endotoxins and bacteria and of septic complications.148,206 In addition, there is some evidence that early enteral feeding favorably influences organ function after hemorrhage and reperfusion, in contrast to (early) parenteral feeding, which may have adverse effects even though earlier meeting caloric requirements.148 The value of selective decontamination of the digestive tract or luminal absorption of endotoxin to prevent sepsis and its harmful sequelae originating from the gut is still controversial in multiple trauma, although such measures may inhibit the cytokine response to hypovolemic shock in animals.357
When treating pain in a patient with extensive trauma, morphinomimetics are cautiously applied because the drugs may have adverse circulatory effects during hypovolemic shock and half-life may be prolonged.11,358 Because many resuscitated patients after trauma or hemorrhage exhibit hypothermia, partly caused by exhausted energy reserves and infusion of substantial amounts of room temperature infusion fluids, and because hypothermia may denote more severe illness, rewarming infusion fluids may be necessary during resuscitation, and this may prevent some organ dysfunction and perhaps promote survival despite the increase in oxygen demand with an elevation in body temperature.56,217,264 In contrast, there also may exist some protective effect of mild hypothermia during bleeding and resuscitation, particularly when accompanied by brain injury, at least in experiments.
Miscellaneous Therapies
A wide array of experimental drugs has been tried in animal experiments to improve the hemodynamics, ameliorate inflammation, and increase survival rates from hypovolemic shock and resuscitation.230 Although many experimental drugs have shown some benefit in animal models of hypovolemic shock, in terms of hemodynamics during shock and after resuscitation and ultimate survival, there are now clinical trials ongoing or showing a benefit of such interventions in humans, including treatment with immune-enhancing factors.24,93,118,230,359 Blockers of NO synthesis (L-arginine analogs) and NO donors, inhibitors of activated K+ channels (oral antidiabetic, sulfonylurea drugs), Na+/H+ exchanger (amiloride, benzamide), and poly(ADP-ribose) polymerase have been tried in animal experiments to overcome vascular unresponsiveness, inflammation, and organ dysfunction early or late after development of hypovolemic shock.* ATP-MgCl2 may provide energy to cells, improve the microcirculation, reduce cell swelling, protect tissues from injury, and promote organ function and survival during hypovolemic shock and resuscitation.24,128,143,144 Ca2+-entry blockers have been used to prevent intracellular accumulation of Ca2+ and further damage of ischemic cells during resuscitation from hypovolemic shock.† Opiate antagonists or inhibitors such as naloxone, ACTH, and thyrotropin-releasing hormone been shown to increase arterial blood pressure, decrease inflammation, and improve survival.‡ Sedatives and analgesic drugs such as dexmedetomidine and ketamine may have anti-inflammatory properties and are tissue protective.360 Other vasoactive agents, including thyroid hormone, glucagon, and angiotensin inhibitors, with a preferential effect on splanchnic blood flow also may have beneficial effects in hypovolemic shock.105,225,226
Experimental models indicate that pretreatment with xanthine oxidase inhibitors (allopurinol) or scavengers of ROS or antioxidants including lazaroids may ameliorate microvascular hemodynamics and membrane injury of organs such as the heart and gut and improve survival after resuscitation from hypovolemic shock, although some of these compounds may have greater effects than others.* A study in trauma patients with elevated lipid peroxidation products in plasma showed that superoxide dismutase ameliorated the inflammatory response and MOF.239 It has been suggested that corticosteroids prevent lysosomal disruption and release of toxic proteases, prevent NO synthesis, and ameliorate the hemodynamic changes and promote survival during hypovolemic shock in animals.92,167,229,268 Cortisol treatment may increase the vascular sensitivity for catecholamines, particularly in patients in whom adrenal cortisol secretion is low relative to severity of disease, the so-called relative adrenal insufficiency. It may also decrease respiratory infections in the hospital course.361 Intravenously administered ACTH fragments may have an adrenal-independent central opioid-inhibiting effect, which may help to prevent vascular decompensation and treat hypotension, even clinically.93 Drugs such as pentoxifylline and complement inhibitors may prevent neutrophil-mediated endothelial injury, dysfunction, and downregulation of NO synthesis after bleeding and resuscitation, diminishing endothelium-dependent vasodilation.118,215 Pentoxifylline may ameliorate not only macrophage cytokine generation, but also adhesion molecule expression and neutrophil activation and aggregation and may improve red blood cell deformability. Administration of pentoxifylline or other methylxanthines may ameliorate reperfusion injury, at least in the rat gut and liver, and survival may be enhanced.79,166,206,230,319
Heparin and nonanticoagulant heparin sulfate or other analogues may have anti-inflammatory effects and may improve the microcirculation, and administration may partly protect various tissues, including the liver and gut, against reperfusion injury after hypovolemic shock by bleeding.250,363 Protease inhibitors, such as aprotinin, may also have beneficial effects. Administration of female hormones or inducers such as dehydroepiandrosterone or testosterone depletion after trauma/hemorrhage and resuscitation partly protects animals from microcirculatory organ dysfunction and immunosuppression, and a wide variety of mechanisms has been implicated.218 The potential of other immunologic and hormonal agents to treat immunosuppression has also been shown.218,250,362 Further drug developments include anticytokine strategies and tissue protective agents interfering with cell stress, apoptosis, or necrosis, such as erythropoietin.216 Mesenchymal stem cells are under investigation.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]26
Hypovolemic Shock
Although hypovolemic shock has been recognized for more than 100 years, Wiggers1 in 1940 first offered a definition of hypovolemic shock that has remained significant until now: “Shock is a syndrome resulting from depression of many functions, but in which the reduction of the effective circulating blood volume is of basic importance, and in which impairment of the circulation steadily progresses until it eventuates in a state of irreversible circulatory failure.” Today, hypovolemic shock can be defined as an acute disturbance in the circulation leading to an imbalance between oxygen supply and demand in the tissues, caused by a decrease in circulating blood volume, mostly caused by trauma and hemorrhage.2 An oxygen debt develops when uptake no longer matches the demand for oxygen and leads to cellular ischemia and ultimately cell death. The condition is life threatening and, if left untreated, becomes irreversible after a certain period. Rapid and adequate resuscitation is mandatory to save lives. Conversely, hypovolemic shock carries a relatively favorable prognosis, if rapidly and adequately recognized and treated.
