15 Hypoplastic Left Heart Syndrome
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, and increased cardiothoracic ratio (0.45).
2. The LV is severely hypoplastic, with atretic mitral and aortic valves.
3. There is evidence of increased echogenicity at the ventricular walls and papillary muscles of the mitral valve. The LV shows endocardial fibroelastosis with decreased function. The RV is dilated, with large tricuspid and pulmonary valves that were not dysplastic and had no significant regurgitation.
4. The transverse and distal aortic arch are of normal size and by color and pulsed Doppler there was flow reversal into the distal arch. Usually, the distal arch is the largest aspect of the arch in a patient with hypoplastic left heart syndrome (HLHS), presumably because it receives the greatest amount of flow.
5. The RV Tei index (myocardial performance index) is normal (0.4).
6. There are no signs of hydrops fetalis.
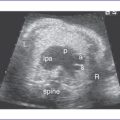
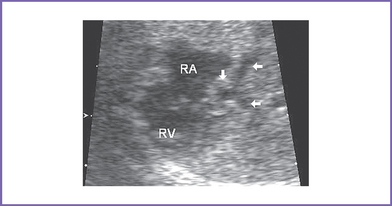
7. The interatrial septum is intact (no flow across the atrial septum) (Fig. 15-1). The left atrium (LA) is mildly enlarged, with bowing of the interatrial septum toward the right atrium (RA).
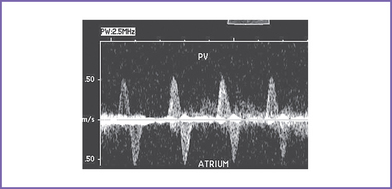
8. The pulmonary veins in the LA are decompressed through a vertical vein, which is obstructed at the site of the left pulmonary artery and left bronchus (mean gradient, 6-7 mm Hg). The pulmonary venous flow is abnormal, with a significant a wave reversal in systole and no forward flow in early ventricular diastole, suggesting severely elevated LA pressure.
9. The pulsatility index is increased in the proximal pulmonary artery, suggesting elevated fetal pulmonary vascular resistance (PVR).
D. Fetal management and counseling
1. Amniocentesis was performed before referral and showed normal fetal karyotype and negative fluorescent in situ hybridization (FISH) analysis for 22q11 microdeletion.
c. Follow-up included serial antenatal studies at weekly intervals initially and then every 2 to 4 weeks.
F. Neonatal management
a. Compassionate care with either no treatment from delivery or withdrawal of treatment once the diagnosis has been confirmed.
c. Staged palliative surgery (Norwood procedure).
d. Cardiac transplantation, particularly in patients with cardiac pathology that can complicate single ventricle palliation, such as significant RV dysfunction or severe tricuspid valve regurgitation.
a. PGE1 infusion to keep the ductus open to provide a pathway to the systemic circulation and improve the body perfusion.
b. Administration of oxygen is normally not advisable in neonates with HLHS because this can cause a decrease in the PVR with increased pulmonary blood flow that further steals from the systemic circulation. However, in a neonate with severe restriction of the atrial septum and LA hypertension, oxygen administration can contribute to an improvement in the systemic oxygen levels.
c. Blood gas revealing severe metabolic acidosis and slightly decreased Po2, a characteristic finding of HLHS, could be due to poor cardiac output. However, babies with severe LA hypertension due to atrial level restriction are both severely cyanotic (Pao2 < 25-30 mm Hg) and metabolically acidotic. In this setting, therapeutic balloon atrial septostomy can help decompress the LA.
d. The hematocrit should be maintained above 45% to increase the oxygen-carrying capacity.
e. Usually, volume and inotropic support (such as milrinone 0.5 μg/kg/min infusion) may be indicated to improve ventricular function.
a. Single ventricle palliation.
G. Follow-up
1. Short-term and medium-term follow up.
2. Long-term follow-up (after Fontan operation).
b. These patients should be excluded from competitive sports.
