Hypoventilation and Respiratory Muscle Dysfunction
Hypercapnic respiratory failure is conventionally defined as a state in which ventilation is insufficient to maintain an arterial tension of carbon dioxide (PaCO2) of less than 45 mm Hg for the level of metabolic activity (measured by CO2 production, 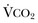 ).1 Under steady-state conditions, the relationship among PaCO2, alveolar ventilation (
).1 Under steady-state conditions, the relationship among PaCO2, alveolar ventilation (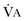 ), and
), and 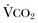 is given by the equation:
is given by the equation:
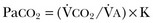
in which K (usually stated as 0.863) is a constant that converts measurement of 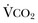 from standard conditions to body temperature conditions. The term
from standard conditions to body temperature conditions. The term 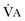 represents the portion of minute ventilation (
represents the portion of minute ventilation (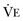 ) that reaches the terminal gas exchange units and is calculated as
) that reaches the terminal gas exchange units and is calculated as
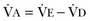
where 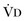 equals dead space ventilation. A reduction in
equals dead space ventilation. A reduction in 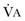 may result from an inadequate
may result from an inadequate 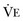 or an increase in
or an increase in 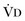 (resulting from an increase in true
(resulting from an increase in true 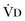 or a functional increase in
or a functional increase in 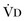 secondary to lung regions with high ventilation-perfusion [
secondary to lung regions with high ventilation-perfusion [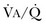 ] relationships).*
] relationships).*
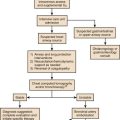
Hypercapnia is synonymous with alveolar hypoventilation for the patient’s level of carbon dioxide production. Alveolar hypoventilation can result from decreased neuromuscular capacity and increased respiratory load (Fig. 40.1).2 Isolated impairments in gas exchange—i.e., conditions in which the alveolar-arterial O2 gradient* is increased—usually do not cause hypercapnia.3 When gas exchange is impaired, however, the magnitude of neuromuscular derangements causing hypercapnia or the magnitude of increased respiratory loads causing hypercapnia is less than the corresponding derangements when gas exchange is normal. Conditions in which gas exchange contributes to hypercapnia are usually characterized by ventilation-perfusion inequality. When gas exchange is impaired total minute ventilation may actually be increased despite a concurrent decrease in alveolar ventilation.
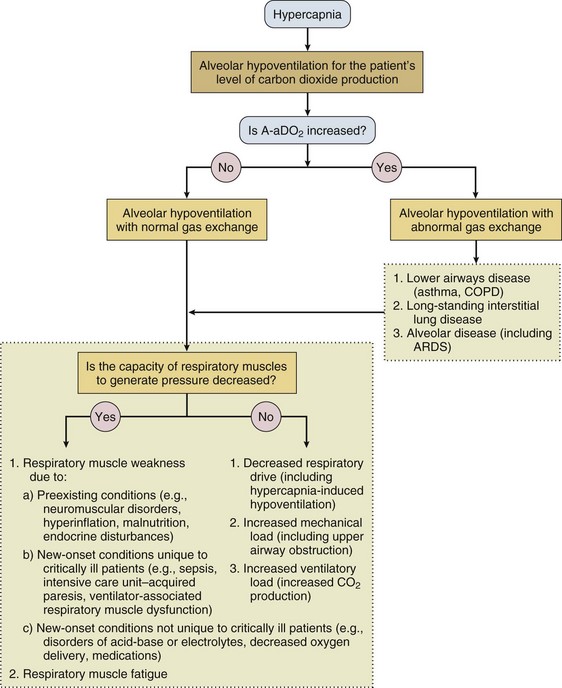
Decreased Neuromuscular Capacity
Decreased Respiratory Center Output
Isolated decreases in respiratory drive can produce ventilatory failure without distress. Potential causes include sedative overdose,1 hypothyroidism,4 metabolic alkalosis,5 semistarvation,6 and central alveolar hypoventilation syndrome. The latter can be either idiopathic (i.e., primary alveolar hypoventilation, or Ondine’s curse) or secondary to neurologic lesions, such as trauma, infection (poliomyelitis), infarction, and obesity hypoventilation syndrome.4 In mechanically ventilated patients, decreases in respiratory drive are suspected when patients develop marked increases in PCO2 despite relatively normal measurements of resistance and elastance and no apparent evidence of lower motor neuron disease.7 Whether sleep deprivation decreases respiratory drive remains controversial.8,9
Respiratory Muscle Weakness
Detection of Respiratory Muscle Weakness in Critically Ill Patients
Measurements of airway pressure during maximal voluntary inspiratory efforts are used to evaluate global inspiratory muscle strength.10 In healthy subjects, maximum inspiratory airway pressure is usually more negative than −80 cm H2O.10 In mechanically ventilated patients recovering from an episode or acute respiratory failure, maximum inspiratory airway pressure can range from less negative than −20 cm H2O to about −100 cm H2O.7,11,12 Values of maximal airway pressure during voluntary maneuvers depend greatly on a level of motivation and comprehension of the maneuver (often not obtainable in critically ill patients).13 Not surprisingly, in patients requiring short-term mechanical ventilation, measurements of maximum inspiratory airway pressure commonly do not differentiate between weaning success and weaning failure patients.12–14
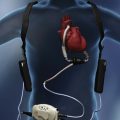
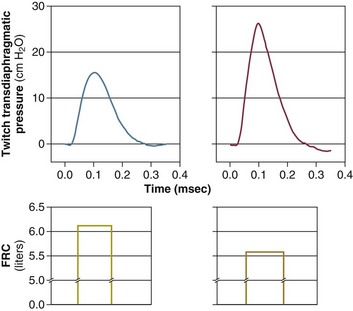
In contrast to the voluntary nature of maximal voluntary inspiratory efforts, transdiaphragmatic pressures elicited by single stimulations of the phrenic nerves—or twitch pressure—are independent of patients’ motivation and eliminate the influence of the central nervous system.10 Activation can be achieved with either an electrical stimulator15 or a magnetic stimulator,15 though the latter is easier to use in mechanically ventilated patients (Fig. 40.2).13,16,17
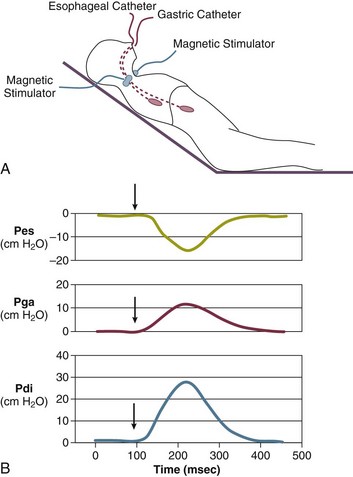
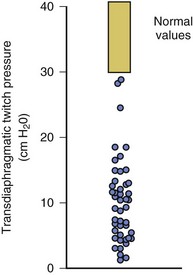
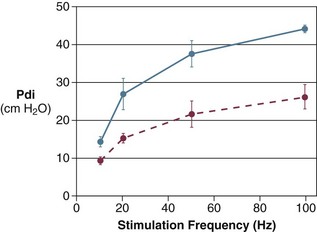
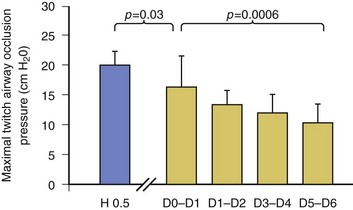
In healthy volunteers, magnetic stimulation elicits twitch pressures that average 31 to 39 cm H2O.10 In patients with severe chronic obstructive pulmonary disease (COPD), twitch pressures average 19 to 20 cm H2O.18,19 The value of transdiaphragmatic twitch pressure in patients recovering from an episode of acute respiratory failure is about one third of that recorded in healthy subjects (Fig. 40.3).20 This marked reduction in twitch pressure16,17 indicates the presence of respiratory muscle weakness in most of these patients. Respiratory muscle weakness in critically ill patients can result from preexisting conditions or from new-onset conditions.
Weakness Due to Preexisting Conditions
Neuromuscular diseases, malnutrition, endocrine disorders, and hyperinflation are some of the preexisting conditions that can cause respiratory muscle weakness. The existence of preexisting conditions can be recognized before the patient develops acute ventilatory failure, at the time the patient develops acute ventilatory failure, or when acute ventilatory failure is already established.4,21–24
Neuromuscular Disorders
The capacity of the respiratory muscles to generate tension can be decreased in certain disorders, such as stroke, amyotrophic lateral sclerosis, spinal cord injuries, poliomyelitis, Guillain-Barré syndrome, neuropathies due to massive intoxications of arsenic25 or thallium,26 chronic inflammatory demyelinating polyneuropathy, axonopathy of acute intermittent porphyria, myasthenia gravis, acid maltase deficiency, and muscular dystrophies such as myotonic dystrophy (i.e., the most frequent adult form of muscular dystrophy).4,27,28
Hypercapnic respiratory failure usually occurs when respiratory muscle strength falls to 39% of the predicted normal value.29 However, Gibson and associates30 described several patients with neuromuscular disease who had a normal partial pressure of CO2 despite decreases in respiratory muscle strength to less than 20% of predicted. Conversely, some patients with only moderate respiratory muscle weakness displayed hypercapnia (Fig. 40.4).30 In other words, reductions in muscle strength do not consistently predict alveolar hypoventilation in this setting.
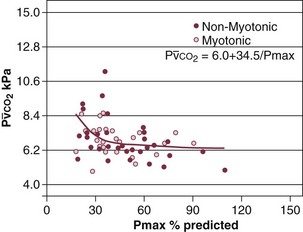
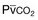 ) in patients with respiratory muscle weakness. Respiratory muscle strength is the arithmetic sum of maximum static inspiratory and expiratory mouth pressures (Pmax = P
) in patients with respiratory muscle weakness. Respiratory muscle strength is the arithmetic sum of maximum static inspiratory and expiratory mouth pressures (Pmax = P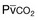 rose, although considerable variability was observed among patients. The regression lines were similar in the myotonic and nonmyotonic patients.
rose, although considerable variability was observed among patients. The regression lines were similar in the myotonic and nonmyotonic patients. Hyperinflation
Hyperinflation is a common preexisting problem in patients with obstructive lung diseases such as COPD,4 cystic fibrosis,31 bronchiolitis,32 and lymphangioleiomyomatosis.4 The severity of preexisting hyperinflation commonly worsens in patients experiencing an exacerbation of COPD.33 Hyperinflation has a number of adverse effects on inspiratory muscle function: the inspiratory muscles operate at an unfavorable position of the length-tension relationship (Fig. 40.5);34 flattening of the diaphragm decreases the size of the zone of apposition with the result that diaphragmatic contraction causes less effective rib cage expansion.4 Hyperinflation also has an adverse effect on the elastic recoil of the thoracic cage.4 This means that the inspiratory muscles must work not only against the elastic recoil of the lungs but also against that of the thoracic cage. The functional consequences of dynamic hyperinflation are probably the main mechanisms of ventilatory failure in patients with COPD.35 Hyperinflation can also occur de novo in patients with pneumonia, chest trauma, and acute respiratory distress syndrome (ARDS).33,36 Impairment of inspiratory muscle function is less likely in patients with ARDS than in patients with obstructive lung disease because patients with ARDS breathe at a low lung volume despite dynamic hyperinflation.36,37
Malnutrition
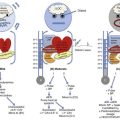
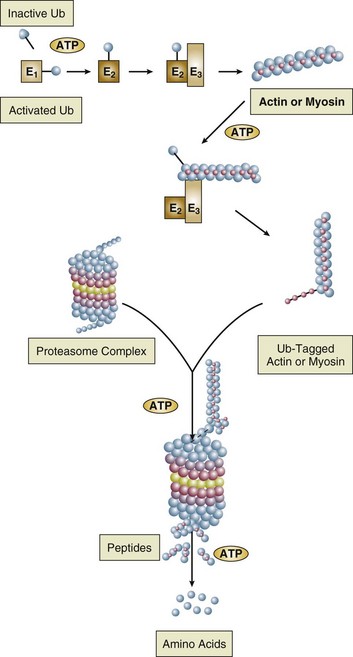
Malnutrition is highly prevalent among critically ill patients requiring mechanical ventilation,38,39 and it is associated with poor prognosis.39 Malnutrition decreases muscle mass and respiratory muscle strength both in humans40,41 and in laboratory animals.42–44 Likely mechanisms contributing to decrease in respiratory muscle mass include proteolysis of myofibrillar proteins by the ubiquitin-proteasome proteolytic system (Fig. 40.6) and apoptosis.4,45 Apoptosis can be triggered by local and circulating tumor necrosis factor-α and by release of cytochrome c from the mitochondria.46,47
In patients with COPD, inspiratory muscle strength is about 30% less in poorly nourished patients than in well-nourished patients with equivalent airway obstruction.48 Similarly, malnourished patients with anorexia nervosa can present with reduced inspiratory muscle strength to 35% to 50% predicted,41 impaired respiratory muscle endurance,49 impaired hypercapnic ventilatory response,49 and occasionally, with hypercapnia at rest.41 In malnourished patients, inspiratory weakness,41,48,49 fatigability,48 and dyspnea48 are partially reversible with nutritional support. The process is slow and, in laboratory animals, can take months of refeeding for muscle mass to return to normal values.50 To date, it remains unclear whether malnutrition by itself can cause sufficient respiratory muscle weakness to produce hypoventilation. It is more likely for malnutrition to be a contributory factor and not a sole cause of hypercapnic respiratory failure.