Hypovolemic shock can occur outside and inside the hospital, in trauma or surgery complicated by excessive loss of blood, but also in the course of burns, gastrointestinal hemorrhage, diarrhea, uncontrolled diabetes mellitus, addisonian crisis, and other conditions (Box 26.1). Some other types of shock, including septic, anaphylactoid, cardiogenic, and burn shock, may be accompanied by hypovolemia. The types of shock not primarily caused by hypovolemia are beyond the scope of this chapter.
Pathogenesis and Pathophysiology
Circulatory Changes
General Changes
Because hypovolemia results in a decrease in preload of the heart and low filling pressures or volumes, the cardiac output decreases.3–9 After unloading of the baroreceptor and activation of the sympathetic nervous system, tachycardia ensues, although some patients may respond with transient sympathetic inhibition and vagal nerve–mediated bradycardia during a sudden, severe loss of circulating blood volume.3,10–16 Tachycardia partially compensates for the decrease in stroke volume. A moderate decrease in cardiac output can be recognized from a decline in pulse pressure, orthostatic hypotension, and fall in regional perfusion indices.8,17,18 Hypovolemia results in wider than usual swings in central venous pressure (CVP) and arterial blood pressure during the respiratory cycle of spontaneous and mechanical ventilation because of increased sensitivity of the underfilled heart in the ascending part of the cardiac function curve to fluctuations in venous return associated with varying intrathoracic pressure.19,20 Although activation of the sympathetic nervous system and resulting arterial vasoconstriction during a moderate decrease in cardiac output prevent a severe reduction in arterial blood pressure, a further decrease in cardiac output leads to hypotension and shock.8,10 Systemic vascular resistance increases early after development of hypovolemic shock but may decrease in the later stages of shock, and this may herald irreversibility and death. The increase in resistance (and heart rate) may be transiently attenuated after an imbalance between sympathetic and vagal activity, possibly associated with release of opioids within the central nervous system and into the systemic circulation.*
Shock is characterized by an oxygen debt in the tissues.9,24–26 In the presence of sufficient oxygen, aerobic combustion of 1 mol of glucose yields 38 mol of energy-rich adenosine triphosphate (ATP), which can be hydrolyzed to provide energy for the vital and metabolic functions of the cell.27 In the absence of oxygen, glucose taken up by cells cannot be combusted because of insufficient uptake of pyruvate into the mitochondrial tricarboxylic acid cycle having a reduced turnover rate. Partly inactivated pyruvate dehydrogenase may play a role in the latter reductions. Pyruvate is converted into lactate, and the lactate-to-pyruvate ratio increases, concomitantly with a reduction in mitochondrial redox potential.24,27–29
Anaerobic glycolysis in the cytosol ultimately yields, per mol of glucose, 2 mol of ATP.27 Hydrolysis of ATP yields hydrogen ions (H+) that lead, when buffers are exhausted, to intracellular and ultimately to extracellular metabolic acidosis.30 These mechanisms form the basis of the so-called lactic acidosis during hypovolemic shock, whereby the lactate level in arterial blood is elevated above the normal 2 mmol/L associated with acidosis, and constitutes a useful measure of the oxygen debt in the tissues.26,27,31–34 Nevertheless, the energy deficit and lactate production in the cells in response to a lack of oxygen can be limited and organ function can be improved by supplying pyruvate and pyruvate dehydrogenase activators, such as dichloroacetate.27,29,35–37 Intracellular acidosis may otherwise protect ischemic cells from dying.36
The specificity of elevated lactate-to-pyruvate levels for an oxygen debt in the tissues has been doubted.27,38 Aerobic glycolysis is probably linked to the membrane Na+/K+-ATPase and stimulation of β2-receptors during sympathetic activation. Catecholamine (epinephrine) secretion may temporarily increase, rather than decrease, ATPase activity, and augment glycolysis and circulating lactate levels in tissues such as skeletal muscle, without a lack of oxygen and reduced ATP resources, during development and resuscitation from hypovolemic shock.38,39 Conversely, adrenergic antagonists may reduce lactic acidosis during hypovolemic shock.38 Epinephrine may increase glycogenolysis. Together, increased glycolytic fluxes independent of oxygen uptake may lead to equal elevations of pyruvate and lactate in the tissues, without the acidosis resulting from ATP hydrolysis with an oxygen debt.27 This situation may partly explain why the extent to which changes in the lactate level parallel changes in the anion gap or bicarbonate/base excess concentration during shock and resuscitation is controversial, and why elevated lactate levels sometimes may fail to predict an increase in oxygen uptake during an increase in oxygen delivery.40–42 This also may explain in part the discrepancies in the course of oxygen-related variables and lactate levels during catecholamine treatment of shock when attempting to boost oxygen delivery.42
The lactate level in blood is determined by production, distribution, and elimination.27 Produced lactic acid in the presence of oxygen may be converted via pyruvate to glucose or oxidized. Bicarbonate is then released. The liver plays a central role in this process, so that the elimination of lactate and clearance from plasma is impaired in case of liver ischemia or prior hepatic disease, even though renal uptake may increase.27,43,44 Nevertheless, changes in the lactate level in blood, rather than absolute values, mainly reflect changes in production and are a fair measure for the course of shock and the response to therapy, even in the presence of liver disease.27,45 Although not beyond doubt, the origin of lactate in hypovolemic shock can be skeletal muscle, lung, and gut, particularly if severe liver ischemia, hypoxia, and acidosis in shock attenuate the hepatic uptake of lactate delivered by the gut through the portal vein.27,43,44,46–48 The respiratory muscles also may contribute to lactic acidosis in a spontaneously breathing patient because, first, the respiratory muscles may demand a share of the cardiac output at the cost of other tissues, and, second, this share may be insufficient to meet oxygen demands of the diaphragm, which may be increased in view of hyperventilation.33,49–52
Notwithstanding the aforementioned limitations, an increase in the lactate level in blood and a decrease in the bicarbonate content/base excess or pH and an increase in the anion gap may be fair predictors of morbidity (multiple organ failure [MOF]) and mortality, whereas clearance of lactic acidosis usually indicates a better outcome. A decrease in the blood lactate level during resuscitation from hypovolemic shock is usually a favorable sign and associated with survival, whereas an increase in the lactate level and progressive acidosis usually are associated with morbidity and mortality, even though successful resuscitation may transiently increase the lactate level because of washout of lactate from ischemic tissues.* The mentioned variables may thus serve as guides for resuscitation.
