[level-membership-for-critical-care-medicine-category]69
Hypothermia, Hyperthermia, and Rhabdomyolysis
Hypothermia
In order to recognize hypothermia, it must be possible to measure a temperature. The first thermometer for clinical use was invented in 1612. It was a foot long and required 20 minutes to register a temperature. The invention of the mercury thermometer by Fahrenheit in 1714 made the tool more practical. However, it was not until the late nineteenth century that the thermometer became a clinical tool. This use was established after the invention of a small thermometer that only required 5 minutes to obtain a temperature (1866) and after Carl Wunderlich’s publication (1868), which presented data on nearly 25,000 patients and analyzed temperature variation in 32 diseases. In the early 1900s, temperature was routinely measured and physiologic experiments started to appear. By the 1930s to 1940s, therapeutic hypothermia for malignancy and anesthesia was investigated. By the middle of the twentieth century, accidental hypothermia was established as a disease entity.1
Epidemiology
Accidental hypothermia continues to be a public health problem. The Centers for Disease Control and Prevention (CDC) reports that between 1979 and 2002, a total of 16,555 deaths in the United States (average of 689 per year, range 417-1021) were attributed to excessive natural cold.2
Approximately 50% of all hypothermia-related deaths occur in persons older than 65. The age-adjusted mortality rate is three times higher for men than it is for women. In blacks and other races, the mortality rate is higher than that of whites.2,3 Blacks have decreased heat production and lower rectal temperatures in response to cold exposure.4 Other populations at risk include the elderly, people with preexisting chronic medical conditions, and people suffering from alcoholism and drug intoxication.2,3,5 Emerging populations include wilderness enthusiasts and winter sport participants.6
Despite advances in medical care, hypothermia remains a challenge to health care providers, with in-hospital mortality rate reaching 40%,7 and an overall mortality rate ranging from 17% to 80%.8 The prompt recognition and appropriate management play a key role in improving outcomes.
Definition
Hypothermia is defined as a core body temperature lower than 35° C (<95° F). It has been further classified by severity according to the degree by which the core temperature has decreased from baseline into mild, moderate, and severe. Mild hypothermia is defined by a core body temperature of 32° to 35° C (90-95° F). Moderate hypothermia reflects a core body temperature of 28° to 32° C (82-90° F). Severe hypothermia is a core body temperature of less than 28° C (less than 82° F). Some reports refer to severe hypothermia below 30° C. In trauma patients, hypothermia carries a poorer prognosis. In this case, the definition is even more conservative, with severe hypothermia starting below 32° C. This subclassification has important implications regarding the anticipated physiologic changes and subsequently the use of appropriate therapeutic modalities.9,10
Heat Regulation
Body temperature is regulated through a balance between heat production and heat dissipation.11 Heat production occurs mostly from the metabolic activity of energy-consuming processes within the viscera, namely, the heart, liver, and brain. Heat production in these organs is estimated to be between 40 and 60 kcal/m2/hour. Normally heat production is increased by food intake and muscle activity. Different stresses such as fever, infection, and cold exposure also increase heat production.
Heat loss occurs through different forms. Radiation cooling (infrared emissions) accounts for approximately 60% of heat loss. It occurs primarily from the head and unisolated parts of the body. Conduction (direct transfer of heat to an adjacent cooler object) and convection (direct transfer of heat to convective air currents) account for around 10% to 15% of heat loss. Evaporation from the skin and respiratory tract accounts for 25% to 30%. Conduction is an important mechanism in immersion accidents because thermal conduction of water is 30 times that of air. Convection is important in windy conditions by removing the warm isolating layer of air around the body.6,10,11
Upon cold exposure, the hypothalamus initiates mechanisms for heat conservation and heat production. Heat conservation is achieved by peripheral vasoconstriction, reducing heat conduction to the skin, and behavioral responses such as wearing more clothes and seeking a warmer environment. Heat production is achieved through stimulation of muscular activity via shivering, which can increase the basal metabolic rate by two to five times. A nonshivering thermogenesis occurs via increased levels of catecholamines and thyroxine.12
This coordinated physiologic response occurs typically for temperatures between 32° and 35° C. It only lasts for few hours, after which muscle fatigue and glycogen depletion ensue. As the core temperature drops below 32° C (90° F), shivering thermogenesis abates, and nonshivering thermogenesis slows down. By temperatures below 20° to 24° C, the different mechanisms of heat production completely fail.13
Clinical Presentation
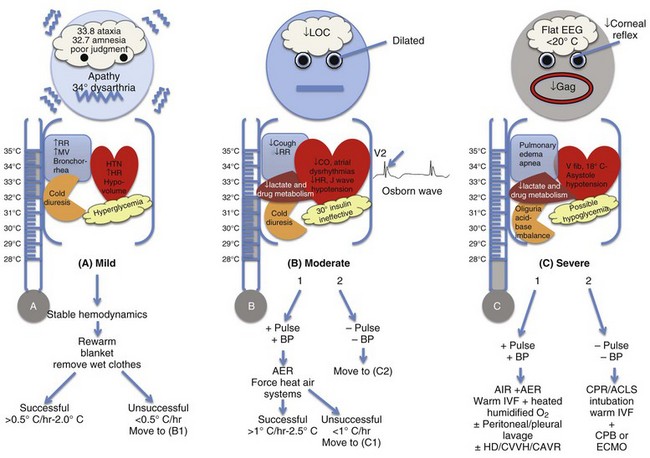
Etiologic Factors (Fig. 69.1)

1. Impaired thermoregulation is of critical importance in the elderly, mentally ill, and patients with chronic medical conditions such as Parkinson’s disease, stroke, multiple sclerosis, and diabetes mellitus. Such populations are often on multiple medications including anxiolytics, antidepressants, phenothiazines, barbiturates, opioids, antipsychotics, oral antihyperglycemics, beta blockers, and alpha blockers. These drugs can cause further impairment in thermoregulation.14 In addition, the elderly and chronically ill may suffer from maladaptive behavior (decreased mobility, and inability to communicate effectively), which increases the chance of remaining in an environment with prolonged cold exposure.14
2. Increased heat loss can be environmental or nonenvironmental.
a. Hypothermia commonly occurs during winter weather and in cold climates. People with inadequate insulation and continued cold exposure are especially at risk.
b. Patients with dermatologic dysfunction such as burns, psoriasis, and exfoliative dermatitis can have increased heat loss.
c. Large amounts of unheated intravenous fluid resuscitation, whether crystalloids or blood, coupled with unheated humidified oxygen, predispose hospital and trauma patients to greater heat loss.
d. Conditions that cause vasodilation should be highlighted in this category and include alcohol, anesthesia, and toxins released in disease entities such as sepsis.
3. Decreased heat production is noted in some specific types of disorders:
a. Endocrine dysfunction such as hypopituitarism, hypothyroidism, hypoadrenalism, and hypoglycemia.
b. Malnutrition and starvation, which cause decreased insulative subcutaneous fat and are often associated with hypoglycemia.
c. Conditions that alter the level of consciousness, causing decreased shivering mechanism. Drugs such as phenothiazines and tricyclic antidepressants as well as alcohol can induce impairment of shivering.6
Diagnosis
Diagnosis should be performed using a thermometer probe capable of measuring temperatures as low as 25° C (77° F). This instrument could be a rectal, esophageal, or bladder probe. It should be noted that standard clinical thermometers do not register below 34.4° C (94.4° F).15 Combined methods for temperature monitoring may allow better detection of further changes.