Endocrine Disturbances
Endocrine disturbances such as hypothyroidism,51 hyperthyroidism,47,52–54 and acromegaly55 can adversely affect respiratory muscle function. Proteolysis of myofibrillar proteins by the ubiquitin-proteasome proteolytic system45 (see Fig. 40.6) is probably responsible for respiratory muscle catabolism and weakness of hyperthyroidism.47 This mechanism is implicated in muscle wasting associated with acidosis, renal failure, denervation, cancer, diabetes, acquired immunodeficiency syndrome (AIDS), trauma, and burns.45 In contrast to other endocrine disturbances, respiratory muscle weakness is unusual in patients with Cushing syndrome.56
Weakness Due to New-Onset Conditions
Ventilator-Associated Respiratory Muscle Dysfunction
Since the late 1980s, several groups have studied the effect of mechanical ventilation on the muscles of laboratory animals.57 A seminal study by Anzueto and colleagues58 showed that 11 days of controlled mechanical ventilation (CMV) together with neuromuscular blocking agents produced a 46% decrease in respiratory muscle strength (Fig. 40.7). Subsequent studies have revealed that complete cessation of diaphragmatic activity with CMV—alone59 or in combination with neuromuscular blocking agents60—results in atrophy and injury of diaphragmatic fibers (Fig. 40.8). Muscle fibers generate less force in response to stimulation, not simply because of their decreased bulk but even when normalized for cross-sectional area.57 The decrease in diaphragmatic force ranges from 20% to more than 50%. The alterations in muscle function occur rapidly, within 12 hours of instituting mechanical ventilation,61 and they appear to increase as ventilator duration is prolonged.62,63

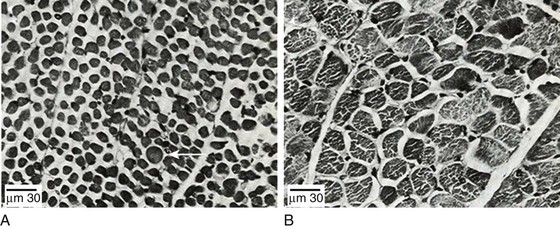
Mechanical ventilation may also harm the respiratory muscles of patients. In 1988, Knisely and coworkers64 reported that the cross-sectional area of diaphragmatic fibers in infants who had died after receiving mechanical ventilation for 12 or more days was much smaller than that in infants who had received mechanical ventilation for 7 days or less (Fig. 40.9). Fibers taken from extradiaphragmatic muscles were similar in the two groups. Twenty years later, Levine and associates65 obtained biopsies of the diaphragm from 14 brain-dead organ donors who were maintained on CMV for 18 to 69 hours. They also obtained intraoperative biopsies of the diaphragms of eight control patients who had received CMV for 2 to 3 hours. Histologic measurements revealed marked diaphragmatic atrophy in the brain-dead patients. Compared with the control group, the mean cross-sectional areas of muscle fibers were decreased by more than 50%. A number of these results have been corroborated by Jaber and colleagues63 and Hermans and coworkers,66 who also reported a progressive decrease in diaphragmatic contractility of mechanically ventilated patients. The decrease in contractility correlated with the duration of ventilator support (Fig. 40.10).63,66
Biochemical and gene-expression studies in animals and humans65,67 suggest that oxidative stress is probably one of the most proximal mechanisms in the biochemical cascade that leads to ventilator-induced muscle injury.68 Oxidative stress decreases contractility by causing protein oxidation and by promoting protein catabolism68—including up-regulation of the autophagy-lysosome pathway.69 The synergism between ventilator-induced muscle injury and oxidative stress has caused investigators to explore whether early administration of antioxidants might minimize muscle injury.70–72 In a study of more than 200 critically ill patients—80% of whom required acute ventilator support—duration of mechanical ventilation was nearly 3 days shorter in those who completed a 10-day antioxidant supplementation protocol (vitamin E and vitamin C) than in those who completed a 10-day course of placebo.73 Similar results have been reported in critically ill surgical patients requiring mechanical ventilation.74 Whether the decrease in duration of mechanical ventilation was, at least in part, due to the potential positive effects of antioxidants on the respiratory muscles remains to be demonstrated. Of concern, however, was the report that administration of the antioxidant N-acetylcysteine (NAC) to critically ill patients with severe sepsis worsened sepsis-induced organ failure.75
In animal models, ventilator settings can affect the extent of ventilator-associated respiratory muscle dysfunction.76–79 In rabbits, assist-control mechanical ventilation causes a nonsignificant decrease in diaphragm muscle contractility, which contrasts with the 48% decrease recorded with CMV.76 Similar results have been obtained when comparing CMV to adaptive support ventilation in piglets78 or when comparing CMV to pressure support in rats79 or CMV to intermittent spontaneous breathing again in rats.77 These observations raise the important question of whether maintenance of partial diaphragmatic activity or intermittent loading of the diaphragm could prevent the harm done to diaphragmatic function by mechanical ventilation.80
Considering that decrease in protein synthesis probably contributes to ventilator-associated respiratory muscle dysfunction,81,82 it would seem biologically plausible that administration of anabolic factors—such as growth hormone—might be of benefit in ventilated patients. Unfortunately, when growth hormone has been administered to patients requiring prolonged mechanical ventilation, duration of mechanical ventilation was not decreased, nor was muscle strength increased.83 Of concern was the report that recombinant growth hormone can increase the mortality rate of critically ill patients.84 An even more fundamental point in regard to ventilator-associated respiratory muscle dysfunction has been raised recently by the preliminary data of Hooijman and associates.85 These investigators measured the contractile properties of single diaphragmatic fibers obtained from nine brain-dead organ donors who had received CMV for 30 to 84 hours (case subjects) and from nine patients undergoing surgery for localized lung cancer (control subjects). In this preliminary study no difference was noted between the muscle fibers of the two groups of patients with respect to maximum force per unit area, calcium sensitivity of contractile force, and rate constant of redevelopment of force (Ktr). How to reconcile the preliminary results of Hooijman and associates85 with previous data obtained on muscle strips from animal models and by phrenic nerve stimulation in patients remains to be determined.
Sepsis-Associated Myopathy
Sepsis, a common occurrence in critically ill patients, can produce ventilatory failure by causing respiratory muscle dysfunction and increased metabolic demands.86 Septic animals develop failure of neuromuscular transmission (due to increased sarcolemmal electric potential)87–89 and failure of excitation-contraction coupling.86,90 Mechanisms responsible for failure of excitation-contraction coupling include the cytotoxic effect of nitric oxide and its metabolites, free radicals, ubiquitin-proteasome proteolysis, and possibly, decrease in nicotinic acetylcholine receptors.91,92 Local dysregulation of the circulation and Krebs cycle may also contribute.86
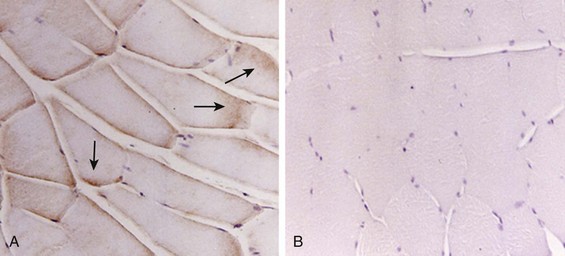
Nitric oxide, a free radical that has a negative inotropic effect in the heart and skeletal muscle, is produced in large amounts during sepsis by a nitric oxide synthase inducible by lipopolysaccharide and several cytokines (Fig. 40.11).93 Increased expression of inducible nitric oxide synthase in the diaphragm during sepsis is associated with morphologic evidence of widespread damage to the myofiber membrane, or sarcolemma (Fig. 40.12).88 Diaphragmatic contractions enhance this sepsis-induced sarcolemmal injury,94 while early mechanical ventilation decreases sarcolemmal injury and the associated diaphragmatic dysfunction.95 The beneficial effects of resting the diaphragm with the use of mechanical ventilation are not coupled with a decrease in oxidative stress or with a decrease in the expression of inducible nitric oxide synthase in the muscle.95 These observations suggest the existence of a detrimental interaction of two independent stressors (oxidative and biomechanical stresses) on the sarcolemma during sepsis.95
To determine whether the inducible nitric oxide synthase pathway contributes to impaired skeletal muscle contractility in humans, Lanone and coworkers96 obtained samples of the rectus abdominis in 16 septic patients and 21 control subjects. The muscles of the patients had lower contractile force, and increases in inducible nitric oxide synthase expression (mRNA and protein) and activity. Immunohistochemical studies revealed the generation of peroxynitrite (a highly reactive oxidant formed by the reaction of nitric oxide with superoxide anion). Exposure of control muscles to the amount of peroxynitrite found in patients caused an irreversible decrease in force generation. These data suggest that sepsis decreases muscle force through the production of nitric oxide and its toxic byproducts.
Production of nitric oxide in sepsis may be protective and not solely deleterious.94,97–99 In mice deficient in inducible97 or constitutive (neuronal) nitric oxide synthase,94 endotoxin caused a greater decline in diaphragmatic contractility than in nondeficient mice. This finding contrasts with the observation that nitric oxide synthase inhibitors prevent muscle dysfunction in septic rats.88,93,100 Although the results may be species-dependent,94 the data underscore that nitric oxide has both antioxidant and pro-oxidant actions.101
In addition to nitric oxide and its derivatives, several other oxygen-derived free radicals (superoxide anion, hydroxyl radicals, hydrogen peroxide) contribute to the decreased contractility of the diaphragm in sepsis.99,102–104 This increased expression of oxygen-derived free radicals in sepsis is accompanied with enhanced activity of the antioxidant enzyme superoxide dismutase102 and with increased expression of the heme oxygenase-1 pathway.104 The heme oxygenase-1 pathway is a powerful cellular system that protects against oxidative stress and contractile fatigue during sepsis.104 Administration of an inhibitor (zinc protoporphyrin IX) or an inducer (hemin) of heme oxygenase activity respectively enhances or reduces the oxidative stress and contractile failure of the diaphragm in a rat model of sepsis.104 In septic rats, decreased diaphragmatic contractility can also be improved by the administration of specific scavengers of superoxide ions, hydrogen peroxide, and hydroxyl radicals.102
Intensive Care Unit–Acquired Paresis
While cared for in the ICU, critically ill patients can develop muscle weakness and, occasionally, paralysis. Some of these patients have evidence for axonal degeneration and denervation atrophy (Fig. 40.13).4 This constellation of findings is known as critical illness polyneuropathy (Table 40.1).105 Cytokines106 and low-molecular-weight neurotoxins107 released during episodes of sepsis or when patients develop multiple organ failure are thought to be responsible for this axonal degeneration. Critical illness polyneuropathy has been considered one of the manifestations of multiple organ failure syndrome. Sepsis and multiple organ failure, though, are not essential prerequisites for the development of critical illness polyneuropathy.108,109 Tight control of hyperglycemia may reduce the risk of polyneuropathy and the duration of mechanical ventilation.110
Table 40.1
Examples of Electromyography (EMG) Findings in Respiratory Muscle Weakness
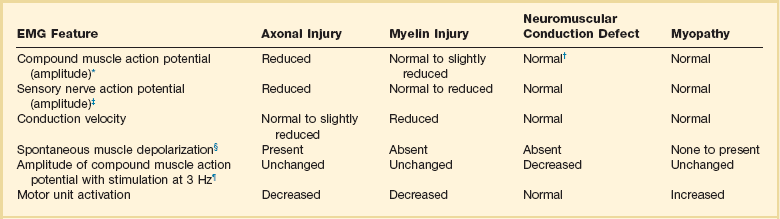
*Elicited by motor nerve stimulation.
†Decreased in Lambert-Eaton syndrome.
‡Elicited by sensory nerve stimulation.
§Spontaneous muscle depolarization (caused by denervation) is detected by presence of fibrillation potentials and positive sharp waves.
¶Repetitive nerve stimulation is performed to exclude neuromuscular transmission defects such as prolonged neuromuscular paralysis.
From Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10.