Oxygen Balance
Because insufficient uptake of oxygen relative to demand in the tissues during shock is central, insight into the factors that determine oxygen uptake in shock is important.25,27 Oxygen delivery is determined by the cardiac output and the content of oxygen in arterial blood, that is, the arterial blood hemoglobin concentration and the saturation of hemoglobin with oxygen. The oxyhemoglobin dissociation curve determines the saturation of hemoglobin with oxygen for a given partial pressure of oxygen (PO2) in blood. During hypovolemic shock, a decrease in hemoglobin concentration, oxygen saturation, or both aggravates the effect of a decrease in cardiac output in compromising oxygen delivery to the tissues. Cardiac output is determined by preload, afterload, contractility, and heart rate.7
During a decrease in oxygen delivery with hypovolemic shock, the body maintains sufficient uptake of oxygen only if the extraction of oxygen increases, and the arteriovenous oxygen content gradient widens, resulting in a decrease in oxygen saturation of venous blood.† Associated with a decrease in oxygen delivery, tissue PO2 declines, and its heterogeneity increases, possibly indicating focal ischemia.‡ The decline in tissue PO2 may be even greater than the decrease in draining venous blood because of some increase in microvascular oxygen shunting at low blood flows.68,69 In animals, it has been shown that the increase in oxygen extraction to compensate for a decrease in oxygen delivery is maximum (but not 100%) if oxygen delivery decreases to less than 8 to 15 mL/kg per minute, that is, the critical oxygen delivery (Fig. 26.1).§ Although the critical oxygen delivery may vary widely among studies, following differences in species, basal oxygen needs, and methods to decrease oxygen delivery, data obtained in patients suggest that the critical oxygen delivery in humans may also amount to approximately 8 mL/kg per minute.58,64 During a decrease in oxygen delivery below this critical value in hypovolemic shock, oxygen uptake decreases to less than tissue demand, cellular ischemia ensues, and the body must rely on anaerobic metabolism to meet energy requirements.¶ Blood lactic acidosis, lacticacidemia, results. Conversely, oxygen uptake is supply-dependent if oxygen delivery is lower than the critical value and blood lactate levels are elevated, whereas oxygen uptake may not be supply-dependent if the lactate level in blood is normal. Treatment of hypovolemic shock, by infusing fluids and blood, is aimed at an increase in cardiac output and the oxygen content of blood and in oxygen delivery above the critical value so that oxygen uptake increases to meet body requirements and the lacticacidemia decreases.*
The critical oxygen delivery is a function of the body oxygen needs and the capability of the body to extract oxygen during a decline in delivery. The body oxygen needs may increase during hypovolemic shock, as a consequence of increased respiratory muscle activity and increased levels of catecholamines in the blood after activation of the sympathetic nervous system, but downregulation of the metabolic stimulant effect of catecholamines has been described.† The critical extraction of oxygen is a function of the adaptation of regional blood flow to tissue needs, the number of perfused capillaries and of diffusion distances, and the exchange surface area for oxygen.63,75 During a reduction in oxygen delivery, however, oxygen uptake is limited by convective transport of oxygen to the tissues, rather than by diffusion of oxygen to respirating mitochondria.76
In experimental animals, a change in hemoglobin affinity for oxygen, by altering the storage duration of reinfused blood, hardly changes the critical oxygen extraction, but changes in acid-base status that affect the position of the oxyhemoglobin dissociation curve may have some effect on the oxygen extraction capabilities of the body.60,76 Acid infusion may increase slightly, and base infusion may reduce oxygen extraction during supply-limited oxygen uptake.76 Nevertheless, hypercapnia may decrease critical oxygen extraction and increase critical oxygen delivery because of blood flow redistribution.77 A leftward shift of the oxyhemoglobin dissociation curve may impair maximum oxygen extraction during a reduction in oxygen delivery and may increase mortality rate in experimental animals with hypovolemic shock.60 Although the oxyhemoglobin dissociation curve may shift to the left in critically ill patients, for example, after transfusion of old, stored blood,78 the effect on oxygen uptake is unclear.
