74 Hyperthermia
Hyperthermia (HT) is the use of elevated temperature for the treatment of cancer, in this case, typically using temperatures in the range of 41° C to 45° C for 1 hour or more. Tumor regression after a high fever caused by erysipelas was first reported in the medical literature in 1866 by the German physician W. Busch.1 This case and others led a New York surgeon, W. B. Coley, to treat cancer patients with bacterial pyrogens (Coley toxin).2 In 1910, Müller described the potential of HT as an adjuvant to radiotherapy (RT).3 However the biologic rationale for applying HT with RT in cancer therapy was not investigated in depth until the 1970s.4–6 Exciting laboratory studies demonstrating the activity of heat against tumor cells in tissue culture and animal models encouraged numerous nonrandomized clinical trials of RT plus HT in small superficial tumors.7 Promising results of these early trials led to premature randomized trials, before adequate thermometry or hyperthermic delivery systems were available, with disappointing results.8,9 Since then, multiple prospective, randomized trials have shown a benefit of adjuvant HT,10–15 and thermal dose-response analyses have predicted appropriate thermal dose prescriptions for superficial and deep tumors.16 New protocols that prescribe higher minimum thermal doses or combine HT simultaneously with RT for increased thermal radiosensitization are nearing completion. Encouraging trials of HT combined with chemotherapy or liposomal chemotherapy have been performed. There is also recent interest in using HT as a modulator for gene therapy or immunotherapy. However, for HT to become an established treatment modality, much work is needed to (1) better understand and exploit the biology of heat in combination with RT or chemotherapy or novel therapies; (2) improve the equipment used for performing, monitoring, and planning hyperthermic treatments; and (3) further define appropriate thermal dose goals and clinical applications. This chapter reviews the biologic rationale for HT, physical principles of techniques used to heat tissues, clinical results, and future directions of HT research.
Biologic Rationale
Heat kills cells as a function of time and temperature and also sensitizes cells to radiation damage. Evidence suggests that protein denaturation is involved in this process, whereas direct damage to deoxyribonucleic acid (DNA) is not.17 Although the exact mechanisms of hyperthermic cell killing are still not clear, much progress has been made in the past decade in understanding the molecular, biochemical, and cellular consequences of thermal stress. Heating induces a variety of intracellular changes: activation of heat shock transcription factors; enhanced synthesis of heat shock proteins (HSPs); and alterations in nuclear and cytoskeletal structures, cellular metabolism, macromolecular synthesis, intracellular signal transduction, and hormone-receptor interactions.18
Cellular Response to Heat
Exposure of mammalian cells to temperatures higher than 40° C leads to reproductive cell death. This effect of hyperthermic treatment on cells’ reproductive capacity depends on both the applied temperature and the duration of the exposure, and can be represented as survival curves resembling x-ray survival curves (Fig. 74-1). When the surviving cell fraction (capable of reproduction) is plotted on a logarithmic scale versus the duration of heating on a linear scale, the survival curve is characterized by an initial shoulder region followed by an exponential decrease in the surviving fraction.5 The time required to reduce the survival in the exponential region to 37% of its initial value is defined as D0.
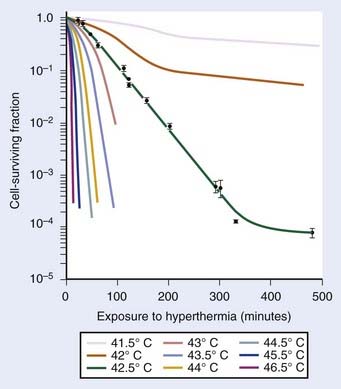
FIGURE 74-1 • Survival curves for asynchronous Chinese hamster ovary cells heated at different temperatures for varying time.
(From Hall EJ: Radiobiology for the radiologist. Philadelphia, 1994, JB Lippincott Company, p 260.)
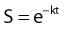
in which S is survival, t is the treatment time, and k is the inactivation rate at the treatment temperature.19 The term k may also be replaced by 1/D0 as used in RT biology.6 This model does not account for the survival curve “shoulder.”
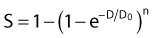
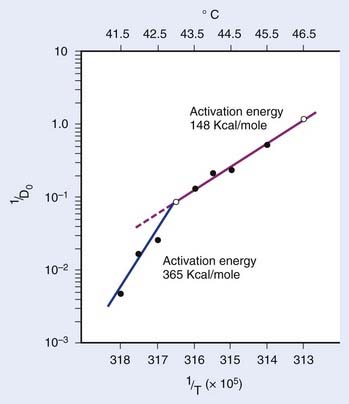
in which n is the number of targets to be inactivated before the cell is killed.6,20 The survival curves for this model are characterized by the parameters n, D0, and Dq (the quasithreshold dose) in which Dq = (ln n)D0. Using this model, one can construct Arrhenius plots to demonstrate the kinetics of heat killing, where 1/D0 is plotted against the inverse of the absolute temperature (Fig. 74-2). For most cell lines studied, the Arrhenius plots of cell inactivation seem to be composed of two segments, with a break at or near 43° C.4 Above the break point, the activation energy is between 110 and 150 kcal/mol, which is the energy required for protein denaturation,21 suggesting that protein damage is mainly responsible for cell death caused by HT. At lower temperatures, the activation energy is higher, approximately 300 to 400 kcal/mol. On the basis of the difference in activation energies, it has been suggested that there may be different primary modes of heat-induced cell death, one dominant higher than 43° C and the other lower than 43° C. However, the change in the slope of Arrhenius plots lower than 43° C could simply be a manifestation of the ability of the cells to develop thermotolerance, the transient resistance of cells to heat killing.22 The Arrhenius plot for a given cell line can be modulated by thermotolerance or environmental parameters such as pH.
Using the activation enthalpy and entropy for hyperthermic cell killing (determined from Arrhenius plots based on D0s), Lepock and colleagues23 introduced a thermodynamic model for hyperthermic cell killing based on the existence of a critical target. The rate of cell killing is thought to correspond with the rate of inactivation and denaturation of this hypothetical critical target. The calculated transition temperature of this critical target appeared to be 46.0° C for V79-WNRE cells when the temperature was raised by 1° C per minute. When the scan rates of 0.1° C per minute or 10° C per minute were used for differential scanning calorimetry (DSC), the calculated transition temperature was 43.5° C or 48.5° C, respectively. Thus, transition temperatures are only meaningful when the scan rate used in the measurements is specified.23 Cellular fractionation to localize the critical target showed that protein denaturation occurred at 46° C in all subcellular fractions.
A fourth model, which nicely describes heat killing under various conditions (single heating, thermotolerance, and step-down heating), was proposed by Jung.24 This model assumes a two-step process. The first step is the production of nonlethal lesions that can be converted into lethal lesions upon further heating in the second step. After the cells are heated for a time t at a certain temperature, the surviving fraction S is given by the equation:
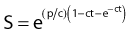
in which p is the rate constant for the production of nonlethal lesions per cell per unit of time and c is the rate constant for the conversion of a nonlethal lesion into a lethal lesion per unit of time. One lethal lesion is sufficient to kill a cell. Jung assumed that the production and conversion of nonlethal lesions were random and the rate constants depended only on the temperature of the heat treatment.
The intrinsic thermal sensitivity of different cell lines varies significantly. So far, no consistent difference has been demonstrated between normal and malignant cells. On the other hand, the extracellular milieu can modify cells’ thermal sensitivity. In general, cells that are at low pH or are nutrient-deprived are more sensitive to heat. These cells may be hypoxic cells that are three times more resistant to radiation than are normally oxygenated cells. However, cells exposed to low pH for long periods adapt to pH changes and lose their increased heat-sensitivity.25,26 Sensitivity to heat shock as a function of cell cycle has also been extensively studied. It is well established that the age response of thermal sensitivity complements that for x-irradiation. The phase of the cell cycle most resistant to x-rays (late S phase), is most sensitive to heat shock response.27
When more than one heat treatment is given, cellular thermal sensitivity to the combined treatments can be modified depending on the order of the heat shock temperatures involved. Step-up heating occurs when the second heat shock is at a higher temperature than the first heat exposure at 39° C to 42° C. Conversely, step-down heating occurs when the first treatment is a short exposure at a high temperature, greater than 43° C, immediately followed by a subsequent exposure to less than 43° C. During step-up heating, thermotolerance may be induced if the first treatment temperature is less than 43° C. Therefore, the cytotoxic effect of combined heat treatments is much less than the effect without preheat treatment. On the other hand, during step-down heating, preheat treatment at greater than 43° C for a short period can sensitize cells to a subsequent heat shock treatment at a lower temperature, which may have been nonlethal by itself without the preheat treatment. However, a high temperature pulse does not abolish any thermotolerance developed earlier.6
Thermal Dose
Because heat killing is a function of both time and temperature, a thermal dose unit is needed to combine both factors and account for the different effect of heat higher and lower than 43° C. Equivalent minutes at 43° C (EM 43° degrees) was proposed as the thermal dose unit by Sapareto and Dewey in 1984:28
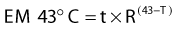
in which t is the time at temperature T, the constant R = 0.5 for temperatures higher than 43° C, and R is 0.25 less than 43° C. Thermal dose for a given treatment can be obtained by summing thermal doses for each minute of treatment, and the thermal dose from multiple treatments can be summed to obtain the cumulative equivalent minutes (CEM) at 43° C for an entire treatment course, assuming that the interval between heat treatments is long enough to allow for decay of thermotolerance. To describe the variations in thermal dose throughout a tumor, minimum, maximum, and average values can be given as well as thermal dose values for the T90 and T50 temperatures (the temperatures exceeded by 90% or 50% of the measured intratumoral points, respectively), and so on.
This concept of thermal dose has been validated in multiple pet animal and human clinical trials. Multivariate analysis of a randomized trial in pet animals with spontaneous tumors treated with RT alone or RT plus HT showed that, along with tumor volume, the non–site-specific average minimum EM 43° was the most important predictor of complete response (CR) (P < 0.001) and local control (P < 0.05). The CR rate was only 32% for 28 tumors with an average minimum CEM 43° (CEM 43° Tmin) of 1.0 minute or less, but 68% for 79 tumors with greater than 1.0 average CEM 43° Tmin.29 A thermal dose analysis of a prospective, randomized trial in patients with recurrent breast cancer showed a significant association between CEM 43° Tmin and response, with CR rates of 43% for CEM 43° Tmin less than or equal to 10 minutes versus 77% for CEM 43° Tmin greater than 10 minutes.30 Retrospective analyses of HT patients treated at Duke University showed that the cumulative minutes of treatment for which T90 exceeded 39.5° C was highly associated with CR of superficial tumors,31 and that the cumulative minutes of treatment for which T50 exceeded 41.5° C was highly associated with greater than 80% necrosis of soft-tissue sarcomas treated preoperatively with thermoradiotherapy.32 Thermal dose-response formulas based on these data showed that T90 and T50 temperatures routinely achieved need to be improved by 1.2° C to 1.5° C, or treatment duration increased threefold to fivefold, to increase thermal dose sufficiently to justify phase III trials.16 There were also significant associations between CEM 43° T90 and response in a study of preoperative regional HT combined with conventional RT and low-dose 5-fluorouracil chemotherapy and leucovorin for 37 locally advanced primary rectal cancers (P = 0.006)33 and between CEM 43° T90 and CR and local control in patients with recurrent breast adenocarcinoma treated at Stanford with RT and HT.34 These parameters, CEM 43° Tmin and CEM 43° T90, have become standard thermal dose descriptors for superficial HT.
Mechanisms of Action of Heat
Unlike ionizing radiation, HT does not result in the localized deposition of high levels of energy in cells. The thermal energy is more or less evenly absorbed by all molecules in the cell. As mentioned previously, the activation energy obtained for cultured cell lines and transplantable tumors in the temperature range from 42.5° C to 47° C is approximately 150 kcal/mol,6,27 suggesting that protein denaturation is the main cause of hyperthermic cell killing.
More direct evidence that protein denaturation is responsible for thermal killing was provided using DSC.23 In eukaryotes, the onset for protein denaturation was found to be approximately 40° C. Thermotolerance, cycloheximide, and D2O all increased the thermostability of proteins and also resulted in increased survival levels after HT.35 These results were further confirmed by studies conducted by Burgman and colleagues36 using electron spin resonance measurements and thermal gel analysis, showing that a correlation existed between protein denaturation and heat killing.
Protein denaturation and subsequent aggregation appear to occur throughout the entire cell, including cytoplasmic, nuclear, and membrane proteins.36 It is unlikely that a single type of protein is the critical target for cell killing by heat. All subcellular structures contain temperature-sensitive proteins that denature and may subsequently aggregate.37 Small changes in temperature can drastically alter the structure of plasma membranes and impair many membrane-related functions that, by themselves or in combination with other protein damage in the cell, could lead to cell death.38,39 Heat has also been shown to alter the mitotic spindle and centrosome organization, resulting in the formation of multinucleated, nonclonogenic cells,40 and to damage mitochondria,41 with heat-induced inhibition of metabolic functions such as glycolysis and respiration. Heat damages polysomes and microsomes and inhibits the synthesis of proteins, ribonucleic acid (RNA), and DNA in a thermal dose–dependent manner.39,41 The inhibition of RNA and DNA synthesis appears to be due to heat-induced changes in chromatin structure caused by the denaturation and aggregation of nuclear proteins. Heat-induced changes in the nuclei of eukaryotic cells include the appearance of actin bundles; increased vesiculation; the reduction of intact nucleoli41; and a dramatic, dose-dependent increase in protein content in isolated chromatin, nuclei, nuclear matrices, and nucleoids.35,42,43 This appears to be at least partially due to decreased leakage of proteins such as DNA and RNA polymerases from the nucleus during isolation as the heat-denatured proteins become aggregated and thus insoluble. This phenomenon is referred to as heat-induced nuclear protein aggregation. It is likely that nuclear protein aggregation, which occurs largely at the nuclear matrix, interferes with DNA replication, transcription complex interactions, and DNA unwinding needed for DNA replication and DNA repair.44
Interestingly, on exposure to heat, a group of proteins called heat shock proteins (HSPs) translocate from the cytoplasm to the nucleus.41,44,45 It has been shown that HSPs protect against protein aggregation or facilitate disaggregation of heat-induced insoluble protein complexes in cell lines and yeast.46–48
Thermotolerance and Heat Shock Proteins
One of the most interesting aspects of thermal biology in the mammalian system is the response of heated cells to subsequent heat challenges. Mammalian cells, when exposed to a nonlethal heat shock, have the ability to acquire a transient and nonheritable resistance to subsequent exposures at elevated temperature. This phenomenon, first shown by Henle and Leeper49 and Gerner and Schneider,50 has been described as induced thermal resistance, thermal tolerance, or, most commonly, thermotolerance. Several excellent reviews have discussed this phenomenon in considerable detail.51–53
In vitro, acute thermotolerance can be induced by a short initial heat treatment at temperatures higher than 43° C followed by a 37° C incubation before the second heat challenge. Thermotolerance can also be induced during a continuous and long exposure (1 to 20 hours) at temperatures lower than 43° C (chronic thermotolerance).54–56 The degree of thermotolerance developed can be dramatic; increases in survival levels by several orders of magnitude are commonplace. The thermal history, heat fractionation interval, and recovery conditions all significantly modify the kinetics of thermotolerance.52,53
Thermotolerance can also be induced by treatment of cells with a variety of chemicals followed by a drug-free period before the heat challenge. Some examples of the thermotolerance-inducing chemicals are heavy metals, ethanol, sodium arsenite, procaine, lidocaine, aliphatic alcohols (C5-C8), dinitrophenol, carbonyl cyanide m-chlorophenyl hydrazone, puromycin, and prostaglandins.56–58 Pretreatment with heat or chemicals induces both thermotolerance and resistance against the cytotoxicity of the tolerance-inducing chemicals. This development of cross-resistance suggests overlap in the mechanisms of induction of tolerance by heat and chemicals.
The amount of thermotolerance expressed depends on the temperature and duration of the first heat treatment and the interval between the two heat treatments (Fig. 74-3).59 At temperatures of 43° C or higher, thermotolerance does not develop during the first heat exposure; a subsequent incubation at 37° C is required for its expression. In contrast, for initial treatment at 41° C, thermotolerance is almost completely developed at the end of the first heat treatment. Based on these data, an operational model for the development of thermotolerance was formulated with three phases: an initial event (trigger), the expression of resistance (development), and the gradual disappearance of resistance (decay).59 First, the triggering event converts normal cells to the triggered state with a rate constant k1. This process very likely involves the activation of the heat shock transcription factor-1 (HSF1)60,61 and probably an additional regulatory factor or factors.62 Second, these triggered cells are converted to thermotolerant cells with a rate constant k2. Higher than 43° C, k2 = 0; the triggered cells remain sensitive, and only after being transferred to 37° C do they convert to their thermotolerant state. This thermotolerant state, in general, is associated with the elevated expression of HSPs, and enhanced protection against and faster recovery from thermal damage. Finally, thermotolerant cells all reconvert to their sensitive state at a slower rate governed by rate constant k3.