H. Risk of recurrence
1. Without a familial component, the risk of recurrence may be as high as 2%.
2. In the current case, the recurrence risk for left-sided heart lesions can be as high as 50% (autosomal dominant inheritance) if both the baby and parents have a normal karyotype and no other abnormalities are detected on physical examination or detailed fetal ultrasound (to rule out conditions such as VACTERL association [vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistula or esophageal atresia, renal or urinary anomalies, limb defect] and Holt–Oram syndrome).
3. Affected children born to healthy parents have been reported, and the possibility of an autosomal recessive mode of inheritance should be considered. In these cases, the recurrence risk can be as high as 25%.
4. There is no way to distinguish between cases with HLHS with multifactorial and autosomal recessive mode of inheritance.
5. Families with left-sided heart lesions associated with NOTCH1 gene mutation and autosomal dominant inheritance with incomplete penetrance and intrafamilial variability of the clinical manifestations have been reported and should be considered in the investigation of such families, including the one in this case.
I. Outcome of this case
1. The baby was delivered at term weighing 2.3 kg. Apgar scores were poor: 4 at 1 minute and 7 at 5 minutes.
2. Pulse oximeter reading was 87% in room air, but the baby had no metabolic acidosis.
3. PGE1 infusion was started at 0.05 μg /kg per minute.
4. There was a small atrial opening after birth (much smaller than following intervention), but there was evidence of severe pulmonary congestion. The baby was ventilated and treated for pulmonary hypertension with inhaled nitric oxide.
5. He had Norwood stage 1 at day 1 of life with ligation of the vertical vein, thus allowing oxygenated blood to channel to the LA.
6. The PVR was normal at catheterization at 3 months after the Norwood, but the baby had persistent pulmonary dysfunction and remained ventilator dependant at 6 months of age.
7. According to the parents’ wish, supportive care was withdrawn at 6 months of age.
8. No autopsy was performed. Death was related to chronic pulmonary parenchymal disease that was not relieved by atrial opening.
II. YOUR HANDY REFERENCE
A. Prevalence
1. Noonan and Nadas (1958) proposed the term hypoplastic left heart syndrome to describe an obstructive lesion on the left side of the heart associated with hypoplasia of the LV and RV hypertrophy.
2. The incidence of HLHS in infants with congenital heart disease varies from 4.8% to 9% in different series. The prevalence is between 0.1 and 0.27 per 1000 live births. It is considered to be the most common cause of cardiac death during the first week of life.
3. Aortic atresia as part of HLHS is one of the commonest forms of cardiac abnormality encountered in utero.
4. In Allan and Sharland’s (1992) study, HLHS accounted for 17% of the abnormalities of their fetal series.
5. Aortic atresia is most commonly associated with an underdeveloped LV. Such anomaly should be easily detectable by examination of the four-chamber view. Sometimes the rarer forms, which can be associated with a normally developed LV, are missed unless the outflow tracts are properly examined.
B. Outcome
1. Surgical palliation of HLHS is via one of two routes: conventional surgery or primary cardiac transplantation.
a. Chromosomal anomalies increase the surgical risk.
b. Surgery is contraindicated in babies with a neurologic malformation.
a. The Norwood procedure (three stages) is the surgical palliation for HLHS. It has been used for about 25 years (with modifications).
b. The outcome of the Norwood procedure has improved with increasing experience with the technique. At the home of the classic operation, the outcome has improved from a 3-year survival of 28% for those operated on between 1984 and 1988 to 66% between 1995 and 1998.
c. The current estimated mortality following the first stage of the Norwood procedure is about 25% and for the second and third stage is less than 10%.
3. Recent comparisons of conventional surgery to primary cardiac transplantation show that identical results can be achieved with either strategy depending on the center’s experience.
a. Of four centers performing a large number of procedures each year, two used reconstructive techniques and two performed primary cardiac transplantation.
b. In both groups, 1-year and 3-year survival was 77% and 61%, respectively.
D. Clues to fetal sonographic diagnosis
2. Echogenic LV (although more critical aortic stenosis).
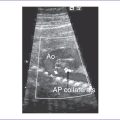
3. Hypoplastic ascending aorta, particularly proximally, with the largest aspect at the distal arch. The ascending aorta may be so hypoplastic that it is not clearly visible in the three-vessel view.