The effect of changes in body temperature is twofold: Changes are accompanied by changes in total body oxygen needs and by changes in critical oxygen extraction, probably by a vascular tone–associated altered distribution of blood flow.61 Hyperthermia increases critical oxygen delivery in hemorrhaged dogs, primarily through an increase in body oxygen needs and despite an increase in critical oxygen extraction, whereas hypothermia, which may be more common in traumatized or hemorrhaged patients, may decrease the critical oxygen delivery.61 Finally, blood viscosity may influence the extent to which a decrease in circulating blood volume affects oxygen uptake by the tissues. Experimental data suggest, however, that prior anemia does not ameliorate the decrease in oxygen uptake during a decrease in oxygen delivery with hypovolemic shock, indicating that the convective transport of oxygen is the major determinant of oxygen uptake when delivery is impaired, even though prior hemodilution may increase oxygen extraction capabilities and decrease critical oxygen delivery.6,70
Taken together, these factors may influence the extent to which oxygen uptake decreases during reduced delivery and how far oxygen delivery should be enhanced during resuscitation from hypovolemic shock. The critical oxygen delivery varies among tissues. The oxygen needs of the kidney may decline during a decrease in renal oxygen delivery because a decrease in renal perfusion may lead to a reduction in glomerular filtration and to a reduction in energy-consuming tubular resorption.63 In contrast, during progressive hypovolemia, the gut may experience supply dependency of oxygen uptake earlier than nongut tissue, partly because of a higher critical oxygen delivery (higher needs and less extraction of oxygen) and partly because of redistribution of blood flow away from the gut mucosa after more intense vasoconstriction in gut than in nongut tissue.* Clinically, this may result in nonocclusive bowel ischemia. Respiratory muscles may also have a higher critical oxygen delivery than the body as a whole during progressive hemorrhage.
Concomitant with an increased arteriovenous oxygen extraction during a decrease in oxygen delivery, the arteriovenous gradient of the carbon dioxide (CO2) content widens.8,52,81 The latter is associated with an increase in tissue and venous partial pressure of carbon dioxide (PCO2) relative to arterial PCO2 and a decrease in venous pH exceeding the decrease in pH in arterial blood.44,52,68 This widening of gradient is caused by the Fick principle and a greater decline in cardiac output than in oxygen uptake and CO2 production in the tissues because of inhibited oxidative metabolism. Nevertheless, the oxygen uptake usually decreases more than CO2 production, leading to an increase in respiratory quotient.49,65 This increase is likely to be caused by buffering of lactic acid by bicarbonate in the tissues and effluent blood, a shift toward glucose instead of fat use for residual oxidation in ischemic tissues, or a combination of both. The end-tidal expiratory CO2 fraction decreases in association with a reduction in oxygen uptake and CO2 production for a given ventilation.65 Conversely, a decrease in arterial PCO2 during a decline in CO2 production versus ventilation may be attenuated by an increase in dead-space ventilation resulting from a decrease in pulmonary blood flow/ventilation ratio.49 An increase in deadspace ventilation leads to widening of the gap between the arterial and expiratory PCO2.49
It has been suggested that the severity and duration of the oxygen debt accumulated during hypovolemic shock is a major determinant of survival in animals3,6 and in patients with trauma/hemorrhage and after major surgery.26,42,82 After trauma and hemorrhage, the defect in circulating blood volume and tissue oxygenation may be greater in patients who develop acute respiratory distress syndrome (ARDS) and MOF than in patients without these complications.* In patients undergoing major surgery, the oxygen debt during and after surgery may relate directly to the development of postoperative organ damage (i.e., MOF) and demise.73,82 Conversely, a high oxygen delivery and uptake during resuscitation may be associated with survival, whereas values that may be too low for elevated tissue demands are believed to contribute to ultimate demise, at least in animals with hypovolemic shock and critically ill patients after trauma or major surgery.† An increase of oxygen delivery and oxygen uptake to supranormal values has been suggested to improve survival further, although the latter debate has not been settled yet.‡ Extensive ischemic mitochondrial damage may limit an increase in oxygen consumption during resuscitation and reperfusion.
Macrocirculation
During loss of blood volume, various mechanisms come into play that may counteract the resultant decrease in cardiac output and tissue oxygenation. First, a decrease in cardiac output during hypovolemic shock results in a redistribution of peripheral blood flow.§ This redistribution is partly the result of regional autoregulation to maintain blood flow, in which endothelial cells and production of endogenous partly gaseous vasodilators, including endothelial nitric oxide synthase–derived nitric oxide (NO), heme oxygenase–derived carbon monoxide, hydrogen sulfide, and metabolic by-products in the tissues including CO2, potassium, and adenosine, may play a central role.86–94 Endothelium-derived NO relaxes underlying smooth muscle in the vessel wall, via stimulation of guanylate cyclase and cyclic guanosine monophosphate (cGMP), which can be inhibited by methylene blue.87,88,95,96 Carbon monoxide also acts via cGMP.91 Some authors describe that inhibition of endothelial NO synthase ameliorates early hypotension and even the mortality risk during bleeding.92 When NO is released, the reactivity to endogenous and exogenous vasoconstrictors may be diminished, even early in hypovolemic shock.92,97 Other authors describe endothelial injury and dysfunction in various organs with diminished endothelium and NO-dependent vasorelaxation, which could be overcome by L-arginine and other NO donors, including ATP-MgCl2, pentoxifylline, or heparin, so that blockade of endothelial NO synthase–derived NO may be detrimental.87–89,97,98
The opposing vasoconstricting factors include catecholamines, liberated by the activated sympathetic nervous system and the adrenal medulla; direct sympathetic stimulation of the vessel wall; angiotensin II, liberated through an activated renin-angiotensin-aldosterone system; and vasopressin, released by the pituitary in hypovolemic shock.* Endothelin is an endothelium-derived potent vasoconstrictor, released on catecholamine stimulation or hypoxia, and its release may contribute to vasoconstriction, particularly in hepatic and renal vascular beds.100,101 Finally, a decrease in cardiac filling may reduce cardiac secretion of atrial natriuretic peptides, reducing the vasodilating and diuretic effect of these factors.102 Levels may also increase as a consequence of diminished renal clearance.103,104