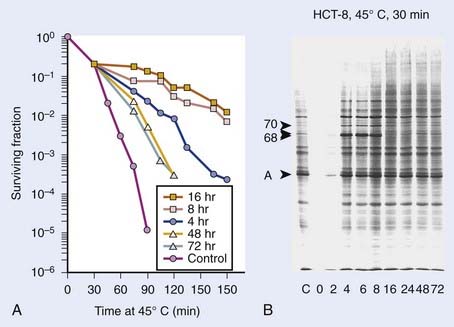
The HSPs are usually identified by their molecular mass; for example, HSP70 is an HSP with a molecular mass of 70,000 daltons. In mammalian systems, HSP110, HSP90, HSP70, HSP60, HSP56, HSP47, HSP40, and HSP27 have been identified. The most extensively studied HSPs with respect to their role in thermotolerance and cellular heat sensitivity are HSP27, HSP70, and HSP90. In mammalian cells, good correlations have been reported for thermotolerance development and (1) HSP70 synthesis, (2) HSP27 synthesis, and (3) HSP27 phosphorylation. Enhanced thermosensitivity has been shown to result from reduction of the level of HSP70, whether by competitive inhibition of HSP70 gene expression or by microinjection of cells with antibodies against HSP70. Reduction of the cellular HSP90 levels has also been shown to increase thermosensitivity in mouse L cells, whereas an increased HSP90 expression resulted in stable heat resistance in Chinese hamster ovary (CHO) cells.63 Transfection of cells with genes coding for HSP2764 or HSP70,65,66 resulting in constitutive expression of the genes, confers stable heat resistance. Transfection of rat cells with a gene coding for a homologue of the mammalian HSP60 also confers heat resistance to the transfected cells (Burgman and colleagues, unpublished results). These data provide direct evidence for a causal relation between the expression of HSPs and heat resistance.
Chronic thermotolerance (thermotolerance that develops during long-duration, mild HT at approximately 41° C) may be different from acute thermotolerance, which occurs after heat shock and incubation before heating at high hyperthermic temperatures at or higher than 43° C). Chronic-type thermotolerance is not always observed in all mammalian cell lines. Studies with a range of human cell lines have shown that, unlike rodent cells, the human cells did not develop chronic thermotolerance during mild HT. In HeLa cells, this absence of thermotolerance may be related to cell-cycle progression.67 In other studies, the lack of thermotolerance did not appear to be linked directly to cell-cycle progression, as shown by cell-cycle analysis and the use of plateau-phase cultures.68,69 More research is needed to better understand the development and decay of acute and chronic thermotolerance in human tumors.
Regulation of Heat Shock Response
As described earlier, the heat shock response refers to the increased transcription of a set of genes, the heat shock genes, when cells are subjected to heat stress. It is a highly conserved process that occurs in all organisms from bacteria to human. In eukaryotes, the response is mediated by a sequence-specific DNA binding protein termed HSF1.70 In its trimeric form, HSF1 binds tightly to multiple copies of a highly conserved sequence motif (nGAAn) in the promoter region of heat shock genes; these HSF-binding sites are termed the heat shock elements (HSEs).71,72 In unstressed mammalian cells, HSF1 is maintained in a monomeric, non-DNA binding form. Upon heat shock, HSF1 assembles into trimers, binds to HSEs with high affinity, becomes hyperphosphorylated, and activates the transcription of the heat shock genes. Transcriptional activation of these genes, in turn, increases the synthesis of HSPs. During post–heat-shock recovery, the HSF1 trimers dissociate from the DNA, and are eventually converted to the inactive, non-DNA binding monomers. Several other models, similar to the previously described model, have also been proposed.18,73
Although the importance of HSF1 in the regulation of mammalian heat shock genes is well-established,70 data from multiple laboratories obtained using various rodent and human cell lines indicate that the activation of HSF1 by itself is not sufficient for the induction of HSP70 messenger RNA (mRNA) synthesis,62,74–80 and that other factors may be involved in the regulation of HSP70 transcription. Subsequent studies of Rat-1, hamster HA-1, and HeLa cells provided evidence of the plausible involvement of a negative regulator,62,81 initially termed constitutive HSE binding factor (CHBF). Upon heat shock, the cellular level of the HSE binding activity of HSF1 rapidly increases (“activation of HSF1”) and that of CHBF rapidly decreases (“inactivation of CHBF”). When HSP70 transcription returns to its pre–heat-shock level during post–heat-shock recovery, the disappearance of the HSF1-HSE binding activity again parallels the reappearance of the CHBF-HSE binding activity.62,82 Interestingly, in rat, mouse, and human cells treated with sodium salicylate or sodium arsenite, which elicits considerable HSF1-HSE binding activity but shows no reduction in CHBF-HSE binding activity, the induction of HSP70 mRNA synthesis was not observed.62,77,80,83 Thus, data from studies of a wide variety of rodent and human cells all support the notion of a negative regulatory role of CHBF in HSP70 induction.
Subsequently, CHBF was purified and identified as the Ku protein, a heterodimer of Ku70 and Ku80,81 allowing more direct testing of the hypothesis that CHBF/Ku acts as a negative regulator in the heat induction of HSP70. To carry out such experiments, Li and colleagues established Rat-1 cells that stably and constitutively express human Ku70 and/or Ku80, and found that overexpression of the human Ku70 by itself, or Ku70 and Ku80 jointly, specifically inhibits heat-induced HSP70 expression without affecting induction of other HSPs or activation of HSF1.84,85 How does Ku70 specifically suppress HSP70 expression? There are at least four non–mutually exclusive possibilities: (1) Ku70 may bind directly to sites located in the HSP70 promoter and repress transcription; (2) Ku may regulate HSF1 binding to the HSP70 promoter; (3) Ku may affect the interaction among other transcription factors and their binding to the HSP70 promoter; (4) overexpression of Ku70 could result in altered chromatin structure, inhibiting the access of positive factors to their respective regulatory elements in the HSP70 promoter. Related to the second possibility is the recent finding that in vitro translated Ku70/Ku can bind to HSF1 and displace it from HSE.86,87 Thus the existing evidence strongly supports a role of Ku in HSP70 gene regulation.
Interaction of Heat With Radiation
Heat interacts with radiation in a more than additive way. This synergistic interaction of heat and radiation is interpreted as a heat-induced sensitization of cells to radiation, termed heat radiosensitization, or thermal radiosensitization. Heat radiosensitization can be quantified using a thermal enhancement ratio (TER), defined as the ratio of radiation doses with and without heat to produce the same biologic effect (isoeffect TER). In some cases, TERs are expressed as a ratio of the biologic effects for the same radiation dose, with and without heat (isodose TER), or as a ratio of the D0’s of the radiation survival curves. Usually, TER increases with increasing heat dose. The maximal interaction is observed when heat and radiation are given simultaneously. The TER decreases with an increasing time interval between heat and radiation (Fig. 74-4). When radiation precedes HT, sensitization is no longer detectable 2 to 3 hours after radiation. When HT precedes radiation, cells can be sensitized for up to several hours.
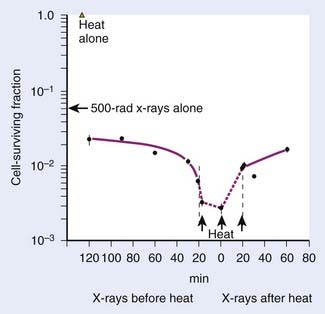
The therapeutic gain factor (TGF) is defined as the ratio of the TER in the tumor to the TER in surrounding normal tissue. Maximal heat radiosensitization is achieved when heat and radiation are applied simultaneously, but a therapeutic gain (more damage to tumor than to normal tissue) may not always be obtained under these conditions, especially if tumor and normal tissue are heated to the same degree.88,89 In some cases, the TGF may increase when time intervals are introduced between radiation and heating.
The combination of heat and radiation does not result in an enhancement of heat lesions; thus, no radiation-induced heat sensitization takes place and the synergistic effect is most likely due to radiosensitization by heat.56 The primary target for radiation is believed to be DNA, whereas protein denaturation and aggregation seem to be involved in thermal cell killing.42,90 DNA damage (e.g., breaks) can be detected at low lethal doses of x-rays (≤1 Gy), whereas no DNA breaks are found at low lethal heat doses.91 The combination of radiation and HT generally does not result in more initial DNA breaks than are observed for radiation alone.90,91 Instead, HT appears to inhibit the rejoining of radiation-induced DNA strand breaks91–93 and the excision of damaged bases94 in a heat-dose–dependent manner.91,93 This effect may be caused by (1) heat-induced inactivation of DNA repair enzymes88,95; or (2) alteration of the chromatin structure resulting from protein denaturation and aggregation, which causes decreased accessibility of the damaged sites to the repair machinery.44,92,94,96 Using cell lines in tissue culture, researchers found a good correlation between the amount of heat-induced nuclear protein aggregates and the extent of thermal radiosensitization.95,97
The effect of thermotolerance on heat radiosensitization has also been a topic of extensive investigation. Studies using tissue-cultured cell lines have shown that thermoradiosensitization could be affected by thermotolerance development.98,99 Because it has been reported that HSPs might be involved in the protection against nuclear protein aggregation as well as in facilitated disaggregation,46,48 the presence of these proteins might indirectly be responsible for the thermotolerance effect on protein aggregation and as such affect DNA repair and cellular radiosensitivity. Rodent cells transfected with human HSP70 showed protection against heat-induced nuclear protein aggregation that was reflected in the extent of thermal radiosensitization.95 Transfection with HSP27 resulted in an accelerated nuclear protein disaggregation and was paralleled by an accelerated decline of thermal radiosensitization. These observations point to the importance of HSPs as a regulator in protein denaturation, aggregation, and disaggregation, thereby modulating the heat effects on DNA repair. As such, the expression of HSPs may explain the effect of thermotolerance on the extent and disappearance of thermal radiosensitization and could have a major effect on the choice of sequence and spacing of heat and radiation in clinical practice. Elevated HSP expression should be either avoided, if occurring in the tumor, or exploited, if occurring in the normal tissue at risk during treatment.
Chronic thermotolerance has been found to affect heat radiosensitization in some cell lines,17,93,99,100 although for other cell lines no effect of thermotolerance was observed at the level of heat radiosensitization.99,101 For example, chronic thermotolerance development in CHO cells inhibited further sensitization to radiation.100 This lack of further radiosensitization was also found in HeLa cells when no chronic thermotolerance developed.67 Interestingly, long-duration, mild HT (as low as 41° C) given concurrently with low-dose-rate irradiation can cause extensive radiosensitization and eliminate the low-dose-rate irradiation sparing effect.69,88,102,103 This thermoradiosensitization to low-dose-rate irradiation occurred for protracted heating times and did not appear to be inhibited for prolonged heating times, supporting the absence of a thermotolerance process in radiosensitization.69,102 The mechanism underlying this low-temperature radiosensitization may very well be different from the one discussed in the previous section on the mechanism of heat radiosensitization because DNA repair rates are often found to be higher at 41° C104–106 and heat-induced protein aggregation in the nucleus is only limited (Stege and associates, unpublished results). Effects of heat on cell-cycle progression or changes in fidelity of repair may be possible explanations of radiosensitization by long-duration, low-temperature HT.
Heat and Chemotherapy
Although the combination of HT with radiation has been the focus of more attention, there is an equally strong rationale for combining HT with chemotherapy. Moderate or even mild HT (such as 39° C) enhances cell killing in vivo of a number of chemotherapeutic agents, such as cyclophosphamide, melphalan, mitomycin C, cisplatin, doxorubicin (DOX), bleomycin, and the nitrosoureas. A variety of mechanisms may account for increased chemotherapeutic effect at elevated temperatures, including altered pharmacokinetics or pharmacodynamics, increased DNA damage, decreased DNA repair, reduced oxygen radical detoxification, and increased membrane damage.107 Concentrations of agents that are not normally toxic at normal body temperature can become cytotoxic at 39° C, and, in some cases, HT may partially overcome some types of drug resistance.
Heat and Liposomes
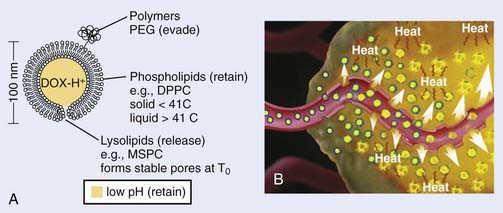
It is well recognized that when systemic chemotherapy is used for the treatment of solid tumors, significant normal tissue toxicities commonly occur when therapeutic levels of drug at the tumor site are achieved.108 Liposomes have been identified as one of the promising carriers for therapeutic agents to improve drug delivery in the treatment of cancer.109,110 Conventional liposomes have been clinically evaluated and approved in a variety of diseases111–113 and HT has been shown to increase extravasation of conventional liposomes within tumor tissue.114 Early in 1978, Yatvin and colleagues proposed the use of HT with temperature-sensitive liposomes to allow increased control over the release of encapsulated drugs at the diseased site,115 and their efforts have stimulated many additional studies.116 In 2000, Dewhirst and collaborators developed a novel thermosensitive, DOX-containing lipid formulation optimized for rapid drug release under mild hyperthermic temperatures (39° C to 40° C) readily achievable in the clinic (Fig. 74-5).113,117,118 Their in vitro studies clearly demonstrate advantages that the new lipid composition has compared with existing liposome formulation; the characteristics of attainable and narrow triggering temperature, rapid release of drug, and high accumulative drug release appear to offer potential advantages that could lead to an increased therapeutic effect over traditional chemotherapy and current liposomal and other drug carrier and delivery systems. Furthermore, in vivo studies demonstrated that this novel liposome, in combination with mild HT, was significantly more effective than free-drug or current liposome formulations at reducing tumor growth in a human squamous cell carcinoma xenograft line (FaDu), producing 100% complete regressions in 11 tumors lasting up to 60 days post-treatment.113,117 To monitor the liposome concentration distribution and drug release in vivo, magnetic resonance imaging (MRI) contrast agent, MnSO(4) and DOX were encapsulated together within this thermosensitive liposomes.119 The MRI clearly showed that the thermally sensitive liposome was only accumulated at the periphery of the tumor, concordant with the release temperature of this formulation (39° C–40° C). Because the liposomal drugs accumulate only in perivascular regions in tumors after intravenous injection, tumor cells in deeper tissue layers cannot be killed. To circumvent this deficiency, Dewhirst and collaborators decided to target tumor microvessels and therefore stop nutrient supply to deeper tumors by constructing a DOX–encapsulated, lysolecithin-containing thermosensitive liposome (LTSL). After DOX-LTSLs in combination with HT treatment, the blood flow of FaDu tumor implanted in dorsal skinfold chamber was significantly decreased, whereas only minor reductions in normal microcirculation in subcutaneous tissues were observed.120 Furthermore, a real-time evaluation of therapeutic protocols in association with outcome on an individual subject basis could be achieved by noninvasively monitoring the temporal and spatial patterns of DOX/MnSO(4)-LTSLs delivery with MRI.121
Heat Shock Proteins and Cancer Immunotherapy
Although HSPs have been intensively studied at the level of transcription and translation for mammalian cells for more than 2 decades, a novel and physiologically important role for these highly conserved gene products has only become evident in the past several years. In the early 1990s, Srivastava and colleagues found that a tumor rejection antigen, isolated by biochemical fractionation of tumor cells, was an HSP (grp94/gp96).122–125 Since then, convincing evidence has accumulated that the HSPs derived from a given cancer, including HSP70, HSP90, and grp94/gp96, can elicit protective immunity specific to that particular cancer. HSPs derived from normal tissues do not have such effect against any cancer tested. In recent years, the immunogenicity of HSP preparations from tumors has been seen in different experimental tumor systems of distinct histologic origins, ranging from chemical- or ultraviolet (UV)-induced tumors to spontaneous tumors.126–128 Immunization of mice and rats with HSP-peptide complexes leads to protective and specific immunity against cancer from which the HSP-peptide complexes are derived.124 This phenomenon has been shown in mice124 and rats129 in a wide array of tumor types, including fibrosarcoma,122 hepatocarcinoma,129 lung carcinoma, melanoma, colon carcinoma,127 squamous carcinoma,130 leukemia,131 and in prophylaxis,122,129 as well as in therapy of pre-existing diseases.127 Two high-molecular weight HSPs, HSP110 and grp170 derived from CT26 and Meth A tumors, were found to induce an antitumor response against these tumors.132–134 Furthermore, it has been demonstrated that the immunization of mice resulted in the induction of memory T cells.130 Taken together, these data suggest that HSPs prepared from tumors can serve as tumor vaccines, and that HSPs may play a central role in the host’s immune system. Currently, one HSP, the endoplasmic reticulum stress-response protein gp96, is undergoing clinical trials for cancer treatment and has yielded promising results, including the induction of antitumor immunity and some benefit for patients when administered as part of a multidose regimen.135 A recent phase III comparison of Vitespen, an autologous tumor-derived HSP gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma, shows that patients with less advanced disease (M1a and M1b) benefit from increasing doses of vaccine treatments.136 Future advances in HSP-based immunotherapy will be aided by an understanding of the mechanisms by which HSP-peptide complexes induce innate and adaptive immunity to tumor cells and target the killing of primary and metastatic cancer cells.137
Hyperthermia and Gene Therapy
Another new application of HT is its use in gene therapy to provide both spatial and temporal control over gene expression governed by heat shock or other stress-inducible promoters.138–141 As one example, Huang and colleagues transfected cells with adenovirus vectors containing an HSP70 promoter and genes for green fluorescence, interleukin 12, or tumor necrosis factor alpha.142 Experiments with heat-induced green fluorescence protein showed detectable expression at 3 hours, reaching a peak at 18 to 24 hours and disappearing by 72 hours after heating. A modest heat shock of 42° C for 30 minutes increased interleukin-12 gene expression by more than 13,000-fold over baseline and increased tumor necrosis alpha gene expression by 6.8 × 105-fold.142 To optimize the application of heat-activated gene-RT in the clinic, techniques are being investigated to noninvasively monitor the transgene activation. Li and colleagues first used micro–positron emission tomography (microPET) imaging to monitor the improvement in viral vector distribution after a mild heat shock 6 hours after administration of virus (Fig. 74-6).143 Rogers and colleagues further used microPET imaging to noninvasively monitor the heat-inducible suicide gene expression in mice bearing head and neck squamous cell carcinoma xenografts.144 Recently, Chrit and colleagues investigated in vivo spatiotemporal control of transgene activation under the regulation of a heat-inducible promoter using magnetic resonance temperature imaging for image-guided, high-intensity focused ultrasound (US).145
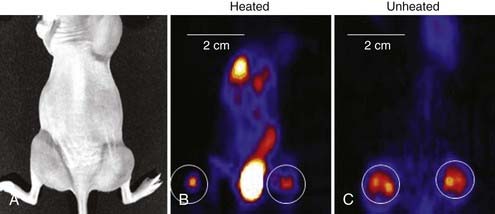
Hyperthermia and Tumor Hypoxia
Tumor hypoxia is not only one of the major factors contributing to RT resistance of solid tumors that leads to adverse treatment outcome, but also is related to disease progression and metastasis.146–149 Mild HT (<42° C) is believed to dilate tumor blood vessels and increase tumor oxygenation, and so enhance tumor radiosensitivity.150 Clinical and experimental studies showed that mild HT could increase the overall tumor pO2 level in rodent,151 canine,152 and human tumors,153,154 and the reoxygenation can even last for 1 to 2 days after heating in some tumor types.150 Recently, the change in tumor hypoxia induced by mild HT was investigated at the microscopic level by dual hypoxia marker immunohistochemistry technique in a human colorectal adenocarcinoma xenograft model (HT29).155 It was found that although the overall hypoxic fraction was significantly decreased during heating, mild HT induced both increases and decreases in tumor hypoxia in different regions of the tumor (Fig. 74-7).155 Specifically, mild HT decreased hypoxia in the regions with relatively well-perfused blood vessels, but increased hypoxia in regions that were poorly perfused. These data implied that the changes in tumor oxygenation depend on the vasodilation ability of tumor microvessels in response to HT.