End-Organ Manifestations (Fig. 69.2)
Although hypothermia is divided into three stages, it is important to note that the physiologic changes follow a continuum. The initial mechanisms mimic intense sympathetic stimulation, which fades as body temperature drops. Almost every organ in the body is affected by hypothermia.16
Cerebral Effects
Hypothermia is associated with progressive decrease in neuronal metabolism between 35° C and 25° C. This protective effect of hypothermia allows better tolerance to periods of cardiocirculatory collapse in contrast with the normothermic state.6,17,18 Neurologic symptoms starts with mild hypothermia. Between 34° and 35° C, loss of fine motor skills, lack of coordination, dysarthria, and amnesia take place. Ataxia and apathy are noted around 33° C.19 Below 32° C, progressive decrease in level of consciousness and pupillary dilation develops. Cerebrovascular autoregulation is lost below 25° C. Deep tendon reflexes diminish and flaccid muscular tone takes place below 27° C. By 23° C, corneal and oculocephalic reflexes are absent. The electroencephalogram (EEG) pattern flattens at 19° to 20° C. The lowest temperature reported for an adult survival with hypothermia is 13.7° C.6,15,20 Therefore, one has to be very careful in assessing brain death when a patient remains hypothermic.8
Cardiovascular Effects
The initial response (34-36° C) includes increased heart rate, blood pressure, and carbon dioxide secondary to elevated catecholamine production and peripheral vasoconstriction.6 This is associated with marked increase in oxygen consumption. A core temperature decrease of as little as 0.3° C is associated with a 7% increase in oxygen consumption. The increase in oxygen requirement results in anaerobic metabolism, acidosis, and significant cardiopulmonary stress, further worsening the abnormal physiology of hypothermia.16 Below 34° C, progressive bradycardia and decrease in cardiac output and blood pressure take place. Below 28° C, 50% of patients show bradycardia with a 50% decrease in heart rate. Bradycardia is chiefly related to progressive decreased spontaneous depolarization of pacemaker cells. The conduction system is also affected, with prolongation initially noted in the PR interval, then QRS complex, then QT intervals.21 Atropine is ineffective. The myocardium becomes irritable. As temperature falls lower than 32° C, atrial fibrillation is common, usually preceding ventricular fibrillation that may occur spontaneously when temperature falls below 28° C (82° F). Asystole usually occurs at temperatures less than 25° C.22
Respiratory Changes
Early in hypothermia (34-36° C) the respiratory drive is stimulated. The minute ventilation increases in response to increased oxygen consumption and CO2 production. Below 33° C, respiratory depression takes place. Minute ventilation decreases. Depressed cough reflex and ciliary action coupled with increased production of mucus (bronchorrhea) increase the risk of atelectasis and aspiration. Noncardiogenic pulmonary edema has also been reported.23,24
Renal Changes
The initial response to hypothermia includes increased renal blood flow secondary to an increase in blood pressure and cardiac output, inducing diuresis. As hypothermia progresses below 32° C and cardiac output declines, renal blood flow and glomerular filtration rate decrease. This response sets the stage for acute renal failure but without affecting urinary output secondary to impaired renal tubular sodium reabsorption, a condition that maintains ongoing diuresis (cold diuresis). This response explains why hypothermia patients may present with profound intravascular volume depletion.6,16,25
Laboratory Evaluation
Arterial Blood Gases
As temperature decreases, the oxyhemoglobin dissociation curve shifts to the left. For every 1° C drop in temperature, pH increases by 0.015, PaO2 decreases by 7.2%, and PaCO2 decreases by 4.4%. Because all arterial blood gas (ABG) samples are warmed to 37° C (98.6° F) when measured, the pH appears lower and the PaO2 and PaCO2 appear higher than the patient’s actual values. Most knowledge on acid-base status during hypothermia comes from cardiovascular surgery reports. Although most literature does not recommend correction of temperature to guide therapy, the use of corrected values should be strictly individualized.26–28
Complete Blood Count
Hemoconcentration, an increase in hematocrit secondary to a decrease in plasma volume, is common (2% for each 1° C decrease in temperature). An initial low hemoglobin suggests acute hemorrhage or preexisting anemia. White blood cells (WBCs) may decrease; therefore, any history suggestive of infection should prompt treatment with antibiotics in high-risk groups such as the elderly, neonates, and the immunocompromised.29
Coagulation Factors
Hypothermia induces inhibition of coagulation factors. It also induces thrombocytopenia secondary to bone marrow suppression and splenic and hepatic sequestration, as well as reduction in platelet adhesions and aggregation. Prolonged bleeding time and clotting times are common. Coagulation tests in the laboratory are performed under 37° C. If normal, they may not negate the presence of underlying hypothermia-induced coagulopathy. The key treatment in this condition is rewarming, not supplementation of coagulation factors.30,31 Rewarming, however, increases the clotting response. Clinicians should use caution during this process, as patients are more susceptible to negative cardiovascular events.4
Blood Urea Nitrogen/Creatinine/Electrolytes
As mentioned earlier, acute renal failure with intravascular volume depletion and decreased renal perfusion is encountered. This is usually associated with higher levels of blood urea nitrogen (BUN) and creatinine (Cr) secondary to decreased clearance. Recurrent evaluation of electrolytes is essential during rewarming because no consistent pattern is present. Either hypo- or hyperkalemia may complicate the course of hypothermia and should be corrected promptly. Hypothermia masks the usual electrocardiogram (ECG) changes seen in normal hyperkalemia.6,8,16
Blood Glucose
Hyperglycemia is initially noted as hypothermia inhibits insulin release and insulin uptake by membrane receptors at temperatures less than 30° C. It is further exacerbated by increased catecholamines. Insulin at these temperatures is ineffective. Exogenous insulin should be avoided, as it may cause rebound hypoglycemia during rewarming.32
Electrocardiography Changes
All intervals—PR, QRS, and QT—are prolonged secondary to hypothermia-induced slowed impulse conduction through potassium channels. When body temperature decreases below 33° C (91.4° F), the J (Osborn) wave may be noted as a positive deflection in the left ventricular leads at the junction of the QRS complex and ST segment in 25% to 30% of patients. The presence of this wave is not pathognomonic and has no prognostic implication. The magnitude of the Osborn wave is associated with the degree of severity of hypothermia.12,21
Management (see Fig. 69.2)
The severity of hypothermia, clinical findings, and comorbid conditions of the patient are important factors for determining the aggressiveness of resuscitation techniques. Once hypothermia is confirmed, assessment and treatment of the critically ill patient should take place simultaneously. Some patients with hypothermia and cardiac arrest achieved an almost complete recovery despite prolonged resuscitation with a temperature as low as 13.7° C. Patients with moderate or severe hypothermia should be resuscitated until adequate rewarming is achieved before declaring efforts as unsuccessful.33–35
Airway/Breathing Support
Chest rigidity and decreased diaphragmatic movement impede adequate ventilation. Oxygenation may be insufficient secondary to underlying aspiration or pulmonary edema. Supplemental oxygen should be provided to alert patients with intact airway reflexes pending full assessment of oxygenation status. Hypothermic patients with apnea, deteriorating mental status, or loss of protective reflexes should undergo endotracheal intubation. The oropharyngeal route is preferred. Jaw maneuvering could be difficult in this category of patients secondary to muscle rigidity. Neuromuscular blocking agents do not have a role as they are ineffective at temperatures below 30° C. Intubation with a smaller endotracheal tube may be attempted. Intubation is unlikely to induce dysrhythmias in hypothermic patients.8
Cardiocirculatory Support
Volume Resuscitation
Most patients with moderate to severe hypothermia suffer from volume depletion. Continuous warmed intravenous fluids (40-42° C) should be administered. Peripheral large-gauge catheters are the preferred route of administration. The femoral vein is the preferred site if a central line is anticipated. Volume status should be monitored continuously during rewarming, as peripheral vasodilation and hypotension are likely to occur.36
Cardiac Resuscitation
Patients with moderate to severe hypothermia should undergo a thorough evaluation, looking for signs of cardiac output or peripheral blood flow. Echocardiography and Doppler ultrasound can be utilized for this purpose. Cardiopulmonary resuscitation should be performed immediately if no pulse or a nonperfusing rhythm (ventricular fibrillation or asystole) is established.37–39
Defibrillation.
The hypothermic heart may be unresponsive to cardioactive drugs, attempted electric pacing, and defibrillation. Most arrhythmias other than ventricular fibrillation tend to convert spontaneously with rewarming. Sinus bradycardia does not require pacing or pharmacologic intervention. Ventricular fibrillation or tachycardia without a pulse requires initial defibrillation with maximum energy. If unsuccessful, rewarming is initiated. Defibrillation is reattempted after every 1° to 2° C increase in core temperature or when temperature rises above 30° to 32° C.37–39
Cardioactive Drugs.
The antiarrhythmic and vasoactive drug efficacy in moderate to severe hypothermia is not well determined. It is mainly based on animal studies. Epinephrine and vasopressin may increase coronary perfusion but not survival. According to the American Heart Association 2010 guidelines, it may be reasonable to consider administration of a vasopressor during cardiac arrest according to the standard ACLS (advanced cardiac life support) algorithm concurrent with rewarming strategies. Isolated use of antiarrhythmics does not appear to be beneficial. In general, these drugs are withheld until rewarming achieves a temperature above 30° C, and then the lowest effective dose should be administered. Caution should be exercised not to administer these drugs repeatedly, as toxic accumulation can occur secondary to decreased metabolism. As normothermia is approached (above 35° C), standard drug protocols are used.37–39
Rewarming Methods
There are several methods of rewarming. Selection depends on the severity of hypothermia and the clinical condition of the patient. Controlled clinical studies and interventional studies are limited, and evidence-based protocols are lacking. Absence of a developed protocol led to the use of a wide variety of treatment strategies, depending on the expertise and previous exposure of the clinical teams and hospital resources.40
Passive External Rewarming
Passive external rewarming is the method of choice for hemodynamically stable patients with mild hypothermia (above 32° C), adequate physiologic reserve, and intact mechanism of shivering. Treatment includes placement of patients in a warm environment, removal of wet clothing, and application of insulating materials, such as blankets, to prevent heat loss. This method is expected to raise core temperature by 0.5° to 2.0° C per hour.6 If unsuccessful, active rewarming is initiated.
Active Rewarming
Active External Rewarming.