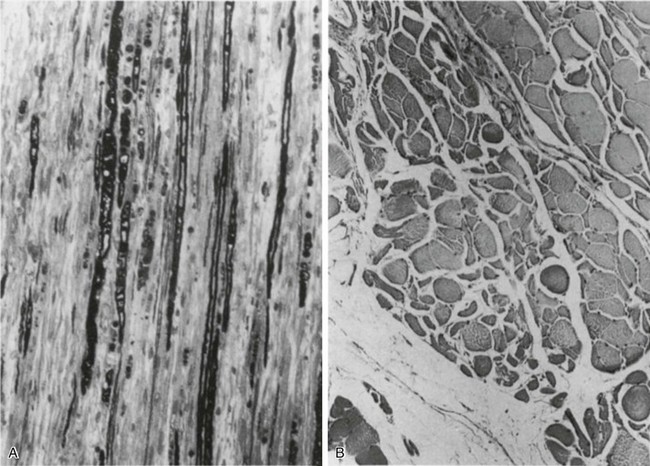
In other patients, rather than axonopathy, there is evidence of isolated myopathy (i.e., critical illness myopathy).4 Patients developing isolated myopathy often have been treated with steroids and neuromuscular blocking agents (e.g., patients with status asthmaticus).4 Muscle biopsies demonstrate a general decrease in myofibrillar protein content and a selective loss of thick filaments (myosin) within type I and type II fibers (Table 40.2 and Fig. 40.14). Animal models of critical illness myopathy suggest that medical denervation with paralytic agents cause an up-regulation of glucocorticoid receptors in the muscle.111 If the animal subsequently receives high-dose corticosteroids, it will develop depletion of thick myosin filaments.111 Although a decrease in thick-filament proteins may be important for prolonged weakness,112 this decrease is probably not the cause of the acute paralysis,113 particularly in patients with compound motor action potentials of low amplitude.114 Impaired muscle membrane excitability is probably more important during the acute stage.113,115
Table 40.2
Characteristics of Types of Muscle Fibers
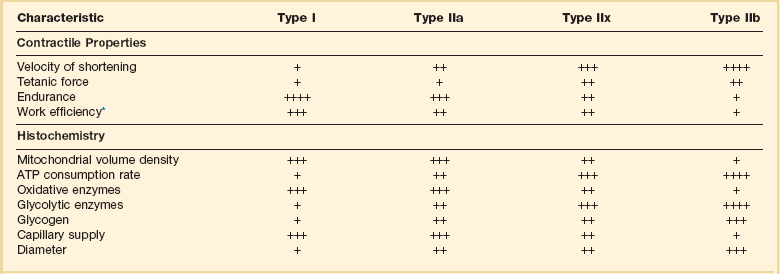
A single myosin heavy chain isoform typically is expressed within an adult skeletal muscle fiber. Fibers classified as type I, IIa, IIx, or IIb express myosin heavy chain isoform I (or slow), IIa, IIx, or IIb, respectively. Type IIx fibers have been reported in peripheral muscles of humans and animals and in the diaphragm of animals. Type IIx fibers have not been reported in the human diaphragm. More than one myosin heavy chain isoform is expressed in a few fibers (about 14% of adult rat diaphragm coexpresses myosin heavy chain isoforms IIb and IIx, and less than 1% coexpresses myosin heavy chain isoforms I and IIa).226 Whereas the velocity of muscle contraction depends primarily on the myosin heavy chain isoform, the velocity of muscle relaxation is mainly determined by troponin C calcium binding and release and by calcium reuptake by the sarcoendoplasmic reticulum calcium-adenosine triphosphatase (SERCA). Several SERCA isoenzymes have been identified: SERCA 1 is expressed in type II fibers (fast calcium reuptake), and SERCA 2a is expressed in type I fibers (slow calcium reuptake).227 The density of pumping sites largely accounts for different rates of calcium uptake in fast- and slow-twitch muscle fibers.227 Despite this separation of tasks, velocity of contraction and velocity of relaxation tend to parallel each other: type II fibers contract and relax with a greater velocity than type I fibers. Slower velocity of relaxation allows fusion of repetitive twitches at lower frequencies of stimulation than with fast relaxations. Impairment of SERCA activity has been implicated in the development of fatigue and in disease states including heart failure and corticosteroid myopathy.
*Amount of work performed per unit of adenosine triphosphate (ATP) consumed.
From Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10.
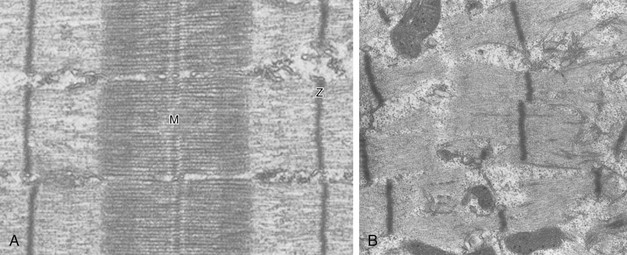
In the last few years it has become increasingly apparent that critical illness neuropathy and myopathy often coexist.106,109,115–118 It has become common to refer to patients who become weak while in the ICU as a result of acquired neuropathy or myopathy (not associated with a known disorder) as simply having ICU-acquired paresis.115,116,118 In patients with ICU-acquired paresis duration of weaning from mechanical ventilation can be two to seven times longer than in patients without ICU-acquired paresis.119
The functional outcome of ICU-acquired paresis is not uniform. Approximately 50% to 60% of patients experience complete recovery (ability to breathe spontaneously and to walk independently) over a period of 2 weeks to 6 months or longer.112,119,120 About 30% experience severe persistent disability with tetraparesis, tetraplegia, or paraplegia.119 Other investigators report even worse outcome: only 2 of 10 patients left the hospital in one study.117 Whether it is possible to prevent ICU-acquired paresis in patients recovering from severe acute illness, and whether that would result in shorter duration of mechanical ventilation remains unknown.
Acid-Base Disorders
Alkalosis, either metabolic or respiratory, does not affect skeletal muscle strength121–123 and might improve endurance.121 Whether acidosis, either metabolic or respiratory, impairs respiratory muscle function remains controversial. Because the contractile response to metabolic and respiratory acidosis is not necessarily the same,124,125 metabolic acidosis and respiratory acidosis will be discussed separately.
Metabolic Acidosis.
Until recently, there was little doubt that metabolic acidosis could decrease muscle contractility.126,127 Purported mechanisms included reduction of actin-myosin cross-bridge activation by H+ competitive inhibition of Ca2+ binding to troponin-C, reduced transition of actin-myosin cross-bridges from low- to high-force state, inhibition of myofibrillar adenosine triphosphatase (ATPase), inhibition of glycolytic rate, inhibition of maximal shortening velocity, and inhibition of sarcoplasmic ATPase with reduction of sarcoplasmic Ca2+ reuptake leading to reduced Ca2+ release from the sarcoplasmic reticulum.128 Despite the biologic plausibility, investigators have reported no effect125,129 or marginal effect130 of metabolic acidosis on respiratory muscle function. Yanos and colleagues125 assessed maximal transdiaphragmatic pressure elicited by tetanic stimulation of the phrenic nerves in seven anesthetized dogs during respiratory acidosis (pH 7.1) and during lactic acidosis (pH 7.1). They observed a fall in maximal diaphragm strength with respiratory acidosis (−18%, p < 0.05), but not with lactic acidosis (+3%). Similarly, when Coast and associates130 exposed isolated diaphragm strips of rats to different concentrations of lactic acid they recorded decreases in force when the pH was lowered to 6.8—but only after the muscle strips were stressed with 75 contractions (25 Hz, 250-ms train duration, one train per second). Contractility was not affected when the pH was decreased to ~7.0 at rest, and following the set of 75 contractions when delivered at a pH of 7.1 and 7.2.130 These negative results are in line with some—but not all131,132—recent investigations questioning the inhibitory role of metabolic acidosis on limb muscle contractility at physiologic temperatures.133–136 For instance, Nielsen and colleagues136 reported that metabolic acidosis (pH 6.80) counteracts the detrimental effects of increased extracellular K+ concentrations (which occur with forceful contractions) on the excitability and force generation of the rat soleus muscle. In humans, Degroot and coworkers134 measured intracellular H+ concentration during sustained isometric foot plantar flexion. In contrast to what would be expected if acidosis caused decrease in contractility, Degroot and coworkers134 reported a decrease in intracellular H+ concentration during the first 10 seconds of exercise when force was declining and a rise in intracellular H+ concentration immediately after exercise, when force partially recovered.134 Whether severe metabolic acidosis in humans might impair skeletal function by causing a reduced central nervous system drive remains to be demonstrated.126
Acute Respiratory Acidosis.
There are contrasting reports on the effect of acute respiratory acidosis on respiratory muscle contractility.122,123,125,137 In anesthetized dogs, diaphragmatic contractility decreases pari passu with the severity of acute respiratory acidosis122 (but has no effect on gastrocnemius muscle contractility).125 Similarly, in healthy volunteers, acute increases of arterial carbon dioxide to 54 mm Hg (corresponding to a pH of about 7.29) reduces the capacity of the unfatigued diaphragm to generate pressure by 10% to 30% (Fig. 40.15).123 Such reduction in pressure generation is even greater when arterial carbon dioxide is increased to 63 mm Hg (corresponding to a pH of about 7.22).123 Acute respiratory acidosis can decrease respiratory muscle endurance,123,138 and it can increase the extent of diaphragmatic fatigue at the conclusion of 2 minutes of maximal voluntary ventilation.139 Despite greater voluntary activation of the diaphragm,140 hypercapnic patients with COPD generate lower maximal static inspiratory pressures than patients with normocapnia.141,142
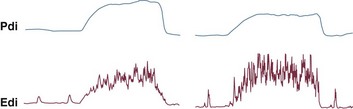
A direct inhibitory effect of acute respiratory acidosis122,123,125,138,139 on respiratory muscle contractility and respiratory muscle endurance could provide a potential mechanism for the rapid clinical deterioration that can occur with severe asthma and during COPD exacerbations.123 Yet, the human data suggesting a direct deleterious effect of acute respiratory acidosis on respiratory muscle function123,138,139,141 is not uniform. Some investigators report no change in diaphragmatic contractility137,139 (but decrease in force of the adductor pollicis)137 when acute respiratory acidosis causes a decrease in pH to about 7.16 to 7.27,137,143 no change in the maximum relaxation rate of the diaphragm,143 and no effect in the extent of diaphragmatic fatigue 20 to 90 minutes after loading.139
For several reasons it is difficult to reconcile the conflicting data on the effects of acute respiratory acidosis on respiratory muscle contractility reported in the literature. First, the nonsignificant decrease in diaphragmatic contractility of 12% (twitch pressure) recorded during acute hypercapnia in 12 healthy subjects in one study137 raises the possibility of type II error. Second, it is impossible to state that more severe acidosis was responsible for the different results in human investigations considering the comparable pH values of the negative137 and positive123 studies. Third, it is unlikely that different frequencies of stimulation used to assess the respiratory muscles123,137,139 are responsible for the contrasting results because, when present, acidosis-associated decrease in contractility is frequency independent.122 Moreover, three additional considerations further cloud our understanding of any interaction (if present) between acute respiratory acidosis and respiratory muscle contractility. First, respiratory acidosis (pH 6.50-6.88) may actually enhance and not depress skeletal muscle contractility.136,144 Second, it is unknown whether comorbid conditions, which often affect hypercapnic patients, such as sepsis, decreased cardiac function, and impaired respiratory mechanics could act synergistically with acute hypercapnia in worsening respiratory muscle function. Third, it is unknown whether equivalent levels of pH occurring as a result of acute- or acute-on-chronic respiratory acidosis may—or may not—have the same effect on respiratory muscle function.
Electrolyte Disturbances
Respiratory muscle function may be impaired by decreased levels of phosphate,145 calcium,146 magnesium,147 and potassium.148 Aubier and associates145 studied the effects of severe hypophosphatemia (0.55 ± 0.18 mmol/L) on diaphragmatic function in eight patients with acute respiratory failure who were mechanically ventilated. Diaphragmatic function was quantified before and after phosphorus replacement therapy by recording transdiaphragmatic twitch pressure elicited by phrenic nerve stimulation. Correction of hypophosphatemia was accompanied by a significant increase in transdiaphragmatic twitch pressure—i.e., from 9.8 ± 3.8 cm H2O before phosphate infusion to 17.3 ± 6.5 cm H2O after phosphate infusion (p < 0.001). Changes in the serum phosphorus level and transdiaphragmatic pressure were well correlated (r = 0.73). These results strongly suggest that hypophosphatemia can impair the contractile properties of the diaphragm during acute respiratory failure.
The same investigators assessed the effects of hypocalcemia on diaphragmatic function in 12 anesthetized dogs.146 A continuous infusion of the chelating agent ethylene glycol-bis(2-aminoethylether)-N,N′-tetraacetic acid (EGTA) was used to produce hypocalcemia. Over the 2 hours of observation, a progressive reduction in diaphragmatic force production paralleled the progressive reduction in ionized serum calcium.
Hypomagnesemia, a common occurrence in the ICU, can also cause respiratory muscle weakness. Dhingra and colleagues147 reported improvements in maximal inspiratory and expiratory pressures in 17 hypomagnesemic patients following magnesium replacement therapy but not following placebo therapy.
Severe hypokalemia can be caused by several disorders including posthypercapnic alkalosis, renal tubular acidosis, primary hyperaldosteronism, gastrointestinal potassium losses, use of diuretics, thyrotoxic periodic paralysis, familial hypokalemic paralysis, β2-adrenergic agonists, and licorice ingestion. When the serum potassium concentration decreases below 2.0 to 2.5 mEq/L, patients may develop muscular weakness (including respiratory muscle weakness) and arrhythmias. Hypokalemia-associated respiratory muscle weakness can result in respiratory failure and death.148
Decreased Oxygen Delivery
Laboratory animals with cardiogenic shock149 or with septic shock86,150 die of respiratory failure. Death is not caused by pulmonary disease per se but by an inability of the respiratory muscles to maintain adequate ventilation. This inability to maintain adequate ventilation is caused by insufficient oxygen delivery to the respiratory muscles.4
Whether the decrease in oxygen delivery to the respiratory muscles—or the respiratory centers—during shock is sufficient to decrease respiratory muscle performance and cause hypoventilation in patients, although likely,151 remains to be determined. A nonrandomized study by Kontoyannis and coworkers152 in 28 patients with cardiogenic shock provides support for the view that hemodynamic instability could decrease respiratory muscle performance in patients with shock. Compared with nonventilated patients, ventilated patients were weaned from an intra-aortic balloon pump more often, and their survival was greater.152 These results are in line with the observation of Viires and associates153 who reported decreased metabolic requirements of the diaphragm following institution of mechanical ventilation in dogs with cardiogenic shock.