Depending on the degree that the mechanisms are operative, the general result of the interplay is that blood flow to intestines, skeletal muscle, and skin is diverted toward vitally more important organs, such as heart and brain, so that the increase of overall peripheral resistance during hypovolemic shock is distributed differently among various organs, with greater increases in gut, skeletal muscle, and skin than in heart and brain.† The kidney also is a target for hypovolemic shock; renal perfusion may be maintained during mild hypotension after hypovolemia, but it rapidly decreases if severe hypotension supervenes, and the decrease may exceed that in other organs.‡ In hypovolemic human volunteers, this redistribution of blood flow accords with the patterns described.74
The redistribution of blood flow results in a greater share of oxygen delivery going to organs with high metabolic demand, such as heart and brain, than tissues with less metabolic demands, including skin, skeletal muscle, kidney, gut, and pancreas.§ The redistribution is probably necessary to optimize the uptake of delivered oxygen to the tissues and partly accounts for the increase in oxygen extraction during a decrease in oxygen delivery.63,75 In dogs, the ability of the body to extract oxygen diminishes with α-receptor blockade of sympathetic activity, suggesting that redistribution of blood flow aided by the sympathetic nervous system is a major determinant of critical oxygen extraction.63
Microcirculation
Vasoconstriction after activation of the sympathetic nervous system during hypovolemia (hemorrhage) occurs in the arteries and medium-sized arterioles but not in terminal arterioles, which may even dilate, as judged from vital microscopy studies in animals.¶ Relatively spared terminal arteriolar blood flow is presumably caused by vasodilating metabolic responses to a decline in nutrient blood flow. Nevertheless, capillary flow usually diminishes, and heterogeneity, both in space and time, increases, particularly in irreversible shock and independent of cardiac output. Traumatic/hypovolemic shock may induce expression of adhesion molecules on primed neutrophils and vascular endothelium and this, together with a reduced flow rate, may promote adherence of neutrophils to endothelium.95,111–119 This adherence may impair red blood cell flow, particularly in capillaries and postcapillary venules. Other authors suggest that capillary leukostasis is pressure-dependent and not receptor-dependent and reversible when perfusion pressure has been restored.121 Finally, endothelial cells may swell and may hamper capillary red and white blood cell flow.95,98,110,122 The microcirculation can be visualized, even in humans, by buccal or sublingual orthogonal polarization spectroscopy and side stream dark-field imaging.123
Vasoconstriction is not confined to arteries, but also occurs in the venous vasculature, more in large than in small venules and particularly in the splanchnic area, and, again, this is largely mediated by increased activity of the sympathetic nervous system and vasopressin and angiotensin II release.5,12,68,108 Because most of the circulating blood volume is located in small venules, splanchnic venoconstriction results in a decrease in compliance and less volume for a given intravascular pressure in the venous system, increasing return of blood to the heart.5,7 Hence, partitioning circulating volume in stressed and unstressed portions now favors the former. During hypovolemic shock, the precapillary to postcapillary resistance increases, resulting in a decrease in capillary hydrostatic pressure and in fluid resorption from the interstitial space as opposed to normal filtration from capillary to interstitium, even though interstitial hydrostatic pressure decreases.4,5 This is accompanied by diminished transport of protein from blood to interstitium.124
Cellular water is mobilized, unless, at a later stage, the cell swells following Na+ overload.21,98,110,125–129 Studies on fluid volumes in hypovolemic shock are not equivocal, but generally suggest that the interstitial and cellular compartments are depleted in defense of the circulating blood volume to promote venous return to the heart.5,7,125,126 Mobilization of fluid from the interstitial and cellular compartment can be promoted by plasma hyperosmolarity, through an increase in the glucose concentration.130,131 Chronically starved rats with depleted glycogen stores more rapidly die of hypovolemic shock than fed ones, and this can be prevented by prior glucose infusion.130 In addition, the lymphatics may show increased pumping ability, increasing return of fluid into the systemic circulation independently of the reduced capillary fluid filtration rate.132 Lymphatic return of interstitial protein and fluid may contribute to repletion of circulating protein and fluid volume.132
Hemorrhage and hypovolemic shock lead to a decrease in hematocrit and a decrease in plasma proteins through transfer of fluid (and protein) from the interstitial to the intravascular space.4,5,124 Refilling of the intravascular space diminishes in time after a sudden decrease in circulating volume, when a decline in colloid osmotic pressure, associated with hypoproteinemia, and an increase in hydrostatic pressure accomplish a new steady state in capillary exchange through readjustment of the pericapillary hydrostatic and colloid osmotic pressures, which determine fluid and protein transport.5 Conversely, hypoproteinemia can promote transcapillary fluid transport and expansion of the interstitial space, if hydrostatic pressure returns toward normal (e.g., during crystalloid fluid resuscitation).126,133–136 During a sudden decrease in circulating blood volume by hemorrhage, some time is needed before the decrease in hematocrit and of proteins in blood is completed, and this decrease is aggravated by nonsanguineous fluid resuscitation.126,134,137 Finally, increased sympathetic discharge results in contraction of the spleen, releasing red blood cells into the circulation and defending a fall in hematocrit.14
Cells
During hypovolemic shock, the oxygen lack in the tissues causes a decline in the mitochondrial production and concentration of high-energy phosphates in the tissues because of greater breakdown than production of these compounds.24,29,46,138,139 This decline is a function of the severity and duration of regional hypoperfusion relative to oxygen demand. The decrease in the redox status and high-energy phosphates during experimental hypovolemic shock is more pronounced in some tissues (diaphragm, liver, kidney, and gut) than in others (heart and skeletal muscle), so regional lactate production may vary.*
