[level-membership-for-basic-science-category]
Chapter 38 Hypothalamic, pituitary and sex hormones
• Many of the pituitary hormones and their hypothalamic releasing factors are used in diagnosis or therapy.
• The main therapeutic use of pituitary hormones is of growth hormone (anterior pituitary) and those from the posterior pituitary: oxytocin and vasopressin.
• Vasopressin (antidiuretic hormone) is used both for its vasoconstrictor effect (in the treatment of oesophageal varices) and for its antidiuretic action.
• The main hypothalamus–pituitary target organ axis for therapeutic intervention is that controlling reproductive hormones, especially in women.
• Suppression of oestrogen and/or androgen production is used in the treatment of tumours stimulated by these: breast and prostate.
• Therapy in women is used to suppress ovulation (contraceptives), to stimulate ovulation (fertility treatment) or to mimic ovarian endocrine function (post-menopausal hormone replacement therapy, HRT).
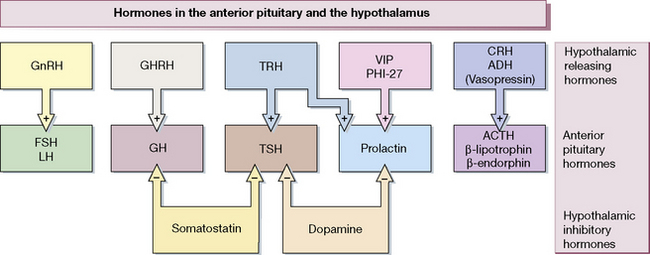
Figure 38.1 shows the hypothalamo-pituitary axes. The hypothalamus and pituitary glands form the centre of the ‘endocrine orchestra’. We will here describe the hypothalamic releasing hormones, and the anterior and posterior pituitary gland hormones, and drugs that are used to manipulate these axes.
These hormones, analogues (agonists) and antagonists can be used:
• To analyse the functional integrity of endocrine control systems.
• As replacement in hormone deficiency states.
• To modify malfunction of endocrine systems.
• To alter normal function where this is inconvenient, e.g. contraception.
The scope of the specialist endocrinologist continues to increase in amount and in complexity and only an outline is appropriate here.
Hypothalamic and anterior pituitary hormones
The hypothalamus releases a number of locally active hormones that stimulate or inhibit pituitary hormone release (see Fig. 38.1).
Octreotide
is a synthetic analogue of somatostatin having a longer action (t½ 1.5 h). It is administered subcutaneously two or three times daily; a depot formulation is available for deep intramuscular injection once a month. Lanreotide is much longer acting than octreotide, and is administered intramuscularly twice a month. Uses of the somatostatin analogues include acromegaly, carcinoid (serotonin-secreting) tumours and other rare tumours of the alimentary tract. An unlicensed use of octeotride is the termination of variceal bleeding (see p. 548). Radiolabelled somatostatin is used to localise, and in higher doses to treat, metastases from neuroendocrine tumours which often bear somatostatin receptors. Both octreotide and lanreotide are now available as a monthly slow-release preparation.
Growth hormone, somatrophin
Growth hormone is approved for treatment of children with short stature due not just to growth hormone deficiency, but also to Turner’s syndrome, renal failure, small size for gestational age, Prader–Willi syndrome1 and, most recently, idiopathic short stature. Treatment is continued until closure of the epiphyses. Subsequent treatment into adulthood is also warranted where UK National Institute for Health and Clinical Excellence (NICE) guidelines are fullfilled. Growth hormone therapy should be confined to specialist clinics.
Posterior pituitary hormones and analogues
Desmopressin
Nephrogenic diabetes insipidus, as is to be expected, does not respond to antidiuretic hormone.
In bleeding oesophageal varices, use is made of the vasoconstrictor effect of vasopressin (as terlipressin, a vasopressin prodrug); see page 564.
In haemophilia, desmopressin can enhance blood concentration of factor VIII.
Felypressin is used as a vasoconstrictor with local anaesthetics.