4. Retrograde flow in the aortic arch.
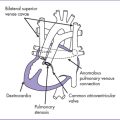
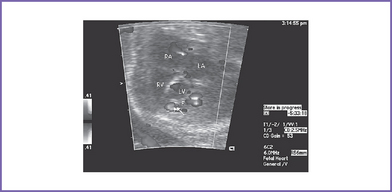
6. Left-to-right atrial shunt (Fig. 15-3).
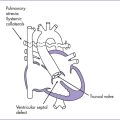
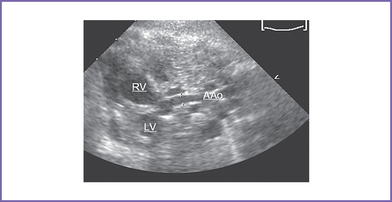
7. Hypoplastic ascending aorta and enlarged pulmonary artery (Fig. 15-4).
8. Retrograde flow in the aortic arch.
F. Immediate postnatal management
1. Management of patients without a prenatal diagnosis of HLHS.
b. Once a patent ductus arteriosus is confirmed, prostaglandins (Box 15-1) are initiated, and maneuvers should be used to minimize systemic vascular resistance and maximize PVR.
c. Observation and monitoring.
d. If the baby is clearly demonstrating problems with either excessive pulmonary blood flow—signs of overcirculation (arterial blood gas showing raised PaO2)—or inadequate systemic perfusion or both, the following measures can be attempted:
e. Surgical treatment has been addressed in the management of the case.
G. New hybrid strategy for palliative management of HLHS
H. Pathophysiology
1. During fetal life, the PVR is higher than systemic vascular resistance (SVR), and the dominant RV maintains normal blood flow to the upper and lower body as well as to the placenta.
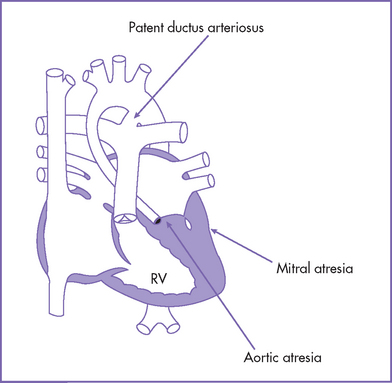
2. The size of the LV is influenced by the morphology of the mitral valve and the presence or absence of a ventricular septal defect. In cases of aortic atresia combined with mitral atresia, the LV cavity is small (Fig. 15-5).
3. If the mitral valve is patent, thus allowing blood inflow, then the LV is usually larger, although still hypoplastic. In such cases, the LV myocardium might show varying degrees of endocardial fibroelastosis.
4. Severe obstruction to the egress of blood from the LV in the presence of an intact ventricular septum is a prerequisite for the presence of endocardial fibroelastosis.
5. Occasionally, the LV cavity can be of normal size with aortic atresia (in 2%-7%). The majority of such cases were associated with a VSD.
6. After birth there normally is a fall in the PVR, with the SVR being higher than the PVR. In addition, the ductus arteriosus typically closes within the first couple of days postnatally.
7. In HLHS, if the lesion is not recognized, there is a progressive increase in pulmonary blood flow that can virtually steal from the systemic circulation (low effective cardiac output).
8. Oxygen content is increased, with large  p/
p/ s (ratio of pulmonary flow to systemic flow). When saturations approach 90%, the systemic blood flow is reduced, causing inadequate tissue perfusion. This can result in severe hypoperfusion, metabolic acidosis, and shock.
s (ratio of pulmonary flow to systemic flow). When saturations approach 90%, the systemic blood flow is reduced, causing inadequate tissue perfusion. This can result in severe hypoperfusion, metabolic acidosis, and shock.
9. In patients with too much pulmonary blood flow, particularly with progressive metabolic acidosis, inotropic support (particularly at doses that cause alpha receptor effect) should be minimized because it can increase the pulmonary blood flow by increasing the SVR. Afterload-reducing agents such as milrinone may be helpful.