Active external rewarming (AER) is applied to patients with a core temperature between 86° and 92° F (30-34° C) without potential compromise of airway or hemodynamic status. It involves one or a combination of techniques, including warming blankets, heating pads, radiant energy, and forced heated air systems. Few complications (such as thermal injuries in the form of skin burns) have been reported using these methods. “After drop” is a phenomenon that was reported as a complication of AER. When the extremities and the trunk are warmed simultaneously, cold acidemic blood returns to the central circulation from the periphery secondary to vasodilation, causing a paradoxical decrease in core temperature. This drop worsens acidemia, precipitating hypotension and arrhythmias that can be fatal. Therefore, it is advised to apply rewarming strategies to the trunk before the extremities.41,42
Estimated rates of rewarming using this method ranges from 0.8° C with heating blankets up to 2.5° C per hour with the forced air rewarming systems. This forced air system is the most widely used by clinicians because it is easy and effective.41,42
Active Internal Rewarming.
Active internal rewarming (AIR) is used for patients with moderate to severe hypothermia (under 86° F or 30° C) with adequate hemodynamics, in conjunction with AER.43 Methods include administration of warmed intravenous fluids (40-42° C) and warmed humidified air (40-45° C) through a mask or endotracheal tube. Intravenous fluids alone are not adequate but prevent further heat loss. It is estimated that an average 1 L crystalloid (40-42° C) for a 70-kg person will increase the temperature by 0.3°. Rewarming by heated oxygen by endotracheal tube is more effective than a facemask. A total rate of 1° to 2.5° C per hour is anticipated by this method.15,43,44 Gastric bladder and colonic irrigation are of limited value secondary to their small surface area.
Peritoneal and Pleural Lavage
If rewarming is inadequate, more aggressive methods include peritoneal and pleural lavage. In pleural irrigation, two large-bore (36F or greater) thoracotomy tubes are used. One tube is placed at the midclavicular line and is connected to saline at 42° C (107.2° F). The other tube is placed at the posterior axillary line and is connected to a chest tube drainage.45
Peritoneal lavage and dialysis are achieved by placement of a peritoneal catheter (8F) into the peritoneum and infusing 10 to 20 mL/kg of warmed 42° C isotonic fluid. The fluid is kept in the peritoneal cavity before it is removed.15
Extracorporeal Methods of Rewarming
This approach is the most effective method in raising core body temperature.
Hemodialysis.
Hemodialysis is appropriate for patients with severe hypothermia and accompanying hyperkalemia and renal failure. The benefit is that hemodialysis is widely available and practical. It is portable and efficient. The rewarming rate ranges between 2° and 4° C per hour, but it requires the absence of circulatory collapse.46
Cardiopulmonary Bypass.
Cardiopulmonary bypass (CPB) is the standard of care for patients with severe hypothermia, apnea, and cardiac arrest or hemodynamic instability. It provides rapid rewarming rates (8-12° C per hour) while maintaining oxygenation, circulation, and rapid correction of metabolic derangements. However, the need for CPB in hypothermic trauma patients may be limited secondary to the need for systemic anticoagulation. In addition, the expert teams required for CPB make the implementation somewhat challenging.6,15,38 Portable CPB systems are available.
Extracorporeal Membrane Oxygenation.
There is a growing body of evidence that extracorporeal membrane oxygenation (ECMO) could be more preferable to CPB. It has the same rate of rewarming (8-12° C per hour).38 In patients undergoing rewarming, ECMO may reduce the risk of multiorgan failure secondary to reperfusion. It can be continued for several days and needs less anticoagulation.
Prognosis
Different reports on the outcome of moderate to severe hypothermic patients have been variable. Multiple confounding factors exist that make it difficult to predict survival. Older age, lower core body temperature, indoor exposure, submersion, trauma, toxin ingestion, prehospital hemodynamic status, BUN and potassium levels, the need for intubation, and presence or absence of asphyxiation are significant predictors of outcome. For patients undergoing CPB for hypothermia and hemodynamic instability, favorable factors for survival are associated with younger age, higher arterial pH, lower PaCO2, lower serum potassium levels, and the absence of asphyxia and suffocation.18 Many patients die even after achieving adequate rewarming secondary to multiorgan failure. Knowing its detrimental outcome, an important approach should pursue prevention. In general, hypothermia is a preventable condition. Public health education and preparedness should be directed toward populations at risk.
Hyperthermia
History and Incidence
Heatstroke and hyperthermia have been identified as the environmental diseases least frequently monitored by epidemiology systems in the United States. Serious heat illness has received considerable recent attention owing to catastrophic heat waves in the United States and Europe, the deaths of high-profile athletes, and military deployments. The exact incidence of heatstroke in the United States is unknown. The CDC reported that between the years 1987 and 1988, 1092 death certificates listed excessive heat as either a primary or a secondary cause.47 It is suspected that a number of other heat-related severe illnesses and deaths occur each year that are attributed to other causes. Guidelines from the National Association of Medical Examiners suggest that in cases in which the measured antemortem body temperature at the time of collapse was 105° F (40.6° C) or higher, the cause of death should be certified as heatstroke or hyperthermia. Deaths also may be certified as heatstroke or hyperthermia with lower body temperatures when cooling has been attempted before arrival at the hospital or when the clinical history includes mental status changes and elevated liver and muscle enzymes.48
Pathogenesis
Heatstroke is a complex syndrome of end-organ dysfunction initiated by hyperthermia that occurs when heat accumulation in the body overwhelms heat-dissipating mechanisms. The diagnosis of heatstroke should be considered in patients with core body temperatures greater than 41° C, or in patients with a core temperature of 40.6° C and concomitant mental status changes. Because anhidrosis is a late finding and sweating may be observed in severely hyperthermic patients, diaphoresis should not be considered a discriminatory finding. Heatstroke traditionally is subdivided into exertional and nonexertional causes. Box 69.1 outlines examples of diseases and conditions within each of these categories. To understand the pathogenesis of heatstroke, the systemic and cellular responses to heat stress must be appreciated. These responses include thermoregulation (with acclimatization), an acute-phase response, and a response that involves the production of heat shock proteins.
Acclimatization
Acclimatization is a physiologic term that collectively describes various adaptive responses to repeated heat stress. Many of these responses are directed at conserving plasma volume. They include decreased total sweat sodium concentration and nonreflexively increased aldosterone secretion. In addition, acclimatized people tend to drink greater volumes of water and have measurable increases in extracellular fluid volume beyond that of nonacclimatized persons. Ultimately, maximum cardiac output and stroke volume increase, while maximum heart rate and O2 consumption decrease (improved efficiency per unit of metabolic work expended).49 Similarly, the net amount of heat generated per unit of work expended also decreases.
Predisposing Factors
The epidemiology of heatstroke parallels that of hypothermia—nonexertional heatstroke is seen in elderly, alcoholic, or otherwise debilitated patients in urban areas. By contrast, exertional heatstroke develops in otherwise healthy people who overexert themselves in exceedingly hot or humid conditions. Like hypothermia, heatstroke most often develops in patients with impaired thermoregulation. Antidopaminergic drugs, alcohol, and primary central nervous system (CNS) disorders are common denominators in many patients.49 A number of environmental and socioeconomic conditions have been associated in recent years with heatstroke.50,51 A lack of air conditioning and a lack of trees or shrubbery around homes are commonly observed in patients presenting with heatstroke. People who are apartment bound, particularly those on higher floors, also appear to be at higher risk during summer heat waves. Finally, elderly patients not only have diminished temperature perception but also do not produce the same volume of sweat, placing them at higher risk for the development of heatstroke.
Presentation and Clinical Manifestations
Nonexertional heatstroke may manifest insidiously.52 An urban setting and the recognized presence of a heat wave may be the best available diagnostic clues. Some form of CNS dysfunction is nearly universal in patients with nonexertional heatstroke. The spectrum of dysfunction ranges from increased irritability and confusion to stupor and coma. Hyperpyrexia often is the most specific physical finding. In many cases, the presentation of an elderly patient with an altered mental status, irritability, and a high fever suggests sepsis. However, epidemiologic associations, concurrent debilitating disease, and the impact of medications that limit thermoregulation should be considered before limiting therapy to empiric antibiotics and antipyretic medications.
Exertional heatstroke typically occurs more suddenly and with a more self-evident clinical history.53 Severe dehydration, anhidrosis, and extreme hyperpyrexia are common in external heatstroke. Affected persons appear hyperdynamic, with tachycardia, increased cardiac output, and peripheral arterial vasodilation. CNS dysfunction is somewhat less common in this patient subset. Initially, thermoregulation is intact in patients with exertional hyperthermia. At high core body temperatures, however, metabolic activity overwhelms normal heat dissipation mechanisms, and clear-cut disruption of normal thermoregulation may then appear. Hyperthermia is further accelerated by the loss of effective sweating for evaporative heat dissipation. Myocardial dysfunction with depression of left ventricular ejection fraction also has been described after heatstroke, which resolves with treatment.54
In addition to alterations of consciousness, hallucinations, focal neurologic deficits, various cranial nerve abnormalities, and opisthotonos are well described. Seizures occur in greater than half of these patients and may be related strictly to core body temperature or to concomitant disorders of free water-electrolyte balance. Decerebrate posturing also is rarely observed in some patients without other known mechanisms of acute brain injury. Many patients may suffer residual defects such as dementia, personality changes, focal deficits, cerebellar changes, or pyramidal findings. Nevertheless, many patients also proceed to complete recovery, suggesting that early findings should not deter acute management.51
Diagnostic Approach
Acute renal failure occurs in up to one third of patients with heatstroke.55 It is considerably more common in patients in whom rhabdomyolysis is a feature (i.e., with myoglobinuria or urate nephropathy). However, direct heat injury and renal hypoperfusion also may contribute. Acute tubular necrosis in heatstroke typically manifests with oliguria, non-nephrotic range proteinuria, and abundant granular cast formation.