Diabetes insipidus: vasopressin deficiency
Sex (gonadal) hormones and antagonists: steroid hormones
Androgens
Preparations and choice of androgens
Testosterone given orally is subject to extensive hepatic first-pass metabolism (see p. 86) and it is therefore usually given by other routes. Androgens are available for oral, buccal, transdermal or depot administration.
Transdermal preparations
Non-scrotal patches are applied to the skin of the upper arms, back, abdomen and thighs.
Antiandrogens (androgen antagonists)
Cyproterone
• Competition with testosterone for receptors in target peripheral organs (but not causing feminisation as do oestrogens); it reduces spermatogenesis even to the level of azoospermia (reversing over about 4 months after the drug is discontinued); abnormal sperm occurs during treatment.
• Competition with testosterone in the CNS, reducing sexual drive and thoughts, and causing impotence.
• Some agonist progestogenic activity on hypothalamic receptors, inhibiting gonadotrophin secretion, which also inhibits testicular androgen production.
Uses
Finasteride and dutasteride (see p. 462), which inhibit conversion of testosterone to dihydrotestosterone, have localised antiandrogen activity in tissues where dihydrotestosterone is the principal androgen; they are therefore useful drugs in the treatment of benign prostatic hypertrophy.
Spironolactone (see p. 563) also has antiandrogen activity and may help hirsutism in women (as an incidental benefit to its diuretic effect). Androgen secretion may be diminished by continued use of a gonadorelin (LHRH) analogue (see p. 597).
Anabolic steroids
They benefit some patients with aplastic anaemia.
None of these agents is free from virilising properties in high doses; acne and greasy skin may be the early manifestation of virilisation (see also, Adverse effects of androgens, p. 593, and Drugs and sport, p. 141).
Oestrogens
Preparations of oestrogens
• Ethinylestradiol (t½ 13 h) is a synthetic agent of first choice for pharmacological uses (mainly contraceptive, female hypogonadism and menstrual disorders); it is effective by mouth, dose 20–50 micrograms/day.
• Estradiol and estriol are orally active mixed natural oestrogens, dose 1–2 mg/day.
• Conjugated oestrogens (Premarin) are orally active mixed natural oestrogens containing 50–65% estrone obtained from the urine of pregnant mares, dose 0.625–1.25 mg.
• Estropipate (piperazine estrone sulphate) is an orally active synthetic conjugate.
• Diethylstilbestrol (stilboestrol) is the first synthetic oestrogen; it is used (rarely) in prostatic cancer, and occasionally in post-menopausal women with breast cancer.
Oestrogen formulations and routes of administration
Indications for oestrogen therapy
Replacement therapy in hypo-oestrogenaemia
This term refers to decreased oestrogen production due to ovarian disease, or to hypothalamic–pituitary disease (hypogonadotrophic hypogonadism). Treatment is by cyclic oestrogen (estradiol 1–2 mg, conjugated oestrogens 0.625/1.25 mg daily or ethinylestradiol 20–30 micrograms continuously) plus a progestogen, medroxyprogesterone 2.5–10 mg daily for the last 10–14 days of oestrogen treatment. An alternative treatment is the oral contraceptive (p. 607).
Post-menopausal hormone replacement therapy (HRT)
Preparations used for HRT
There are three types of regimen:
1. Women without a uterus take continuous oestrogen alone.
2. Women with a uterus require oestrogen combined with progestogen to prevent endometrial proliferation and risk of endometrial cancer.
Anti-oestrogens
Tamoxifen
is a non-steroidal competitive oestrogen antagonist on target organs. Although available for anovulatory infertility (20 mg daily on days 2, 3, 4 and 5 of the cycle), its main use now is in the treatment of oestrogen-dependent breast cancer (see p. 518). Treatment with tamoxifen delays the growth of metastases and increases survival; if tolerated it should be continued for 5 years.
Progesterone and progestogens
• Progesterone and its derivatives: dydrogesterone, hydroxyprogesterone, medroxyprogesterone (t½ 28 h).