10. The volume-overloaded single ventricle also suffers from myocardial dysfunction and AV valve regurgitation
I. Risk of recurrence
1. In general, risk of recurrence of some form of left-sided heart lesions ranges from 8% to 16%.
2. Specifically for HLHS, the risk of recurrence may be as high as 2%.
3. In practice, a recessive mode of inheritance should be considered in families with two affected children or in cases of parental consanguinity.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. Management of HLHS starts prenatally with in utero diagnosis.
2. The foramen ovale is usually patent and shows left-to-right shunt (reverse of normal). A clue to the presence of restrictive atrial septum and significant left atrial hypertension can be the abnormal pulmonary venous pulsed Doppler flow.
3. In HLHS, the interatrial septum is usually fibrotic and thick.
B. Treatment
1. Stage 1 Norwood preoperative care should include resuscitation with prostaglandin to maintain ductal patency and careful management of ventilation, avoiding overventilation and accepting a high normal carbon dioxide level.
2. Optimally, a neonate with HLHS should be left unventilated, trying to preserve the physiologic pulmonary and systemic balance and avoiding overventilation or overcirculation.
3. With increasing experience of the technique, the 3-year survival of the Norwood procedure has improved from 28% to 66%.
4. Comparing conventional surgery to primary cardiac transplantation, recent results show that identical results can be achieved with either strategy so long as the center has an adequate throughput.
5. Further developments in the technology of fetal intervention in the very small heart could alter the otherwise poor prognosis of this subgroup of HLHS.
C. Prognosis
1. Morbidity and mortality are significantly better with prenatal diagnosis: 100% survival as opposed to 66% for those diagnosed postnatally (P = 0.009).
2. Limited data are available about long-term survival of the staged Norwood procedure.
3. Critical aortic stenosis can progress to the HLHS by term due to failure of growth of the LV and the development of complete aortic atresia.
Allan LD, Sharland GK, Milburn A, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-1458.
Bash SE, Huhta JC, Vick GW3rd, et al. Hypoplastic left heart syndrome: Is echocardiography accurate enough to guide surgical palliation? J Am Coll Cardiol. 1986;7(3):610-616.
Baumgart S, Paul JJ, Huhta JC. Left heart hypoplasia in neonates having congenital diaphragmatic hernia and treated with ECMO. J Pediatr Surg. 1998;33(12):1848.
Chang AC, Huhta JC, Yoon GY, et al. Diagnosis, transport, and outcome in fetuses with left ventricular outflow tract obstruction. J Thorac Cardiovasc Surg. 1991;102(6):841-848.
Freedom RM, Benson LN, Smallhorn JF. Hypoplastic left heart syndrome. In: Freedom RM, Benson LN, Smallhorn JF, editors. Neonatal Heart Disease. New York: Springer-Verlag; 1992:333-357.
Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270-274.
Huhta JC. Neonatal hemodynamics in patients with hypoplastic left heart syndrome. Cardiol Young. 2004;14(Suppl 1):22-26.
Huhta J, Quintero RA, Suh E, Bader R. Advances in fetal cardiac intervention. Curr Opin Pediatr. 2004;16(5):487-493.
Jackson GM, Ludmir J, Castelbaum AJ, et al. Intrapartum course of fetuses with isolated hypoplastic left heart syndrome. Am J Obstet Gynecol. 1991;165(4 Pt 1):1068-1072.
Jacobs ML, Blackstone EH, Bailey LL. Intermediate survival in neonates with aortic atresia: A multi-institutional study. The Congenital Heart Surgeons Society. J Thorac Cardiovasc Surg. 1998;116(3):417-431.
Noonan JA, Nadas AS. The hypoplastic left heart syndrome; an analysis of 101 cases. Pediatr Clin North Am. 1958;5(4):1029-1056.
Taketazu M, Barrea C, Smallhorn JF, et al. Intrauterine pulmonary venous flow and restrictive foramen ovale in fetal hypoplastic left heart syndrome. J Am Coll Cardiol. 2004;43(10):1902-1907.
Tworetzky W, McElhinney DB, Reddy VM, et al. Improved surgical outcome after fetal diagnosis of hypoplastic left heart syndrome. Circulation. 2001;103(9):1269-1273.