Evidence of hepatocellular injury is present to some degree in essentially all patients with heatstroke and is attributed principally to a direct toxic effect of hyperthermia on hepatocytes.56 Elevation of serum transaminases is a cardinal feature of this toxicity and often is observed within 30 minutes of syndrome onset. So prevalent is this finding that the absence of transaminitis should cast serious doubt on the diagnosis of heatstroke. Histologic changes including centrilobular necrosis have been observed within 24 hours of heat injury and evolve over the ensuing days. Transaminase and lactate dehydrogenase levels typically peak on the third or fourth hospital day, and depending upon the extent of injury, these changes may be accompanied by increased alkaline phosphatase levels, hyperbilirubinemia, and a prolonged prothrombin time. Overall, fulminant hepatic necrosis is rare; however, hepatic insufficiency contributes to late morbidity and death in patients who survive the initial resuscitation.57 Prolongation of coagulation times also may be due to disseminated intravascular coagulation (DIC). Although DIC also is rare, it is a marker of poor prognosis when present. Clinical manifestations of DIC range from isolated laboratory abnormalities to generalized bleeding. In patients with heatstroke, DIC may exacerbate hepatic injury and is associated with the development of acute respiratory distress syndrome (ARDS). In fact, the codevelopment of DIC and ARDS in patients with heatstroke is predictive of a high mortality rate (>75%). Qualitative coagulation function (including platelet function) also may be impaired by heat injury. The clinical effects of these impairments are variable.
A range of electrolyte abnormalities related to renal failure and dehydration in heatstroke victims has been well described and includes hyponatremia, hypocalcemia, hypokalemia (hyperkalemia in patients with acute tubular necrosis or rhabdomyolysis), hypophosphatemia, and hypomagnesemia. Of these, hyponatremia and free water excess may produce severe neurologic effects including central pontine myelinolysis (CPM). CPM has been reported in patients with heatstroke who receive aggressive prehospital resuscitation with hypotonic solutions. Serum osmolality should be carefully monitored during volume resuscitation, and intravenous solutions should be modified accordingly. Severe lactic acidosis is especially common in patients with exertional heat injury.58 Finally, arterial blood gas results are influenced by temperature in a fashion opposite that described with hypothermia (Box 69.2).
Approach to Management
Duration of extreme hyperpyrexia is the single most important determinant of morbidity and fatality in all patients with heatstroke (both exertional and nonexertional). Therefore, expeditious cooling should be initiated simultaneously with other basic and advanced life support modalities. Indeed, a review of outcome data for heatstroke victims during the Chicago heat wave of 1995 suggested that computed tomography imaging of the brain contributed little to patient management and simply delayed efforts to rapidly cool the patient.59 General supportive measures and specific measures for cooling the patient are discussed next.
Supportive Measures
Intravascular volume restoration must be individualized. In general, patients with exertional hypothermia are severely hypovolemic. Hemodynamic monitoring has demonstrated that patients may manifest two distinct responses to heatstroke: (1) a hyperdynamic response with an elevated cardiac index (CI) and depressed systemic vascular resistance (SVR) and (2) a hypodynamic response with depressed CI and elevated SVR.55 Although the hypodynamic response may be more common in older patients with preexisting medical problems and classic heatstroke, severe hyperthermia can lead to myocardial dysfunction even in young, healthy adults without preexisting cardiac disease. Overzealous volume resuscitation in this setting can produce significant pulmonary congestion. The rapid restoration of an adequate perfusion pressure without pressor support is a reasonable goal, and if this is achieved, further volume resuscitation should proceed at a more measured pace. Pulmonary artery catheterization is recommended early in the clinical course for patients with unclear cardiovascular physiology. Finally, isoproterenol traditionally has been considered the inotrope of choice for patients with severely depressed myocardial performance. Its lack of α-activity ensures that heat dissipation is not impeded. Dobutamine may be a more rational choice, however, because it has considerably fewer myocardial irritant and arrhythmogenic properties.
Cooling Measures
Evaporative techniques are quite effective and allow the ICU patient to be placed on a bed or other supportive surface and treated accordingly. The patient is repeatedly wetted with tepid water (not alcohol) or sprayed with water mist while warm air from a fan is blown across the body surface. Evaporative cooling by this method has shown a faster cooling rate in a canine model that is as effective as aggressive peritoneal lavage cooling, reaching rates as high as 0.32° C per minute.60 An aggressive approach to cooling the heatstroke victim, whatever the approach, is most important. Other adjuncts to cooling include rectal, intraperitoneal, and gastric lavage with iced saline. These techniques are effective at rapidly lowering core temperature but are cumbersome and generally unnecessary and may lead to unwanted iatrogenesis.
Efforts to cool a patient should be discontinued when the core temperature falls to 38° to 39° C. At that point, a continued fall by 1° or 2° C is expected. Dantrolene sodium has been used in some patients but was found to be ineffective in a double-blinded randomized study.61,62 Chlorpromazine may be useful in patients in whom shivering develops during active cooling efforts; however, if neuroleptic malignant syndrome (NMS) is in the differential diagnosis (and it often is), meperidine may be a better option than an antidopaminergic agent in patients with normal renal function.
Malignant Hyperthermia
MH develops in approximately 1 of 15,000 patients who undergo surgical procedures requiring general anesthesia. The vast majority of cases are associated with either halothane or succinylcholine use; however, a variety of other drugs also have been implicated. Box 69.3 outlines these agents by type. Unlike in heatstroke, endogenous heat production is solely responsible for the observed hyperpyrexia. MH constitutes a true medical emergency because of the rapid tempo at which severe sequelae evolve. Better recognition of this syndrome and modern treatment techniques, however, have reduced mortality rates from 70% to approximately 10% in recent years.
Pathogenesis
MH results from a rapid, sustained increase in myoplasmic Ca2+ levels in response to halogenated anesthetics and depolarizing neuromuscular blocking agents. Susceptibility to MH results from mutations in calcium channel proteins that mediate excitation-contraction coupling, with the ryanodine receptor calcium release channel (RyR1) representing the major locus. The mode of inheritance appears to be autosomal dominant, with variable penetrance. The phenomenon occurs in persons who have a mutation in the ryanodine type 1 receptor, resulting in a defective protein in the skeletal muscle sarcoplasmic reticulum membrane.63 The uncontrolled rise in the myoplasmic Ca2+ concentration disables the troponin inhibition of actin and myosin, resulting in uncontrolled muscle tetany. This surge in muscle metabolic activity causes thermogenesis to increase exponentially and is rapidly followed by total body rigidity and extreme hyperpyrexia.
Numerous familial myopathies (such as Evans myopathy, King-Denborough syndrome, and central core disease) have been associated with the reaction.64,65 Curiously, this genetic predisposition does not translate into the predictable development of MH. That is, an initial challenge with an anesthetic drug that leads to MH does not consistently predict whether or not MH will occur with future exposures to the same drug. Measured contraction in caffeine or halothane preparations of muscle biopsy specimens is the most reliable predictor of risk for MH.
Presentation
MH is characterized by the triad of (1) severe hyperthermia, (2) muscle rigidity, and (3) metabolic acidosis. MH usually is an acute and rapidly progressive process; however, many reports suggest that the syndrome may vary considerably in severity and rate of progression. In the anesthesia setting, capnography may provide the earliest clue of impending MH, as end-tidal CO2 rises in response to the increased metabolic rate. Clinical harbingers of impending MH include masseter muscle rigidity or trismus and tachycardia, which is reported to occur in greater than 95% of patients.64 Cyanosis, increased blood pressure, and increased respiratory rate all may be observed with the onset of the reaction as the hypermetabolic response is triggered.