• Testosterone derivatives: norethisterone and its prodrug ethynodiol (t½ 10 h), levonorgestrel, desogestrel, gestodene, gestronol, norgestimate.
Drospirenone is a derivative of the synthetic aldosterone antagonist, spironolactone (see p. 563). It therefore has antimineralocorticoid activity, reducing salt retention and blood pressure. It also exhibits partial antiandrogenic activity, about 30% of that of cyproterone acetate. It is available as a combination with ethinylestradiol for use as a contraceptive.
Most progestogens can virilise directly or by metabolites (except progesterone and dydrogesterone), and fetal virilisation to the point of sexual ambiguity has occurred with vigorous use during pregnancy (see also Contraception, p. 608).
Megestrol is used only in cancer; it causes tumours in the breasts of beagle dogs.
Preparations
Available progestogens (some used only in combined formulations) include:
• Oral: norethisterone, dydrogesterone, gestodene, desogestrel, levonorgestrel, megestrol, medroxyprogesterone.
• Suppositories or pessaries: progesterone.
• Injectable: progesterone, hydroxyprogesterone, medroxyprogesterone.
Antiprogestogens
Mifepristone
Guidelines may vary in detail and the following are general regimens:
• For gestation of up to 1 week where the fetus is deemed viable, mifepristone 600 mg by mouth followed 36–48 h later by gemeprost 1 mg by vagina.
• For mid-trimester medical abortion (13–24 weeks), mifepristone 600 mg by mouth followed 36–48 h later by gemeprost 1 mg every 3 h by vagina to a maximum of 5 mg.
Fertility regulation
Polycystic ovary syndrome (PCOS)
The diagnosis requires at least two of the following features:
• Treatment of infertility. Induction of ovulation can be accomplished in 75–80% of women with PCOS by the use of anti-oestrogens, typically clomifene citrate. More recent data indicate that metformin (see below) may improve ovulation rates in women with PCOS when given alone or in combination with clomifene.
• Menstrual regulation in those who do not desire pregnancy. A low-dose combined oral contraceptive (containing ethinylestradiol, 20–35 micrograms) may be the most convenient form of treatment, although cyclical progestogen is a reasonable alternative. Norgestimate and desogestrel are the preferred progestins, having virtually no androgenic properties.
• Treatment of associated symptoms of hyperandrogenism. Management of hirsutism usually involves cosmetic treatment to remove unwanted hair and, in more severe cases, antiandrogen therapy. The most commonly used antiandrogen is cyproterone acetate. This also has progestogenic activity and can be combined with ethinylestradiol to provide cycle control in addition to management of hyperandrogenic symptoms. Drospirenone (see above) is ideal in PCOS because of its antiandrogen and antimineralocorticoid properties. Spironolactone can be used at high doses, 100–200 mg. Flutamide is a potent non-steroidal antiandrogen that is effective in the treatment of hirsutism. Concern about inducing hepatocellular dysfunction has limited its use.
• Prevention of the possible long-term consequences of the metabolic disturbance characteristic of anovulatory women with PCOS.
Contraception by drugs and hormones
• It must be extremely safe as well as highly effective.
• Its action must be quick in onset and quickly and completely reversible, even after years of continuous use.
The fact that alternative methods are less reliable implies that their use will lead to more unwanted pregnancies with their attendant inconvenience, morbidity and mortality, and this must be taken into account in deciding what risks of hormonal contraception are acceptable.
Combined contraceptives (the ‘pill’)
Important aspects
of the breast and cervix are slightly increased in incidence;2 the incidence of hepatoma (very rare) is increased. The risk to life seems to be less than that of moderate smoking (10 cigarettes/day) and than that of a normal pregnancy, as the risks of pulmonary embolus are higher in a normal pregnancy. The risk of carcinoma of the ovary and endometrium is substantially reduced. The overall incidence of cancer is unaltered.
