[level-membership-for-pediatrics-category]
Chapter 645 Hypopigmented Lesions
Albinism
Several types of congenital oculocutaneous albinism (OCA) consist of partial or complete failure of melanin production in the skin, hair, and eyes despite the presence of normal number, structure, and distribution of melanocytes. They may be divided into two major classes: those with abnormal protein function involved in the formation and transfer of melanin, and those with defects in melanosomes (Table 645-1). Tyrosinase is the copper-containing enzyme that catalyzes at multiple steps in melanin biosynthesis (Chapter 79.2). Tyrosinase-positive variants are characterized by darkening of the hair bulb on incubation with tyrosine.
| DISORDER | GENE DEFECT |
|---|---|
| Oculocutaneous albinism: | |
| OCA1 | Tyrosinase |
| OCA2 | P protein |
| OCA3 | TRP-1 |
| OCA4 | MATP |
| Hermansky-Pudlak: | |
| Type 1 | HPS-1 Mouse (pale ear) |
| Type 2 | HPS-2 b3A subunit of AP3 |
| Type 3 | HPS-3 Mouse (cocoa) |
| Type 4 | HPS-4 Mouse (light ear) |
| Type 5 | HPS-5 KIAA107 |
| Type 6 | HPS-6 Mouse (ruby eye) |
| Type 7 | HPS-7 DTNBP1 |
| Type 8 | HPS-8 Bloc153 |
| Chédiak-Higashi | CHS1/LYST |
| Piebaldism | C-KIT receptor |
| Heterozygous SLUG | |
| Waardenburg: | |
| Type 1 | Heterozygous PAX-3 |
| Type 2a | MITF |
| Type 2b | Chromosome 1p |
| Type 2c | Chromosome 8p23 |
| Type 2d | SNAIL |
| Type 2e | Sox 10 |
| Type 3 | Homozygous PAX-3 |
| Type 4 | SOX 10 |
| Endothelin 3 | |
| Endothelin B receptor | |
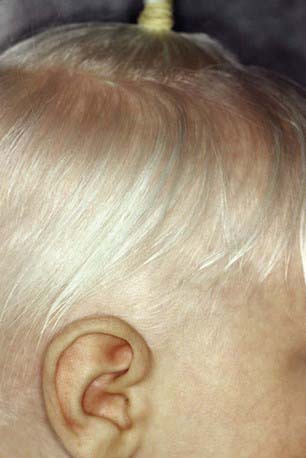
Oculocutaneous albinism type 1 (OCA1) is characterized by great reduction in or absence of tyrosinase activity. OCA1A, the most severe form, is characterized by a lack of visible pigment in hair, skin, and eyes (Fig. 645-1). This manifests as photophobia, nystagmus, defective visual acuity, white hair, and white skin. The irises are blue-gray in oblique light and prominent pink in reflected light. OCA1B, or yellow mutant albinism, manifests at birth as white hair, pink skin, and gray eyes. This type is particularly prevalent in Amish communities. Progressively the hair becomes yellow-red, the skin tans lightly on exposure to the sun, and the irises may accumulate some brown pigment, with a resultant improvement in visual acuity. Photophobia and nystagmus are present but mild. OCATS is a temperature-sensitive type of albinism. The abnormal tyrosinase has decreased activity at 35-37°C. Therefore, cooler regions of the body such as the limbs and head pigment to some degree, whereas other areas remain depigmented.
OCA4 is a rare OCA with clinical findings similar to those in OCA2.
Because of the absence of normal protection by adequate amounts of epidermal melanin, persons with albinism are predisposed to development of actinic keratoses and cutaneous carcinoma secondary to skin damage by ultraviolet light. Protective clothing and a broad-spectrum sunscreen preparation (Chapter 648) should be worn during exposure to sunlight.
Oculocutaneous Albinism with Melanosomal Abnormalities (See Table 645-1)
Chédiak-Higashi syndrome (CHS; Chapter 124) is another genetic abnormality associated with dysfunction of lysosome-related organelles. Patients with CHS have hypopigmentation of the skin, eyes, and hair; prolonged bleeding times and easy bruising; recurrent infections; abnormal natural killer cell function; and peripheral neuropathy. CHS is caused by mutations in the CHS1/LYST gene, which is a lysosomal trafficking regulatory gene.