Diagnosis
Susceptibility to MH can be identified unequivocally only by an in vitro muscle test. Fascicles of muscle obtained from the thigh by biopsy are exposed to halothane and separately to increasing concentrations of caffeine in vitro. The muscle contractures are increased in persons who are susceptible to MH. This test is now widely used to identify susceptibility to MH in patients who have had a reaction to inhaled halogenated anesthetics or suxamethonium that is suggestive of MH. If the test results in the propositus are positive, testing can be offered to first-degree relatives for genetic counseling. Patients whose biopsied muscle demonstrates a contracture to 3% halothane of 0.5 g or more and a contracture to 2 mmol/L caffeine of 0.2 g or more are considered to be susceptible to MH.66 An in vitro contracture test is the gold standard for diagnosing susceptibility to MH. Two different protocols are used. European laboratories use the protocol devised by Ellis in Leeds with incremental doses of halothane up to 2% and incremental doses of caffeine. In the United States, a single dose of 3% halothane and incremental doses of caffeine are used. The two protocols give essentially the same results.67
Neuroleptic Malignant Syndrome
NMS is an idiosyncratic reaction to neuroleptic drugs and other antidopaminergic agents. A number of drugs that are known to be associated with NMS are listed in Box 69.3. NMS is characterized by muscle rigidity and altered mental status, followed by autonomic instability and hyperthermia. However, temperature elevations generally are less extreme than in MH and are not as life-threatening.
Pathophysiology
Some investigators suspect that the genetic predisposition to MH also defines patients at risk for NMS, but most work has failed to demonstrate a relationship between NMS and altered sarcolemmal membrane calcium permeability. Unlike in MH, patients with NMS appear to have both increased heat production and impaired central thermoregulation, compromising heat dissipation. In these patients, a decrease in central dopaminergic tone also results in extrapyramidal signs of skeletal muscle rigidity and tremor and may account for alterations in mental status due to effects in the mesolimbic system and mesocortical pathways.68 Finally, even though altered central thermoregulation may contribute to hyperthermia in NMS, rapid resolution of fever after administration of neuromuscular relaxing agents or dantrolene suggests that hyperthermia is due primarily to increased heat generation by skeletal muscle rigidity.
Presentation
NMS should be considered when fever, muscular rigidity, catatonia, or dystonia occur in the setting of neuroleptic drug administration. Recent reviews have suggested the following criteria for the diagnosis of NMS: treatment with neuroleptics within 7 days; hyperthermia with core temperatures greater than 38° C; muscle rigidity; exclusion of other systemic or drug-related illness; and any five of the following: altered mental status, tachycardia, hypertension or hypotension, tachypnea or hypoxia, diaphoresis or sialorrhea, tremor, incontinence, CK elevation or myoglobinuria, leukocytosis, or metabolic acidosis.68,69
Treatment
Dopamine agonists such as bromocriptine, amantadine, and levodopa-carbidopa have been successfully employed in NMS.68,70 These agents act to directly increase central dopaminergic tone, thereby antagonizing the various effects of neuroleptic dopamine blockade. Regimens include bromocriptine or amantidine for up to 10 days before tapering, If a levodopa-carbidopa combination is used, it should be dosed four times a day owing to its shorter half-life. Some authors advocate avoiding central dopamine agonists if the diagnosis is unclear or if psychosis remains in the differential diagnosis, to avoid acutely exacerbating the psychotic state or contributing to a lethal catatonia.
Sodium dantrolene has been successfully used in numerous small series. The muscle relaxation that occurs helps to reduce temperature and decrease the sequelae of prolonged rigidity. Neuromuscular blockers also are effective at decreasing skeletal muscle tone, and in patients with high fever and autonomic instability, these agents offer a rapid means of establishing therapeutic effects. Combinations of central dopamine agonists and peripheral-acting agents have not as yet demonstrated a significant advantage over either treatment alone. Electroconvulsive therapy (ECT) has been used in a number of reports, but its advantages over supportive care alone have not been adequately assessed.70 Furthermore, the need for anesthesia presents an additional hazard that may complicate management in these patients.
Rhabdomyolysis
Rhabdomyolysis is a pathologic state in which the necrosis of muscle cells, through trauma, toxins, medications, or other causes, leads to an excess of intracellular solutes in the extracellular compartments. Target organ damage can range from minimal to severe depending on the degree of necrosis; the main consequences are metabolic abnormalities and acute kidney injury (AKI). Our understanding of the pathophysiology of rhabdomyolysis has evolved with an emphasis on early diagnosis and treatment, often in the setting of major catastrophes. The first report in the English literature, describing four cases of crush injury in Britain during World War II, was recently republished.71
Epidemiology
The incidence of rhabdomyolysis is dependent on the population undergoing analysis and the diagnostic criteria being used. It is recognized as a major cause of fatality following severe musculoskeletal injury from trauma.72 Following the 1999 Marmara earthquake in northwestern Turkey, 8.9% of hospitalized patients required renal replacement therapy for crush-related injuries.73 Recently, a particular focus has been the ability of the HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase) inhibitor class of medications to cause rhabdomyolysis. Though likely a class phenomenon and rare overall, the incidence of rhabdomyolysis is not equivalent for all medications and is increased with the addition of other specific agents, in particular gemfibrozil, other fibrates, or cyclosporine.74,75 Additional causes of rhabdomyolysis are listed in Box 69.4.76–81 A recent review noted exogenous toxins, both prescribed and illicit, to be responsible for nearly 50% of cases.82
Two additional scenarios that have received significant attention include “propofol infusion syndrome” and the potential for patients with sickle cell trait (SCT) to develop rhabdomyolysis. Both of these situations occur in younger patients. Propofol is rarely associated with a syndrome of metabolic acidosis and rhabdomyolysis. Risk factors include high-dose infusions over a prolonged period and a young age.83–86 SCT with subsequent rhabdomyolysis due to exertional sickling is now recognized as a cause for sudden deaths in young patients during strenuous workouts, in particular, football. 87–93
The epidemiology of rhabdomyolysis is limited by a lack of uniformity regarding the critical value in CK elevation needed to make the diagnosis. Threshold values above which rhabdomyolysis may occur vary and have ranged from 1000 to 10,000 U/L, though 5000 to 10,000 U/L are most common.94 From an intensivist’s perspective, CK elevations less than 5000 U/L should not be associated with myoglobin-induced renal failure unless clear additive causes such as volume depletion, sepsis, or radiocontrast are also present.
Pathophysiology
The primary event in rhabdomyolysis is muscle cell necrosis resulting in the accumulation of sodium and calcium intracellularly; disruption of the cell membrane and excessive requirements for adenosine triphosphate accelerate this insult.95,96 As muscle cells undergo necrosis, intracellular substances are released and include potassium, uric acid, phosphorus, lactic acid, and myoglobin. Under normal circumstances, haptoglobin binds any free myoglobin, thus prohibiting filtration across the glomerulus. As the haptoglobin binding is overwhelmed, free myoglobin is filtered and injures renal tubular cells. This process involves free radical toxicity. Additional causes include volume depletion, renal vasoconstriction, and tubular obstruction.76,97 Myoglobin and urinary tubular-protein casts are more prone to precipitation at a lower pH in experimental models. As a result, inducing an alkaline diuresis has been a traditional part of therapy for cases of rhabdomyolysis.98
Research has suggested that it is the reperfusion of ischemic muscle as much as the ischemia itself that produces a high burden of oxygen-derived free radicals with subsequent cellular lipid membrane injury.99 As renal function worsens, particularly when accompanied by intravascular volume depletion and renal failure, patients cannot excrete the high load of metabolites associated with muscle death, especially potassium.
Myoglobin release occurs across a spectrum of physical injuries from moderate exercise to lethal crush injuries. Myoglobinemia was noted in 39% of a cohort of Marine recruits, illustrating the frequency of mild muscle cell necrosis with strenuous exercise.100 Obviously, not all such injuries lead to AKI. Beyond the actual amount of muscle necrosis, the development of AKI is most dependent on the degree of volume depletion present during the initial insult and the rapidity with which it is restored. Many of the syndromes that lead to the development of rhabdomyolysis, such as very strenuous exercise, crush injuries, or intoxications, are accompanied by volume depletion.
Clinical Presentation
Patients with rhabdomyolysis present with a variety of symptoms. Often there is a history of injury, weakness, or muscle pain. Dark red or brown urine may or may not be present, depending on the amount of muscle necrosis and delay from injury. Frequently, the history is limited because of altered sensorium; clues include unexplained acidosis, hyperkalemia, hyperuricemia, hypocalcemia, elevations in CK not consistent with myocardial infarction, and unexplained renal failure. Recurrent presentations warrant an evaluation for a metabolic myopathy.101 Screening for rhabdomyolysis, through serial CK measurements, may be worthwhile in high-risk obese patients (body mass index over 56 kg/m2) undergoing gastric bypass surgery.102
In most instances, serum CK will increase during the initial 48 hours of hospitalization. In a retrospective review of patients admitted to an ICU with rhabdomyolysis, defined as a CK greater than 10,000 U/L, significantly higher CK levels were associated with renal failure.103 In the group with renal failure, the mean CK levels were 47,194 and 55,366 U/L at admission and peak, respectively; in the group without renal failure, the same levels were 17,531 and 28,643 U/L.