Other adverse effects
The above account gives rise to guidelines for use:
• A personal history of thromboembolic venous, arterial or cardiac disease, or severe or multiple risk factors for these.
• Transient cerebral ischaemic attacks without headache.
• Infective hepatitis, until 3 months after liver function test results have become normal, and other liver disease including disturbances of hepatic excretion, e.g. cholestatic jaundice, Dubin–Johnson and Rotor syndromes.
• Migraine, if there is a typical aura, focal features, or if it is severe and lasts for more than 72 h despite treatment, or is treated with an ergot derivative (use with caution is acceptable if there is no aura, focal features, or if it is controlled with a 5-HT1 receptor agonist).
• Carcinoma of the breast or genital tract, past or present.
• Other conditions including: systemic lupus erythematosus, porphyria, following evacuation of a hydatidiform mole (until urine and plasma gonadotrophin concentrations are normal), undiagnosed vaginal bleeding.
or uses with caution, include:
• Family history of venous thromboembolism, arterial disease or a known prothrombotic condition, e.g. factor V Leiden (pretreatment coagulation investigation is advised).
• Diabetes mellitus, which may be precipitated or become more difficult to control (avoid if there are diabetic complications).
• Hypertension (avoid if blood pressure exceeds 160/100 mmHg).
• Smoking more than 40 cigarettes per day (15 cigarettes/day enhances the risks of circulatory disease three-fold, and constitutes an absolute contraindication for women over 35 years).
• Age over 35 years (avoid if older than 50 years).
• Obesity (avoid if body mass exceeds 39 kg/m2).
• Long-term immobility, e.g. due to leg plaster, confinement to bed.
• Breast feeding (until weaning or for 6 months after birth).
Common problems
The following refers to the combined pill (see later for the progestogen-only pill):
• If an omitted dose is remembered within 12 h, it should be taken at once and the next dose at the usual time, and all should be well.
• If more than 12 h have elapsed, the above procedure should be followed but an additional barrier method of contraception should be used for 7 days. Although the protective effect of cervical mucus returns within 48 h, this 7-day period is needed to ensure effective suppression of an ovulation that may have been initiated by the missed pill.
Progestogen-only contraception
Progestogen-only pills (POPs) are indicated where oestrogen is contraindicated (see above, p. 603) and in lactating women. Progestogens render cervical mucus less easily penetrable by sperm and induce a premature secretory change in the endometrium so that implantation does not occur. Older POPs became unreliable if not taken at the same time of day, because their effect on cervical mucus wears off after 3 h and their additional action to inhibit ovulation occurs in only 40% of cycles. There is also liability to breakthrough bleeding.
Drug interaction with steroid contraceptives
Antiepileptics (phenytoin and carbamazepine but not sodium valproate) create a similar risk. Indeed, all drugs that induce metabolising enzymes (see p. 93), whether prescribed or self-administered (alcohol, tobacco smoking), constitute a risk to contraceptive efficacy and prescribing should be specifically reviewed for the effect.
Menstrual disorders
Myometrium
Oxytocics
Oxytocin is structurally close to vasopressin and it is no surprise that it also has antidiuretic activity (see p. 454). Serious water intoxication can occur with prolonged intravenous infusions, especially where accompanied by large volumes of fluid. The association of oxytocin with neonatal jaundice appears to be due to increased erythrocyte fragility causing haemolysis.
is used to contract the uterus. It is an α-adrenoceptor and dopamine receptor agonist and acts almost immediately when injected intravenously. The uterus is stimulated at all times, but is much more sensitive in late pregnancy (see also ergotamine, p. 297).
There are advantages in a mixture of oxytocin and ergometrine (Syntometrine).
Prostaglandins
(For a general account of the prostaglandins see Ch. 16.)
Induction of abortion
Gemeprost, administered vaginally as pessaries, is the preferred prostaglandin for the medical induction of late therapeutic abortion. Gemeprost ripens and softens the cervix before surgical abortion, particularly in primigravida. Misoprostol by mouth or by vaginal administration, or gemeprost, may be given to induce medical abortion (an unlicensed indication in the UK). Pretreatment with mifepristone (see p. 606) can facilitate the process, by sensitising the uterus to the prostaglandin so that abortion occurs in a shorter time and with a lower dose of prostaglandin.