Some hemodynamic situations that are less extreme than shock could still affect respiratory muscle performance, including failed attempts of spontaneous respiration during weaning from mechanical ventilation. Jubran and colleagues154 investigated the importance of hemodynamic performance in determining weaning outcome in 8 ventilator-supported patients who failed a trial of spontaneous breathing and 11 patients who tolerated a trial and were successfully extubated. Immediately before the trial, mixed oxygen venous saturation was not different between the two groups. Mixed venous oxygen saturation progressively decreased over the course of the trial in the failure group, whereas it remained unchanged in the success group (Fig. 40.16, upper panel). Although the calculated oxygen demand was similar in the two groups, the manner in which it was met differed between them. In the success group, oxygen transport increased, mainly resulting from an increase in cardiac index; in the failure group, the increase in demand was met by an increase in oxygen extraction, resulting in a decrease in mixed venous oxygen saturation (Fig. 40.16, lower panel). Although increased, the oxygen extraction ratio at the end of the trial in the failure group was close to the ratio reported to signify the onset of anaerobic metabolism (0.60)155 in only two patients, who had ratios of 0.50 and 0.56, respectively. The ability of the failure group to deal with respiratory muscle energy demands through aerobic pathways is probably related to the capacity of the diaphragm to achieve higher blood flow than most other skeletal muscles.156 During loading, the diaphragm rapidly increases oxygen extraction to a plateau of about 55% to 65%; further increases in oxygen demand are achieved by increases in blood flow.157,158 The diaphragmatic musculature appears to be extremely resistant to hypoxic stress, and animals can maintain a ventilation that is sufficient to avoid hypercapnia until phrenic vein PO2 falls to 12 mm Hg.156 Likewise, investigators have found that an oxygen tension of about 10 mm Hg in the phrenic vein is the threshold associated with the onset of diaphragmatic lactate production159 and the development of fatigue.160 The lowest mixed venous oxygen tension recorded by Jubran and colleagues154 was 26 mm Hg, which is above the threshold for onset of diaphragmatic lactate production. This association needs to be interpreted with caution, however, because mixed venous blood contains effluents from many tissue beds other than the diaphragm. The investigation of Jubran and colleagues154 is the first documentation that, when challenged by an increase in mechanical load,7 the respiratory muscles of critically ill patients do not appear to switch from aerobic to anaerobic metabolism.
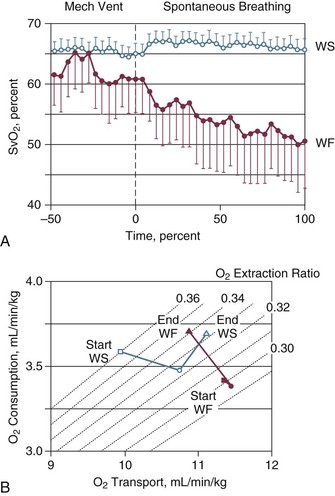
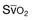 ) during mechanical ventilation and a trial of spontaneous breathing in 11 weaning success patients (WS, open symbols) and in 8 weaning failure patients (WF, closed symbols). During mechanical ventilation,
) during mechanical ventilation and a trial of spontaneous breathing in 11 weaning success patients (WS, open symbols) and in 8 weaning failure patients (WF, closed symbols). During mechanical ventilation, 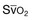 was similar in the two groups (p = 0.28). Between the onset (dashed line) and the end of the trial,
was similar in the two groups (p = 0.28). Between the onset (dashed line) and the end of the trial,  decreased in the failure group (p < 0.01), whereas it remained unchanged in the success group (p = 0.48). Over the course of the trial,
decreased in the failure group (p < 0.01), whereas it remained unchanged in the success group (p = 0.48). Over the course of the trial,  was lower in the failure group than in the success group (p < 0.02). Bars indicate SE (standard error). B, Oxygen transport, oxygen consumption, and isopleths of oxygen extraction ratio in the success (WS, open symbols) and failure (WF, closed symbols) groups during mechanical ventilation (squares) and at the onset (circles) and end (triangles) of a spontaneous breathing trial. See text for details.
was lower in the failure group than in the success group (p < 0.02). Bars indicate SE (standard error). B, Oxygen transport, oxygen consumption, and isopleths of oxygen extraction ratio in the success (WS, open symbols) and failure (WF, closed symbols) groups during mechanical ventilation (squares) and at the onset (circles) and end (triangles) of a spontaneous breathing trial. See text for details. High variability in hemodynamic response during failure to wean has been reported by Zakynthinos and coworkers.14 Similar to Jubran and colleagues,154 Zakynthinos and coworkers14 recorded a decrease in mixed venous oxygen saturation and an increase in oxygen consumption (met mainly by an increase in oxygen extraction) in 9 of 18 weaning failure patients. In the remaining 9 failures, however, mixed venous oxygen saturation and oxygen consumption were not affected by the weaning trial. It is unclear whether the absent interaction between weaning failure and oxygen consumption was due to depression of the respiratory centers, limited capacity to extract oxygen, or limited cardiac reserve.161
Medications
Weakness can result from medications that have a direct myotoxic effect, such as blockade of myocyte glycoprotein synthesis and electron transport caused by inhibitors of the hydroxymethylglutaryl coenzyme A reductase or nucleoside analogs used in patients with human immunodeficiency virus.162–165 Weakness can also result with neuromuscular blocking agents and aminoglycosides, which interfere with neuromuscular transmission.166,167
Paralysis, including the respiratory muscles, can persist after discontinuation of neuromuscular blocking agents.167–169 Prolonged neuromuscular blockade has been defined as 2,167 4,169 or 6 hours168 of paralysis after discontinuation of neuromuscular blocking agents. Prolonged blockade is estimated to occur in 12% to 44% of patients receiving pancuronium or vecuronium for 1 or more days.167–169 The risk with vecuronium is increased in patients with renal failure.167,169 Accumulation of metabolites of the neuromuscular blocking agents is responsible for the prolonged blockade.167 Recovery from prolonged neuromuscular blockade begins within 2 days of the last dose,167,168 and this contrasts with the prolonged course of critical illness myopathy or neuropathy.112,120,170,171 Train-of-four monitoring of the dose of a neuromuscular blocking agent with a peripheral nerve stimulator may hasten recovery.169
Limitations in the Current Classification of Respiratory Muscle Weakness
1. The distinction between preexisting conditions and new-onset conditions can be arbitrary.
2. Conditions that are preexisting (malnutrition and hyperinflation) can worsen during the course of an unrelated critical illness.
3. The nosologic designation is often unsatisfactory—see the nebulous distinction between ICU-acquired paresis and sepsis-associated myopathy or between ICU-acquired paresis and ventilator-associated respiratory muscle dysfunction.
4. Conditions in which respiratory muscle weakness is associated with muscle damage can display also some degree of muscle atrophy—see diaphragmatic atrophy in cases of ventilator-associated respiratory muscle dysfunction.
5. Laboratory specificity to differentiate the various conditions causing weakness in the ICU is limited.
6. In any given patient more than one mechanism may be responsible for respiratory muscle weakness.
7. Respiratory muscle weakness can be combined with depressed drive—for example, in the setting of hypercapnia-induced hypoventilation.
Respiratory Muscle Fatigue
Contractile fatigue occurs when a sufficiently large respiratory load is applied over a sufficiently long period.15,172–180 Contractile fatigue can be brief or prolonged. Short-lasting fatigue results from accumulation of inorganic phosphate,135,181,182 failure of the membrane electrical potential to propagate beyond T tubules,183 and to a much lesser extent intramuscular acidosis.184–186 Short-lasting fatigue appears to have a protective function, because it can prevent injury to the sarcolemma caused by forceful muscle contractions.187 Long-lasting fatigue173 is consistent with the development of, and recovery from, muscle injury (Fig. 40.17).187,188
Whether critically ill patients develop short-lasting or long-lasting contractile fatigue of the respiratory muscles is not clear. Patients who fail a trial of weaning from mechanical ventilation are at particular risk of developing fatigue because they experience marked increases in respiratory load.7,11,33 The addition of a new injury to the respiratory muscles (secondary to the development of contractile fatigue) might be the ultimate determinant of whether or not some patients are ever successfully weaned. Circumstantial evidence of contractile fatigue in patients experiencing respiratory distress has been reported.7,189–191 Because of technical limitations,189–191 these early data did not provide proof of contractile fatigue.10,192
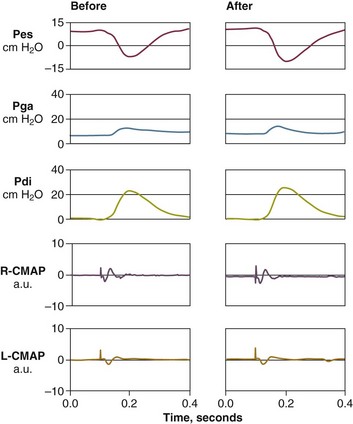
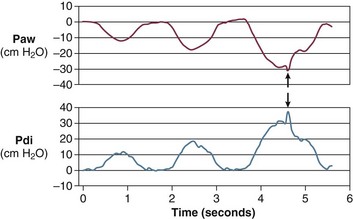
The most direct method for detecting fatigue in patients is to measure the transdiaphragmatic pressure (Pdi) elicited by phrenic nerve stimulation over time (see Fig. 40.2).15,173 When employing this technique in critically ill patients, it is especially challenging to ensure that successive stimulations are all generated at the same end-expiratory lung volume, to ensure a constant degree of neural depolarization by the stimulator, and ensure that twitch potentiation (the transient increase in pressure that occurs with a recent forceful contraction) does not occur.15,173 Controlling for these factors, Laghi and associates13 studied 11 weaning-failure patients and 8 weaning-success patients before and after a T-tube trial. Twitch Pdi was 8.9 ± 2.2 H2O before and 9.4 ± 2.4 H2O after the trial in the weaning-failure patients (Fig. 40.18). The respective values in the weaning-success patients were 10.3 ± 1.5 and 11.2 ± 1.8 cm H2O. Not a single patient developed a decrease in twitch Pdi. The absence of fatigue was surprising because 7 of the 9 weaning-failure patients had a tension-time index (the product of two fractions: [mean pressure per breath/maximum transdiaphragmatic pressure] × [inspiratory time/total time of respiratory cycle]) above the threshold reported to lead to task failure and fatigue (0.15).193
One of the most likely reasons that patients did not develop fatigue is that physicians reinstituted mechanical ventilation before there was enough time for its development. The relationship between tension-time index and the length of time that a load can be sustained until task failure follows an inverse-power function. Bellemare and Grassino193 expressed the relationship as follows:
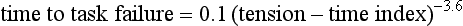
The increase in tension-time index over the course of the weaning trial13 and predicted time to task failure193 are shown in Figure 40.19. At the point that the physician reinstituted mechanical ventilation, patients were predicted to be an average of 13 minutes away from task failure. Moreover, the time to task failure was underestimated because diaphragmatic recruitment during maximal voluntary contractions was incomplete (Fig. 40.20).13 In other words, patients display clinical manifestations of severe respiratory distress for a substantial time before they would develop fatigue. In an ICU setting, these clinical signs will lead attendants to reinstitute mechanical ventilation before fatigue has time to develop. Other factors that might have protected the respiratory muscles from contractile fatigue include development of dynamic hyperinflation (susceptibility to fatigue is greater when fatiguing protocols are conducted at optimal muscle length rather than when a muscle is shortened)194 and activation of extradiaphragmatic muscles of respiration.195
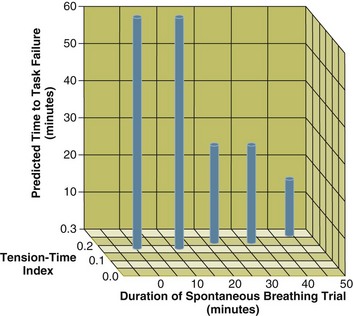
Figure 40.19 The interrelationship between the duration of a spontaneous breathing trial, tension-time index of the diaphragm, and predicted time to task failure in nine patients who failed a trial of weaning from mechanical ventilation. The patients breathed spontaneously for an average of 44 minutes before a physician terminated the trial. At the start of the trial, tension-time index was 0.17, and the formula of Bellemare and Grassino193 (see text for details) predicted that patients could sustain spontaneous breathing for another 59 minutes before developing task failure. As the trial progressed, tension-time index increased and predicted time to the development of task failure decreased. At the end of the trial, tension-time index reached 0.26; that patients were predicted to sustain spontaneous breathing for another 13 minutes before developing task failure clarifies why patients did not develop a decrease in diaphragmatic twitch pressure. In other words, physicians interrupted the trial based on clinical manifestations of respiratory distress before patients had sufficient time to develop contractile fatigue. (From Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10-48.)