A decrease in high-energy phosphates heralds irreversible cell injury during ischemia, whereas a less severe decline may result only in prolonged programmed cell death—apoptosis. In animals with hypovolemic shock and in critically ill patients, the circulating levels of ATP can be diminished, and ATP degradation products, including adenosine, inosine, hypoxanthine, and xanthine, can be elevated, suggesting breakdown of ATP following a lack of oxygen in the tissues.28,29,56,141,142 Conversely, reperfusion is associated with restoration of energy charge, depending on the effect of ischemia, the oxygen demand, and the level of reperfusion. The intravenous administration of energy in the form of ATP-MgCl2 may help tissues (kidney, liver, heart, gut) to recover from ischemia and resume function, independently of the vasodilating effects of the compound.24,128,143,144 Also, pretreatment with coenzyme Q10, involved in the respiratory chain reactions in mitochondria, has a beneficial effect during hypovolemic shock and resuscitation, at least in dogs.81 Nevertheless, part of the mitochondrial dysfunction after trauma and hypovolemic shock has been suggested to be independent of a lack of oxygen.53 Near-infrared spectroscopy, which can be applied in animals and patients, may indeed reveal normal absorption spectra for tissue oxyhemoglobin and low mitochondrial cytochrome aa3 redox status.53,123,145
About 60% of the energy produced by respirating mitochondria is needed to fuel the Na+/K+ pump of the cell, through which the gradient in electrolyte concentrations and electrical potential over the cell membrane are controlled.24 When ATP becomes insufficient because of a decline in production associated with lack of oxygen and production of protons increases, the Na+/K+ pump is inhibited and the Na+/H+ exchanger is activated, and this results, together with a possibly selective increase in cell membrane permeability for ions, in an influx of Na+ into and efflux of H+ and K+ out of the cell, leading to cellular uptake of fluid.† Measurement of membrane potentials of skeletal muscle and liver in experimental animals has shown that hypovolemic shock rapidly decreases the transmembrane potential (a less negative inner membrane potential), associated with electrolyte and fluid shifts across the cell membrane.‡ A decrease in activity of the Na+/K+ pump may contribute to hyperkalemia because of potassium exchange between cells, interstitial fluid, and vascular space.38,46,111,125 Finally, calcium (Ca2+) influx into cells and their mitochondria inhibits cellular respiration and ultimately contributes to cellular damage and swelling, particularly during resuscitation, and this can be prevented by administration of Ca2+ antagonists.* Because of cellular influx, the plasma-free Ca2+ levels may decrease in experimental and human hypovolemic shock.127,149,150 Intracellular lysosomes lose their integrity so that proteolytic enzymes are released and contribute to cell death.4,24,107,151 These enzymes eventually may reach the systemic circulation and may damage remote organs.4,24,107,151
As has become apparent in past years, the cellular response to stress, such as heat and tissue hypoxia, involves the expression of certain genes, coding for synthesis of the so-called heat-shock proteins, which play an important role in protecting the cells against stress.152–155 The clinical significance of these molecular cellular changes is unknown. The response may be partially responsible, however, for the decreased susceptibility to and tissue injury by hemorrhagic shock in animals with a prior challenge by endotoxin or other forms of preconditioning.113,156
Organ Perfusion and Function in Shock
Heart
According to Starling’s law of the heart, a change in preload, approximated by the end-diastolic volume and determined by the venous return of blood to the heart, directly results in a change in stroke volume, defining myocardial function.7 The relationship between end-diastolic filling pressure and volume reflects compliance. Apart from preload, cardiac output also depends on afterload, which is approximated by the end-systolic volume of the heart, and contractility, reflected by the peak systolic pressure-to-volume relationship (maximal elastance).7,55,157 A diminished response of the stroke work by the heart, that is, the product of stroke volume and arterial blood pressure, to an increase in preload during resuscitation from hypovolemic shock may indicate diminished cardiac contractility that is associated with a worse outcome (e.g., caused by preexisting cardiac disease, hypovolemic shock itself, myocardial contusion, or combinations).4,55,158,159 The effect of hypovolemic shock on myocardial function in animal models is controversial. Depending on models, methods, and definitions of cardiac dysfunction, some authors describe a decrease, but others describe an unchanged function of the left side of the heart.† The latter can be explained if a decrease in contractility of the heart is masked by the inotropic effect of catecholamines and other positive inotropic substances, such as endothelin, liberated during hypovolemic shock, even though receptor-mediated catecholamine responses may decline.4,23,100,160
Although coronary blood flow may be defended, and the oxygen demands of the heart may decrease associated with a decrease in filling (preload) and arterial blood pressure (afterload) during initial hypovolemic shock, hypotension may become so severe that coronary vasodilation to compensate for a decline in perfusion pressure becomes exhausted, so that myocardial oxygen delivery decreases to less than the oxygen needs of the heart and ischemia ensues, particularly if tachycardia is present.3,21,140,161–163 This sequence leading to ischemia may occur primarily in endocardium because of more rapidly exhausted vasodilation in endocardium than epicardium and redistribution of blood flow from the inner to the outer layer of the heart.140 The subendocardium may become ischemic, and patchy necrosis may ensue. Because of regional transmural and intramural differences in vasodilator reserve, myocardial ischemia may be heterogeneously distributed and associated with a diminished redox state, lactate production, and creatine phosphate breakdown.140,164 Ischemia ultimately may contribute to a decrease in myocardial contractility during hypovolemic shock. Smooth muscle–dependent and, particularly, endothelium-dependent coronary vasomotion may be impaired after hypovolemic shock.89,165 Myocardial edema and compression of capillaries with resultant impairment of diffusion and extraction of oxygen may also contribute to a decrease in regional coronary blood flow, regional myocardial ischemia, and decreased myocardial function in hemorrhaged animals.127,131,140,161