Although myoglobin is the most important protein in terms of nephrotoxicity, serum CK is routinely used in clinical practice as a marker of disease severity. This inconsistency is due to the widespread availability of the CK assay. Myoglobin clearance is not dependent on the kidneys and thus levels peak before CK levels.104 Consequently, the CK levels being monitored for disease activity may not correlate with the timing of the renal injury. AKI due to peak CK levels less than 5000 U/L should not be attributed to rhabdomyolysis unless the diagnosis was made substantially after the peak period of muscle cell necrosis; in this scenario, the CK levels will have decreased from a previously undocumented level.
The change of serum Cr in nonrhabdomyolysis oliguric AKI is usually not more than 1 mg/dL/24 hours. In rhabdomyolysis, Cr enters the intravascular compartment at a higher rate due to cell lysis, yielding an unusually rapid rise in serum Cr. In an early report, Grossman and associates noted a rise in Cr frequently greater than 2.5 mg/dL/day.105 Speculation exists that this change in Cr may, in part, be due to the larger than average size in patients affected with rhabdomyolysis.106 These same authors noted the impressive rate of renal recovery from rhabdomyolysis-induced AKI seen in the majority of patients; this high rate of recovery is now well recognized.
Specific Treatment Requirements
The most important intervention to limit renal complications of crush-related injuries is early and aggressive volume resuscitation. Delays in initiation of intravenous fluid are associated with a higher frequency of patients requiring renal replacement therapy.107 The damaged muscle cells provide a large reservoir for fluid sequestration; as a result large volumes of intravenous fluids have to be given and, ideally, should be instituted as early as possible, even on-site before victims of crush injuries are freed.78,108 A debate exists in the medical literature regarding the ideal type of volume resuscitation. As previously mentioned, alkaline diuresis has been shown in animal models to decrease the formation of myoglobin casts and subsequent AKI.98 Similarly, mannitol is an agent favored for its proposed ability to scavenge free radicals and to reduce tissue swelling due to osmotically induced fluid removal from damaged muscle.109,110 These treatments have not been validated in well-designed, prospective trials. Retrospective studies suggest no benefit with the addition of mannitol or alkaline diuresis over normal saline alone.111 In addition, there are potential risks to volume expansion with bicarbonate-based solutions or mannitol. They include the increased risk of calcium-phosphate complex formation and hypocalcemia from an increase in pH and the development of a hyperosmolar state when renal failure limits mannitol excretion. Finally, mannitol may increase the risk of renal failure in certain populations.112 Risk factors for mannitol-induced AKI include kidney dysfunction, high-dose mannitol therapy, and use of cyclosporin A. In summary, volume expansion irrespective of the type of agent is the most important treatment modality. The focus on adequate diuresis is appropriate but presumes myoglobin-induced renal damage is ongoing. Intensivists often encounter patients whose myoglobin load is falling and whose renal insult has already occurred. Though the Cr may continue to rise, the onset of oliguria suggests limited benefit from further aggressive volume resuscitation, presuming adequate intravascular volume has been restored. In fact, given the good prognosis of AKI, overzealous volume resuscitation risks pulmonary compromise.
In the setting of AKI requiring renal replacement therapy, the ideal modality is the one most easily accessible, assuming hemodynamic stability. Options include hemodialysis, peritoneal dialysis, and continuous modalities. Though continuous renal replacement modalities provide the best theoretical protection from ongoing metabolic and volume complications, its superiority has not been verified in rigorous trials. In cases associated with large-scale injuries such as earthquakes, the remaining infrastructure often dictates which dialytic modality can and should be provided.72,113 Historically, peritoneal dialysis has not been considered adequate in its ability to remove the large solute load present with rhabdomyolysis.114 Nonetheless, if it is the only available modality its potential role in managing rhabdomyolysis may be improved with more frequent exchanges. It should be noted that no traditional renal replacement method has the capability of removing myoglobin because of its size. However, considering the rapid extrarenal clearance of myoglobin it is unclear if therapy focused on myoglobin clearance will provide any superiority over current care. A novel approach recently evaluated the use of super high-flux (SHF) membranes to clear myoglobin during continous venovenous hemofiltration.115,116 The authors demonstrated a fivefold increase in myoglobin removal but did acknowledge the potential for the removal of albumin, clotting factors, and protein-bound medications.
Hypocalcemia is common in rhabdomyolysis and may provide challenging management choices. Calcium entry into necrotic muscle cells often causes profound hypocalcemia. Calcium supplementation should be reserved for those patients with symptomatic hypocalcemia as hypercalcemia is a frequent finding during disease recovery.117 In addition, rapid increases in serum pH through the administration of intravenous bicarbonate may worsen hypocalcemia. Rasburicase has been reported to reduce hyperuricemia in young patients with rhabdomyolysis.118
Additional issues include close monitoring for limb ischemia due to compartment syndromes induced by muscle damage and subsequent fluid sequestration. Intracompartmental pressure monitoring may predict the need for intervention though research has suggested additional factors, most importantly hypotension and the difference between mean arterial pressure and intracompartmental pressures, influence whether compartment syndrome develops.119 The history, logic, and disappointing results of early and aggressive fasciotomy have recently been described.120 Mannitol is postulated to “decompress” intracompartmental edema in acute muscle compartment syndromes. Animal data support an acute reduction in intracompartmental pressures121; long-term tissue protection and human outcomes have not been validated. High-dose corticosteroids have been proposed as a treatment option for refractory cases of rhabdomyolysis but prospective studies are lacking.122
References
1. Guly, H. History of accidental hypothermia. Resuscitation. 2011; 82:122–125.
2. Centers for Disease Control and Prevention. Hypothermia related deaths, United States 2003-2004. MMWR. 2005; 54(7):173–175.
3. Centers for Disease Control and Prevention. Hypothermia related deaths, Virginia, November 1996-April 1997. MMWR. 1997; 46:1157.
4. Koutsavlis, AT, Kostasky, T. Environmental-temperature injury in a Canadian metropolis. J Environ Health. 2003; 66:40.
5. Farnell, GS, Pierce, KE, Collinsworth, TA, et al. The influence of ethnicity on thermoregulation after acute cold exposure. Wilderness Environ Med. 2008; 19:238–244.
6. Ulrich, AS, Rathlev, NK. Hypothermia and localized cold injuries. Emerg Med Clin North Am. 2004; 22:281–298.
7. Vassal, T, Benoit-Gonnin, B, Carrat, F, et al. Severe accidental hypothermia treated in an ICU: Prognosis and outcome. Chest. 2001; 120:1998.
8. Danzl, D, Pozos, R, Auerback, P, et al. Multicenter hypothermia survey. Ann Emerg Med. 1987; 16:1042–1055.
9. Giesbrecht, GG. Cold stress, near drowning, and accidental hypothermia: A review. Aviat Space Environ Med. 2000; 71:733.
10. Jolly, BT, Ghezzi, KT. Accidental hypothermia. Emerg Med Clin North Am. 1992; 10:311.
11. Schneider, SM, Danzl, DR. Hypothermia from recognition to re-warming. Emerg Med Rep. 1992; 13:1.
12. McCulough, L, Arora, S. Diagnosis and treatment of hypothermia. Am Fam Physician. 2004; 70(12):2325–2332.
13. Tikuisis, P, Eyolfson, DA, Xu, X. Shivering endurance and fatigue during cold water immersion in humans. Eur J Appl Physiol. 2002; 87:50–58.
14. Guatam, P, Ghosh, S, Mandal, A, Vargas, E. Hypothermia: Sociomedical characteristics and the outcome in 86 patients. Public Health. 1989; 103:15.
15. Danzl, DF, Pozos, RS. Accidental hypothermia. N Engl J Med. 1994; 331:1756.
16. Jurkovich, GJ. Environmental cold induced injury. Surg Clin North Am. 2007; 87:247–267.
17. Ginsberg, MD, Globus, MY, Dietrich, WD, et al. Temperature modulation of ischemic brain injury: A synthesis of recent advances. Prog Brain Res. 1993; 96:13.
18. Silfvaot, T, Pettila, V. Outcome from severe accidental hypothermia in southern Finland—A 10 year review. Resuscitation. 2003; 59:285.
19. Ozaki, H, Nagai, Y, Tochihara, Y. Physiological responses and manual responses in humans following repeated exposures to cold at night. Eur J Appl Physiol. 2001; 84(4):343–349.
20. Gilbert, M, Busund, R, Skagseth, A, et al. Resuscitation from accidental hypothermia of 13. 7 degrees C with circulatory arrest. Lancet. 2000; 355(9201):375–376.
21. Aslam, AF, Aslam, AK, Vasarada, BC, Khan, IA. Hypothermia: Evaluation, electrocardiographic manifestations and management. Am J Med. 2006; 119(4):297–401.
22. Bartley, B, Crnkovich, DJ, Usman, AR, Carlson, RW. How to recognize hypothermia in critically ill patients. J Crit Care Illness. 1996; 11:118.