Physical Principles
Mechanisms of Heating
Thermal Conduction
In the simplest form of HT, tissue may be heated by circulating externally preheated blood through the tissue, by placing a heated surface (e.g., water bolus pad) on the skin or in natural body cavities, or by interstitially implanting hot sources in wires, needles, or catheters into the target tissue. It is generally not possible to heat well-perfused, normal tissue more than 3 to 5 millimeters from a hot surface.156
A good approximation for describing heat transfer in biologic tissue is given by the bioheat transfer equation (BHTE), derived in 1948 by Penne157:
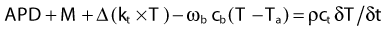
in which Δ is the vector gradient operator; tissue thermal conductivity (kt) quantifies the tissue’s ability to conduct heat; ωb and cb quantify the perfusion rate and specific heat capacity of blood; ct and ρ are the specific heat and mass density of tissue; APD represents the absorbed power density (APD) or external heat input to the tissue; M is the heat generated from tissue metabolism (generally negligible compared with other heat inputs); and T and Ta are the tissue and arterial blood temperatures, respectively, at time t. The first two terms represent heat inputs to the tissue volume; the third and fourth terms describe heat transfer kinetics caused by thermal conduction and convection (blood flow), respectively; and the right side of the equation specifies the net heating effect in terms of the rate of change of temperature with time (δT/δt). Alternative theoretical models have been studied to describe the heat transfer characteristics of tumors more accurately by accounting for “thermally significant” blood vessels,156,158 but the BHTE serves as a good starting point. Review articles are available on thermal modeling and HT treatment-planning issues.159–161
Electromagnetic Power Deposition
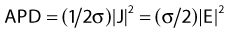
in watts per cubic meter, in which J = σE is the induced current density. This APD is closely related to the more commonly used power deposition rate or specific absorption rate (SAR) by the formula:
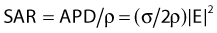
in which ρ is the mass density of tissue in kilograms per cubic meter.
For both RF and MW radiation, APD and SAR decrease with depth in tissue. The APD for the simplest case of plane-wave radiation is plotted as a function of penetration depth into muscle tissue in Fig. 74-8. At higher frequencies, power is deposited more superficially, whereas lower frequencies provide deeper penetration. However, for practical HT applicators, the tissue penetration may be much less than this because of coupling problems at the applicator-tissue interface, tissue heterogeneities, and nonuniform beam profile in the near-field region of HT applicators that are small compared with the wavelength.
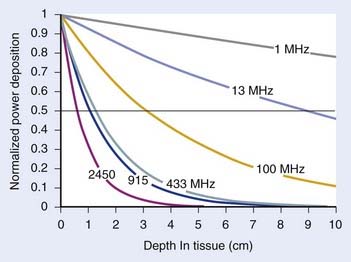
For frequencies used in HT, the wavelength (λ) of EM energy in soft tissue varies from approximately 3.4 cm at 10 MHz to less than 4 cm at 1000 MHz. The minimum spot size for focusing power deposition is approximately λ/2, or approximately 20 cm at the lowest RF frequencies typically used (100 MHz) and approximately 2 cm at the higher MW frequencies. Thus, lower RF frequencies penetrate well but affect large regions of the body, whereas higher MW frequencies may be localized effectively in tumor-sized tissue volumes. Compilations of electrical conductivity, dielectric constants, wavelengths, and penetration depths of EM waves in air, muscle, and fat are readily available.161,162
Ultrasound Power Deposition
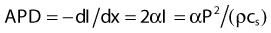
in watts per cubic meter, and again SAR is APD/ρ in watts per kilogram.
Fig. 74-9 shows the penetration depth characteristics of US in homogenous muscle tissue as a function of frequency, assuming well-behaved plane wave radiation. Higher frequencies result in more superficial localization of power and lower frequencies result in deeper penetration. For practical HT transducers, the target volume usually appears in the transducer near field, where there are marked fluctuations in beam intensity both longitudinally in the direction of propagation as well as across the wavefront, as opposed to much smoother inverse-square-law longitudinal decay of intensity in the far field. Thus, reflections, scattering, and thermal conduction within the tissue are relied on to smooth the temperature distribution that results from the peaky SAR. Nonfocused applicators operating at approximately 3.5 MHz can heat tumors up to 4 to 6 cm in depth.163 Nonfocused applicators operating at lower frequencies generally produce increased pain because of unavoidable absorption in underlying bone. Deeper penetration is made possible by directing the US beams to avoid bone and air interfaces, using a larger acoustic window at the surface, and focusing the US beams to increase the power density at depth relative to that at the skin surface and critical normal tissues outside the focal volume.
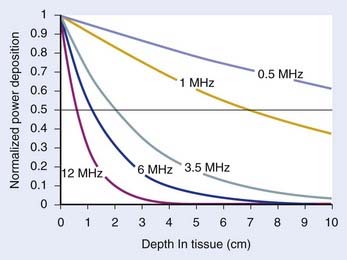
The characteristic acoustic impedance Z = ρcs is an important parameter in determining the behavior of US at tissue interfaces. The Z’s of most soft tissues are quite similar, so there is little reflective loss during the propagation of US from one soft tissue to another. However, the Z values for bone, air, and lung are considerably different from those of soft tissues, causing almost complete reflection at soft tissue–gas interfaces and both reflection and rapid absorption of the transmitted portion of the wave at soft tissue–bone interfaces.164 A tabulation of acoustic properties of tissues and more in-depth coverage of US interactions with tissue are available.161,162,165
Heating Equipment and Techniques
Local Heating—External Sources
A large development effort has gone into producing local heating devices with improved control of power deposition pattern. EM techniques are generally used for superficial tumors less than approximately 3 cm in depth, such as chest-wall recurrence of breast carcinoma, whereas US beams are useful for somewhat deeper tumors up to approximately 6 cm deep.161,166–169
Electromagnetic Techniques for Superficial Heating
The simplest, most reliable applicator used in clinical HT has been the single-aperture MW waveguide. A MW radiator mounted within a waveguide (a tubular or rectangular structure for guiding EM waves) radiates EM waves out the waveguide opening (aperture). Commercial applicators have been available in a variety of sizes from 7.5 to 24 cm on a side, normally operated at 915 MHz or approximately 430 MHz as these are the Industrial, Scientific, and Medical band frequencies allowed for use with no power restrictions in the United States, and Europe and Asia respectively. The effective heating area, or the area under the applicator producing 50% or greater of the maximal SAR, typically covers only 30% to 60% of the applicator face, normally with high-SAR “hot spots” located centrally, surrounded by a “cool” periphery.170,171 These single-aperture waveguide devices are useful for only small, superficial tumors up to approximately 3 cm in diameter.8 One group has placed variable absorption bolus pads in front of the aperture to reduce SAR centrally and achieve a larger area of effective heating.172 The HTS-100 Lens Applicator has movable metal plate “vanes” within the concave opening of a single waveguide horn aperture to form convergent 430-MHz beams for heating small tumors up to 3 to 5 cm deep.173
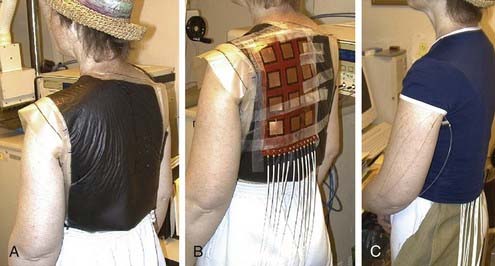
A variety of approaches have been taken to build applicators capable of heating larger areas, such as computer-controlled motorized scanning of one or two spiral microstrip MW antennas repetitively over the target surface174–176 with thermal feedback control systems. Alternatively, several groups have developed multiaperture array waveguide applicators for large-area superficial disease such as the Microtherm 1000 (Labthermics Technologies, Urbana, IL)—a 16-element, 915-MHz waveguide planar array,177 and a somewhat deeper heating array of 433-MHz Lucite Cone Applicators.178,179 Other more flexible tissue-conforming, large-area MW array applicators have been constructed and used clinically, including a 433-MHz contact-flexible microwave applicator180,181; a hinged array of 433-MHz current sheet applicators182; a 915-MHz, 25-element spiral microstrip conformal array183; and subsequent commercial implementations (Fig. 74-10)184; and the largest 915-MHz conformal microwave array (CMA) applicator.168,185 Fig. 74-11 shows a 6-mm thick water bolus vest and flexible printed circuit board CMA applicator being set up on a patient with chest-wall recurrence of breast carcinoma extending across the left upper back and shoulder. The patient is shown at right with an elastic overgarment to secure the applicator in place, allowing the patient freedom to stand, sit, or lie down during heat treatment while providing effective temperature control of the skin surface.186 This applicator is essentially transparent to photon and electron radiation and thus may be used to apply heat simultaneously with radiation for potentially much higher TER.187–189 Recent developments are beginning to produce EM superficial heating applicators with the capability of noninvasive thermal monitoring of surface190 and subsurface191–194 temperature distributions for feedback control of multiple antenna arrays.
Ultrasound Techniques
Early efforts to apply US for superficial heating applications used single-round disk “piston” style transducers operating at 0.5 to 3.5 MHz.195 These devices were capable of heating only small, superficial lesions less than 3 cm in diameter.8 Later, multitransducer arrays were constructed to increase the lateral extent and control of heating. The Sonotherm 1000 (Labthermics Technologies, Urbana, IL) is one such device with 16 individual transducers arranged in a 15-cm square 4 × 4 element planar array coupled to the patient with an attached, degassed water bolus bag.163 This device has been used successfully to heat tumors up to 4 to 6 cm in depth.
Another approach uses arrays of focused transducers. The scanned focused US system uses a small number of low-frequency transducers (≤2 MHz) that produce a small focal spot at depth that is mechanically scanned rapidly around the tumor volume.196 The addition of less-penetrating 4-MHz transducers and a patient pain feedback button to turn off power for short segments of the scan path improved the heating uniformity of this scanning US technique and yielded higher average tumor temperatures.197 Another innovative US system has a computer-controlled scanning reflector for distributing US energy at two different frequencies (e.g., 1 and 5 MHz) over large surface areas.198 With only water and plexiglas over the tumor surface, this system allows simultaneous HT and RT for potentially much higher TER.199–201 Development of high-intensity focused US for heating small- to medium-size regions at depth continues, with most commercial development supporting thermal ablation applications.202,203
Local Heating—Interstitial Sources
For applications requiring precise localization of heat in regions that are too difficult to access externally, there are at least nine distinctly different interstitial heating technologies. Excellent reviews are available.166,204–206
Microwave Antennas
Interstitial MW antennas are usually constructed from flexible or semirigid coaxial cable less than 1.5 mm in diameter that fits snugly inside afterloading plastic implant catheters. The most common types are dipole antennas, which can be used in phased arrays to enlarge the heated volume,207–209 and helical coil antennas, which tend to restrict heating more effectively to the region immediately surrounding the active coil length of the antenna.210,211 Frequencies between 433 and 2450 MHz are normally used for interstitial MW heating. Radial penetration of heating may be extended by several millimeters with air or water cooling of the implanted antenna surface, but generally multiple antennas required for heating larger tumors up to 5 cm across should be placed within 1.5 to 2 cm from each other.212 Implantable MW antennas have been most useful in high-perfusion tissues or when nonparallel or flexible catheter implants are required. Recent developments have produced interstitial MW antennas with integrated MW radiometry for real-time monitoring of tissue temperature around each antenna.213,214
Radiofrequency Electrodes
RF electrode systems heat tissue with resistively or capacitively coupled electric currents between implanted metal electrodes. Initial work used arrays of implant needles up to 20 cm long spaced 1 to 1.25 cm apart connected to a single RF power amplifier operating at less than 1 MHz. Subsequent systems used time-sequenced computer controlled localized current field (LCF) sources that distributed power to multiple electrode pairs around the implant array.204 Systems with segmented RF electrodes,215,216 printed circuit board implant templates to simplify power connections, and computer-control programs allow simultaneous long-duration heat and brachytherapy.204,217 At frequencies above 10 MHz, current may be coupled capacitively through a thin layer of insulating plastic, resulting in a more uniform distribution of heating current, as in the 64-channel, 27.12-MHz capacitively coupled RF (CC-RF) electrode system.216,218,219 For both LCF and CC-RF systems, the radial falloff of power deposition is extremely steep so that implant spacing is limited to less than 1.25 to 1.5 cm for HT applications. Methods of cooling the electrode surface and driving multiple electrode sets have proven useful for extending the effective heating volume, especially for thermal ablation applications in which higher temperatures can be tolerated near the sources.212
Hot Source Techniques
Because the penetration of thermally conducted heat energy is limited, hot source techniques are appropriate only for tissues with low to moderate perfusion that can be implanted with closely spaced arrays of implants—generally no more than 1.0 to 1.25 cm apart. However, heating length is almost unlimited. Hot source techniques include hot water perfusion through implanted catheters,204,217 resistive wire implants heated by computer-controlled direct-current voltage sources,220 and ferromagnetic implants (FMIs) heated by an externally applied magnetic field at less than 1 MHz.221–223 An advantage of the FMI approach is that the implants can be made of special high-permeability alloys that operate near their Curie point transition from magnetic to nonmagnetic such that they automatically thermoregulate over a narrow range of temperature.224,225 The ferromagnetic material providing this controlled temperature can be a needle or rod, 5- to 10-mm long cylindrical seeds interspersed with 5-mm long or less radioactive brachytherapy sources for simultaneous thermobrachytherapy, or ferromagnetic fluids or nanoparticles that are either inserted directly in the tumor in one or more needle tracks or are delivered systemically.226,227 The FMI techniques simplify reheating of permanently implanted seeds,228 rods,229 or nanoparticles230 because no externalized connections are required for either power or thermal monitoring of the thermoregulating implant material.
Ultrasound Radiators
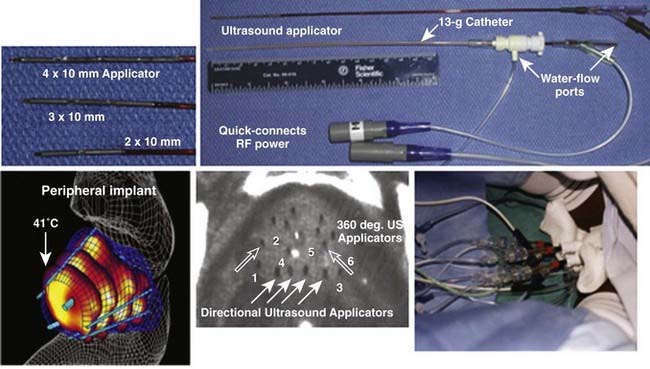
Interstitial US applicators appear close to revolutionizing interstitial HT (IHT) delivery for many sites. Linear arrays of 0.5- to 1-cm long tubular transducers have been constructed either with water cooling231 or direct coupled sources with an open central lumen to accommodate brachytherapy sources for simultaneous thermobrachytherapy.232 Implant spacing up to 2.5 cm apart should be possible for the water-cooled version. These applicators allow precise power control along the implant length as well as radially directional heating capability. Fig. 74-12 shows an example of a clinical treatment of prostate with an array of different-length US array applicators that fit inside 13-g brachytherapy catheters for highly steerable power deposition. Implantable US arrays have been used for treatment with real-time, noninvasive MRI monitoring of both temperature and tissue property changes during heating.233–235
Local Heating—Intracavitary Sources
A number of site-specific EM- and US-powered devices have been developed for inducing local HT around natural body cavities such as the esophagus, vagina, urethra, and rectum for esophageal and pelvic tumors using dipole MW antennas,236 tapered diameter dipole antennas,237 helical coil antennas,238 and segmented multielement intracavitary US applicators such as the eight-element transrectal US applicator239 and tubular phased array intracavitary applicators with up to 64 US transducers driven at 0.5 to 1 MHz.240,241
Regional Heating
Early regional heating devices included the single large 27-MHz ridged waveguide aperture242 and the single 82-MHz torso-sized helix coil.243 Using a 70-MHz coaxial transverse electromagnetic (TEM) applicator, the entire patient is positioned inside a hollow 60-cm diameter chamber filled with coupling water, and partial steering of SAR is possible by shifting the patient’s position within the chamber.244,245 Another single-power-source, deep-heating technique uses 8 to 27 MHz RF capacitive heating, such as the Thermotron RF-8.246,247 This device creates an electric field between two saline bolus–coupled metal plate electrodes on opposite sides of the patient. Power deposition is maximum in the high-resistance fat layer, but this problem can be overcome with aggressive skin cooling in patients with a fat layer less than 1 to 2 cm thick.248 Limited steering of power deposition is possible through adjustment of water temperature, electrode placement, and by using a smaller electrode on one side to concentrate heating.