Studies in animals184,196–198 support the finding that patients do not develop long-lasting contractile fatigue of the respiratory muscles. Inspiratory loading in animals causes respiratory failure and acidosis before force output decreases or substrate is depleted in the diaphragm,184,196–198 suggesting that central199 and reflex mechanisms200,201 affect the breathing pattern185 and α-motor neuron firing rates202 in response to loading. Two neural pathways may convey information from the respiratory muscles to the central nervous system.203,204 One pathway transmits information from mechanoreceptors (Golgi tendon organs and muscle spindles)200,205 in the dorsal column, relaying it to the brainstem and thalamus, before reaching the sensomotor cortex.206 This pathway may participate in proprioceptive control of the respiratory muscles, integrating movements originating in the motor cortex.199 The second pathway consists of vagal198 and possibly phrenic nerve afferents (group IV phrenic afferent fibers)200 that reach the amygdala after relaying in the brainstem and then projecting to the mesocortex (cingulated gyrus). This pathway may deal with respiratory nociception,199,207 such as dyspnea (through the relay in the amygdala),208 and with the ventilatory response to carbon dioxide (through the relay in the brainstem, ventral cerebellum, and limbic system).199,207,209 In addition to these two pathways projecting to the central nervous system, there is evidence for the existence of a spinal pathway responsible for phrenic-to-phrenic reflex inhibition.201 An increase in carbon dioxide during loading may also protect the respiratory muscles by decreasing reactive oxygen species production.210
Resistive breathing causes hypercapnia without affecting diaphragmatic force output or aerobic metabolism (assessed by 31P nuclear magnetic resonance spectroscopy) in spontaneously breathing piglets.7,185,211 The hypoxia combined with resistive breathing caused a decrease in diaphragmatic force and inadequate oxidative metabolism, as reflected by accumulation of inorganic phosphorus and decreased phosphocreatine.185 Force output and oxidative metabolism returned to baseline values with normoxia despite persistent loading.185 The investigators speculated that loading produces a decrease in central activation (to the point of ventilatory failure), which decreases metabolic demands and prevents peripheral fatigue; additional stress of hypoxia may overwhelm this defense mechanism and cause peripheral fatigue.185 Hypoxemia can also induce degradation of myofibrillar proteins.212
Hypoxia impairs the respiratory centers at a higher oxygen tension than it impairs the respiratory muscles,160,213 making it difficult to decipher the effects of hypoxemia on the respiratory muscles. Animals develop severe alveolar hypoventilation and respiratory arrest before the tension-generating ability of the diaphragm has begun to decrease.160,213 As previously stated, it is unknown whether, in patients with shock,151 decreases in oxygen delivery to the respiratory muscles154,214 or respiratory centers can be sufficient to cause hypoventilation.
Increased Load
Increased Mechanical Load
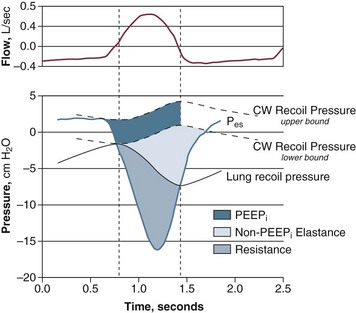
Patients in acute respiratory failure usually experience an increased mechanical load.* The patients typically have a 30% to 50% greater inspiratory resistance,7,13,33 100% greater dynamic elastance,7,11 and 100% to 200% greater intrinsic positive end-expiratory pressure (PEEP)7,11,13 than do similar patients who are not in acute respiratory failure. Inspiratory effort—quantified by calculation of the pressure time product of the inspiratory muscles—is almost equally divided in offsetting intrinsic PEEP, elastic recoil, and inspiratory resistance (Fig. 40.21).7,211 Abnormal mechanics arise from bronchoconstriction, bronchial edema, pulmonary edema,7 and lung inflammation.215,216 Rapid shallow breathing can aggravate the abnormalities in lung elastance, intrinsic PEEP, and carbon dioxide clearance.7,33 Expiratory muscle recruitment can also increase intrinsic PEEP and breathing effort.218 In some patients, increased mechanical load results from upper airway obstruction. The upper airway, which encompasses the passage between the nares and carina, can be obstructed by functional or anatomic causes. Among the first are vocal cord paralysis and laryngospasm. Among the second are trauma, burn, infections, foreign bodies, and tumors. Functional and anatomic obstruction can occur postoperatively in patients with redundant pharyngeal soft tissue (sleep apnea) and loss of muscle tone related to postanesthetic state.1 Upper airway obstruction is one of the most urgent and potentially lethal medical emergencies.1 Complete airway obstruction lasting for as little as 4 to 6 minutes can cause irreversible brain damage.1
Increased Ventilatory Requirements
Increased ventilatory requirements can result from increased carbon dioxide production, increased dead space ventilation, and elevated respiratory drive. Carbon dioxide production can increase as a result of sepsis,86 fever, shivering, drugs (salicylates), lipogenesis,219,220 and a shift in utilization of fuels from lipids (RQ = 0.7) to carbohydrates (RQ = 1.0).221 An increase in carbon dioxide production can only be a contributory factor and not a sole cause of hypercapnic respiratory failure.3
The ratio of dead space to tidal volume is normally 0.30. The ratio increases up to 0.65 in patients with severe COPD,222 ARDS,223 and other severe lung diseases.3 Patients can compensate for such an increase in dead space by doubling minute ventilation.3 Such an increase in minute ventilation poses a minor challenge when respiratory mechanics and respiratory muscles are normal; for example, hypercapnia is uncommon with pulmonary vascular disease.3 Accordingly, an increase in dead space ventilation should never be considered the primary mechanism responsible for hypercapnic respiratory failure unless there is a concurrent abnormality in the control of breathing, or in the mechanical load of the respiratory muscles or in their contractile performance.3
Hypercapnia-Induced Hypoventilation
Severe hypercapnia depresses the central nervous system and decreases respiratory motor output.220 A vicious circle can arise, whereby hypercapnia causes depressed drive leading to more hypercapnia.224,225 The possibility of hypercapnia-induced hypoventilation is supported by reports of successful resolution of hypercapnic respiratory failure in some patients following short-term infusion of the respiratory stimulant, doxapram.224,225
Summary
A relative or absolute decrease in respiratory muscle output is responsible for alveolar hypoventilation and hypercapnic respiratory failure. The number of distinct clinical entities that result in hypercapnia is vast and often more than one disease process may coexist—e.g., hyperinflation-associated respiratory muscle weakness and ventilation-perfusion inequality in patients with COPD. The relative or absolute decrease in respiratory muscle output in patients with hypercapnia may stem from a decrease in respiratory drive, excess mechanical load on the respiratory muscles, and respiratory muscle weakness (see Fig. 40.1). Whether respiratory muscle fatigue is responsible for hypercapnic respiratory failure is an important issue that has yet to be resolved.
References
1. Laghi, F, Tobin, MJ. Indications for mechanical ventilation. In: Tobin MJ, ed. Principles and Practice of Mechanical Ventilation. 3rd ed. New York: McGraw-Hill; 2013:99–136.
2. West, JB. Pulmonary Pathophysiology: The Essentials, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2003.
3. Younes, M. Mechanisms of ventilatory failure. Curr Pulmonol. 1993; 14:243–292.
4. Laghi, F, Tobin, MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med. 2003; 168(1):10–48.
5. Webster, NR, Kulkarni, V. Metabolic alkalosis in the critically ill. Crit Rev Clin Lab Sci. 1999; 36(5):497–510.
6. Baier, H, Somani, P. Ventilatory drive in normal man during semistarvation. Chest. 1984; 85(2):222–225.
7. Tobin, MJ, Laghi, F, Jubran, A, Ventilatory failure, ventilator support and ventilator weaning. Compr Physiol; 2. 2012:2871–2921.
8. Cooper, KR, Phillips, BA. Effect of short-term sleep loss on breathing. J Appl Physiol. 1982; 53(4):855–858.
9. Spengler, CM, Shea, SA. Sleep deprivation per se does not decrease the hypercapnic ventilatory response in humans. Am J Respir Crit Care Med. 2000; 161(4 Pt 1):1124–1128.
10. Tobin, MJ, Laghi, F. Monitoring respiratory muscle function. In: Tobin MJ, ed. Principles and Practice of Intensive Care Monitoring. New York: McGraw-Hill; 1998:497–544.
11. Purro, A, Appendini, L, De Gaetano, A, et al. Physiologic determinants of ventilator dependence in long-term mechanically ventilated patients. Am J Respir Crit Care Med. 2000; 161(4 Pt 1):1115–1123.
12. Yang, KL, Tobin, MJ. A prospective study of indexes predicting the outcome of trials of weaning from mechanical ventilation. N Engl J Med. 1991; 324(21):1445–1450.
13. Laghi, F, Cattapan, SE, Jubran, A, et al. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med. 2003; 167(2):120–127.
14. Zakynthinos, S, Routsi, C, Vassilakopoulos, T, et al. Differential cardiovascular responses during weaning failure: Effects on tissue oxygenation and lactate. Intensive Care Med. 2005; 31(12):1634–1642.
15. Laghi, F, Harrison, MJ, Tobin, MJ. Comparison of magnetic and electrical phrenic nerve stimulation in assessment of diaphragmatic contractility. J Appl Physiol. 1996; 80(5):1731–1742.
16. Cattapan, SE, Laghi, F, Tobin, MJ. Can diaphragmatic contractility be assessed by airway twitch pressure in mechanically ventilated patients? Thorax. 2003; 58(1):58–62.
17. Watson, AC, Hughes, PD, Louise, HM, et al. Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med. 2001; 29(7):1325–1331.
18. Polkey, MI, Kyroussis, D, Hamnegard, CH, et al. Diaphragm strength in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1996; 154(5):1310–1317.
19. Laghi, F, Jubran, A, Topeli, A, et al. Effect of lung volume reduction surgery on diaphragmatic neuromechanical coupling at 2 years. Chest. 2004; 125(6):2188–2195.
20. Laghi, F. Ventilator-induced diaphragmatic dysfunction: Is there a dim light at the end of the tunnel? Crit Care Med. 2011; 39(4):903–905.
21. Oomman, A, Gurtoo, A. Acute intermittent porphyria as a cause of acute respiratory failure. J Indian Med Assoc. 2002; 100(1):44–46.
22. Tyagi, A, Chawla, R, Sethi, AK, et al. Respiratory failure in acute intermittent porphyria. J Assoc Physicians India. 2002; 50:443–445.
23. Vodoff, MV, Cremer, R, Martinot, A, et al. [Acute respiratory insufficiency revealing myasthenia gravis: Apropos of 3 cases]. Arch Pediatr. 1997; 4(9):845–848.
24. Vaidya, H. Case of the month: Unusual presentation of myasthenia gravis with acute respiratory failure in the emergency room. Emerg Med J. 2006; 23(5):410–413.
25. Greenberg, C, Davies, S, McGowan, T, et al. Acute respiratory failure following severe arsenic poisoning. Chest. 1979; 76(5):596–598.
26. Vergauwe, PL, Knockaert, DC, Van Tittelboom, TJ. Near fatal subacute thallium poisoning necessitating prolonged mechanical ventilation. Am J Emerg Med. 1990; 8(6):548–550.
27. Henderson, RD, Sandroni, P, Wijdicks, EF. Chronic inflammatory demyelinating polyneuropathy and respiratory failure. J Neurol. 2005; 252(10):1235–1237.
28. Trend, PS, Wiles, CM, Spencer, GT, et al. Acid maltase deficiency in adults. Diagnosis and management in five cases. Brain. 1985; 108(Pt 4):845–860.
29. Braun, NM, Arora, NS, Rochester, DF. Respiratory muscle and pulmonary function in polymyositis and other proximal myopathies. Thorax. 1983; 38(8):616–623.
30. Gibson, GJ, Gilmartin, JJ, Veale, D, et al. Respiratory muscle function in neuromuscular disease. In: Jones NL, Killian KJ, eds. Breathlessness. The Campbell Symposium. Hamilton, Ontario: Boehringer-Ingelheim; 1992:66–73.
31. Alison, JA, Regnis, JA, Donnelly, PM, et al. End-expiratory lung volume during arm and leg exercise in normal subjects and patients with cystic fibrosis. Am J Respir Crit Care Med. 1998; 158(5 Pt 1):1450–1458.
32. Bloch, KE, Weder, W, Boehler, A, et al. Successful lung volume reduction surgery in a child with severe airflow obstruction and hyperinflation due to constrictive bronchiolitis. Chest. 2002; 122(2):747–750.
33. Vassilakopoulos, T, Zakynthinos, S, Roussos, C. The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med. 1998; 158(2):378–385.
34. Laghi, F, Jubran, A, Topeli, A, et al. Effect of lung volume reduction surgery on neuromechanical coupling of the diaphragm. Am J Respir Crit Care Med. 1998; 157(2):475–483.
35. Coussa, ML, Guerin, C, Eissa, NT, et al. Partitioning of work of breathing in mechanically ventilated COPD patients. J Appl Physiol. 1993; 75(4):1711–1719.