23. Cohen, D, Cline, J, Lepinski, S. Resuscitation of the hypothermic patient. Am J Emerg Med. 1998; 6:475–478.
24. Ledinham, I, Mone, J. Treatment of accidental hypothermia: A prospective clinical study. BMJ. 1980; 1:102–105.
25. Granberg, PO. Human physiology under cold exposure. Arctic Med Res. 1991; 50(Suppl 6):23–27.
26. Shapiro, BA. Temperature correction of blood gas values. Respir Care Clin North Am. 1995; 1:69.
27. Zander, R. Optimization of the acid-base status in hypothermia. Anaesthetist. 2007; 55(9):912–916.
28. Brieva, J, McFadyen, B, Rowley, M. Severe hypothermia. Anaesth Intensive Care. 2005; 33:662–664.
29. Levin, S, Brettman, LR, Holzman, RS. Infections in hypothermic patients. Arch Internal Med. 1981; 141:920–925.
30. Rohrer, MJ, Jatale, AM. Effect of hypothermia on the coagulation cascade. Crit Care Med. 1992; 20:1402.
31. Bokenes, L, Alexandersen, TE, Osterud, B, et al. Physiological and haematological responses to cold exposure in the elderly. Int J Circumpolar Health. 2000; 59:216–221.
32. Helman, A, Gilbert, M, Pfisler-Lemaire, N, et al. Glucagon and insulin secretion and their biological activities in hypothermic rats. Endocrinology. 1984; 115(5):1722–1728.
33. Weinberg, AD. Hypothermia. Ann Emerg Med. 1993; 22(Pt 2):370.
34. Braun, R, Krishel, S. Environmental emergencies. Emerg Med Clin North Am. 1997; 15:1451.
35. Lazar, HL. The treatment of hypothermia. N Engl J Med. 1997; 337:1545.
36. Sheaff, C, Fildes, J, Keogh, P, et al. Safety of 65 degrees intravenous fluid for the treatment of hypothermia. Am J Surg. 1996; 172:52–55.
37. American Heart Association. 2005 Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care: Hypothermia. Circulation. 2005; 112(24 Suppl):IV136.
38. Soar, J, Perkins, GD, Abbas, G, et al. European Resuscitation Council guidelines for resuscitation section 8—Cardiac arrest in special circumstances: Section 8d. Hypothermia. Resuscitation. 2010; 81:1408–1410.
39. Wira, CR, Becker, JV, Martin, G, Donnino, MW. Antiarrhythmic and vasopressor medications for the treatment of ventricular fibrillation in severe hypothermia: A systemic review of literature. Resuscitation. 2008; 78:21–29.
40. Vanderploeg, GJ, Goslings, JC, Walpoth, BH, Bierens, JJLM. Accidental hypothermia: Rewarming treatments, complications, and outcomes from university medical centre. Resuscitation. 2010; 81:1550–1555.
41. Keller, R, Schnider, TW, Nerdhart, P. Deep accidental hypothermia and cardiac arrest—Rewarming with forced air. Acta Anesthesiol Scand. 1997; 41:1359.
42. Steele, MT, Nelson, MJ, Sessler, DI, et al. Forced air speeds rewarming in accidental hypothermia. Ann Emerg Med. 1996; 27:479.
43. Lloyd, EL. Accidental hypothermia. Resuscitation. 1996; 32:111–124.
44. Slouis, CM, Bachvarov, HL. Heated inhalation treatment of hypothermia. Am J Emerg Med. 1984; 2:533.
45. Kjaerguard, B, Back, P. Warming of patients with accidental hypothermia using warm water lavage. Resuscitation. 2006; 68:203.
46. Sultan, N, Theakston, KD, Butler, R, Suri, RS. Treatment of severe accidental hypothermia with intermittent hemodialysis. Can J Emerg Med. 2009; 11(2):174–177.
47. Centers for Disease Control and Prevention. Monitoring environmental disease—United States, 1997. JAMA. 1998; 280:688–689.
48. Heat-related deaths—Philadelphia and the United States. MMWR Morb Mortal Wkly Rep. 1994; 43:453–455.
49. Kilbourne, EM, Choi, K, Jones, TS, et al. Risk factors for heat stroke. A case control study. JAMA. 1982; 247:3332.
50. Donoghue, ER, Graham, MA, Jentzen, JM, et al. Criteria for the diagnosis of heat-related deaths: National Association of Medical Examiners. Position paper. National Association of Medical Examiners Ad Hoc Committee on the Definition of Heat-Related Fatalities. Am J Forensic Med Pathol. 1997; 18:11–14.
51. Richards, D, Richards, R, Schofield, J. Management of heat exhaustion in Sydney’s the Sun City-to-Surf fun runners. Med J Aust. 1979; 2:457.
52. Nadel, ER. Temperature regulation and hyperthermia during exercise. Clin Chest Med. 1984; 5:13–20.
53. Schvartz, E, Shapiro, Y, Magazanik, A, et al. Heat acclimatization, physical fitness and response to exercise in temperate and hot environment. J Appl Physiol. 1977; 43:678.
54. Rousseau, JM, Villevieille, T, Schiano, P, et al. Reversible myocardial dysfunction after exertional heat stroke. Intensive Care Med. 2001; 27:328–329.
55. Tucker, LE, Stanford, J, Graves, B, et al. Classical heatstroke: Clinical and laboratory assessment. South Med J. 1985; 78:20.
56. Hashim, IA. Clinical biochemistry of hyperthermia. Ann Clin Biochem. 2010; 47:516–523.
57. Jacobsen, TD, Krenzelok, EP, Shicker, L, et al. Environmental injuries. Dis Mon. 1997; 43:809–907.
58. Bouchama, A, Devol, EB. Acid-base alterations in heatstroke. Intensive Care Med. 2001; 27:680–685.
59. Dematte, JE, O’Mara, K, Buescher, J, et al. Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med. 1998; 129:179–181.
60. White, DJ, Kamath, R, Nucci, R, et al. Evaporation versus iced peritoneal lavage treatment of heatstroke: Comparative efficacy in a canine model. Am J Emerg Med. 1993; 11:1–3.
61. Bouchama, A, Cafege, A, Devol, EB, et al. Ineffectiveness of dantrolene sodium in the treatment of heatstroke. Crit Care Med. 1991; 19:176–180.
62. Zucherman, GB, Singer, LP, Rubin, DH, et al. Effects of dantrolene on cooling times and cardiovascular parameters in an immature porcine model of heatstroke. Crit Care Med. 1997; 25:135–139.
63. Quane, KA, Healy, JM, Keating, KE, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. 1993; 5:51–55.
64. Brandon, BW. The genetics of malignant hyperthermia. Anesthesiol Clin North Am. 2005; 23:615–619.
65. Denborough, M. Malignant hyperthermia. Lancet. 1998; 352:1131–1136.
66. Moulds, RFW, Denborough, MA. Identification of susceptibility to malignant hyperthermia. BMJ. 1974; 2:245–247.
67. European Malignant Hyperpyrexia Group. A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. Br J Anaesth. 1984; 56:1267–1269.
68. Chan, TC, Evans, SD, Clark, RF. Drug-induced hyperthermia. Crit Care Clin. 1997; 13:785–808.
69. Caroff, SN, Mann, SC. Neuroleptic malignant syndrome. Med Clin North Am. 1993; 77:185.
70. Balzan, MV. The neuroleptic malignant syndrome: A logical approach to the patient with temperature and rigidity. Postgrad Med J. 1998; 74:72–76.
71. Bywaters, EG, Beall, D. Crush injuries with impairment of renal function. 1941. J Am Soc Nephrol. 1998; 9(2):322–332.
72. Sever, MS, Vanholder, R, Lameire, N. Management of crush-related injuries after disasters [see comment]. N Engl J Med. 2006; 354(10):1052–1063.
73. Sever, MS, Erek, E, Vanholder, R, et al. Renal replacement therapies in the aftermath of the catastrophic Marmara earthquake. Kidney Int. 2002; 62(6):2264–2271.
74. Ballantyne, CM, Corsini, A, Davidson, MH, et al. Risk for myopathy with statin therapy in high-risk patients [see comment]. Arch Internal Med. 2003; 163(5):553–564.
75. Amend, KL, Landon, J, Thyagarajan, V, et al. Incidence of hospitalized rhabdomyolysis with statin and fibrate use in an insured US population. Ann Pharmacother. 2011; 45(10):1230–1239.
76. Huerta-Alardin, AL, Varon, J, Marik, PE. Bench-to-bedside review: Rhabdomyolysis—An overview for clinicians. Crit Care. 2005; 9(2):158–169.
77. Bosch, X, Poch, E, Grau, JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009; 361(1):62–72.
78. Vanholder, R, Sever, MS, Erek, E, Lameire, N. Rhabdomyolysis. J Am Soc Nephrol. 2000; 11(8):1553–1561.
79. Berney-Meyer, L, Putt, T, Schollum, J, Walker, R. Nephrotoxicity of recreational party drugs. Nephrology (Carlton). 2012; 17(2):99–103.
80. Liang, WC, Nishino, I. State of the art in muscle lipid diseases. Acta Myol. 2010; 29(2):351–356.
81. Cervellin, G, Comelli, I, Lippi, G. Rhabdomyolysis: Historical background, clinical, diagnostic and therapeutic features. Clin Chem Lab Med. 2010; 48(6):749–756.