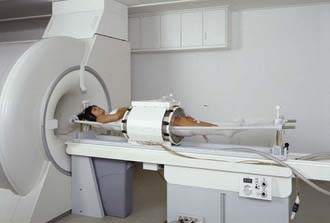
It is possible to localize heating in a region at depth by using an array of phase-focused radiating antennas. The first commercial device built for this approach was the Annular Phased Array (BSD Medical Corp, Salt Lake City UT), which consisted of an array of four equal-power, equal-phase twin dipole radiators mounted on the surface of a large cylinder coupled tightly around the body torso with a refillable distilled water bolus.249 Subsequent development of the 75- to 120-MHz Sigma 30, 60, and Ellipse applicators improved the patient interface and increased control flexibility via four independent relative phase and amplitude controls for the eight radiators.250,251 A similar device, the Matched Phased Array system, has four large 70-MHz waveguide sources with flexible positioning as well as phase and amplitude steering of power deposition within the body.252 Treatment planning systems have been developed to take advantage of this control flexibility and improve the quality of regional heating.253,254 Although these regional heating devices are still in clinical use, newer systems are now available, such as the 100-MHz SigmaEye applicator (BSD Medical Corp, Salt Lake City UT), with 12 independently-controlled twin dipole radiators mounted in three rings to allow phase and amplitude steering both longitudinally and radially within the body.255 More recently, the SigmaEye system has been integrated with both MRI and treatment planning systems. These hybrid SigmaEye/MRI systems (Fig. 74-13) allow MRI-based preplanning as well as noninvasive monitoring of temperature rise during HT to facilitate beam steering into the tumor.256,257 Smaller versions of the Sigma series annular phased array applicators have been implemented for MRI guided HT of tumors in the extremities.258–261 Application of MRI-guided heating of breast is already available for thermal surgery applications262,263and should be available for deep-breast HT soon.260
Whole-Body Heating
Whole-body heating produces the most uniform tumor heating, regardless of tumor location within the body. The disadvantages are the significant systemic stress exerted by whole-body heating, the lack of preferential tumor heating, and the fact that temperatures are limited to approximately 41.8° C to 42° C,264,265 because of thermosensitivity of critical tissues such as the heart, lung, liver, and brain. Thus the thermal goals of systemic therapy are usually more modest than local or regional heating techniques, and intended for activation of drugs266 or enhancement of immunologic response.267 Systemic heating has generally been accomplished by noninvasive means such as immersion in hot fluids like water or molten wax, enclosure of the patient in a radiant heat chamber with infrared or water-RF heat input, or entirely wrapping the patient in hot-water blankets. Other approaches use supplemental RF regional heating devices, extracorporeal circulation of heated blood through an exteriorized arterial-venous shunt, or intraperitoneal irrigation with heated fluid. For all of these techniques, normal body-cooling mechanisms such as respiration and contact of skin with room-temperature air must be blocked by preheating the patient’s breathing circuit and thermally insulating the patient. The patient is often anesthetized or sedated and physiologic conditions must be carefully monitored and controlled throughout the treatment, which often extends over many hours.161,166
Thermometry and Quality Assurance
Appropriate monitoring of tissue temperature during HT is an essential component of therapy, and successful analyses correlating tumor response with thermal parameters require detailed knowledge of tumor temperature distributions. Excellent reviews of thermometry techniques for treatment monitoring and control are available.160,161,166,168,268
Invasive Thermometry Techniques
Thermocouple probes are one of the primary monitoring techniques for clinical HT because of their low cost, simple operation, interchangeability, small size, and accuracy within 0.3° C, which remains stable for many years. Multiple sensor probes in needle, catheter, and bare-wire configurations are available commercially. The most common copper-constantan probes can produce significant (>2° C) thermal conduction smearing of high thermal gradients, especially in multisensor probes,269 so that many users have switched to manganin-constantan thermocouples that have lower thermal smearing errors.270,271 Small-diameter bare wire, fused silica, or metal needle-encased thermocouples are preferred for thermometry in US fields because of minimal self-heating and field-perturbation effects. With appropriate filtering, shielding, and orientation of wires perpendicular to the electric field, thermocouples may be used with caution in RF fields,272 but are generally not recommended for use in MW fields.273
Fiberoptic probes consist of plastic or glass fibers with a nonmetallic, temperature-sensing element at one to four locations along the fibers. Current systems provide essentially artifact-free readout of temperature even in the most intense EM fields, but care must be taken to maintain proper calibration within 0.3° C accuracy. Because the fiberoptic probes are fragile, they are generally inserted into soft plastic catheters that may be left in the tumor between heat treatments. For use in US fields, fiberoptic probes can be inserted inside small-gauge metal needles to minimize probe self-heating.163
Although early HT trials of the 1980s were performed with limited thermometry, usually one to four fixed-location implanted temperature probes, later studies generally report use of an increasing number of sensors to document the temperature distribution. One common approach involves pulling a temperature sensor through catheters located both at depth in tumor and on the skin surface to record thermal maps of temperature at 0.25- to 1-cm spaced increments.161,168,274 These temperature profiles may be obtained manually or via automated thermal mapping systems.275 Radiation Therapy Oncology Group (RTOG) thermometry guidelines recommend that temperatures be mapped along each probe track at least once every 10 minutes, in increments of no more than 5 mm for tumors less than 5 cm and 1 cm for tumors greater than 5 cm across.276 Additional quality assurance guidelines specific to interstitial,277 US,278 and deep regional heating246,279,280 provide guidance for recommended number and locations of thermometry probes. Updated guidelines for clinical thermometry are currently being assembled and should appear in a special issue of the International Journal of Hyperthermia in 2010.
Noninvasive Thermometry Techniques
A number of noninvasive thermometry techniques are under investigation to allow both improved patient comfort and quantification of more complete temperature distributions. Infrared thermography has been available for years for monitoring complete surface temperature distributions.281 Although spatial resolution and accuracy of surface temperature measurements are excellent, the method requires open access to visualize the surface, so it is not compatible with most heating technologies. A novel approach is currently under investigation to monitor surface temperature distributions with high-density arrays of fiberoptic sensors mounted in large surface conforming thermal monitoring sheet arrays.190,282 Other methods are under development to determine temperatures within tissue, including computerized tomography; US time-of-flight tomography; electrical impedance tomography, sometimes called applied potential tomography268,283; active MW imaging284; MW radiometry191,193,213,285,286; and real-time, multislice magnetic resonance thermal imaging (MRTI).233,257,287 Multiple investigators report that MRI should be capable of 0.5° C resolution in regions less than or equal to 1 cm3, with scan-acquisition times less than 1 minute throughout small or large tumor volumes at depth in the body.202,256,288 MRTI has been shown practical for real-time temperature feedback to help the operator steer power deposition into a tumor at depth233,257,260 and potentially for use as an integral part of automatic power control algorithms.261,289,290
Clinical Results
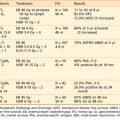
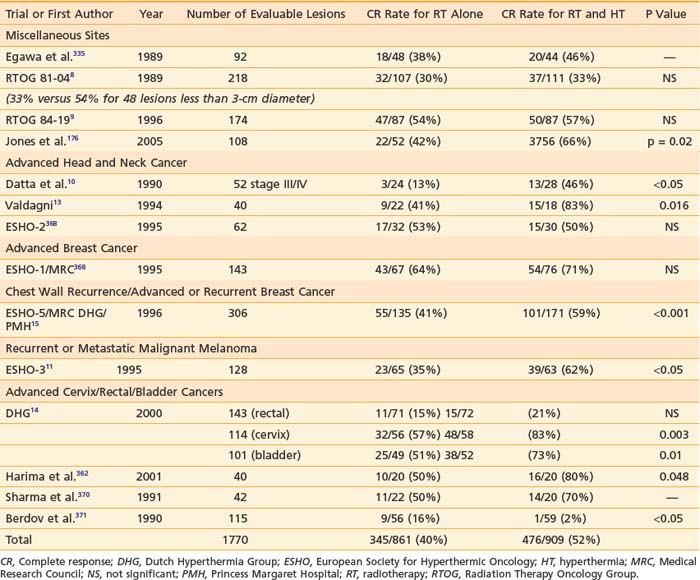
This section reviews applicator appropriateness standards and thermal-dose targets for clinical HT, followed by a discussion of clinical experience with HT alone, HT combined with RT, HT combined with chemotherapy, and trimodality therapy. Selected studies are described individually in the text; Table 74-1 and Table 74-2 provide a more complete listing.
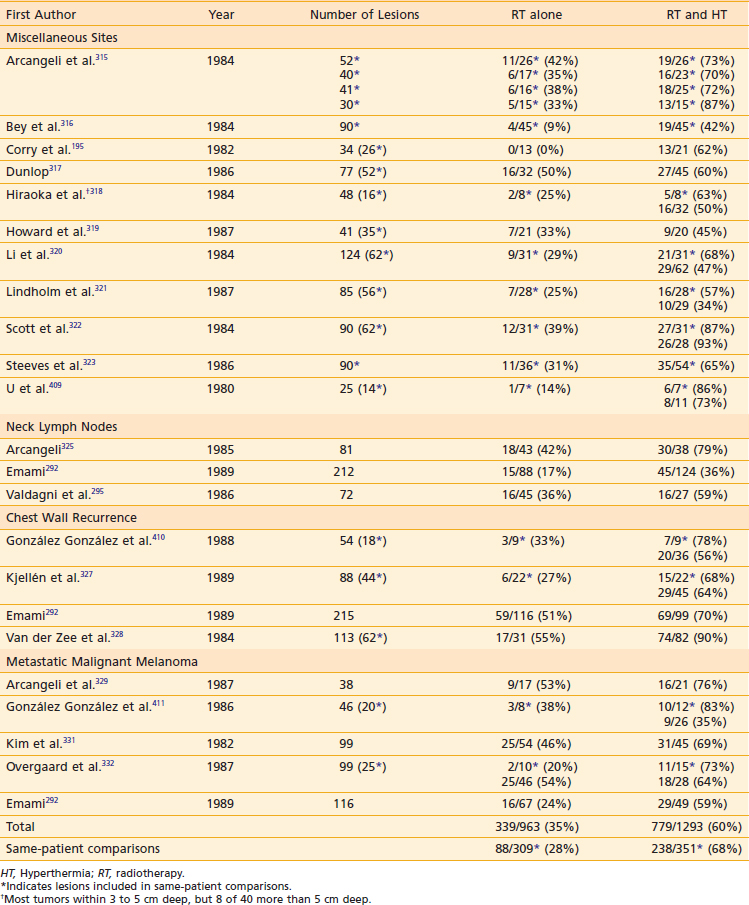
Table 74-1 Nonrandomized Trials Comparing Radiotherapy to Radiotherapy and Hyperthermia in Superficial Malignancies
Thermal Dose and Applicator Adequacy Standards in Clinical Hyperthermia
Applicator Adequacy
Several groups have evaluated the effect of tumor size.291–295 In an analysis of the experience with superficial HT at Washington University, Perez and Emami292 reported 70% of tumors less than 3 cm in depth achieved temperatures higher than 42° C, whereas tumors larger than 3 cm in depth achieved this “therapeutic temperature” in only 20% of the lesions. The CR rate for tumors less than 3 cm in diameter was 77.6% compared with 63.6% for tumors larger than 3 cm in diameter. Similar observations were reported by Kapp and colleagues34 and Valdagni and associates.295 In RTOG protocol 81-04, 78% of lesions in both arms were larger than 3 cm in diameter.296 This is considered a major contributing factor to the negative overall results of that study. Thus, large tumor size is currently a significant adverse factor in the response of lesions to thermoradiotherapy, as a result of inability to heat larger tumors with the then-available HT technology.
Myerson and co-workers297 proposed an SAR-based standard of applicator adequacy for superficial (915-MHz) MW HT. They reviewed the long-term control of superficial tumors and found that lesions covered by the 25% iso-SAR contour had significantly better long-term control than those not adequately covered. They found that the best results were obtained for lesions that satisfied both the applicator standard and that achieved adequate minimum monitored temperatures. The interpretation was that thermometry was essential, but because it is impossible to monitor temperature in 100% of the tumor volume, the best way to minimize the chances of undetected “cold spots” occurring at unmonitored locations was to make certain that the lesion was treated with an adequate applicator. An SAR-based applicator adequacy standard may not necessarily be appropriate for all HT devices. For example, US or MW array systems can have very small regions of high- or low-energy deposition rates that are clinically unimportant because they do not lead to significant temperature changes. These make it very difficult to normalize an SAR distribution and to define SAR-based adequacy criteria. Similarly, for low-frequency EM HT, the adequacy standards defined for 915 MHz may not necessarily be appropriate. Specifically, the 25% SAR for 915-MHz applicators lies within approximately 1 cm of the 50% SAR contour, a distance that can be heated by thermal conduction. The SAR gradients are more gradual with lower-frequency EM devices, so it is less likely that thermal conduction from higher SAR regions will bring the 25% SAR volume up to target temperature. Therefore Myerson and associates298 re-evaluated their 915-MHz data using model temperature distributions in a simple uniform perfusion model. The maximum allowed temperature was set at 47° C. They demonstrated that the 25% SAR volume corresponded to the volume heated to at least 42° C, with a perfusion rate ωb typical for superficial tissue of approximately 1.5 × 10-4 sec-1. Therefore an applicator adequacy standard based on thermal modeling using realistic perfusion estimates could be used in place of the 25% SAR criterion for devices other than 915-MHz MW applicators.
Thermal Dosimetry
A variety of measures have been used to characterize achieved thermal-dose distributions. The older literature often used measures based on steady-state temperature distribution. Minimum and average temperatures and T90 (temperature exceeded by 90% of monitored intratumoral points)299 have been correlated with tumor response. Maximum temperatures have correlated with frequency of thermal injury.29,300 One drawback to this approach is that “steady state” itself is an imperfect construct: Although some monitored locations may display very stable temperature after the first few minutes of heating, others may drift up or down as perfusion and tissue coupling vary over time. In addition, characterizations based solely on a single point in time ignore the very important contribution that duration at temperature makes to both heat-induced radiosensitization and thermal cytotoxicity.
It is better to characterize the achieved temperature distribution by a measure that includes duration. The Sapareto-Dewey equivalent minutes at an index temperature (e.g., EM43)28,301 is the most commonly used method for reducing time/temperature dose to a single number. It is probably the best way to compare the potential for cellular injury among different time/temperature histories. Myerson and colleagues302 characterized time/temperature histories at an average of six monitored locations in three to six sessions in 47 lesions. Times at or longer than index temperatures of 40° C, 41° C, 42° C, and 43° C each correlated well with Sapareto-Dewey equivalent minutes at 43° C. The times at or longer than the four index temperatures did not all correlate strongly with each other. Thus, the Sapareto-Dewey measure was a more robust way to characterize time/temperature histories than time at or longer than a specific index temperature. In other words, even if another measure might ultimately prove to be biologically more appropriate, cases with high Sapareto-Dewey equivalent minutes would still be expected to show greater thermal effect than those with low equivalent minutes.
The target minimum thermal dose depends on the biologic objective. Direct thermal cell kill probably requires per-session thermal doses in excess of the equivalent of 1 hour at 42° C. Heat-induced radiosensitivity becomes substantial if the thermal dose exceeds 1 hour at 41° C, particularly if the HT can be delivered simultaneously with radiation.303 Thermal modification of tumor physiology (e.g., enhanced perfusion/oxygenation and immune stimulus may develop with much lower thermal doses.151,152,267,304–306
The optimal number of HT sessions is controversial. Heating the same area with less than 72 hours between sessions may be unproductive, because residual thermotolerance may render the second session biologically ineffective. For this reason, heating the same region more than twice a week is not commonly done. There remains debate regarding the total number of sessions within this constraint. Kapp and colleagues307 reported the results of a randomized trial comparing two versus six HT treatments for superficial tumors. There was no significant difference either in response rate or in long-term control. Several other institutions have conducted trials comparing once-weekly with twice-weekly HT for superficial tumors.308,309 In one study, as with the Kapp study, there was no benefit to increasing the number of HT sessions.308 On the other hand, Duke University compared once-weekly with twice-weekly HT preoperatively for soft-tissue sarcomas of the extremities.309 This was administered in conjunction with 50 Gy conventionally fractionated. There was a statistically significant increase in pathologic response in the group receiving twice-weekly HT. Similarly, Arcangeli and co-workers310 randomized multiple lesions in the same patient to receive either one or four HT treatments and demonstrated an advantage to four treatments. More recently, Jones and colleagues311 published the results of a prospective, randomized trial in which patients superficial tumors received an initial, test HT session delivering a thermal dose of 0.5 T90 43° C equivalent minutes (equals 16 × 0.5 = 8 equivalent minutes at 41° C). Patients were randomized to receive either no further treatment or up to 10 additional twice-weekly treatments, to achieve a cumulative CEM43T90 thermal dose 10 equivalent minutes or more. Local control and response rates were significantly enhanced with the higher thermal dose.
One possible benefit to increasing the number of HT sessions is that it may increase the likelihood that all portions of the tumor will receive at least one or two adequate HT sessions. Vascular perfusion may change from session to session, and the treatment physicist may make adjustments from session to session. Thus a monitored location that was inadequately heated in the first session may eventually be well heated in the second or third session. Myerson and colleagues302,312 reported thermal dosimetric results from a prospective trial in which care was taken to monitor temperature at the same locations during a series of superficial HT sessions. They computed the per session average of the total equivalent time at 41° C for the least heated tumor location (Min Sum teq 41° C / # sessions). After one session, 36% of cases had a minimum tumor per session average teq 41° C greater than 60 minutes, 48% after two sessions, 50% after three sessions, and 63% after four sessions. Thus increasing the number of sessions not only increased total equivalent time (as might be expected), but also increased the per-session average time in the least effectively heated tumor location. Note that a per-session average of 60 equivalent minutes at 41° C, a thermal dose capable of producing heat-induced radiosensitization, is equivalent to only 60/4 = 15 minutes at 42° C and 60/16 = 3.75 equivalent minutes at 43° C. These thermal doses would be unlikely to produce substantial direct thermal cell kill, thus underlining the observation that HT (in contrast to thermal ablation) works best as an adjunct to another modality. These thermal doses also indicate that a reasonable objective cumulative minimum tumor dose over a course of approximately five HT sessions would be a total of approximately 10 to 20 equivalent minutes at 43° C.