Hypovolemic shock may induce a decrease in left ventricular compliance and relaxation.160,161 The diastolic dysfunction may be particularly pronounced during resuscitation from hypovolemic shock.157,160,161 Postischemic failure (stunning) also may play a role during resuscitation, at least temporarily. Ischemia-reperfusion of the heart results in accumulation of intracellular Ca2+.127 This may impair mitochondrial and sarcoplasmic reticulum function and contribute to impaired cardiac function after hypovolemic shock.127,160 In dogs, the administration of Ca2+ blockers may prevent such deterioration during resuscitation from hypovolemic shock.127 Finally, systemic release or intramyocardial production of negative inotropic substances and inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL) 6 and platelet activating factor, oxidant damage, metabolic acidosis, diminished adrenoreceptor density, and resultant diminished sensitivity of the heart to circulating catecholamines may contribute to myocardial dysfunction during hypovolemic shock.* Pentoxifylline may improve endothelial and myocardial function.166 Reversibility of dysfunction is associated with survival.161
The clinical evidence for myocardial dysfunction during hypovolemic shock is scarce.46,55,163 Nevertheless, it is conceivable that severe hypotension reduces the balance between oxygen delivery and demand of the heart because many patients with hypovolemic shock may be elderly with coronary artery disease, compromising coronary vasodilation. Some may have preexisting impaired function while on beta blockers. For a patient with hypovolemic shock, a decrease in left ventricular compliance, contractility, or both may imply that a relatively high filling pressure would be needed to restore cardiac output during fluid resuscitation.55,71,161,163,171 The averaged optimal pulmonary capillary wedge pressure (PCWP), that is, the pressure above which cardiac output does not increase further, may not be elevated in patients with hypovolemic shock (i.e., 12 to 15 mm Hg), although in some patients, abnormally elevated filling pressures may be needed to increase cardiac output, or cardiac output does not increase at all during fluid resuscitation.55,71,171,172 A diminished function of the heart may hamper restoration of oxygen delivery to the tissues during resuscitation necessary for survival.9,45,55,159,160 Myocardial dysfunction may thus be greater in nonsurvivors than in survivors. There may be some electrocardiographic or enzymatic evidence for myocardial ischemia and injury, and some patients may experience a myocardial infarction as a complication of severe hypovolemic shock after hemorrhage.163,173
Lung
Hypovolemic shock often induces an increase in ventilatory minute volume, resulting in tachypnea or hyperventilation and a decrease in arterial PCO2.33,49,50,52,174 Unless complicated by pulmonary abnormalities, these changes are, at least initially, not the result of hypoxemia but an increase in dead-space ventilation following a decrease in pulmonary perfusion so that a higher minute ventilation is necessary for a given CO2 production to eliminate CO2 from the blood and to maintain a normal PCO2 in arterial blood.33,49,50 Minute ventilatory volume may increase further if a decrease in PCO2 is necessary to compensate for metabolic acidosis after accumulation of lactate in the blood.* The imbalance between increased demands of the diaphragm and reduced blood flow in shock may finally lead to respiratory muscle fatigue and a subsequent decline in ventilatory minute volume.50
Hypovolemic shock caused by trauma and hemorrhage and followed by extensive transfusion therapy of red blood cell concentrates can be complicated by pulmonary edema and impaired gas exchange.51,175–180 In some patients, fluid overloading, overtransfusion, and an elevated filtration pressure (PCWP) may be responsible: transfusion-associated circulatory overload (TACO). In others, pulmonary edema may be due to a pulmonary vascular injury, however, and increased vascular permeability at a relatively low PCWP, indicating noncardiogenic permeability edema or ARDS.51,174,177,178 The reaction to diuretics may help to differentiate between hydrostatic and permeability edema of the lungs. The latter seems relatively rare in polytransfused, polytraumatized patients unless associated with complications, but other studies suggest that about 30% of patients with severe trauma/hemorrhage, particularly if polytransfused, may develop ARDS.177,178,180,181
Experimental studies are at variance concerning alterations in capillary permeability of the lungs during hypovolemic shock and resuscitation.4,133,174,182,183 According to some investigators, hypovolemic shock following bleeding and transfusion mildly increases transvascular filtration of fluid and proteins and results in accumulation of interstitial fluid as a consequence of increased permeability,182 but other authors do not observe such changes.131,133,182,184 In other animal studies, however, traumatic/hypovolemic shock resulted in extensive morphologic changes of the lung, with endothelial and interstitial edema, accumulation of degranulated neutrophils, and scattered fat emboli, which may resemble the pulmonary changes after traumatic/hypovolemic shock in humans.156,174,185–187 As measured by the transvascular albumin flux in the lungs, almost 80% of patients with multiple trauma may show increased pulmonary vascular permeability in the disease course.51 This leak ultimately may contribute to pulmonary edema, impaired mechanics, and gas exchange.51 As suggested by animal experiments, among others, several factors may play a role, including release of proinflammatory mediators (TNF-α) and priming and activation of blood neutrophils after ischemia-reperfusion, contusion or ischemia-reperfusion of the lungs themselves, pulmonary microemboli of neutrophils, platelets and fat particles from the medulla of fractured long bones and pelvis, and neutrophilic antibodies or humoral or cellular breakdown products and released cytokines in long-stored and transfused blood products (transfusion-related acute lung injury, TRALI).* Translocated endotoxin may also play a role.185 Finally, aspiration of foreign material or gastric contents and posttraumatic pneumonia and sepsis may contribute to the development of ARDS in trauma patients. When pulmonary edema has developed, active resorption by alveolar cells becomes necessary for clearance. This process is cylic AMP–dependent and can be disturbed by inducible nitric oxide synthase (iNOS)–derived NO and peroxynitrite and enhanced by expression of heme oxygenase, which may mitigate lung injury in animal models.156 How this translates clinically is unclear.