Induction and augmentation of labour
The UK National Institute for Health and Clinical Excellence has recommended that:
• Dinoprostone is preferable to oxytocin for induction of labour in women with intact membranes, regardless of parity or cervical favourability.
• Dinoprostone and oxytocin are equally effective for the induction of labour in women with ruptured membranes, regardless of parity or cervical favourability.
• Intravaginal dinoprostone preparations are preferable to intracervical preparations.
• Oxytocin should not be started for 6 h following administration of vaginal prostaglandins.
• When used to induce labour, the recommended dose of oxytocin by intravenous infusion is initially 0.001–0.002 units/min, increased at intervals of at least 30 min until a maximum of three to four contractions occurs every 10 min (0.012 units/min is often adequate); the maximum recommended rate is 0.032 units/min (licensed max. 0.02 units/min).
Amin A. New drugs for hyponatraemia: evidence is lacking that they are better than cheaper standard treatment. Br. Med. J.. 2011;342:559–560.
Banerjee A., Wynne K., Tan T., et al. High dose cabergoline therapy for resistant macroprolactinoma during pregnancy. Clin. Endocrinol. (Oxf.). 2009;70:812–813.
Barrett-Connor E.L., Mosca L., Collins P., et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N. Engl. J. Med.. 2006;355:125–137.
Coombs N.J., Taylor R., Wilcken N., Boyages J. Hormone replacement therapy and breast cancer: estimate of risk. Br. Med. J.. 2005;331:347–349.
Jayasena C.N., Wujanto C., Donaldson M., et al. Measurement of basal growth hormone (GH) is a useful test of disease activity in treated acromegalic patients. Clin. Endocrinol. (Oxf).. 2008;68:36–41.
Martin N.M., Dhillo W.S., Banerjee A., et al. Comparison of the dexamethasone-suppressed corticotropin-releasing hormone test and low-dose dexamethasone suppression test in the diagnosis of Cushing’s syndrome. J. Clin. Endocrinol. Metab.. 2006;91:2582–2586.
Martin N.M., Tan T., Meeran K. Dopamine agonists and hyperprolactinaemia. Br. Med. J.. 2009;338:b381.
Melmed S. Medical progress: acromegaly. N. Engl. J. Med.. 2006;355:2558–2573.
Peterson H.B., Curtis K.M. Long-acting methods of contraception. N. Engl. J. Med.. 2005;353:2169–2175.
1 An inherited condition that gives rise to obesity, decreased muscle tone, poor large muscle strength, decreased mental capacity, and sex glands that produce few or no hormones.
2 A meta-analysis of 54 studies concluded that use of the COCP was associated with a relative risk of 1.24, which disappears over 10 years after stopping the COCP. A higher relative risk in women who started taking the COCP at a young age is explained by the lower background rate and there is little added effect from long-term COCP ingestion. (Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 1996; 347:1713–1727).
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 38 Hypothalamic, pituitary and sex hormones
• Many of the pituitary hormones and their hypothalamic releasing factors are used in diagnosis or therapy.
• The main therapeutic use of pituitary hormones is of growth hormone (anterior pituitary) and those from the posterior pituitary: oxytocin and vasopressin.
• Vasopressin (antidiuretic hormone) is used both for its vasoconstrictor effect (in the treatment of oesophageal varices) and for its antidiuretic action.
• The main hypothalamus–pituitary target organ axis for therapeutic intervention is that controlling reproductive hormones, especially in women.
• Suppression of oestrogen and/or androgen production is used in the treatment of tumours stimulated by these: breast and prostate.
• Therapy in women is used to suppress ovulation (contraceptives), to stimulate ovulation (fertility treatment) or to mimic ovarian endocrine function (post-menopausal hormone replacement therapy, HRT).