For the larger tumors treated in the Duke sarcoma study, the need for multiple sessions to achieve an adequate coverage of the full tumor volume may be more important clinically than in the superficial tumors. For these lesions, Oleson and associates313 proposed a measure of how well the full tumor volume was heated in any one session. Lesions that achieved at least two HT sessions that satisfied this measure were more likely to respond to treatment. The data showed that the likelihood of achieving at least two sessions satisfying the volume coverage criterion increased from 33% (4 of 12 lesions) treated with fewer than six HT treatments to 88% (7 of 8 lesions) treated with more HT treatments.
Results With Hyperthermia Alone
Early clinical studies using HT alone for the treatment of small, superficial neoplasms showed that HT alone (using multiple 30-60 minute HT sessions) has very limited efficacy. Overgaard314 summarized the results of 18 such studies including 380 tumors treated with 2 to 22 heat sessions with a CR rate of 13% and a partial response (PR) rate of 36%. The duration of HT responses was generally brief.
Hyperthermia and Radiation
Superficial Hyperthermia
Miscellaneous Sites
In the 1970s and 1980s, numerous nonrandomized studies in superficial malignancies showed higher CR rates for RT plus HT in comparison with RT alone (see Table 74-1).195,292,295,315–332 These trials typically used 30- to 60-minute HT sessions one, two, or three times weekly. Some of the lesions included in these early studies were paired lesions used for same-patient comparisons (indicated with asterisks in Table 74-1). Results were surprisingly similar among these studies despite a broad range of histologies, RT dose and fractionation, and number of HT treatments. Overall, the CR rate was 35% for 963 lesions treated with RT alone compared with 60% for 1293 lesions treated with RT plus HT. In the same-patient comparisons, the CR rate was 28% for 309 lesions treated with RT alone compared with 68% for 351 lesions treated with RT plus HT, representing more than a doubling of the CR rate. The most frequent toxicity of HT was blisters in approximately 10% to 23% of patients and rare thermal burns.292,333 Most studies showed no increase in late effects.
Encouraging results of early nonrandomized trials prompted the RTOG to mount a randomized phase II trial (RTOG 81-04) comparing RT alone (32 Gy in eight fractions over 4 weeks) with RT plus twice-weekly HT (nominally 43° C for 60 minutes) in measurable advanced primary, recurrent, or metastatic epithelial or mesenchymal malignancies. Overall, there was no difference in the CR rate for the 107 single lesions treated with RT alone (30%) and for the 111 single lesions treated with RT plus HT (33%) (see Table 74-2).8 For 48 lesions less than 3 cm in diameter, amenable to adequate HT with the heating equipment available at the time, there was a trend toward improved CR rate for RT plus HT in comparison with RT alone (54% versus 33%), and the actuarial probability of local control at 1 year was 80% for RT plus HT compared with 15% for RT alone (P = 0.02). However, with 78% of lesions entered on the study measuring 3 cm or greater in diameter, the study was negative overall. The resulting report cited poor thermometry and quality assurance as major factors influencing the outcome of the trial. A large number of the lesions entered were too large or too deep, or both, to be adequately heated by the existing HT equipment. Furthermore, the thermometry was too limited in many cases to allow for adequate control of treatment delivery or for meaningful subsequent evaluation of the temperature distributions achieved.8 One positive outcome was the formulation of RTOG quality assurance guidelines for future clinical trials, which improved the outcome of subsequent clinical trials of HT.334
In another non–site-specific, prospective, randomized trial comparing RT with RT plus HT for 92 evaluable superficial lesions, the CR rates were 38% versus 45%, and overall response rates of 63% versus 82% (P < 0.05) for RT versus RT plus HT, respectively. No improvement in the CR rate was observed for patients with head and neck cancer (45% versus 44%), whereas HT was associated with a doubling of the CR rate for patients with chest-wall recurrence of breast cancer (67% versus 33%).335
A trial incorporating careful quality assurance guidelines was recently completed at Duke University in which patients underwent a low-thermal-dose test HT treatment prior to randomization. Only patients whose tumors were deemed to be heatable were allowed to proceed to randomization into RT-alone or RT-plus-HT arms to rule out inadequate heating in the full-dose HT arm. Results of this study demonstrated the value of effective heating as the CR for HT plus RT was 66% versus 42% for RT alone (p = 0.02).176 Local control was also significantly improved in the full-dose HT arm: 48% versus 25% with RT alone (p = 0.02).176
Advanced or Recurrent Neck Nodes
Results of RT with or without HT for superficially located large, fixed, or recurrent lymph nodes have been reported by Perez and Emami,292 Valdagni and colleagues,295 and Arcangeli and colleagues,325 with CR rates of 17%, 36%, and 42% for RT alone versus 36%, 59%, and 79% for RT plus HT, respectively (see Table 74-1). By plotting dose-response curves for RT and RT plus HT from available published and unpublished observations, Overgaard7 estimated a TER of 1.6 for advanced neck nodes.
Several prospective, randomized trials have been performed for head and neck cancers (see Table 74-2). Datta and colleagues10 reported on 65 patients with head and neck carcinomas treated with 60 to 65 Gy at 2.0 Gy per daily fraction with or without twice-weekly HT. Overall, the CR rate was 10 of 32 (31%) for RT versus 18 of 33 (55%) for RT plus HT. HT appeared to benefit only stage III and stage IV patients, in whom the CR rate was 3 of 24 (13%) for RT alone versus 13 of 28 (46%) for RT plus HT. Another prospective, randomized trial compared RT alone (64-70 Gy at 2-2.5 Gy per fraction) to RT plus HT in 44 fixed, inoperable squamous cell carcinoma neck nodes. The CR rate at 3 months was 9 of 22 (41%) for RT alone versus 15 of 18 (83%) for RT plus HT, without increased toxicity.12 Long-term follow-up of the study showed significantly improved 5-year actuarial nodal control (P = 0.015) and 5-year survival (P = 0.02).13 On the other hand, a separate European Society for Hyperthermic Oncology (ESHO) trial for advanced neck nodes showed no difference in CR rate or local control for RT versus RT plus HT (53% versus 50% and 33% versus 31%, respectively).336
Chest-Wall Recurrence and Locally Advanced Breast Cancer
Local control of chest-wall recurrences of breast cancer with local HT and RT probably does not affect survival, but it can profoundly improve quality of life. Promising results were obtained in many nonrandomized trials by combining HT with RT for the treatment of chest-wall lesions with or without prior RT (see Table 74-1). González González and colleagues,326 Kjellén and colleagues,327 Lindholm and colleagues,321 Perez and Emami,292 Scott and colleagues,322 and van der Zee and colleagues328 reported CR rates of 27% to 55% for RT versus 60% to 94% for RT plus HT. By plotting dose-response curves for RT versus RT plus HT from available published and unpublished observations, Overgaard7 estimated the TER to be 1.5 for breast cancer chest-wall recurrence.
A multi-institutional prospective, randomized experience has been reported from the Medical Research Council (MRC) of Great Britain, ESHO, the Dutch Hyperthermia Group, and Princess Margaret Hospital in Toronto (see Table 74-2).15 Each institution had planned independent prospective, randomized trials for patients with chest-wall recurrence of breast cancer or T3 to T4 primary breast cancer, but because the goals were similar and better accrual was desired, the studies were combined into one joint trial. The RT schemes used included 28.8 Gy at 3.6 Gy per fraction for 2 weeks with HT three times weekly, 32 Gy at 4 Gy per fraction for 4 weeks with HT once or twice weekly, 32 Gy at 1.8 Gy per fraction with 2 HT treatments, and 40 to 50 Gy at 2 to 3 Gy per fraction with a total of two to eight HT treatments. Of 306 evaluable lesions in 306 patients, 216 (71%) were on the chest wall, 79 (26%) were in breast tissue, and 11 (3%) were in lymph nodes. Thirty (10%) were primary breast cancers, 66 (21%) were recurrences without prior RT, and 210 (70%) were recurrences in the setting of previous RT. Adjuvant HT resulted in significantly improved response, with a CR rate of 41% in 135 lesions treated with RT alone versus 59% in 171 lesions treated with RT plus HT, and an odds ratio for CR of 2.3 overall after stratification by trial. Patients with recurrent lesions in previously irradiated areas appeared to benefit the most from HT.15 An analysis of 120 of the HT patients with recurrent breast cancer demonstrated a significant association between response and the cumulative minimum thermal dose over all HT treatments; the CR rate was 77% for CEM 43° Tmin greater than 10 minutes versus 43% for CEM 43° C Tmin less than or equal to 10 minutes.337 A separate multivariate analysis of 101 patients treated on the MRC trials for intact or recurrent breast cancers showed that the most important nonthermal parameters associated with a decreased probability of CR included greater tumor depth (P = 0.001), the use of a palliative as opposed to a curative RT regimen (P = 0.005), and presence or history of disease outside the treated area (P = 0.010).338 Tumor area was not a significant prognostic covariate. Higher CEM 43° C Tmin during the first, second, and third HT treatments was consistently significantly associated with a greater probability of CR in various models tested. Among 74 recurrent tumors, the CR rates were 44%, 69%, 75%, and 80% for CEM 43° C Tmin less than 10, 10 to 20, 20 to 30, and at least 30 minutes, respectively, demonstrating consistently improving response with increasing thermal dose.
Metastatic Malignant Melanoma
Melanoma recurrent in the skin or metastatic to skin, subcutaneous sites, or superficial lymph nodes is often amenable to HT, and control rates are generally inadequate with RT alone. Results of nonrandomized trials are shown in Table 74-1. Arcangeli and associates,329 González González and associates,330 Kim and associatess,331 Overgaard and Overgaard,332 and Perez and Emami292 reported CR rates of 24% to 53% for RT alone versus 50% to 76% for RT plus HT. Overgaard7 estimated the TER to be 1.6 for malignant melanoma.
A prospective, randomized trial comparing RT to RT plus HT for metastatic or recurrent melanoma (ESHO-3) randomized 134 lesions to receive 24 or 27 Gy in three fractions for 8 days with or without HT (with a goal of 43° C for 60 minutes) after each RT fraction. Of the 128 evaluable lesions, the CR rate was 35% for RT alone versus 62% for RT plus HT (P < 0.05) (see Table 74-2), and the 2-year actuarial local control was 28% for RT versus 46% for RT plus HT (P = 0.008). A stepwise logistic regression analysis showed that HT (P = 0.0015), smaller tumor size (P = 0.0048), and higher RT dose (P = 0.049) were significantly associated with CR.339
Subclinical Disease
HT has been used postoperatively in settings with no gross tumor but with a high risk for residual microscopic local disease. Its most common use has been for patients with a past history of full-dose irradiation who have undergone surgical removal of recurrent neck nodes or chest-wall recurrences of breast carcinoma. It has also been used postoperatively for locally advanced melanoma and sarcoma. Emami and Perez340 reported on 23 postoperative cases treated with 30 to 40 Gy and HT. With a follow-up of 1 to 5 years (median, 18 months) 20 of 23 cases have local control. Kapp and associates341 described a large series of cases treated for recurrent breast carcinoma, consisting of 38 fields treated electively after local excision and 224 fields adjacent to fields containing macroscopic disease. The thermoradiotherapy was well tolerated, and 3-year actuarial local control was 68%.
Interstitial Hyperthermia
Miscellaneous Sites
For the major nonrandomized IHT trials for head and neck, breast, pelvic, and other sites, CR rates have ranged from 38% to 88%.342 The IHT was generally given for 1 hour immediately before brachytherapy, and usually immediately after brachytherapy as well. Excellent review articles on IHT are available.204,206,343
A prospective, randomized RTOG trial (RTOG 84-19) for recurrent or advanced tumors comparing IRT with IRT plus IHT accrued 173 evaluable patients from 1986 to 1992. No differences between the two arms were observed in terms of CR rates (54% versus 57%) (see Table 74-2) or survival (2-year survival 34% versus 35%). It was noted that only one patient met minimal criteria for “adequate” HT and that substantial technical advances to improve heat delivery were needed.9 Lesions less than 4 cm in diameter showed a trend toward improved CR rate in the HT arm (74% for IRT plus IHT versus 58% for IRT; P = 0.11). The trial did have a positive effect in terms of prompting the RTOG to develop quality assurance guidelines for IHT.277
Malignant Gliomas
For a long time, there was considerable interest in combining IHT with brachytherapy to improve local control of malignant gliomas. Although normal brain tolerance to HT is very limited, patient tolerance to HT is excellent because of the lack of pain sensation in the brain. Phase I and II brain HT trials are summarized elsewhere.344 Because of the difficulty in distinguishing radiation necrosis from tumor, survival results are reported rather than response rates.
Sneed and colleagues345,346 performed multivariate analyses to determine parameters associated with improved freedom from local tumor progression and survival among 25 patients with recurrent glioblastoma and 16 with recurrent anaplastic astrocytoma treated with thermoradiotherapy. They found that minimum tumor temperature and T90 were the only significant factors predicting freedom from local tumor progression. The median freedom from local tumor progression was 38 weeks for gliomas heated to a T90 of at least 41.5° C versus 18 weeks for those heated to a T90 less than 41.5° C (P < 0.001). A multivariate analysis stratifying by histologic type showed that age (P = 0.04) and T90 (P = 0.04) were also significant prognostic factors for survival. For patients with glioblastoma, the median survival was 63 weeks for a T90 of at least 41.2° C versus 38 weeks for a T90 less than 41.2° C (P = 0.008).
Stea and colleagues retrospectively compared implant alone with implant with IHT in patients with primary or recurrent malignant gliomas treated with brachytherapy boost or brachytherapy for recurrence.347 On multivariate analysis, three parameters were significantly associated with improved survival: lower patient age (P < 0.0001), anaplastic astrocytoma versus glioblastoma (P = 0.023), and adjuvant HT (P = 0.0487). The median survival from the date of diagnosis was 35.1 months for patients who received HT versus 22.3 months for those who did not.
The largest prospective, randomized phase II and III trial was performed at the University of California, San Francisco, for 112 patients with newly-diagnosed glioblastoma.274 Patients were registered postoperative, treated with focal RT (59.4 Gy at 1.8 Gy per daily fraction) with oral hydroxyurea, and then randomized to a 60 Gy brachytherapy boost with or without IHT (42.5° C for 30 minutes immediately before and after brachytherapy). Primarily owing to tumor progression, 33 patients were never randomized and 11 randomized patients never underwent brachytherapy. Comparing the 33 brachytherapy and 35 brachytherapy plus IHT patients, the median time to progression was 33 versus 49 weeks (P = 0.045), the median survival times were 76 versus 85 weeks (P = 0.02), and the 2-year survival probabilities were 15% versus 31%, respectively. A multivariate analysis adjusting for age and Karnofsky performance status showed significantly improved survival probability for the IHT arm (P = 0.008 with a hazard ratio of 0.51). Toxicity was acceptable, with six grade 3 toxicities on the IHT arm, one grade 3 toxicity on the brachytherapy-alone arm, and one grade 4 toxicity on each arm.
Long-Duration, Simultaneous Thermoradiotherapy
Because the greatest radiosensitization occurs when HT and RT are delivered simultaneously, several groups are investigating simultaneous external beam RT and superficial HT348 or simultaneous brachytherapy and IHT. Forty-four patients with recurrent superficial tumors were treated on phase I and II studies with simultaneous superficial 41° C HT and RT at Mallinckrodt Institute of Radiology.302 The CR rate was 51% and slow-healing soft tissue ulcers were noted in 10 of 47 sites treated (21%), perhaps suggesting increased toxicity compared with sequential HT and RT. In vitro studies suggest that low-temperature (41° C), continuous, long-duration heat applied simultaneously with continuous low-dose-rate radiation or pulsed high-dose-rate radiation may maximize radiosensitization.103,349 Preliminary results for long-duration interstitial thermoradiotherapy of advanced, bulky pelvic malignancies have been promising. At William Beaumont Hospital, 20 patients with persistent or recurrent pelvic tumors had simultaneous low-dose-rate brachytherapy with long-duration continuous 41° C IHT using a computer-controlled RF-LCF system. The CR and overall response rates were 74% and 95% (versus 50% and 57% in patients at the same institution who had standard sequential interstitial RT [IRT] plus HT) (Corry, personal communication).
Regional Hyperthermia
Regional HT treatments are sometimes limited by local pain or systemic stress or both, frequently resulting in tumor temperatures less than the desired range, although treatment tolerance and heating quality improve with each successive generation of regional heating devices and treatment planning sophistication. In an early trial, the average Tmin and Tmax were 39.3° C and 41.4° C, respectively, in 119 patients treated with RT and 8-MHz RF capacitive heating. Moving from the initial annular phased array system to the phase and amplitude controllable BSD 2000 (BSD Medical Corp, Salt Lake City, UT) with Sigma 60 applicator, the average Tmin, T90, and Tmax tumor temperatures improved from 39.3° C to 39.5° C, 39.9° C to 40.1° C, and 42.5° C to 42.9° C, respectively.350,351 In a subsequent trial of 772 BSD 2000 regional HT sessions in 190 patients with pelvic tumors, the mean T90 and Tmax were 40.2° C and 41.6° C, respectively.352 More recently, Fatehi and colleagues reviewed data from 444 cervix patients treated with 75- to 120-MHz RF phased-array Sigma applicators, demonstrating T90 temperatures of 40° C to 40.5° C in this disease site.353
Response rates have varied widely in the major nonrandomized regional HT trials. One large multi-institutional series reported 10% CRs and 17% PRs.354 A later RTOG trial of RT plus regional HT, including 66 patients treated predominantly with the BSD 2000 system, achieved CR in 34% of patients and PR in 16% of patients.355 Response was strongly associated with radiation dose but not with maximum tumor temperature.