Brain
Classically, brain perfusion and microcirculation are considered to be relatively spared during progressive hypovolemia because of the extensive autoregulatory capacity of cerebral arteries.15,189 In case of autoregulation impairment after neurotrauma, however, brain perfusion may decrease, and subsequent reperfusion may contribute to secondary cerebral damage during hypovolemic shock and resuscitation. Hemorrhagic shock and resuscitation per se may also impair autoregulatory capacity of brain vessels, however, because of endothelial dysfunction and diminished NO-dependent vasodilator reactivity, so that the brain may experience an oxygen debt and subsequent metabolic and functional deterioration.29,88
Kidney
Hypovolemic hypotension is an important risk factor for acute kidney injury and failure after trauma.138 During a decrease in cardiac output following progressive hemorrhage, renal blood flow can be maintained because of renal vasodilation, so that the kidneys may not participate in the systemic vasoconstriction that characterizes hypovolemic shock.3 Vasodilating prostaglandins are released in the kidney through activation of the cyclooxygenase pathway of arachidonic acid metabolism in response to ischemia, increased sympathetic activity, and angiotensin II, so that renal vasodilation during the early phase of hemorrhage can be blocked by prostaglandin synthesis inhibition, resulting in a profound decrease in blood flow even if accompanied by an increase in arterial blood pressure.3 When blood pressure decreases during progressive hypovolemia, the renal vessels constrict, impairing blood flow to the kidneys more than to other organs.* This is partly caused by a baroreflex-mediated increase in sympathetic activity; activation of the renin-angiotensin-aldosterone system; and release of catecholamines, angiotensin II, endothelin, and vasopressin.13,14,74 During prolonged hypovolemic shock, sympathetic inhibition may protect against renal ischemia.14 This propensity for vasoconstriction is thus partly offset if NO and other factors with vasodilatory actions are released intrarenally.4,86 Inhibition of NO synthesis increases blood pressure, however, and increases renal perfusion and glomerular filtration during hypovolemic shock.86 In another study, endothelium-dependent renal vasodilation was impaired after hypovolemic shock.87
Renal ischemia results in a decrease in glomerular filtration (prerenal renal failure) that is less than the decline in blood flow so that the filtration fraction often increases.138 The latter is caused by greater constriction of efferent than of afferent arterioles in glomeruli, in which high levels of circulating angiotensin II are probably involved. The decrease in glomerular filtration together with an increase in tubular resorption of electrolytes and fluids, mediated by increased levels of antidiuretic hormone released by the pituitary and decreased levels of atrial natriuretic peptides through low atrial filling, results in oliguria or anuria (<0.3 mL/kg/hour) and a low sodium content of urine.138
The decrease in renal perfusion during hypovolemic shock is often accompanied by redistribution of blood flow from outer to inner cortex and medulla, which is already borderline hypoxic even in the normal state.127 If long-lasting and severe, the cortical kidney becomes ischemic, despite a decrease in oxygen needs associated with fewer energy needs for tubular resorption in the presence of less filtration, so that the levels of high-energy phosphates decline.138,139 Severe and prolonged renal ischemia and metabolic deterioration finally result in acute kidney injury and failure with morphologic changes, particularly in proximal tubules and medullary segments (acute tubular necrosis) when an increase in renal perfusion does not immediately restore filtration and diuresis, but rather injures renal structures (reperfusion injury), limiting a return of blood flow and glomerular filtration during resuscitation.129,138,185,186 This is often recognized by a persistent oliguria and a gradual increase in creatinine and urea levels in blood. In addition, the plasma levels and urinary excretion of biomarkers of injury and dysfunction may increase.104
Gut
During hypovolemic shock, blood flow from stomach to colon is redistributed to other organs, and this may be primarily mediated by elevated sympathetic activity and increased levels of vasopressin and angiotensin II even though vascular reactivity to the latter may diminish.† Vasoconstriction may overwhelm NO and other vasodilating mechanisms, and endothelium-dependent vasodilation may be impaired after oxidant endothelial injury.194 Gut ischemia is aggravated further by the countercurrent mechanism in mucosal (villous) blood flow, promoting diffusional shunting of oxygen from arteries to veins, bypassing tissues. Other studies reported that gut mucosal blood flow may be relatively spared during hypovolemia, however.75,105 Portal blood flow decreases, and portal blood levels of lactate increase after gut ischemia.27,48,195
Gastric mucosal ischemia may result in diminished energy-consuming acid production and may predispose to mucosal stress ulceration.192,196 Microscopic studies in experimental animals show damage of gastric mucosa, villous epithelium in small bowel, and mucosa of the large bowel after hypovolemic shock.114,192,197–199 Gastric mucosal ischemia-reperfusion injury after bleeding may be aggravated by gastric acid itself, neutrophils, inflammatory mediators, endothelin, reactive oxygen species (ROS), and proteases.198,200 Bowel ischemia and mucosal damage during hypovolemic shock in the dog may ultimately lead to leakage of fluid from the bloodstream to the bowel lumen, instead of normal resorption of luminal fluids.115,197 Diarrhea may contribute to intravascular volume depletion during severe and prolonged hypovolemic shock, at least in animals.
Gut mucosal ischemia, energy depletion, injury, and inflammation may compromise the barrier function of the mucosa, enhancing the likelihood that bacteria and endotoxins in intestinal lumen (large bowel) translocate through the damaged gut wall to lymph nodes, portal venous blood, or both.* The gut epithelial (lumen to plasma) permeability for small molecules also is increased. Indigenous flora, generated toxic ROS, cytokines, Ca2+ overload, iNOS, peroxynitrite, phospholipase A2 activation, and activated and adhering neutrophils during ischemia and reperfusion probably all play a role in the injury, promoting hyperpermeability and translocation.199,203 Mucosal injury and translocation can be inhibited by compounds targeted against these factors.199 Impaired detoxifying capacity of the Kupffer cells of the liver because of ischemia or preexistent liver disease may contribute further to bacteria and endotoxins reaching the systemic circulation and contributing to progression of shock by triggering an inflammation cascade, ultimately resulting in release of vasoactive substances.4,204,205 This translocation has been shown to contribute to the lethality of hypovolemic shock in experimental animals because clearance or blockade of translocated bacteria and endotoxins is associated with survival, and germ-free animals survive an episode of bleeding more often and longer than ones with normal intestinal flora.4,143,203,205 Finally, it has been shown that the absorptive capacity of the gut for carbohydrates, amino acids, and lipids decreases during hypovolemic shock.148,195 Although enteral feeding during hypovolemic shock and after resuscitation may increase metabolic demands of the gut, there is experimental evidence that luminal application of nutrients, particularly of enterocyte-fueling glutamine, induces an increase in small vessel blood flow, ameliorates damage, and diminishes the likelihood for translocation of endotoxins and bacteria during resuscitation from hemorrhage.206
[/not-level-membership-for-critical-care-medicine-category]