Melanoblast Migration Abnormalities (See Table 645-1)
Piebaldism
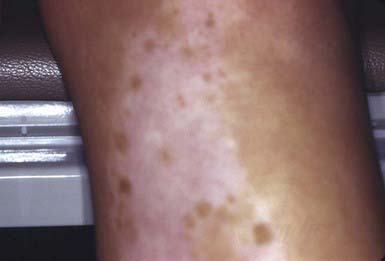
A congenital autosomal dominant disorder, piebaldism is characterized by sharply demarcated amelanotic patches that occur most frequently on the forehead, anterior scalp (producing a white forelock), ventral trunk, elbows, and knees. Islands of normal or darker than normal pigmentation may be present within the amelanotic areas (Fig. 645-2). The plaques are a result of a permanent localized absence of melanocytes. The pattern of depigmentation arises from defective melanoblast migration from the neural crest during development. The reason that piebaldism is a localized and not a generalized process remains unknown. Piebaldism must be differentiated from vitiligo, which may be progressive and is not usually congenital, nevus depigmentosus, and Waardenburg syndrome.
Hypomelanosis of Ito
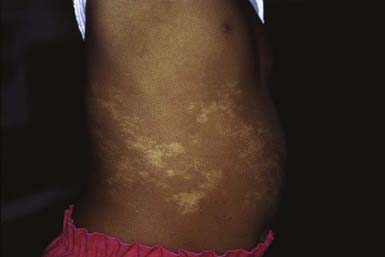
The skin lesions of hypomelanosis of Ito are generally present at birth but may be acquired in the first 2 years of life. The lesions are similar to a negative image of those present in incontinentia pigmenti, consisting of bizarre, patterned, hypopigmented macules arranged over the body surface in sharply demarcated whorls, streaks, and patches that follow the lines of Blaschko (Fig. 645-3). The palms, soles, and mucous membranes are spared. The hypopigmentation remains unchanged throughout childhood but fades during adulthood. The degree of depigmentation varies from hypopigmented to achromic. Neither inflammatory nor vesicular lesions precede the development of the pigmentary changes as in incontinentia pigmenti. The hypopigmented areas demonstrate fewer and smaller melanocytes and a decreased number of melanin granules in the basal cell layer than normal. Inflammatory cells and pigment incontinence are lacking.
Vitiligo
Clinical Manifestations
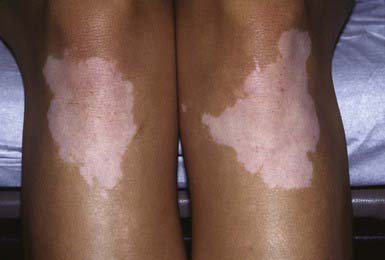
There are two subtypes of vitiligo, generalized (nonsegmental) and segmental, which probably are distinctly different diseases (Table 645-2). Generalized vitiligo (85-90% of cases) may be divided into widespread (type A) and localized (type B). About 50% of all patients with vitiligo have onset before 18 yr of age, and 25% demonstrate depigmentation before age 8. Most children have the generalized form, but the segmental type is more common among children than among adults. Patients with the generalized form usually present with a remarkably symmetric pattern of white macules and patches (Fig. 645-4); the margins may be somewhat hyperpigmented. The patches tend to be acral and/or periorificial. Occasionally, almost the entire skin surface becomes depigmented.
Table 645-2 TYPICAL FEATURES OF SEGMENTAL AND NONSEGMENTAL VITILIGO
| SEGMENTAL VITILIGO | NONSEGMENTAL VITILIGO |
|---|---|
| Often begins in childhood | Can begin in childhood, but later onset is more common |
| Has rapid onset and stabilizes | Is progressive, with flare-ups |
| Involves hair compartment soon after onset | Involves hair compartment in later stages |
| Is usually not accompanied by other autoimmune diseases | Is often associated with personal or family history of auto-immunity |
| Often occurs in the face | Commonly occurs at sites sensitive to pressure and friction and prone to trauma |
| Is usually responsive to autologous grafting, with stable repigmentation | Frequently relapses in situ after autologous grafting |
| Can be difficult to distinguish from nevus depigmentosus, especially in cases with early onset |
From Taïeb A, Picardo M: Vitiligo, N Engl J Med 360:160–168, 2009.
Berker N, Ozdamar Y, Soykan E, et al. Vogt-Koyanagi-Harada syndrome in children: a report of a case and a review of the literature. Ocul Immunol Inflamm. 2007;15:351-357.
Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007;2:43.
Huggins RH, Schwartz RA, Janniger CJ. Childhood vitiligo. Cutis. 2007;79:277-280.
Hurford MT, Sebastiano C. Hermansky-Pudlak syndrome: report of a case and review of the literature. Int J Clin Exp Pathol. 2008;1:550-554.
Janiua SA, Khachemoune A, Guidbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
Karaman A, Aliagaoglu C. Waardenbur syndrome type 1. Dermatol Online J. 2006;30:21.
LePoole IC, Luiten RM. Autoimmune etiology of generalized vitiligo. Curr Dir Autoimmun. 2008;10:237-243.
Lio PA. Little white spots: an approach to hypopigmented macules. Arch Dis Child Educ Pract Ed. 2008;93:98-102.
Spritz RA. The genetics of generalized vitiligo. Curr Dir Autoimmun. 2008;10:244-257.
Taïeb A, Picardo M. Vitiligo. N Engl J Med. 2009;360:160-168.
Westerhof W, d’Ischia M. Vitiligo puzzle: the pieces fall in place. Pigment Cell Res. 2007;20:345-359.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 645 Hypopigmented Lesions
Albinism
Several types of congenital oculocutaneous albinism (OCA) consist of partial or complete failure of melanin production in the skin, hair, and eyes despite the presence of normal number, structure, and distribution of melanocytes. They may be divided into two major classes: those with abnormal protein function involved in the formation and transfer of melanin, and those with defects in melanosomes (Table 645-1). Tyrosinase is the copper-containing enzyme that catalyzes at multiple steps in melanin biosynthesis (Chapter 79.2). Tyrosinase-positive variants are characterized by darkening of the hair bulb on incubation with tyrosine.
| DISORDER | GENE DEFECT |
|---|---|
| Oculocutaneous albinism: | |
| OCA1 | Tyrosinase |
| OCA2 | P protein |
| OCA3 | TRP-1 |
| OCA4 | MATP |
| Hermansky-Pudlak: | |
| Type 1 | HPS-1 Mouse (pale ear) |
| Type 2 | HPS-2 b3A subunit of AP3 |
| Type 3 | HPS-3 Mouse (cocoa) |
| Type 4 | HPS-4 Mouse (light ear) |
| Type 5 | HPS-5 KIAA107 |
| Type 6 | HPS-6 Mouse (ruby eye) |
| Type 7 | HPS-7 DTNBP1 |
| Type 8 | HPS-8 Bloc153 |
| Chédiak-Higashi | CHS1/LYST |
| Piebaldism | C-KIT receptor |
| Heterozygous SLUG | |
| Waardenburg: | |
| Type 1 | Heterozygous PAX-3 |
| Type 2a | MITF |
| Type 2b | Chromosome 1p |
| Type 2c | Chromosome 8p23 |
| Type 2d | SNAIL |
| Type 2e | Sox 10 |
| Type 3 | Homozygous PAX-3 |
| Type 4 | SOX 10 |
| Endothelin 3 | |
| Endothelin B receptor | |
Oculocutaneous albinism type 1 (OCA1) is characterized by great reduction in or absence of tyrosinase activity. OCA1A, the most severe form, is characterized by a lack of visible pigment in hair, skin, and eyes (Fig. 645-1). This manifests as photophobia, nystagmus, defective visual acuity, white hair, and white skin. The irises are blue-gray in oblique light and prominent pink in reflected light. OCA1B, or yellow mutant albinism, manifests at birth as white hair, pink skin, and gray eyes. This type is particularly prevalent in Amish communities. Progressively the hair becomes yellow-red, the skin tans lightly on exposure to the sun, and the irises may accumulate some brown pigment, with a resultant improvement in visual acuity. Photophobia and nystagmus are present but mild. OCATS is a temperature-sensitive type of albinism. The abnormal tyrosinase has decreased activity at 35-37°C. Therefore, cooler regions of the body such as the limbs and head pigment to some degree, whereas other areas remain depigmented.
OCA4 is a rare OCA with clinical findings similar to those in OCA2.
Because of the absence of normal protection by adequate amounts of epidermal melanin, persons with albinism are predisposed to development of actinic keratoses and cutaneous carcinoma secondary to skin damage by ultraviolet light. Protective clothing and a broad-spectrum sunscreen preparation (Chapter 648) should be worn during exposure to sunlight.