In general, regional HT is better tolerated and produces better response rates for pelvic disease than for tumors located higher in the thorax, presumably because of restricted breathing from a water bolus tight around the thorax. Even so, regional heating has been applied with some apparent success for advanced lung cancers,356 especially those in contact with the chest wall,357 using the RF capacitive plate approach which is somewhat less confining. Major improvements, including a larger number of heating elements, better phase and amplitude control of the heating elements,253 flexible patient positioning, and sophisticated three-dimensional computed tomography-based358 or MRI-based256 HT treatment-planning software, should allow higher tumor temperatures and even better patient tolerance in the future. Comprehensive quality assurance guidelines have also been published covering treatment planning, the treatment process, treatment documentation, equipment, and safety.359,360
Despite the moderate temperatures achieved, regional HT significantly improved local control and survival in patients with advanced cervical cancer in a phase III prospective, randomized multicenter trial performed by the Dutch Deep Hyperthermia Group.14 From 1990 through 1996, 358 evaluable patients with T2 through T4 bladder cancer; stage IIB, IIIB, or IV cervical cancer; or locally advanced or recurrent rectal cancer were randomized to conventional RT (including brachytherapy boost for cervical cancer patients when feasible) with or without weekly regional HT for a total of five treatments. The respective CR rates for RT compared with RT plus HT were 39% versus 55% overall (P = 0.0003), 15% versus 21% among 143 rectal cancer patients (not significant), 57% versus 83% among 114 cervical cancer patients (P = 0.003), and 51% versus 73% among 101 bladder cancer patients (P = 0.01). The 3-year local control rates for RT versus RT plus HT were 26% and 38% overall, 8% and 16% for rectal cancer, 41% and 61% of cervical cancer, and 33% and 42% for bladder cancer, respectively. Survival was not significantly different between RT and RT plus HT groups except in the case of cervical cancer, for which 3-year survival probabilities were 27% after RT alone versus 51% after RT plus HT (P = 0.009). An associated economic evaluation of the Rotterdam experience indicated that long-term outcome of treatment with adjuvant HT was cost-effective in cervical cancer patients.361
A smaller prospective, randomized trial showed that regional HT using 8-MHz RF capacitive regional HT significantly improved local control in patients with stage IIIB cervical cancer treated with external-beam RT plus high-dose-rate brachytherapy.362 The CR rate was 50% for 20 patients treated with RT alone versus 80% for 20 patients treated with RT plus HT. The 3-year local control probabilities for RT versus RT plus HT were 58% versus 80% (P = 0.048). There was a suggestion of improved overall survival and overall freedom from disease, but neither achieved statistical significance.
In 2005, Vasanthan and colleagues363 published pooled results from five facilities in India, Ukraine, China, and South Korea from separate phase III trials for stage IIB through IVA cancer of the cervix. The trials all compared RT alone with RT plus regional HT. There were a total of 55 patients in each arm. No thermometry standards were set and many patients did not have intratumoral temperature measured in every session. Some patients never had temperature measured. In two of the institutions, no information is provided about the HT device; in the others, RF capacitive HT was used. This trial showed no local control benefit to HT, and response rates were not reported.
Whole-Body Hyperthermia
Whole-body temperatures of approximately 41.8° C to 42.0° C for 2 hours can be tolerated by mildly sedated patients with acceptable toxicity, commonly including acute thrombocytopenia and granulocytosis, mild hypotension, and occasional pulmonary edema.364 There has also been interest in fever-range whole-body hyperthermia (WBH) using temperatures of 39° C to 40° C for 3 to 6 hours.365 In either case, WBH is generally combined with chemotherapy for treatment of advanced systemic disease, as discussed in the section on “Hyperthermia and Chemotherapy,” and there is a rationale for combining WBH with immunotherapy.267,366
Summary of Randomized Human Studies of Radiotherapy and Hyperthermia
Table 74-2 summarizes major published prospective, randomized human HT trials of RT with or without HT.8–12,335,362,367–371 Considering all trials, more than 1700 patients were treated in 14 trials with a CR rate of 40% for RT alone versus 52% for RT plus HT. Overgaard372 performed a meta-analysis of 22 known trials, comparing the risk of failure for patients treated with RT plus HT versus RT alone. Although a few of the individual trials were inconclusive, the combined data indicated a significantly reduced risk of failure in patients who received adjuvant HT, with an odds ratio of 1.77 within 0.20 (95% confidence interval) and a P-value of less than 0.00001. These data are grouped by disease site and presented in graphical form in Fig. 74-14, showing clear evidence for a benefit of adjuvant HT for melanomas and head and neck, chest wall, cervical, rectal, and bladder cancers, but no benefit for prostate or intact breast cancers. Results of the thermal dosimetry studies suggest that improved heating of breast and prostate tissue possible with developing equipment should improve clinical response.
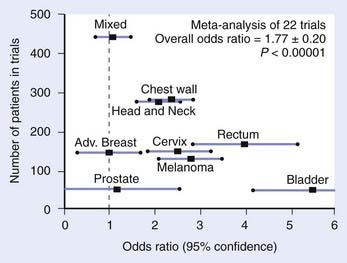
Hyperthermia and Chemotherapy
Multiple, mostly small, phase II trials have been performed combining local, endoluminal, regional, or WBH with chemotherapy for treating esophageal cancer preoperatively, stomach cancer, pancreatic cancer, metastatic or high-risk soft-tissue sarcomas, recurrent cervical cancer, advanced rectal cancer, miscellaneous advanced or metastatic cancers,373,374 small cell lung cancer,375 disseminated melanoma,376 and ovarian cancer.377,378 A phase III randomized trial comparing preoperative chemotherapy to preoperative chemotherapy plus HT was performed in 40 esophageal cancer patients showing higher rates of subjective and radiographic improvement (70% versus 40% and 50% versus 25%, respectively) and a higher proportion of necrosed cancer cells on histologic examination in the combined therapy arm.379 Following up on promising phase II chemothermotherapy trials in patients with high-risk soft-tissue sarcomas, ESHO and the European Organization for Research and Treatment of Cancer are conducting a phase III trial of neoadjuvant chemotherapy (etoposide, ifosfamide, and DOX) with or without regional hyperthermia (RHT) prior to resection and RT.380
Preoperative hyperthermic isolated limb perfusion (HILP) with melphalan, tumor necrosis factor, or other agents has been used extensively in patients with high-risk or recurrent extremity melanomas. One prospective, randomized trial was closed early because of significantly higher local control and survival in patients treated with preoperative melphalan HILP compared with those who underwent surgery alone.381 A large phase III, multicenter, multicontinent randomized trial in 832 assessable patients showed a trend toward longer disease-free interval but no benefit in terms of survival or time-to-distant metastasis for HILP prior to surgery compared with surgery alone.382
HT has also been used in combination with drug-containing liposomes to improve drug delivery. Kong and Dewhirst have provided an excellent review of preclinical data116 and Park has reviewed early clinical data and rationale for liposome-based breast cancer treatment.383 A phase I and II trial was performed in 25 patients with chest-wall recurrence of breast cancer using superficial HT immediately before intravenous administration of DOX-containing liposomes (Doxil) at 45 mg/m2 every 28 days for 4 to 6 cycles.384,385 The response rate in heated fields was 57% with 1 CR and 12 PRs. Furthermore, HT was associated with significantly improved response in 18 patients with both heated and nonheated disease (P < 0.04). Another phase I and II trial was performed in 15 patients with advanced, chemotherapy-resistant cancers using fever-range WBH with DOX-containing liposomes, 5-fluorouracil, and low-dose metronymic interferon-α repeated every 28 days for one to nine cycles (median, three cycles).386 Six patients had objective responses, five had stable disease for more than 12 weeks, and four had progressive disease.
Hyperthermia, Radiation, and Chemotherapy
There is also growing literature on trimodality studies combining HT, RT, and chemotherapy for head and neck, breast, esophageal, and rectal cancers, including promising phase III studies for resectable or unresectable esophageal cancer.373 In one trial, 66 patients with resectable squamous cell carcinoma of the esophagus were randomized to preoperative chemoradiotherapy or thermochemoradiotherapy using twice weekly RF capacitive HT with an endoluminal applicator, cisplatin or bleomycin, and 30 Gy in 15 fractions for 3 weeks.387 Operative specimens showed absence of viable cancer cells in 25% of the trimodality patients versus 6% of the chemoradiotherapy patients, and 3-year survival rates were 50% versus 24%, respectively. Kouloulias and colleagues388 have reported a promising initial study of trimodality approach in recurrent breast cancer. Colombo and colleagues389 compared intravesical mitomycin C chemotherapy alone versus chemotherapy plus transurethral MW HT for 52 patients with superficial bladder tumors. There was a significant improvement in response and disease-free survival. To date, experience with thermochemotherapy and thermochemoradiotherapy is too limited to draw conclusions, although preliminary results are encouraging.
Heat Shock Proteins and Immunotherapy
The fact that vaccines against tumor HSPs are applicable to the treatment of cancer has opened up a new perspective in cancer immunotherapy. A number of preliminary clinical trials have been carried out to test the use of HSPs derived from autologous human tumors in immunotherapy of human cancer: Janetzki and colleagues prepared individual gp96 vaccines derived from the tumors of 16 patients with advanced malignancies390; Amato and colleagues have treated patients with renal cell carcinoma with gp96 vectors.391 Eton and colleagues also used gp96 vaccines in metastatic melanoma using a similar dosing regimen as described for renal cancers.392 However, all of these clinical trials are nonrandomized and the clinical data generated thus far can only be suggestive. To the extent that one may draw any inferences from them, the results so far are consistent with the murine data that showed that the autologous gp96 vaccine was effective in the adjuvant setting in which most treated animals remained disease-free for their entire life span, and less effective in animals with progressive tumors, in which only disease stabilization was achieved.127 Most recently, double-blind phase III clinical trials using gp96-peptide complex for the treatment of stage III renal cell carcinoma in the adjuvant setting are being conducted at numerous centers worldwide.393
The advantages and disadvantages of various vaccination approaches are discussed in a recent review article by Manjili and colleagues.394 In addition, several other excellent reviews of this field emphasize the development, analysis, and application of HSP-related vaccination approaches.127,395–399
Randomized Animal Studies
In comparison with human trials, studies in pet animals often allow for better acceptance of novel therapies and randomization, more detailed thermometry, better control over the heat treatment, and better tolerance resulting from the use of general anesthesia, resulting in higher minimum tumor temperatures. Four prospective, randomized trials performed in pet animals with spontaneous tumors all demonstrated significant benefit for HT in combination with RT over RT alone.29,400–402 A University of Arizona trial included 236 dogs and cats with a variety of spontaneous cancers stratified by histologic type and randomized to receive RT alone (36.8 Gy in eight fractions over 4 weeks) or RT plus once-weekly HT for 30 minutes. For 217 evaluable animals, the CR rate was 35% in the RT arm and 59% in the RT plus HT arm, with no increase in acute or late effects.29
Denman and colleagues400 performed a phase III randomized trial in spontaneous canine malignancies stratified by histologic type and anatomic site and randomized to receive RT (49.0 Gy in 14 fractions given on Mondays, Wednesdays, and Fridays) with or without once-weekly HT for 30 minutes. HT significantly increased the CR rate for sarcomas (33% for RT alone versus 73% for RT plus HT), tumors of the trunk and extremities (45% versus 83%), and tumors less than 10 cm3 in volume (50% versus 84%).
Gillette and associates401 randomized oral carcinomas in 38 dogs to graded radiation doses (25-45 Gy in 10 fractions for 22 days) with or without seven 30-minute HT treatments to determine the dose that would result in a 50% probability of tumor control (TCD50) and then to determine the TER as the ratio of the TCD50 for RT alone to the TCD50 for RT plus HT. To limit normal tissue temperatures, the tumor margins did not exceed 41.5° C. The TER was 1.15 and the slope of the dose-response curve was much steeper for RT and HT than for RT alone. Late complications were not increased in the animals receiving heat.
A similar study was performed in 64 canines with spontaneous soft tissue sarcomas.402 The animals were stratified by tumor volume and randomized to receive graded doses of RT (35.0-55.0 Gy in 10 fractions on a Monday-Wednesday-Friday schedule) with or without US HT for 30 minutes 3 hours after RT twice weekly on a Monday-Friday schedule. The HT goal was a minimum tumor temperature of 42° C. Freedom from local tumor progression was significantly increased in animals treated with RT plus HT (Wilcoxon P = 0.046; log rank P = 0.064). There was no significant difference between the two study arms in terms of late normal-tissue complications or distant-metastatic rate.
A fifth phase III study of RT (36 Gy at 9 Gy per fraction) with or without local HT for spontaneous head and neck tumors in 145 dogs showed no difference between the treatment arms in terms of CR rate (47% for RT versus 43% for RT and HT)403 or progression-free survival (median, 26 weeks for RT versus 27 weeks for RT and HT). The authors cited inadequate thermal dose as a probable cause for the negative results.
WBH has also been investigated in combination RT or chemotherapy in dogs. A phase III trial of RT with or without WBH performed in 45 dogs with spontaneous brain tumors resulted in 1-year survival probabilities of 44% for RT versus 40% for RT and WBH, and six dogs had severe toxicity from WBH.404 In another study, 64 dogs with spontaneous soft-tissue sarcomas were randomized to RT with localized HT with or without WBH to improve Tmin and T90 temperatures. The addition of WBH significantly increased CEM 43° C T90, but also resulted in more toxicity and significantly shorter time to metastasis (P = 0.02).405 However, WBH was associated with a trend toward improved disease-free survival among 46 dogs with high-grade lymphoma achieving a CR with DOX and then randomized to five additional DOX treatments with or without WBH.406,407
Conclusions and Future Directions
There is a strong radiobiologic rationale for combining heat and RT: heat radiosensitizes cells and kills cells as a function of time and temperature. HT appears to be especially effective against cells that are radioresistant, and mild or moderate HT may allow increased reoxygenation of tumors during the course of RT.152,408 As we come to better understand the mechanisms and kinetics of thermal effects, we should be able to devise strategies to enhance the therapeutic gain for adjuvant HT, taking advantage of pH, response modifiers, and the relative thermotolerance of normal tissues and tumors. Further phase I and II trials are needed to help define the optimal thermal dose and sequencing of HT with RT, including investigation of long-duration, simultaneous RT plus HT; and to evaluate HT with chemotherapy, conventional liposomes, or thermosensitive liposomes, with or without RT. Phase I and II trials are also in progress exploring entirely new applications of HT in immunotherapy or as a promoter for gene therapy.
1 Busch W. Uber den einfluss, welchen heftigere erysipelin zuweilig auf organisierte neubildungen ausüben. Verhandlugen des naturhistorischen vereines der preussischen rheinlande und Westphalens. 1866;23:28-30.
2 Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;105:486-511.
3 Müller C. Eine neue behandlungsmethode bosartiger geschwulste. Munchener Medizinische Wochenschrift. 1910;28:1490-1493.
4 Dewey WC, et al. Cellular responses to combinations of hyperthermia and radiation. Radiology. 1977;123(2):463-474.
5 Dewey WC, et al. Cell biology of hyperthermia and radiation. In: Meyn RE, Withers HR, editors. Radiation biology in cancer research. New York: Raven Press; 1980:589-621.
6 Hahn GM. Hyperthermia and cancer. New York: Plenum Press; 1982.
7 Overgaard J. The current and potential role of hyperthermia in radiotherapy. Int J Radiat Oncol Biol Phys. 1989;16(3):535-549.
8 Perez CA, et al. Quality assurance problems in clinical hyperthermia and their impact on therapeutic outcome: a report by the Radiation Therapy Oncology Group. Int J Radiat Oncol Biol Phys. 1989;16(3):551-558.
9 Emami B, et al. Phase III study of interstitial thermoradiotherapy compared with interstitial radiotherapy alone in the treatment of recurrent or persistent human tumors: a prospectively controlled randomized study by the Radiation Therapy Oncology Group. Int J Radiat Oncol Biol Phys. 1996;34(5):1097-1104.
10 Datta NR, et al. Head and neck cancers: results of thermoradiotherapy versus radiotherapy. Int J Hyperthermia. 1990;6(3):479-486.
11 Overgaard J, et al. Randomised trial of hyperthermia as adjuvant to radiotherapy for recurrent or metastatic malignant melanoma. Lancet. 1995;345(8949):540-543.
12 Valdagni R, Amichetti M, Pani G. Radical radiation alone versus radical radiation plus microwave hyperthermia for N3 (TNM-UICC) neck nodes: a prospective randomized clinical trial. Int J Radiat Oncol Biol Phys. 1988;15(1):13-24.
13 Valdagni R, Amichetti M. Report of long-term follow-up in a randomized trial comparing radiation therapy and radiation therapy plus hyperthermia to metastatic lymph nodes in stage IV head and neck patients. Int J Radiat Oncol Biol Phys. 1994;28(1):163-169.
14 Van der Zee J, et al. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Lancet. 2000;355(9210):1119-1125.
15 Vernon CC, et al. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: results from five randomized controlled trials. Int J Radiat Oncol Biol Phys. 1996;35(4):731-744.
16 Oleson JR, et al. Sensitivity of hyperthermia trial outcomes to temperature and time: implications for thermal goals of treatment. Int J Radiat Oncol Biol Phys. 1993;25(2):289-297.
17 Jorritsma JBM, et al. DNA polymerase activity in heat killing and hyperthermia radiosensitization of mammalian cells as observed after fractionated heat treatments. Radiat Res. 1986;105(3):307-319.