Figure 38.1 shows the hypothalamo-pituitary axes. The hypothalamus and pituitary glands form the centre of the ‘endocrine orchestra’. We will here describe the hypothalamic releasing hormones, and the anterior and posterior pituitary gland hormones, and drugs that are used to manipulate these axes.
These hormones, analogues (agonists) and antagonists can be used:
• To analyse the functional integrity of endocrine control systems.
• As replacement in hormone deficiency states.
• To modify malfunction of endocrine systems.
• To alter normal function where this is inconvenient, e.g. contraception.
The scope of the specialist endocrinologist continues to increase in amount and in complexity and only an outline is appropriate here.
Hypothalamic and anterior pituitary hormones
The hypothalamus releases a number of locally active hormones that stimulate or inhibit pituitary hormone release (see Fig. 38.1).
Octreotide
is a synthetic analogue of somatostatin having a longer action (t½ 1.5 h). It is administered subcutaneously two or three times daily; a depot formulation is available for deep intramuscular injection once a month. Lanreotide is much longer acting than octreotide, and is administered intramuscularly twice a month. Uses of the somatostatin analogues include acromegaly, carcinoid (serotonin-secreting) tumours and other rare tumours of the alimentary tract. An unlicensed use of octeotride is the termination of variceal bleeding (see p. 548). Radiolabelled somatostatin is used to localise, and in higher doses to treat, metastases from neuroendocrine tumours which often bear somatostatin receptors. Both octreotide and lanreotide are now available as a monthly slow-release preparation.
Growth hormone, somatrophin
Growth hormone is approved for treatment of children with short stature due not just to growth hormone deficiency, but also to Turner’s syndrome, renal failure, small size for gestational age, Prader–Willi syndrome1 and, most recently, idiopathic short stature. Treatment is continued until closure of the epiphyses. Subsequent treatment into adulthood is also warranted where UK National Institute for Health and Clinical Excellence (NICE) guidelines are fullfilled. Growth hormone therapy should be confined to specialist clinics.
Posterior pituitary hormones and analogues
Desmopressin
Nephrogenic diabetes insipidus, as is to be expected, does not respond to antidiuretic hormone.
In bleeding oesophageal varices, use is made of the vasoconstrictor effect of vasopressin (as terlipressin, a vasopressin prodrug); see page 564.
In haemophilia, desmopressin can enhance blood concentration of factor VIII.
Felypressin is used as a vasoconstrictor with local anaesthetics.
Diabetes insipidus: vasopressin deficiency
Sex (gonadal) hormones and antagonists: steroid hormones
Androgens
Preparations and choice of androgens
Testosterone given orally is subject to extensive hepatic first-pass metabolism (see p. 86) and it is therefore usually given by other routes. Androgens are available for oral, buccal, transdermal or depot administration.
Transdermal preparations
Non-scrotal patches are applied to the skin of the upper arms, back, abdomen and thighs.
Antiandrogens (androgen antagonists)
Cyproterone
• Competition with testosterone for receptors in target peripheral organs (but not causing feminisation as do oestrogens); it reduces spermatogenesis even to the level of azoospermia (reversing over about 4 months after the drug is discontinued); abnormal sperm occurs during treatment.
• Competition with testosterone in the CNS, reducing sexual drive and thoughts, and causing impotence.
• Some agonist progestogenic activity on hypothalamic receptors, inhibiting gonadotrophin secretion, which also inhibits testicular androgen production.
Uses
Finasteride and dutasteride (see p. 462), which inhibit conversion of testosterone to dihydrotestosterone, have localised antiandrogen activity in tissues where dihydrotestosterone is the principal androgen; they are therefore useful drugs in the treatment of benign prostatic hypertrophy.
Spironolactone (see p. 563) also has antiandrogen activity and may help hirsutism in women (as an incidental benefit to its diuretic effect). Androgen secretion may be diminished by continued use of a gonadorelin (LHRH) analogue (see p. 597).
[/not-level-membership-for-basic-science-category]