36. Koutsoukou, A, Armaganidis, A, Stavrakaki-Kallergi, C, et al. Expiratory flow limitation and intrinsic positive end-expiratory pressure at zero positive end-expiratory pressure in patients with adult respiratory distress syndrome. Am J Respir Crit Care Med. 2000; 161(5):1590–1596.
37. Pelosi, P, Cereda, M, Foti, G, et al. Alterations of lung and chest wall mechanics in patients with acute lung injury: Effects of positive end-expiratory pressure. Am J Respir Crit Care Med. 1995; 152(2):531–537.
38. Laaban, JP, Kouchakji, B, Dore, MF, et al. Nutritional status of patients with chronic obstructive pulmonary disease and acute respiratory failure. Chest. 1993; 103(5):1362–1368.
39. Faisy, C, Rabbat, A, Kouchakji, B, et al. Bioelectrical impedance analysis in estimating nutritional status and outcome of patients with chronic obstructive pulmonary disease and acute respiratory failure. Intensive Care Med. 2000; 26(5):518–525.
40. Kim, J, Heshka, S, Gallagher, D, et al. Intermuscular adipose tissue-free skeletal muscle mass: Estimation by dual-energy X-ray absorptiometry in adults. J Appl Physiol. 2004; 97(2):655–660.
41. Murciano, D, Rigaud, D, Pingleton, S, et al. Diaphragmatic function in severely malnourished patients with anorexia nervosa. Effects of renutrition. Am J Respir Crit Care Med. 1994; 150(6 Pt 1):1569–1574.
42. Ameredes, BT, Watchko, JF, Daood, MJ, et al. Growth hormone restores aged diaphragm myosin composition and performance after chronic undernutrition. J Appl Physiol. 1999; 87(4):1253–1259.
43. Lewis, MI, Li, H, Huang, ZS, et al. Influence of varying degrees of malnutrition on IGF-I expression in the rat diaphragm. J Appl Physiol. 2003; 95(2):555–562.
44. Lewis, MI, Lorusso, TJ, Zhan, WZ, et al. Interactive effects of denervation and malnutrition on diaphragm structure and function. J Appl Physiol. 1996; 81(5):2165–2172.
45. Mitch, WE, Goldberg, AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med. 1996; 335(25):1897–1905.
46. Lewis, MI. Apoptosis as a potential mechanism of muscle cachexia in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002; 166(4):434–436.
47. Tawa, NE, Jr., Odessey, R, Goldberg, AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997; 100(1):197–203.
48. Schols, AM, Soeters, PB, Mostert, R, et al. Physiologic effects of nutritional support and anabolic steroids in patients with chronic obstructive pulmonary disease. A placebo-controlled randomized trial. Am J Respir Crit Care Med. 1995; 152(4 Pt 1):1268–1274.
49. Ryan, CF, Whittaker, JS, Road, JD. Ventilatory dysfunction in severe anorexia nervosa. Chest. 1992; 102(4):1286–1288.
50. Lanz, JK, Jr., Donahoe, M, Rogers, RM, et al. Effects of growth hormone on diaphragmatic recovery from malnutrition. J Appl Physiol. 1992; 73(3):801–805.
51. Martinez, FJ, Bermudez-Gomez, M, Celli, BR. Hypothyroidism. A reversible cause of diaphragmatic dysfunction. Chest. 1989; 96(5):1059–1063.
52. Norrelund, H, Hove, KY, Brems-Dalgaard, E, et al. Muscle mass and function in thyrotoxic patients before and during medical treatment. Clin Endocrinol (Oxford). 1999; 51(6):693–699.
53. Goswami, R, Guleria, R, Gupta, AK, et al. Prevalence of diaphragmatic muscle weakness and dyspnoea in Graves’ disease and their reversibility with carbimazole therapy. Eur J Endocrinol. 2002; 147(3):299–303.
54. Siafakas, NM, Milona, I, Salesiotou, V, et al. Respiratory muscle strength in hyperthyroidism before and after treatment. Am Rev Respir Dis. 1992; 146(4):1025–1029.
55. Iandelli, I, Gorini, M, Duranti, R, et al. Respiratory muscle function and control of breathing in patients with acromegaly. Eur Respir J. 1997; 10(5):977–982.
56. Mills, GH, Kyroussis, D, Jenkins, P, et al. Respiratory muscle strength in Cushing’s syndrome. Am J Respir Crit Care Med. 1999; 160(5 Pt 1):1762–1765.
57. Tobin, MJ, Laghi, F, Jubran, A. Narrative review: Ventilator-induced respiratory muscle weakness. Ann Intern Med. 2010; 153(4):240–245.
58. Anzueto, A, Tobin, MJ, Moore, G. Effect of prolonged mechanical ventilation on diaphragmatic function: A preliminary study of a baboon model. Am Rev Respir Dis. 1987; 135:A201.
59. Sassoon, CS, Ciaozzo, VJ, Manka, A, et al. Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol. 2002; 92(6):2585–2595.
60. Yang, L, Luo, J, Bourdon, J, et al. Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med. 2002; 166(8):1135–1140.
61. McClung, JM, Kavazis, AN, DeRuisseau, KC, et al. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med. 2007; 175(2):150–159.
62. Powers, SK, Shanely, RA, Coombes, JS, et al. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol. 2002; 92(5):1851–1858.
63. Jaber, S, Petrof, BJ, Jung, B, et al. Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med. 2011; 183(3):364–371.
64. Knisely, AS, Leal, SM, Singer, DB. Abnormalities of diaphragmatic muscle in neonates with ventilated lungs. J Pediatr. 1988; 113(6):1074–1077.
65. Levine, S, Nguyen, T, Taylor, N, et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008; 358(13):1327–1335.
66. Hermans, G, Agten, A, Testelmans, D, et al. Increased duration of mechanical ventilation is associated with decreased diaphragmatic force: A prospective observational study. Crit Care. 14(4), 2010.
67. DeRuisseau, KC, Shanely, RA, Akunuri, N, et al. Diaphragm unloading via controlled mechanical ventilation alters the gene expression profile. Am J Respir Crit Care Med. 2005; 172(10):1267–1275.
68. Powers, SK, Kavazis, AN, Levine, S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med. 2009; 37(10 Suppl):S347–S353.
69. Hussain, SN, Mofarrahi, M, Sigala, I, et al. Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am J Respir Crit Care Med. 2010; 182(11):1377–1386.
70. Betters, JL, Criswell, DS, Shanely, RA, et al. Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med. 2004; 170(11):1179–1184.
71. Whidden, MA, Smuder, AJ, Wu, M, et al. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol. 2010; 108(5):1376–1382.
72. Agten, A, Maes, K, Smuder, A, et al. N-Acetylcysteine protects the rat diaphragm from the decreased contractility associated with controlled mechanical ventilation. Crit Care Med. 2011; 39(4):777–782.
73. Crimi, E, Liguori, A, Condorelli, M, et al. The beneficial effects of antioxidant supplementation in enteral feeding in critically ill patients: A prospective, randomized, double-blind, placebo-controlled trial. Anesth Analg. 2004; 99(3):857–863.
74. Nathens, AB, Neff, MJ, Jurkovich, GJ, et al. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg. 2002; 236(6):814–822.
75. Spapen, HD, Diltoer, MW, Nguyen, DN, et al. Effects of N-acetylcysteine on microalbuminuria and organ failure in acute severe sepsis: Results of a pilot study. Chest. 2005; 127(4):1413–1419.
76. Sassoon, CS, Zhu, E, Caiozzo, VJ. Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med. 2004; 170(6):626–632.
77. Gayan-Ramirez, G, Testelmans, D, Maes, K, et al. Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med. 2005; 33(12):2804–2809.
78. Jung, B, Constantin, JM, Rossel, N, et al. Adaptive support ventilation prevents ventilator-induced diaphragmatic dysfunction in piglet: An in vivo and in vitro study. Anesthesiology. 2010; 112(6):1435–1443.
79. Futier, E, Constantin, JM, Combaret, L, et al. Pressure support ventilation attenuates ventilator-induced protein modifications in the diaphragm. Crit Care. 12(5), 2008.
80. Sassoon, CS. Ventilator-associated diaphragmatic dysfunction. Am J Respir Crit Care Med. 2002; 166(8):1017–1018.
81. Nguyen, T, Friscia, M, Kaiser, LR, et al. Ventilator-induced proteolysis in human diaphragm myofibers. Proc Am Thorac Soc. 2006; 3:A259.
82. Shanely, RA, Van Gammeren, D, DeRuisseau, KC, et al. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med. 2004; 170(9):994–999.
83. Pichard, C, Kyle, U, Chevrolet, JC, et al. Lack of effects of recombinant growth hormone on muscle function in patients requiring prolonged mechanical ventilation: A prospective, randomized, controlled study. Crit Care Med. 1996; 24(3):403–413.
84. Takala, J, Ruokonen, E, Webster, NR, et al. Increased mortality associated with growth hormone treatment in critically ill adults. N Engl J Med. 1999; 341(11):785–792.
85. Hooijman, PE, van Hess, HW, Paul, MA, et al. Preservation of diaphragm muscle fiber contractility in mechanically ventilated humans. Am J Respir Crit Care Med. 2012; 185:A6871.
86. Hussain, SN. Respiratory muscle dysfunction in sepsis. Mol Cell Biochem. 1998; 179:125–134.
87. Leon, A, Boczkowski, J, Dureuil, B, et al. Effects of endotoxic shock on diaphragmatic function in mechanically ventilated rats. J Appl Physiol. 1992; 72(4):1466–1472.
88. Lin, MC, Ebihara, S, El Dwairi, Q, et al. Diaphragm sarcolemmal injury is induced by sepsis and alleviated by nitric oxide synthase inhibition. Am J Respir Crit Care Med. 1998; 158(5 Pt 1):1656–1663.
89. Aarli, JA, Skeie, GO, Mygland, A, et al. Muscle striation antibodies in myasthenia gravis. Diagnostic and functional significance. Ann N Y Acad Sci. 1998; 841:505–515.
90. Callahan, LA, Nethery, D, Stofan, D, et al. Free radical-induced contractile protein dysfunction in endotoxin-induced sepsis. Am J Respir Cell Mol Biol. 2001; 24(2):210–217.
91. Tsukagoshi, H, Morita, T, Takahashi, K, et al. Cecal ligation and puncture peritonitis model shows decreased nicotinic acetylcholine receptor numbers in rat muscle: Immunopathologic mechanisms? Anesthesiology. 1999; 91(2):448–460.
92. Callahan, LA, Supinski, GS. Sepsis-induced myopathy. Crit Care Med. 2009; 37(10 Suppl):S354–S367.
93. Boczkowski, J, Lanone, S, Ungureanu-Longrois, D, et al. Induction of diaphragmatic nitric oxide synthase after endotoxin administration in rats: Role of diaphragmatic contractile dysfunction. J Clin Invest. 1996; 98(7):1550–1559.
94. Comtois, AS, Barreiro, E, Huang, PL, et al. Lipopolysaccharide-induced diaphragmatic contractile dysfunction and sarcolemmal injury in mice lacking the neuronal nitric oxide synthase. Am J Respir Crit Care Med. 2001; 163(4):977–982.
95. Ebihara, S, Hussain, SN, Danialou, G, et al. Mechanical ventilation protects against diaphragm injury in sepsis: Interaction of oxidative and mechanical stresses. Am J Respir Crit Care Med. 2002; 165(2):221–228.
96. Lanone, S, Mebazaa, A, Heymes, C, et al. Muscular contractile failure in septic patients: Role of the inducible nitric oxide synthase pathway. Am J Respir Crit Care Med. 2000; 162(6):2308–2315.
97. Comtois, AS, El Dwairi, Q, Laubach, VE, et al. Lipopolysaccharide-induced diaphragmatic contractile dysfunction in mice lacking the inducible nitric oxide synthase. Am J Respir Crit Care Med. 1999; 159(6):1975–1980.
98. Afulukwe, IF, Cohen, RI, Zeballos, GA, et al. Selective NOS inhibition restores myocardial contractility in endotoxemic rats; however, myocardial NO content does not correlate with myocardial dysfunction. Am J Respir Crit Care Med. 2000; 162(1):21–26.
99. Javesghani, D, Magder, SA, Barreiro, E, et al. Molecular characterization of a superoxide-generating NAD(P)H oxidase in the ventilatory muscles. Am J Respir Crit Care Med. 2002; 165(3):412–418.
100. El Dwairi, Q, Comtois, A, Guo, Y, et al. Endotoxin-induced skeletal muscle contractile dysfunction: Contribution of nitric oxide synthases. Am J Physiol. 1998; 274(3 Pt 1):C770–C779.
101. Wink, DA, Mitchell, JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998; 25:434–456.
102. Fujimura, N, Sumita, S, Aimono, M, et al. Effect of free radical scavengers on diaphragmatic contractility in septic peritonitis. Am J Respir Crit Care Med. 2000; 162(6):2159–2165.
103. Supinski, G, Nethery, D, DiMarco, A. Effect of free radical scavengers on endotoxin-induced respiratory muscle dysfunction. Am Rev Respir Dis. 1993; 148(5):1318–1324.