82. Melli, G, Chaudhry, V, Cornblath, DR. Rhabdomyolysis: An evaluation of 475 hospitalized patients. Medicine (Baltimore). 2005; 84(6):377–385.
83. Amrein, S, Amrein, K, Amegah-Sakotnik, A, et al. Propofol infusion syndrome—A critical incident report highlighting the danger of reexposure. J Neurosurg Anesthesiol. 2011; 23(3):265–266.
84. Wong, JM. Propofol infusion syndrome. Am J Ther. 2010; 17(5):487–491.
85. Roberts, RJ, Barletta, JF, Fong, JJ, et al. Incidence of propofol-related infusion syndrome in critically ill adults: A prospective, multicenter study. Crit Care. 2009; 13(5):R169.
86. Kam, PC, Cardone, D. Propofol infusion syndrome. Anaesthesia. 2007; 62(7):690–701.
87. Eichner, ER. Sickle cell considerations in athletes. Clin Sports Med. 2011; 30(3):537–549.
88. Eichner, ER. Sickle cell trait in sports. Curr Sports Med Rep. 2010; 9(6):347–351.
89. Eichner, ER. Pearls and pitfalls: Exertional sickling. Curr Sports Med Rep. 2010; 9(1):3–4.
90. Eichner, ER. Exertional rhabdomyolysis. Curr Sports Med Rep. 2008; 7(1):3–4.
91. Anzalone, ML, Green, VS, Buja, M, et al. Sickle cell trait and fatal rhabdomyolysis in football training: A case study. Med Sci Sports Exerc. 2010; 42(1):3–7.
92. Tsaras, G, Owusu-Ansah, A, Boateng, FO, Amoateng-Adjepong, Y. Complications associated with sickle cell trait: A brief narrative review. Am J Med. 2009; 122(6):507–512.
93. Makaryus, JN, Catanzaro, JN, Katona, KC. Exertional rhabdomyolysis and renal failure in patients with sickle cell trait: Is it time to change our approach? Hematology. 2007; 12(4):349–352.
94. Allison, RC, Bedsole, DL. The other medical causes of rhabdomyolysis. Am J Med Sci. 2003; 326(2):79–88.
95. Knochel, JP. Mechanisms of rhabdomyolysis. Curr Opin Rheumatol. 1993; 5(6):725–731.
96. Malinoski, DJ, Slater, MS, Mullins, RJ. Crush injury and rhabdomyolysis. Crit Care Clin. 2004; 20(1):171–192.
97. Boutaud, O, Roberts, LJ, 2nd. Mechanism-based therapeutic approaches to rhabdomyolysis-induced renal failure. Free Radic Biol Med. 2011; 51(5):1062–1067.
98. Zager, RA. Studies of mechanisms and protective maneuvers in myoglobinuric acute renal injury. Lab Invest. 1989; 60(5):619–629.
99. Harris, K, Walker, PM, Mickle, DA, et al. Metabolic response of skeletal muscle to ischemia. Am J Physiol. 1986; 250(2 Pt 2):H213–H220.
100. Olerud, JE, Homer, LD, Carroll, HW. Incidence of acute exertional rhabdomyolysis. Serum myoglobin and enzyme levels as indicators of muscle injury. Arch Internal Med. 1976; 136(6):692–697.
101. Lofberg, M, Jankala, H, Paetau, A, et al. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand. 1998; 98(4):268–275.
102. Youssef, T, Abd-Elaal, I, Zakaria, G, Hasheesh, M. Bariatric surgery: Rhabdomyolysis after open Roux-en-Y gastric bypass: A prospective study. Int J Surg. 2010; 8(6):484–488.
103. de Meijer, AR, Fikkers, BG, de Keijzer, MH, et al. Serum creatine kinase as predictor of clinical course in rhabdomyolysis: A 5-year intensive care survey. Intensive Care Med. 2003; 29(7):1121–1125.
104. Lappalainen, H, Tiula, E, Uotila, L, Manttari, M. Elimination kinetics of myoglobin and creatine kinase in rhabdomyolysis: Implications for follow-up. Crit Care Med. 2002; 30(10):2212–2215.
105. Grossman, RA, Hamilton, RW, Morse, BM, et al. Nontraumatic rhabdomyolysis and acute renal failure. N Engl J Med. 1974; 291(16):807–811.
106. Woodrow, G, Brownjohn, AM, Turney, JH. The clinical and biochemical features of acute renal failure due to rhabdomyolysis. Renal Fail. 1995; 17(4):467–474.
107. Gunal, AI, Celiker, H, Dogukan, A, et al. Early and vigorous fluid resuscitation prevents acute renal failure in the crush victims of catastrophic earthquakes. J Am Soc Nephrol. 2004; 15(7):1862–1867.
108. Better, OS, Stein, JH. Early management of shock and prophylaxis of acute renal failure in traumatic rhabdomyolysis [see comment]. N Engl J Med. 1990; 322(12):825–829.
109. Odeh, M. The role of reperfusion-induced injury in the pathogenesis of the crush syndrome [see comment]. N Engl J Med. 1991; 324(20):1417–1422.
110. Zager, RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int. 1996; 49(2):314–326.
111. Brown, CV, Rhee, P, Chan, L, et al. Preventing renal failure in patients with rhabdomyolysis: Do bicarbonate and mannitol make a difference? J Trauma. 2004; 56(6):1191–1196.
112. Visweswaran, P, Massin, EK, Dubose, TD, Jr. Mannitol-induced acute renal failure. J Am Soc Nephrol. 1997; 8(6):1028–1033.
113. Sever, MS, Erek, E, Vanholder, R, et al. Marmara Earthquake Study G: Treatment modalities and outcome of the renal victims of the Marmara earthquake. Nephron. 2002; 92(1):64–71.
114. Nolph, KD, Whitcomb, ME, Schrier, RW. Mechanisms for inefficient peritoneal dialysis in acute renal failure associated with heat stress and exercise. Ann Internal Med. 1969; 71(2):317–336.
115. Naka, T, Jones, D, Baldwin, I, et al, Myoglobin clearance by super high-flux hemofiltration in a case of severe rhabdomyolysis: A case report. Crit Care. 2005(2):R90–R95.
116. Cruz, DN, Bagshaw, SM. Does continuous renal replacement therapy have a role in the treatment of rhabdomyolysis complicated by acute kidney injury? Semin Dial. 2011; 24(4):417–420.
117. Akmal, M, Goldstein, DA, Telfer, N, et al. Resolution of muscle calcification in rhabdomyolysis and acute renal failure. Ann Internal Med. 1978; 89(6):928–930.
118. Lin, PY, Lin, CC, Liu, HC, et al. Rasburicase improves hyperuricemia in patients with acute kidney injury secondary to rhabdomyolysis caused by ecstasy intoxication and exertional heat stroke. Pediatr Crit Care Med. 2011; 12(6):e424–e427.
119. Heppenstall, RB, Sapega, AA, Izant, T, et al. Compartment syndrome: A quantitative study of high-energy phosphorus compounds using 31P-magnetic resonance spectroscopy. J Trauma. 1989; 29(8):1113–1119.
120. Better, OS, Rubinstein, I, Reis, DN. Muscle crush compartment syndrome: Fulminant local edema with threatening systemic effects. Kidney Int. 2003; 63(3):1155–1157.
121. Better, OS, Zinman, C, Reis, DN, et al. Hypertonic mannitol ameliorates intracompartmental tamponade in model compartment syndrome in the dog. Nephron. 1991; 58(3):344–346.
122. Antoon, JW, Chakraborti, C. Corticosteroids in the treatment of alcohol-induced rhabdomyolysis. Mayo Clin Proc. 2011; 86(10):1005–1007.
123. Brumback, RA, Feeback, DL, Leech, RW. Rhabdomyolysis in childhood. A primer on normal muscle function and selected metabolic myopathies characterized by disordered energy production. Pediatr Clin North Am. 1992; 39(4):821–858.
124. Knochel, JP, Barcenas, C, Cotton, JR, et al. Hypophosphatemia and rhabdomyolysis. J Clin Invest. 1978; 62(6):1240–1246.
125. Knochel, JP, Carter, NW. The role of muscle cell injury in the pathogenesis of acute renal failure after exercise. Kidney Int Suppl. 1976; 6:S58–S64.
126. Knochel, JP, Schlein, EM. On the mechanism of rhabdomyolysis in potassium depletion. J Clin Invest. 1972; 51(7):1750–1758.
127. Trimarchi, H, Gonzalez, J, Olivero, J. Hyponatremia-associated rhabdomyolysis. Nephron. 1999; 82(3):274–277.
128. Mor, A, Wortmann, RL, Mitnick, HJ, Pillinger, MH. Drugs causing muscle disease. Rheum Dis Clin North Am. 2011; 37(2):219–231.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]<
[/not-level-membership-for-critical-care-medicine-category]