18 Morimoto RI, Tissieres A, Georgopoulos C. The biology of heat shock proteins and Molecular Chaperones. Plainview: Cold Spring Harbor Laboratory Press; 1994.
19 Landry J, Marceau N. Rate limiting events in hyperthermic cell killing. Radiat Res. 1978;75(3):573-585.
20 Alper T. Cellular radiobiology. Cambridge: Cambridge University Press; 1979.
21 Privalov P. Stability of proteins: small globular proteins. Adv Protein Chem. 1979;33:167-241.
22 Lepock JR, Kruuv J. Thermotolerance as a possible cause of the critical temperature at 43° C in mammalian cells. Cancer Res. 1980;40(12):4485-4488.
23 Lepock JR, et al. Increased thermostability of thermotolerant CHL V79 cells as determined by differential scanning calorimetry. J Cell Physiol. 1990;142(3):628-634.
24 Jung HA. A generalized concept for cell killing by heat. Effect of chronically induced thermotolerance. Radiat Res. 1991;127(3):235-242.
25 Hahn GM, Shiu EC. Adaptation to low pH modifies thermal and thermo-chemical responses of mammalian cells. Int J Hyperthermia. 1986;2(4):379-387.
26 Hofer KG, Mivechi NF. Tumor cell sensitivity to hyperthermia as a function of extracellular and intracellular pH. J Natl Cancer Inst. 1980;65(3):621-625.
27 Westra A, Dewey WC. Variation in sensitivity to heat shock during the cell cycle of Chinese hamster cells in vitro. Int J Radiat Biol. 1971;19(5):467-477.
28 Sapareto SA, Dewey WC. Thermal dose determination in cancer therapy. Int J Radiat Oncol Biol Phys. 1984;10(6):787-800.
29 Dewhirst MW, Sim DA. The utility of thermal dose as a predictor of tumor and normal tissue responses to combined radiation and hyperthermia. Cancer Res. 1984;44(Suppl 10):4772s-4780s.
30 Sherar M, et al. Relationship between thermal dose and outcome in thermoradiotherapy treatments for superficial recurrences of breast cancer: data from a Phase III trial. Int J Radiat Oncol Biol Phys. 1997;39(2):371-380.
31 Leopold KA, et al. Cumulative minutes with T90 greater than tempindex is predictive of response of superficial malignancies to hyperthermia and radiation. Int J Radiat Oncol Biol Phys. 1993;25(5):841-847.
32 Leopold KA, et al. Relationships among tumor temperature, treatment time, and histopathological outcome using preoperative hyperthermia with radiation in soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 1992;22(5):989-998.
33 Rau B, et al. Preoperative radiochemotherapy in locally advanced or recurrent rectal cancer: regional radiofrequency hyperthermia correlates with clinical parameters. Int J Radiat Oncol Biol Phys. 2000;48(2):381-391.
34 Kapp DS, Cox RS. Thermal treatment parameters are most predictive of outcome in patients with single tumor nodules per treatment field in recurrent adenocarcinoma of the breast. Int J Radiat Oncol Biol Phys. 1995;33(4):887-899.
35 Borrelli MJ, et al. Reduction of levels of nuclear-associated protein in heated cells by cycloheximide, D2O, and thermotolerance. Radiat Res. 1992;131(2):204-213.
36 Burgman PWJJ, Kampinga HH, Konings AWT. Possible role of localized protein denaturation in the induction of thermotolerance by heat, sodium-arsenite, and ethanol. Int J Hyperthermia. 1993;9(1):151-162.
37 Kampinga HH, et al. Thermal protein denaturation and protein aggregation in cells made thermotolerant by various chemicals: role of heat shock proteins. Exp Cell Res. 1995;219(2):536-546.
38 Konings AWT. Membranes are targets for hyperthermic cell killing. Recent Results Cancer Res. 1988;109:9-21.
39 Laszlo A. The effects of hyperthermia on mammalian cell structure and function. Cell Prolif. 1992;25(2):59-87.
40 Vidair CA, Doxsey SJ, Dewey WC. Heat shock alters centrosome organization leading to mitotic dysfunction and cell death. J Cell Physiol. 1993;154(3):443-455.
41 Welch WJ, Suhan JP. Morphological study of the mammalian stress response: characterization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance of intranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol. 1985;101(4):1198-1211.
42 Kampinga HH, et al. The relationship of increased nuclear protein content induced by hyperthermia to killing of HeLa S3 cells. Radiat Res. 1989;117(3):511-522.
43 Roti Roti JL, Henle KJ, Winward RT. The kinetics of increase in chromatin protein content in heated cells: a possible role in cell killing. Radiat Res. 1979;78(3):522-531.
44 Kampinga HH, et al. The interaction of heat and radiation affecting the ability of nuclear DNA to undergo supercoiling changes. Radiat Res. 1988;116(1):114-123.
45 Hayashi Y, et al. Translocation of hsp-70 and protein synthesis during continuous heating at mild temperatures in HeLa cells. Radiat Res. 1991;125(1):80-88.
46 Kampinga HH, et al. Cells overexpressing hsp27 show accelerated recovery from heat-induced nuclear protein aggregation. Biochem Biophys Res Commun. 1994;204(3):1170-1177.
47 Parsell DA, et al. Protein disaggregation mediated by heat-shock protein hsp104. Nature. 1994;372(6505):475-478.
48 Stege GJJ, et al. On the role of hsp72 in heat-induced intranuclear protein aggregation. Int J Hyperthermia. 1994;10(5):659-674.
49 Henle KJ, Leeper DB. Interaction of hyperthermia and radiation in CHO cells: recovery kinetics. Radiat Res. 1976;66(3):505-518.
50 Gerner EW, Schneider MJ. Induced thermal resistance in HeLa cells. Nature. 1975;256(5517):500-502.
51 Field SB, Anderson RL. Thermotolerance: a review of observations and possible mechanisms. Cancer therapy by hyperthermia, drugs and radiation. Natl Cancer Inst Monogr. 1982;61:193-201.
52 Gerner EW. Thermotolerance. In: Storm FK, editor. Hyperthermia in cancer therapy. Boston: G. K. Hall; 1983:141-162.
53 Henle KJ, Dethlefsen LA. Heat fractionation and thermotolerance: a review. Cancer Res. 1978;38(7):1843-1851.
54 Sapareto SA, et al. Effects of hyperthermia on survival and progression of Chinese hamster ovary cells. Cancer Res. 1978;38(2):393-400.
55 Gerweck LE. Modification of cell lethality at elevated temperatures: the pH effect. Radiat Res. 1977;70(1):224-235.
56 Henle KJ. Thermotolerance in cultured mammalian cells. In: Henle KJ, editor. Thermotolerance. Boca Raton: CRC Press; 1987:13-71.
57 Hahn GM, et al. Mechanistic implications of the induction of thermotolerance in Chinese hamster cells by organic solvents. Cancer Res. 1985;45(9):4138-4143.
58 Lee YJ, Dewey WC. Induction of heat shock proteins in Chinese hamster ovary cells and development of thermotolerance by intermediate concentrations of puromycin. J Cell Physiol. 1987;132(1):1-11.
59 Li GC, Hahn GM. A proposed operational model of thermotolerance based on effects of nutrients and the initial treatment temperature. Cancer Res. 1980;40(12):4501-4508.
60 Lis J, Wu C. Protein traffic on the heat shock promoter: parking, stalling, and trucking along. Cell. 1993;74(1):1-4.
61 Morimoto RI, Sarge KD, Abravaya K. Transcriptional regulation of heat shock genes: a paradigm for inducible genomic responses. J Biol Chem. 1992;267(31):21987-21990.
62 Liu RY, et al. Dual control of heat shock response: involvement of a constitutive heat shock element-binding factor. Proc Natl Acad Sci U S A. 1993;90(7):3078-3082.
63 Burgman P, Nussenzweig A, Li GC. Thermotolerance. In: Seegenschmiedt MH, Fessenden P, Vernon CC, editors. Principles and practice of thermoradiotherapy and thermochemotherapy, vol. 1. Berlin, New York: Springer-Verlag; 1995:75-87.
64 Landry J, et al. Heat shock resistance conferred by expression of the human HSP27 gene in rodent cells. J Cell Biol. 1989;109(1):7-15.
65 GC Li, et al. Thermal response of rat fibroblasts stably-transfected with the human 70 kDa heat shock protein-encoding gene. Proc Natl Acad Sci U S A. 1991;88(5):1681-1685.
66 Angelidis CE, Lazaridis I, Pagoulatos GN. Constitutive expression of heat-shock protein 70 in mammalian cells confers thermoresistance. Eur J Biochem. 1991;199:35-39.
67 Mackey MA, Anolik SL, Roti Roti JL. Cellular mechanisms associated with the lack of chronic thermotolerance expression in HeLa S3 cells. Cancer Res. 1992;52(5):1101-1106.
68 Armour EP, et al. Sensitivity of human cells to mild hyperthermia. Cancer Res. 1993;53(12):2740-2744.
69 Raaphorst GP, et al. Comparison of thermoradiosensitization in two human melanoma cell lines and one fibroblast cell line by concurrent mild hyperthermia and low-dose-rate irradiation. Radiat Res. 1994;137(3):338-345.
70 Morimoto RI, Tissieres A, Georgopoulos C. Stress proteins in biology and medicine. Plainview: Cold Spring Harbor Laboratory Press; 1990.
71 Maresca B, Lindquist S. Heat shock. New York: Springer-Verlag; 1991.
72 Wu C, et al. Transcriptional regulation of heat shock genes. In: Morimoto RI, Tissierres A, Georgopoulos C, editors. Stress proteins in biology and medicine. Plainview: Cold Spring Harbor Laboratory Press; 1990:429-442.
73 Sorger PK. Heat shock factor and the heat shock response. Cell. 1991;65(3):363-366.
74 Hensold JO, et al. DNA binding of heat shock factor to the heat shock element is insufficient for transcriptional activation in murine erythroleukemia cells. Mol Cell Biol. 1990;10(4):1600-1608.
75 Gorzowski JJ, et al. Methylation-associated transcriptional silencing of the major histocompatibility complex-linked hsp70 genes in mouse cell lines. J Biol Chem. 1995;270(45):26940-26949.
76 Bruce JL, et al. Oxidative injury rapidly activates the heat shock transcription factor but fails to increase levels of heat shock proteins. Cancer Res. 1993;53(1):12-15.
77 Liu RY, Corry PM, Lee YJ. Regulation of chemical stress-induced hsp70 gene expression in murine L929 cells. J Cell Sci. 1994;107(Pt 8):2209-2214.
78 Leppa S, et al. Differential induction of hsp70-encoding genes in human hematopoietic cells. J Biol Chem. 2001;276(34):31713-31719.
79 Mathur SK, et al. Deficient induction of human hsp70 heat shock gene transcription in Y79 retinoblastoma cells despite activation of heat shock factor 1. Proc Natl Acad Sci U S A. 1994;91(18):8695-8699.
80 Jurivich DA, et al. Effect of sodium salicylate on the human heat shock response. Science. 1992;255(5049):1243-1245.
81 Kim D, et al. A constitutive heat shock element-binding factor is immunologically identical to the Ku-autoantigen. J Biol Chem. 1995;270(25):15277-15284.
82 Kim D, Ouyang H, Li GC. Heat shock protein hsp70 accelerates the recovery of heat-shocked mammalian cells through its modulation of heat shock transcription factor HSF1. Proc Natl Acad Sci U S A. 1995;92(6):2126-2130.
83 Liu RY, Corry PM, Lee YJ. Potential involvement of a constitutive heat shock element binding factor in the regulation of chemical stress-induced hsp70 gene expression. Mol Cell Biochem. 1995;144(1):27-34.
84 Li GC, et al. Suppression of heat-induced hsp70 expression by the 70-kDa subunit of the human Ku-autoantigen. Proc Natl Acad Sci U S A. 1995;92(10):4512-4516.
85 Yang S-H, et al. Modulation of thermal induction of hsp70 expression by Ku autoantigen or its individual subunits. Mol Cell Biol. 1996;16(7):3799-3806.
86 Tang D, et al. Repression of the HSP70B promoter by NFIL6, Ku70, and MAPK involves three complementary mechanisms. Biochem Biophys Res Commun. 2000;280:280-285.
87 Huang J, et al. Heat shock transcription factor 1 binds selectively in vitro to Ku protein and the catalytic subunit of the DNA-dependent protein kinase. J Biol Chem. 1997;272(41):26009-26016.
88 Konings AWT. Interaction of heat and radiation in vitro and in vivo. In: Seegenschmiedt MH, Fessenden P, Vernon CC, editors. Thermoradiotherapy and Thermochemotherapy, vol. 1: Biology, Physiology and Physics. Heidelberg: Springer-Verlag; 1995:89-102.
89 Overgaard J, Nielsen OS, Lindegaard JC. Biological basis for rational design of clinical treatment with combined hyperthermia and radiation. In: Field SB, Franconi C, editors. Physics and technology of hyperthermia. Amsterdam: Martinus Nijhoff; 1987:54-79.
90 Warters RL, Roti Roti JL. Hyperthermia and the cell nucleus. Radiat Res. 1982;92(3):458-462.
91 Jorritsma JBM, Konings AWT. Inhibition of repair of radiation-induced strand breaks by hyperthermia and its relation to cell survival after hyperthermia alone. Int J Radiat Biol. 1983;43(5):506-516.
92 Mills HD, Meyn RE. Hyperthermic potentiation of unrejoined DNA strand breaks following irradiation. Radiat Res. 1981;87(2):314-328.
93 Dikomey E, Franzke J. Effect of heat on induction and repair of DNA strand breaks in X-irradiated CHO cells. Int J Radiat Biol. 1992;61(2):221-234.
94 Warters RL, Roti Roti JL. Excision of X-ray-induced thymine damage in chromatin from heated cells. Radiat Res. 1979;79(1):113-121.
95 Stege GJJ, Kampinga HH, Konings AWT. Heat-induced intranuclear protein aggregation and thermal radiosensitization. Int J Radiat Biol. 1995;67(2):203-209.
96 Wynstra JH, Wright WD, Roti Roti JL. Repair of radiation-induced DNA damage in thermotolerant and nonthermotolerant HeLa cells. Radiat Res. 1990;124(1):85-89.
97 Kampinga HH, et al. Interaction of hyperthermia and radiation in tolerant and nontolerant HeLa S3 cells: role of DNA polymerase inactivation. Int J Radiat Biol. 1989;55(3):423-433.
98 Henle KJ, Nagle WA, Moss AJ. Effect of thermotolerance on the cellular heat-radiation response. In: Urano M, Douple E, editors. Hyperthermia and oncology, vol. 2. Boca Raton: VSP; 1989:53-81.
99 Raaphorst GP, Azzam EI. Thermal radiosensitization in Chinese hamster (V79) and mouse C3H 10T 1/2 cells. The thermotolerance effect. Br J Cancer. 1983;48(1):45-54.
100 Freeman ML, Raaphorst GP, Dewey WC. The relationship of heat killing and thermoradiosensitization to the duration of heating at 42° C. Radiat Res. 1979;78(1):172-175.
101 Konings AWT. Effects of heat and radiation on mammalian cells. Radiat Phys Chem. 1987;30:339-349.
102 Heller DP, Raaphorst GP. Low dose-rate irradiation of human glioma cells and thermoradiosensitization by mild hyperthermia. Radiat Oncol Investig. 1993;1:218-226.
103 Armour EP, et al. Sensitization of rat 9L gliosarcoma cells to low dose rate irradiation by long duration 41° C hyperthermia. Cancer Res. 1991;51(12):3088-3095.
104 Dikomey E. Effect of hyperthermia at 42 and 45° C on repair of radiation-induced DNA strand breaks in CHO cells. Int J Radiat Biol. 1982;41(6):603-614.
105 Ben Hur E, Elkind MM. Thermally enhanced radioresponse of cultured Chinese hamster cells: Damage and repair of single-stranded DNA and a DNA complex. Radiat Res. 1974;59(2):484-495.
106 Warters RL, Lyons BW, Axtell-Bartlett J. Inhibition of repair of radiation-induced DNA damage by thermal shock in Chinese hamster ovary cells. Int J Radiat Biol. 1987;51(3):505-517.
107 Dahl O, Mella O. Hyperthermia and chemotherapeutic agents. In: Field SB, Hand JW, editors. An Introduction to the practical aspects of clinical hyperthermia. London, New York: Taylor & Francis; 1990:108-142.
108 Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1997;271(1):58-65.
109 Lasic DD. Liposomes from Physics to Applications. Amsterdam: Elsevier; 1993.
110 Massing U. Cancer therapy with liposomal formulations of anticancer drugs. Int J Clin Pharmacol Ther. 1997;35(2):87-90.
111 Campos D, Shapiro C. Clinical evaluation of liposomal anthracyclines. In: Janoff A, editor. Liposomes: Rational Design. New York: Marcel Dekker, Inc; 1999:363-377.
112 Gabizon A, Barenholtz Y. Liposomal anthracyclines. From basics to clinical approval of PEGylated liposomal doxorubicin. In: Janoff A, editor. Liposomes: rational design. New York: Marcel Dekker, Inc; 1999:343-362.
113 Needham D, et al. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 2000;60(5):1197-1201.
114 Ning S, et al. Hyperthermia induces doxorubicin release from long-circulating liposomes and enhances their anti-tumor efficacy. Int J Radiat Oncol Biol Phys. 1994;29(4):827-834.
115 Yatvin MB, et al. Design of liposomes for enhanced local release of drugs by hyperthermia. Science. 1978;202(4374):1290-1293.
116 Kong G, Dewhirst MW. Review: hyperthermia and liposomes. Int J Hyperthermia. 1999;15(5):345-370.
117 Kong G, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer Res. 2000;60(24):6950-6957.
118 Ponce AM, et al. Hyperthermia mediated liposomal drug delivery. Int J Hyperthermia. 2006;22(3):205-213.
119 Viglianti BL, et al. In vivo monitoring of tissue pharmacokinetics of liposome/drug using MRI: illustration of targeted delivery. Magn Reson Med. 2004;51(6):1153-1162.