104. Taille, C, Foresti, R, Lanone, S, et al. Protective role of heme oxygenases against endotoxin-induced diaphragmatic dysfunction in rats. Am J Respir Crit Care Med. 2001; 163(3 Pt 1):753–761.
105. Bolton, CF, Laverty, DA, Brown, JD, et al. Critically ill polyneuropathy: Electrophysiological studies and differentiation from Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 1986; 49(5):563–573.
106. Faragher, MW, Day, BJ. A practical approach to weakness in the intensive care unit. In: Cros D, ed. Peripheral Neuropathy: A Practical Approach to Diagnosis and Management. Philadelphia: Lippincott Williams & Wilkins; 2001:370–386.
107. Druschky, A, Herkert, M, Radespiel-Troger, M, et al. Critical illness polyneuropathy: Clinical findings and cell culture assay of neurotoxicity assessed by a prospective study. Intensive Care Med. 2001; 27(4):686–693.
108. Hund, EF, Fogel, W, Krieger, D, et al. Critical illness polyneuropathy: Clinical findings and outcomes of a frequent cause of neuromuscular weaning failure. Crit Care Med. 1996; 24(8):1328–1333.
109. Latronico, N, Fenzi, F, Recupero, D, et al. Critical illness myopathy and neuropathy. Lancet. 1996; 347(9015):1579–1582.
110. Van den Berghe, G, Wouters, P, Weekers, F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001; 345(19):1359–1367.
111. Rouleau, G, Karpati, G, Carpenter, S, et al. Glucocorticoid excess induces preferential depletion of myosin in denervated skeletal muscle fibers. Muscle Nerve. 1987; 10(5):428–438.
112. Larsson, L, Li, X, Edstrom, L, et al. Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: Mechanisms at the cellular and molecular levels. Crit Care Med. 2000; 28(1):34–45.
113. Rich, MM, Teener, JW, Raps, EC, et al. Muscle is electrically inexcitable in acute quadriplegic myopathy. Neurology. 1996; 46(3):731–736.
114. Rich, MM, Bird, SJ, Raps, EC, et al. Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve. 1997; 20(6):665–673.
115. Lefaucheur, JP, Nordine, T, Rodriguez, P, et al. Origin of ICU-acquired paresis determined by direct muscle stimulation. J Neurol Neurosurg Psychiatry. 2006; 77(4):500–506.
116. De Jonghe, B, Sharshar, T, Lefaucheur, JP, et al. Paresis acquired in the intensive care unit: A prospective multicenter study. JAMA. 2002; 288(22):2859–2867.
117. Faragher, MW, Day, BJ, Dennett, X. Critical care myopathy: An electrophysiological and histological study. Muscle Nerve. 1996; 19(4):516–518.
118. De Jonghe, B, Bastuji-Garin, S, Sharshar, T, et al. Does ICU-acquired paresis lengthen weaning from mechanical ventilation? Intensive Care Med. 2004; 30(6):1117–1121.
119. Latronico, N, Shehu, I, Seghelini, E. Neuromuscular sequelae of critical illness. Curr Opin Crit Care. 2005; 11(4):381–390.
120. Leatherman, JW, Fluegel, WL, David, WS, et al. Muscle weakness in mechanically ventilated patients with severe asthma. Am J Respir Crit Care Med. 1996; 153(5):1686–1690.
121. Roberts, PA, Loxham, SJ, Poucher, SM, et al. Bicarbonate-induced alkalosis augments cellular acetyl group availability and isometric force during the rest-to-work transition in canine skeletal muscle. Exp Physiol. 2002; 87(4):489–498.
122. Schnader, JY, Juan, G, Howell, S, et al. Arterial CO2 partial pressure affects diaphragmatic function. J Appl Physiol. 1985; 58(3):823–829.
123. Juan, G, Calverley, P, Talamo, C, et al. Effect of carbon dioxide on diaphragmatic function in human beings. N Engl J Med. 1984; 310(14):874–879.
124. Jackson, DC, Arendt, EA, Inman, KC, et al. 31P-NMR study of normoxic and anoxic perfused turtle heart during graded CO2 and lactic acidosis. Am J Physiol. 1991; 260(6 Pt 2):R1130–R1136.
125. Yanos, J, Wood, LD, Davis, K, et al. The effect of respiratory and lactic acidosis on diaphragm function. Am Rev Respir Dis. 1993; 147(3):616–619.
126. Cairns, SP. Lactic acid and exercise performance: Culprit or friend? Sports Med. 2006; 36(4):279–291.
127. Keeton, RB, Binder-Macleod, SA. Low-frequency fatigue. Phys Ther. 2006; 86(8):1146–1150.
128. Gladden, LB. Lactate metabolism: A new paradigm for the third millennium. J Physiol. 2004; 558(Pt 1):5–30.
129. Vogiatzis, I, Georgiadou, O, Giannopoulou, I, et al. Effects of exercise-induced arterial hypoxaemia and work rate on diaphragmatic fatigue in highly trained endurance athletes. J Physiol. 2006; 572(Pt 2):539–549.
130. Coast, JR, Shanely, RA, Lawler, JM, et al. Lactic acidosis and diaphragmatic function in vitro. Am J Respir Crit Care Med. 1995; 152(5 Pt 1):1648–1652.
131. Knuth, ST, Dave, H, Peters, JR, et al. Low cell pH depresses peak power in rat skeletal muscle fibres at both 30 degrees C and 15 degrees C: Implications for muscle fatigue. J Physiol. 2006; 575(Pt 3):887–899.
132. Kristensen, M, Albertsen, J, Rentsch, M, et al. Lactate and force production in skeletal muscle. J Physiol. 2005; 562(Pt 2):521–526.
133. Posterino, GS, Dutka, TL, Lamb, GD. L(+)-lactate does not affect twitch and tetanic responses in mechanically skinned mammalian muscle fibres. Pflugers Arch. 2001; 442(2):197–203.
134. Degroot, M, Massie, BM, Boska, M, et al. Dissociation of [H+] from fatigue in human muscle detected by high time resolution 31P-NMR. Muscle Nerve. 1993; 16(1):91–98.
135. Westerblad, H, Allen, DG, Lannergren, J. Muscle fatigue: Lactic acid or inorganic phosphate the major cause? News Physiol Sci. 2002; 17:17–21.
136. Nielsen, OB, de Paoli, F, Overgaard, K. Protective effects of lactic acid on force production in rat skeletal muscle. J Physiol. 2001; 536(Pt 1):161–166.
137. Mador, MJ, Wendel, T, Kufel, TJ. Effect of acute hypercapnia on diaphragmatic and limb muscle contractility. Am J Respir Crit Care Med. 1997; 155(5):1590–1595.
138. Schnader, J, Howell, S, Fitzgerald, RS, et al. Interaction of fatigue and hypercapnia in the canine diaphragm. J Appl Physiol. 1988; 64(4):1636–1643.
139. Rafferty, GF, Lou, HM, Polkey, MI, et al. Effect of hypercapnia on maximal voluntary ventilation and diaphragm fatigue in normal humans. Am J Respir Crit Care Med. 1999; 160(5 Pt 1):1567–1571.
140. Topeli, A, Laghi, F, Tobin, MJ. The voluntary drive to breathe is not decreased in hypercapnic patients with severe COPD. Eur Respir J. 2001; 18(1):53–60.
141. Begin, P, Grassino, A. Inspiratory muscle dysfunction and chronic hypercapnia in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1991; 143(5 Pt 1):905–912.
142. Braun, NMT, Rochester, DF. Respiratory muscle function in chronic obstructive pulmonary disease (COPD). Am Rev Respir Dis. 1977; 115:A91.
143. Vianna, LG, Koulouris, N, Moxham, J. Lack of effect of acute hypoxia and hypercapnia on muscle relaxation rate in man. Rev Esp Fisiol. 1993; 49(1):7–15.
144. Ranatunga, KW. Effects of acidosis on tension development in mammalian skeletal muscle. Muscle Nerve. 1987; 10(5):439–445.
145. Aubier, M, Murciano, D, Lecocguic, Y, et al. Effect of hypophosphatemia on diaphragmatic contractility in patients with acute respiratory failure. N Engl J Med. 1985; 313(7):420–424.
146. Aubier, M, Viires, N, Piquet, J, et al. Effects of hypocalcemia on diaphragmatic strength generation. J Appl Physiol. 1985; 58(6):2054–2061.
147. Dhingra, S, Solven, F, Wilson, A, et al. Hypomagnesemia and respiratory muscle power. Am Rev Respir Dis. 1984; 129(3):497–498.
148. Stedwell, RE, Allen, KM, Binder, LS. Hypokalemic paralyses: A review of the etiologies, pathophysiology, presentation, and therapy. Am J Emerg Med. 1992; 10(2):143–148.
149. Aubier, M, Trippenbach, T, Roussos, C. Respiratory muscle fatigue during cardiogenic shock. J Appl Physiol. 1981; 51(2):499–508.
150. Hussain, SN, Simkus, G, Roussos, C. Respiratory muscle fatigue: A cause of ventilatory failure in septic shock. J Appl Physiol. 1985; 58(6):2033–2040.
151. Ronco, JJ, Fenwick, JC, Tweeddale, MG, et al. Identification of the critical oxygen delivery for anaerobic metabolism in critically ill septic and nonseptic humans. JAMA. 1993; 270(14):1724–1730.
152. Kontoyannis, DA, Nanas, JN, Kontoyannis, SA, et al. Mechanical ventilation in conjunction with the intra-aortic balloon pump improves the outcome of patients in profound cardiogenic shock. Intensive Care Med. 1999; 25(8):835–838.
153. Viires, N, Sillye, G, Aubier, M, et al. Regional blood flow distribution in dog during induced hypotension and low cardiac output. Spontaneous breathing versus artificial ventilation. J Clin Invest. 1983; 72(3):935–947.
154. Jubran, A, Mathru, M, Dries, D, et al. Continuous recordings of mixed venous oxygen saturation during weaning from mechanical ventilation and the ramifications thereof. Am J Respir Crit Care Med. 1998; 158(6):1763–1769.
155. Weber, KT, Kinasewitz, GT, Janicki, JS, et al. Oxygen utilization and ventilation during exercise in patients with chronic cardiac failure. Circulation. 1982; 65(6):1213–1223.
156. Reid, MB, Johnson, RL, Jr. Efficiency, maximal blood flow, and aerobic work capacity of canine diaphragm. J Appl Physiol. 1983; 54(3):763–772.
157. Robertson, CH, Jr., Foster, GH, Johnson, RL, Jr. The relationship of respiratory failure to the oxygen consumption of, lactate production by, and distribution of blood flow among respiratory muscles during increasing inspiratory resistance. J Clin Invest. 1977; 59(1):31–42.
158. Rochester, DF, Bettini, G. Diaphragmatic blood flow and energy expenditure in the dog. Effects of inspiratory airflow resistance and hypercapnia. J Clin Invest. 1976; 57(3):661–672.
159. Rochester, DF, Briscoe, AM. Metabolism of the working diaphragm. Am Rev Respir Dis. 1979; 119(2 Pt 2):101–106.
160. Bark, H, Supinski, G, Bundy, R, et al. Effect of hypoxia on diaphragm blood flow, oxygen uptake, and contractility. Am Rev Respir Dis. 1988; 138(6):1535–1541.
161. Richard, C, Teboul, JL. Weaning failure from cardiovascular origin. Intensive Care Med. 2005; 31(12):1605–1607.
162. Rodriguez, JA, Crespo-Leiro, MG, Paniagua, MJ, et al. Rhabdomyolysis in heart transplant patients on HMG-CoA reductase inhibitors and cyclosporine. Transplant Proc. 1999; 31(6):2522–2523.
163. Masters, BA, Palmoski, MJ, Flint, OP, et al. In vitro myotoxicity of the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, pravastatin, lovastatin, and simvastatin, using neonatal rat skeletal myocytes. Toxicol Appl Pharmacol. 1995; 131(1):163–174.
164. Sugiyama, S. HMG CoA reductase inhibitor accelerates aging effect on diaphragm mitochondrial respiratory function in rats. Biochem Mol Biol Int. 1998; 46(5):923–931.
165. Cote, HC, Brumme, ZL, Craib, KJ, et al. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N Engl J Med. 2002; 346(11):811–820.
166. Hasfurther, DL, Bailey, PL. Failure of neuromuscular blockade reversal after rocuronium in a patient who received oral neomycin. Can J Anaesth. 1996; 43(6):617–620.
167. Segredo, V, Caldwell, JE, Matthay, MA, et al. Persistent paralysis in critically ill patients after long-term administration of vecuronium. N Engl J Med. 1992; 327(8):524–528.
168. de Lemos, JM, Carr, RR, Shalansky, KF, et al. Paralysis in the critically ill: Intermittent bolus pancuronium compared with continuous infusion. Crit Care Med. 1999; 27(12):2648–2655.
169. Rudis, MI, Sikora, CA, Angus, E, et al. A prospective, randomized, controlled evaluation of peripheral nerve stimulation versus standard clinical dosing of neuromuscular blocking agents in critically ill patients. Crit Care Med. 1997; 25(4):575–583.
170. de Seze, M, Petit, H, Wiart, L, et al. Critical illness polyneuropathy. A 2-year follow-up study in 19 severe cases. Eur Neurol. 2000; 43(2):61–69.
171. Hirano, M, Ott, BR, Raps, EC, et al. Acute quadriplegic myopathy: A complication of treatment with steroids, nondepolarizing blocking agents, or both. Neurology. 1992; 42(11):2082–2087.
172. Laghi, F, Topeli, A, Tobin, MJ. Does resistive loading decrease diaphragmatic contractility before task failure? J Appl Physiol. 1998; 85(3):1103–1112.
173. Laghi, F, D’Alfonso, N, Tobin, MJ. Pattern of recovery from diaphragmatic fatigue over 24 hours. J Appl Physiol. 1995; 79(2):539–546.
174. Mador, JM, Rodis, A, Diaz, J. Diaphragmatic fatigue following voluntary hyperpnea. Am J Respir Crit Care Med. 1996; 154(1):63–67.
175. Travaline, JM, Sudarshan, S, Roy, BG, et al. Effect of N-acetylcysteine on human diaphragm strength and fatigability. Am J Respir Crit Care Med. 1997; 156(5):1567–1571.
176. McKenzie, DK, Bigland-Ritchie, B, Gorman, RB, et al. Central and peripheral fatigue of human diaphragm and limb muscles assessed by twitch interpolation. J Physiol. 1992; 454:643–656.
177. Moxham, J, Morris, AJ, Spiro, SG, et al. Contractile properties and fatigue of the diaphragm in man. Thorax. 1981; 36(3):164–168.
178. Bellemare, F, Bigland-Ritchie, B. Central components of diaphragmatic fatigue assessed by phrenic nerve stimulation. J Appl Physiol. 1987; 62(3):1307–1316.
179. Similowski, T, Straus, C, Attali, V, et al. Cervical magnetic stimulation as a method to discriminate between diaphragm and rib cage muscle fatigue. J Appl Physiol. 1998; 84(5):1692–1700.
180. Yan, S, Lichros, I, Zakynthinos, S, et al. Effect of diaphragmatic fatigue on control of respiratory muscles and ventilation during CO2 rebreathing. J Appl Physiol. 1993; 75(3):1364–1370.
181. Dahlstedt, AJ, Katz, A, Westerblad, H. Role of myoplasmic phosphate in contractile function of skeletal muscle: Studies on creatine kinase-deficient mice. J Physiol. 2001; 533(Pt 2):379–388.
182. Dahlstedt, AJ, Katz, A, Wieringa, B, et al. Is creatine kinase responsible for fatigue? Studies of isolated skeletal muscle deficient in creatine kinase. FASEB J. 2000; 14(7):982–990.
183. Westerblad, H, Lee, JA, Lannergren, J, et al. Cellular mechanisms of fatigue in skeletal muscle. Am J Physiol. 1991; 261(2 Pt 1):C195–C209.
184. Nichols, DG, Buck, JR, Eleff, SM, et al. Diaphragmatic fatigue assessed by 31P-magnetic resonance spectroscopy in vivo. Am J Physiol. 1993; 264(5 Pt 1):C1111–C1118.
185. Radell, PJ, Eleff, SM, Nichols, DG. Effects of loaded breathing and hypoxia on diaphragm metabolism as measured by 31P-NMR spectroscopy. J Appl Physiol. 2000; 88(3):933–938.
186. Westerblad, H, Bruton, JD, Lannergren, J. The effect of intracellular pH on contractile function of intact, single fibres of mouse muscle declines with increasing temperature. J Physiol. 1997; 500:193–204.
187. Zhu, E, Comtois, AS, Fang, L, et al. Influence of tension time on muscle fiber sarcolemmal injury in rat diaphragm. J Appl Physiol. 2000; 88(1):135–141.
188. Jiang, TX, Reid, WD, Road, JD. Delayed diaphragm injury and diaphragm force production. Am J Respir Crit Care Med. 1998; 157(3 Pt 1):736–742.
189. Efthimiou, J, Fleming, J, Spiro, SG. Sternomastoid muscle function and fatigue in breathless patients with severe respiratory disease. Am Rev Respir Dis. 1987; 136(5):1099–1105.
190. Cohen, CA, Zagelbaum, G, Gross, D, et al. Clinical manifestations of inspiratory muscle fatigue. Am J Med. 1982; 73(3):308–316.
191. Goldstone, JC, Green, M, Moxham, J. Maximum relaxation rate of the diaphragm during weaning from mechanical ventilation. Thorax. 1994; 49(1):54–60.
192. Tobin, MJ, Walsh, JM, Laghi, F. Monitoring of respiratory neuromuscular function. In: Tobin MJ, ed. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill; 1994:945–966.
193. Bellemare, F, Grassino, A. Effect of pressure and timing of contraction on human diaphragm fatigue. J Appl Physiol. 1982; 53(5):1190–1195.
194. Fitch, S, McComas, A. Influence of human muscle length on fatigue. J Physiol. 1985; 362:205–213.
195. Parthasarathy, S, Jubran, A, Laghi, F, et al. Sternomastoid, rib-cage and expiratory muscle activity during weaning failure. J Appl Physiol. 2007; 103:140–147.
196. Radell, PJ, Eleff, SM, Traystman, RJ, et al. In vivo diaphragm metabolism: Comparison of paced and inspiratory resistive loaded breathing in piglets. Crit Care Med. 1997; 25(2):339–345.
197. Sassoon, CS, Gruer, SE, Sieck, GC. Temporal relationships of ventilatory failure, pump failure, and diaphragm fatigue. J Appl Physiol. 1996; 81(1):238–245.
198. Adams, JM, Farkas, GA, Rochester, DF. Vagal afferents, diaphragm fatigue, and inspiratory resistance in anesthetized dogs. J Appl Physiol. 1988; 64(6):2279–2286.
199. Straus, C, Zelter, M, Derenne, JP, et al. Putative projection of phrenic afferents to the limbic cortex in humans studied with cerebral evoked potentials. J Appl Physiol. 1997; 82(2):480–490.
200. Hill, JM. Increase in the discharge of muscle spindles during diaphragm fatigue. Brain Res. 2001; 918(1-2):166–170.
201. Speck, DF, Revelette, WR. Attenuation of phrenic motor discharge by phrenic nerve afferents. J Appl Physiol. 1987; 62(3):941–945.
202. Marsden, CD, Meadows, JC, Merton, PA. Isolated single motor units in human muscle and their rate of discharge during maximal voluntary effort. J Physiol. 1971; 217(1):12P–13P.
203. Attali, V, Mehiri, S, Straus, C, et al. Influence of neck muscles on mouth pressure response to cervical magnetic stimulation. Am J Respir Crit Care Med. 1997; 156(2 Pt 1):509–514.
204. Peiffer, C, Poline, JB, Thivard, L, et al. Neural substrates for the perception of acutely induced dyspnea. Am J Respir Crit Care Med. 2001; 163(4):951–957.
205. Macefield, G, Hagbarth, KE, Gorman, R, et al. Decline in spindle support to alpha-motoneurons during sustained voluntary contractions. J Physiol. 1991; 440:497–512.
206. Zifko, UA, Young, BG, Remtulla, H, et al. Somatosensory evoked potentials of the phrenic nerve. Muscle Nerve. 1995; 18(12):1487–1489.
207. Corfield, DR, Fink, GR, Ramsay, SC, et al. Evidence for limbic system activation during CO2 breathing in man. J Physiol. 1995; 488:77–84.
208. Bernard, JF, Huang, GF, Besson, JM. The parabrachial area: Electrophysiological evidence for an involvement in visceral nociceptive processes. J Neurophysiol. 1994; 71(5):1646–1660.
209. Gozal, D, Hathout, GM, Kirlew, KA, et al. Localization of putative neural respiratory regions in the human by functional magnetic resonance imaging. J Appl Physiol. 1994; 76(5):2076–2083.
210. Stofan, DA, Callahan, LA, DiMarco, AF, et al. Modulation of release of reactive oxygen species by the contracting diaphragm. Am J Respir Crit Care Med. 2000; 161(3 Pt 1):891–898.
211. Appendini, L, Purro, A, Patessio, A, et al. Partitioning of inspiratory muscle workload and pressure assistance in ventilator-dependent COPD patients. Am J Respir Crit Care Med. 1996; 154(5):1301–1309.
212. Simpson, JA, van Eyk, JE, Iscoe, S. Hypoxemia-induced modification of troponin I and T in canine diaphragm. J Appl Physiol. 2000; 88(2):753–760.
213. Nava, S, Bellemare, F. Cardiovascular failure and apnea in shock. J Appl Physiol. 1989; 66(1):184–189.
214. Maldonado, A, Bauer, TT, Ferrer, M, et al. Capnometric recirculation gas tonometry and weaning from mechanical ventilation. Am J Respir Crit Care Med. 2000; 161(1):171–176.
215. Zakynthinos, SG, Vassilakopoulos, T, Roussos, C. The load of inspiratory muscles in patients needing mechanical ventilation. Am J Respir Crit Care Med. 1995; 152(4 Pt 1):1248–1255.
216. D’Angelo, E, Calderini, E, Robatto, FM, et al. Lung and chest wall mechanics in patients with acquired immunodeficiency syndrome and severe Pneumocystis carinii pneumonia. Eur Respir J. 1997; 10(10):2343–2350.
217. Gattinoni, L, Pelosi, P, Suter, PM, et al. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med. 1998; 158(1):3–11.
218. Parthasarathy, S, Jubran, A, Tobin, MJ. Cycling of inspiratory and expiratory muscle groups with the ventilator in airflow limitation. Am J Respir Crit Care Med. 1998; 158(5 Pt 1):1471–1478.
219. Covelli, HD, Black, JW, Olsen, MS, et al. Respiratory failure precipitated by high carbohydrate loads. Ann Intern Med. 1981; 95(5):579–581.
220. Kellog, RH. Central chemical regulation of respiration. In: Fenn WO, Rahn H, eds. Handbook of Physiology. Washington, DC: American Physiological Society; 1964:513.
221. Efthimiou, J, Mounsey, PJ, Benson, DN, et al. Effect of carbohydrate rich versus fat rich loads on gas exchange and walking performance in patients with chronic obstructive lung disease. Thorax. 1992; 47(6):451–456.
222. Pitcher, WD, Cunningham, HS. Oxygen cost of increasing tidal volume and diaphragm flattening in obstructive pulmonary disease. J Appl Physiol. 1993; 74(6):2750–2756.
223. Nuckton, TJ, Alonso, JA, Kallet, RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med. 2002; 346(17):1281–1286.
224. Costello, R, Deegan, P, Fitzpatrick, M, et al. Reversible hypercapnia in chronic obstructive pulmonary disease: A distinct pattern of respiratory failure with a favorable prognosis. Am J Med. 1997; 102(3):239–244.
225. Moser, KM, Luchsinger, PC, Adamson, JS, et al. Respiratory stimulation with intravenous doxapram in respiratory failure. A double-blind co-operative study. N Engl J Med. 1973; 288(9):427–431.
226. Sieck, GC, Prakash, YS. Cross-bridge kinetics in respiratory muscles. Eur Respir J. 1997; 10(9):2147–2158.
227. Aubier, M, Viires, N. Calcium ATPase and respiratory muscle function. Eur Respir J. 1998; 11(3):758–766.
*Terminology of dead space is confusing. Anatomic dead space is made up of the conducting airways (nose, mouth, pharynx, larynx, trachea bronchi, and bronchioles). Alveolar dead space is made up of alveoli that receive some or no blood flow, which does not match ventilation (units with very high 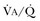 ratio). Physiologic dead space is the sum of anatomic dead space and alveolar dead space.1
ratio). Physiologic dead space is the sum of anatomic dead space and alveolar dead space.1
*The alveolar-arterial O2 gradient (A-aDO2) is calculated as PAO2 – PaO2, in which PAO2 (alveolar O2 tension) can be estimated according to the simplified alveolar gas equation:
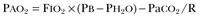
where FIO2 = fractional concentration of inspired O2 (about 0.21 when breathing room air), PB = barometric pressure (about 760 mm Hg at sea level), PH2O = water vapor pressure (usually taken as 47 mm Hg at 37° C), and R = respiratory exchange ratio of the whole lung. The respiratory exchange ratio (R) = CO2 production/O2 consumption (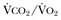 ). R is normally about 0.8. In steady state, R is determined by the relative proportions of free fatty acids, protein, and carbohydrate consumed by the tissues. In this equation, it is assumed that alveolar PCO2 and PaCO2 are the same (usually they nearly are). In healthy young subjects (≤30 years old) breathing air at sea level, A-aDO2 is usually less than 10 mm Hg, but it increases to as much as 28 mm Hg in healthy 60-year-old subjects.1
). R is normally about 0.8. In steady state, R is determined by the relative proportions of free fatty acids, protein, and carbohydrate consumed by the tissues. In this equation, it is assumed that alveolar PCO2 and PaCO2 are the same (usually they nearly are). In healthy young subjects (≤30 years old) breathing air at sea level, A-aDO2 is usually less than 10 mm Hg, but it increases to as much as 28 mm Hg in healthy 60-year-old subjects.1