120 Chen Q, et al. Targeting tumor microvessels using doxorubicin encapsulated in a novel thermosensitive liposome. Mol Cancer Ther. 2004;3(10):1311-1317.
121 Ponce AM, et al. Magnetic resonance imaging of temperature-sensitive liposome release: drug dose painting and antitumor effects. J Natl Cancer Inst. 2007;99(1):53-63.
122 Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci U S A. 1986;83(10):3407-3411.
123 Srivastava PK, Maki RG. Stress-induced proteins in immune response to cancer. Curr Top Microbiol Immunol. 1991;167:109-123.
124 Srivastava PK. Peptide-binding heat shock proteins in the endoplasmic reticulum: role in immune response to cancer and in antigen presentation. Adv Cancer Res. 1993;62:153-177.
125 Ullrich SJ, et al. A mouse tumor-specific transplantation antigen is a heat shock-related protein. Proc Natl Acad Sci U S A. 1986;83(10):3121-3125.
126 Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol. 1994;152(11):5398-5403.
127 Tamura Y, et al. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 1997;278(5335):117-120.
128 Basu S, Srivastava PK. Calreticulin, a peptide-binding chaperone of the endoplasmic reticulum, elicits tumor- and peptide-specific immunity. J Exp Med. 1999;189(5):797-802.
129 Srivastava PK, Das MR. The serologically unique cell surface antigen of Zajdela ascitic hepatoma is also its tumor-associated transplantation antigen. Int J Cancer. 1984;33(3):417-422.
130 Janetzki S, Blachere NE, Srivastava PK. Generation of tumor-specific cytotoxic T lymphocytes and memory T cells by immunization with tumor-derived heat shock protein gp96. J Immunother. 1998;21(4):269-276.
131 Jimbo J, et al. Induction of leukemia-specific antibodies by immunotherapy with leukemia-cell-derived heat shock protein 70. Cancer Sci. 2008;99(7):1427-1434.
132 Wang X-Y, et al. Characterization of heat shock protein 110 and glucose-regulated protein 170 as cancer vaccines and the effect of fever-range hyperthermia on vaccine activity. J Immunol. 2001;166(1):490-497.
133 Wang X-Y, et al. Hsp110 over-expression increases the immunogenicity of the murine CT26 colon tumor. Cancer Immunol Immunother. 2002;51(6):311-319.
134 Manjili MH, et al. Development of a recombinant HSP110-HER-2/neu vaccine using the chaperoning properties of HSP110. Cancer Res. 2002;62(6):1737-1742.
135 Murshid A, Gong J, Calderwood SK. Heat-shock proteins in cancer vaccines: agents of antigen cross-presentation. Expert Rev Vaccines. 2008;7(7):1019-1030.
136 Testori A, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: The C-100–21 Study Group. J Clin Oncol. 2008;26(6):955-962.
137 Liu B, et al. A novel therapeutic fusion protein vaccine by two different families of heat shock proteins linked with HPV16 E7 generates potent antitumor immunity and antiangiogenesis. Vaccine. 2008;26(10):1387-1396.
138 Gerner EW, et al. Heat-inducible vectors for use in gene therapy. Int J Hyperthermia. 2000;16(2):171-181.
139 Lohr F, et al. Enhancement of radiotherapy by hyperthermia-regulated gene therapy. Int J Radiat Oncol Biol Phys. 2000;48(5):1513-1518.
140 Vekris A, et al. Control of transgene expression using local hyperthermia in combination with a heat-sensitive promoter. J Gene Med. 2000;2(2):89-96.
141 Borrelli MJ, et al. Heat-activated transgene expression from adenovirus vectors infected into human prostate cancer cells. Cancer Res. 2001;61(3):1113-1121.
142 Huang Q, et al. Heat-induced gene expression as a novel targeted cancer gene therapy strategy. Cancer Res. 2000;60(13):3435-3439.
143 Li GC, He F, Ling CC. Hyperthermia and gene therapy: potential use of microPET imaging. Int J Hyperthermia. 2006;22(3):215-221.
144 Parry JJ, et al. PET imaging of heat-inducible suicide gene expression in mice bearing head and neck squamous cell carcinoma xenografts. Cancer Gene Ther. 2009;16(2):161-170.
145 Deckers R, et al. Image-guided, noninvasive, spatiotemporal control of gene expression. Proc Natl Acad Sci U S A. 2009;106(4):1175-1180.
146 Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93(4):266-276.
147 Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38-47.
148 Brizel DM, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56(5):941-943.
149 Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol. 2004;14(3):198-206.
150 Song CW, Park H, Griffin RJ. Improvement of tumor oxygenation by mild hyperthermia. Radiat Res. 2001;155(4):515-528.
151 Shakil A, Osborn JL, Song CW. Changes in oxygenation status and blood flow in a rat tumor model by mild temperature hyperthermia. Int J Radiat Oncol Biol Phys. 1999;43(4):859-865.
152 Vujaskovic Z, et al. Temperature-dependent changes in physiologic parameters of spontaneous canine soft tissue sarcomas after combined radiotherapy and hyperthermia treatment. Int J Radiat Oncol Biol Phys. 2000;46(1):179-185.
153 Brizel DM, et al. Radiation therapy and hyperthermia improve the oxygenation of human soft tissue sarcomas. Cancer Res. 1996;56:5347-5350.
154 Vujaskovic Z, et al. Ultrasound guided pO2 measurement of breast cancer reoxygenation after neoadjuvant chemotherapy and hyperthermia treatment. Int J Hyperthermia. 2003;19(5):498-506.
155 Sun X, et al. Changes in tumor hypoxia induced by mild temperature hyperthermia as assessed by dual-tracer immunohistochemistry. Radiother Oncol. 2008;88(2):269-276.
156 Lagendijk JJW. Thermal models: principles and implementation. In: Field SB, Hand JW, editors. An Introduction to the practical aspects of clinical hyperthermia. London, New York: Taylor & Francis; 1990:478-512.
157 Pennes HH. Analysis of tissue and arterial blood temperatures in the resting human forearm. J Appl Physiol. 1948;85(1):5-34.
158 Lagendijk JJ. Hyperthermia treatment planning. Phys Med Biol. 2000;45(5):R61-R76.
159 Arkin H, Xu LX, Holmes KR. Recent developments in modeling heat transfer in blood perfused tissues. IEEE Trans Biomed Eng. 1994;41(2):97-107.
160 Gautherie M, editor; Thermal dosimetry and treatment planning. Clinical thermology: subseries thermotherapy; 1990; Springer-Verlag, Berlin:216.
161 Seegenschmiedt MH, Fessenden P, Vernon CC. Thermoradiotherapy and thermochemotherapy, vol. 1: Biology, Physiology, and Physics. Berlin: Springer-Verlag; 1995.
162 Fessenden P, Hand JW. Hyperthermia therapy physics. In: Smith AR, editor. Medical radiology: radiation therapy physics. Berlin: Springer-Verlag; 1995:315-363.
163 Samulski TV, et al. Clinical experience with a multi-element ultrasonic hyperthermia system: analysis of treatment temperatures. Int J Hyperthermia. 1990;6(5):909-922.
164 Hynynen K. Hot spots created at skin-air interfaces during ultrasound hyperthermia. Int J Hyperthermia. 1990;6(6):1005-1012.
165 Goss SA, Johnston RL, Dunn F. Comprehensive compilation of empirical ultrasonic properties of mammalian tissues. J Acoust Soc Am. 1978;64(2):423-457.
166 Field SB, Hand JW. An introduction to the practical aspects of clinical hyperthermia. London: Taylor & Francis; 1990.
167 Gautherie M, editor; Methods of External hyperthermic heating. Clinical thermology: subseries thermotherapy; 1990; Springer-Verlag, Berlin:143.
168 Stauffer PR. Thermal therapy techniques for skin and superficial tissue disease. In: Ryan TP, editor. Critical reviews: matching the energy source to the clinical need. Bellingham, WA: SPIE Optical Engineering Press; 2000:327-367.
169 Stauffer PR. Evolving technology for thermal therapy of cancer. Int J Hyperthermia. 2005;21(8):731-744.
170 Chou CK, et al. Evaluation of captive bolus applicators. Med Phys. 1990;17(4):705-709.
171 Straube WL, et al. SAR patterns of external 915 MHz microwave applicators. Int J Hyperthermia. 1990;6(3):665-670.
172 Sherar MD, et al. Beam shaping for microwave waveguide hyperthermia applicators. Int J Radiat Oncol Biol Phys. 1993;25(5):849-857.
173 Nishimura Y, et al. Thermoradiotherapy of superficial and subsurface tumours: analysis of thermal parameters and tumour response. Int J Hyperthermia. 1995;11(5):603-613.
174 Fessenden P, et al. Review of the Stanford experience developing non-focusing scanning and array surface microwave (MW) applicators. In: Gerner EW, Cetas TC, editors. Hyperthermic oncology, 1992, vol. 2. Tucson: Arizona Board of Regents; 1993:183-186.
175 Arabe OA, et al: A 400 MHz hyperthermia system using rotating spiral antennas for uniform treatment of large superficial sub-surface tumors. Proceedings of microwave theory and techniques society international microwave symposium, Honolulu, HI, 2007, IEEE Press, pp 1333-1336
176 Jones EL, et al. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol. 2005;23:3079-3085.
177 Diederich CJ, Stauffer PR. Pre-clinical evaluation of a microwave planar array applicator for superficial hyperthermia. Int J Hyperthermia. 1993;9(2):227-246.
178 Van Rhoon GC, Rietveld PJ, van der Zee J. A 433 MHz Lucite cone waveguide applicator for superficial hyperthermia. Int J Hyperthermia. 1998;14(1):13-27.
179 Rietveld PJM, et al. Quantitative evaluation of 2 × 2 arrays of Lucite cone applicators in flat layered phantoms using Gaussian-beam-predicted and thermographically measured SAR distributions. Phys Med Biol. 1998;43(8):2207-2220.
180 Gelvich EA, Mazokhin VN. Contact flexible microstrip applicators (CFMA) in a range from microwaves up to short waves. IEEE Trans Biomed Eng. 2002;49:1015-1023.
181 Lee WM, et al. Assessment of the performance characteristics of a prototype 12-element capacitive contact flexible microstrip applicator (CFMA-12) for superficial hyperthermia. Int J Hyperthermia. 2004;20(6):607-624.
182 Gopal MK, et al. Current sheet applicator arrays for superficial hyperthermia of chestwall lesions. Int J Hyperthermia. 1992;8(2):227-240.
183 Lee ER, et al. Body conformable 915 MHz microstrip array applicators for large surface area hyperthermia. IEEE Trans Biomed Eng. 1992;39(5):470-483.
184 Johnson JE, et al. Evaluation of a dual-arm Archimedean spiral array for microwave hyperthermia. Int J Hyperthermia. 2006;22(6):475-490.
185 Stauffer P, et al. Progress on conformal microwave array applicators for heating chestwall disease. In: Ryan T, editor. Proceedings of SPIE. San Jose: SPIE Press; 2007:1-13.
186 Juang T, et al. Multilayer conformal applicator for microwave heating and brachytherapy treatment of superficial tissue disease. Int J Hyperthermia. 2006;22(7):527-544.
187 Taschereau R, et al. Radiation dosimetry of a conformal heat-brachytherapy applicator. Tech Cancer Res Treat. 2004;3(4):347-358.
188 Stauffer PR, Diederich CJ, Pouliot J. Thermal therapy for cancer. In Thomadsen B, Rivard M, Butler W, editors: Brachytherapy physics, ed 2, Madison WI: Medical Physics Publishing, 2005. Joint AAPM/ABS Summer School, Med Phys Monograph No 31
189 Stauffer PR, et al. Progress on system for applying simultaneous heat and brachytherapy to large-area surface disease. In: SPIE BIOS. San Jose: SPIE Press; 2005.
190 Arunachalam K, Maccarini PF, Stauffer PR. A thermal monitoring sheet with low influence from adjacent waterbolus for tissue surface thermometry during clinical hyperthermia. IEEE Trans Biomed Eng. 2008;55(10):2397-2406.
191 Stauffer PR, Jacobsen S, Neuman DG. Microwave array applicators for radiometry controlled superficial hyperthermia. In: Ryan TP, editor. Thermal Treatment of tissue: energy delivery and assessment. Bellingham, WA: SPIE Press; 2001:19-29.
192 Chive M. Use of microwave radiometry for hyperthermia monitoring and as a basis for thermal dosimetry. In: Gautherie M, editor. Methods of hyperthermia control, Heidelberg. Berlin: Springer-Verlag; 1990:113-128.
193 Jacobsen S, Stauffer PR, Neuman DG. Dual-mode antenna design for microwave heating and noninvasive thermometry of superficial tissue disease. IEEE Trans Biomed Eng. 2000;47(11):1500-1509.
194 Novak P, et al. Ultrasonic approach to noninvasive temperature monitoring during microwave thermotherapy. Radioengineering. 2001;10(1):32-37.
195 Corry PM, et al. Combined ultrasound and radiation therapy treatment of human superficial tumors. Radiology. 1982;145(1):165-169.
196 Hynynen K, et al. Temperature distributions during clinical scanned, focused ultrasound hyperthermia treatments. Int J Hyperthermia. 1990;6(5):891-908.
197 Anhalt DP, et al. Scanned ultrasound hyperthermia for treating superficial disease. In: Gerner E, Cetas T, editors. Hyperthermic oncology 1992, vol. 2. Tucson: Arizona Board of Regents; 1993:191-192.
198 Moros EG, Fan X, Straube WL. Experimental assessment of power and temperature penetration depth control with a dual frequency ultrasonic system. Med Phys. 1999;26(5):810-817.
199 Novak P, et al. SURLAS: A new clinical grade ultrasound system for sequential or concomitant thermoradiotherapy of superficial tumors: applicator description. Med Phys. 2005;32(1):230-240.
200 Novak P, et al. Influence of the SURLAS applicator on radiation dose distributions during simultaneous thermoradiotherapy with helical tomotherapy. Phys Med Biol. 2008;53(10):2509-2522.
201 Penagaricano JA, et al. Feasibility of concurrent treatment with the scanning ultrasound reflector linear array system (SURLAS) and the helical tomotherapy system. Int J Hyperthermia. 2008;24(5):377-388.
202 Hynynen K, et al. MR imaging-guided focused ultrasound surgery of fibroadenomas in the breast: a feasibility study. Radiology. 2001;219(1):176-185.
203 McDannold N, et al. Uterine leiomyomas: MR imaging-based thermometry and thermal dosimetry during focused ultrasound thermal ablation. Radiology. 2006;240(1):263-272.
204 Seegenschmiedt MH, Sauer R, editors. Interstitial and intracavitary thermoradiotherapy. Medical Radiology. Berlin: Springer-Verlag, 1993.
205 Stauffer PR, Diederich CJ, Seegenschmiedt MH. Interstitial heating technologies. In: Seegenschmiedt MH, Fessenden P, Vernon CC, editors. Thermoradiotherapy and Thermochemotherapy: Volume 1, Biology, Physiology and Physics. Berlin, New York: Springer-Verlag; 1995:279-320.
206 Seegenschmiedt MH, et al. Clinical practice of interstitial thermoradiotherapy. In: Seegenschmiedt MH, Fessenden P, Vernon CC, editors. Thermoradiotherapy and thermochemotherapy: volume 2, clinical applications. Berlin, New York: Springer-Verlag; 1996:207-320.
207 Strohbehn JW. Interstitial techniques for hyperthermia. In: Field SB, Franconi C, editors. Physics and technology of hyperthermia, Dordrecht. Boston, Lancaster: Martinus Nijhoff Publishers; 1987:211-240.
208 Camart JC, et al. 915 MHz microwave interstitial hyperthermia. Part II: array of phase-monitored antennas. Int J Hyperthermia. 1993;9(3):445-454.
209 Trembly BS, et al. The effect of phase modulation on the temperature distribution of a microwave hyperthermia antenna array in vivo. Int J Hyperthermia. 1994;10(5):691-705.
210 Satoh T, Stauffer PR, Fike JR. Thermal distribution studies of helical coil microwave antennas for interstitial hyperthermia. Int J Radiat Oncol Biol Phys. 1988;15(5):1209-1218.
211 Ryan TP. Comparison of six microwave antennas for hyperthermia treatment of cancer: SAR results for single antennas and arrays. Int J Radiat Oncol Biol Phys. 1991;21(2):403-413.
212 Haemmerich D, Lee FTJr. Multiple applicator approaches for radiofrequency and microwave ablation. Int J Hyperthermia. 2005;21(2):93-106.
213 Fabre JJ, et al. 915 MHz microwave interstitial hyperthermia. Part I: Theoretical and experimental aspects with temperature control by multifrequency radiometry. Int J Hyperthermia. 1993;9(3):433-444.
214 Camart JC, et al. New 434 MHz interstitial hyperthermia system monitored by microwave radiometry: theoretical and experimental results. Int J Hyperthermia. 2000;16(2):95-111.
215 Prionas SD, et al. Interstitial radiofrequency-induced hyperthermia. In: Gerner EW, Cetas TC, editors. Hyperthermic Oncology 1992, vol. 2. Tucson: Arizona Board of Regents; 1993:249-253.
216 Lagendijk JJW, et al: Interstitial hyperthermia & treatment planning: the 27 MHz multi-electrode current source method. Nucletron-Odelft Activity Report No. 6, 1995:83–90.
217 Handl-Zeller L, editor. Interstitial hyperthermia. Wien, New York: Springer-Verlag, 1992.
218 Visser AG, et al. An interstitial hyperthermia system at 27 MHz. Int J Hyperthermia. 1989;5(2):265-276.
219 Marchal C, et al. Practical interstitial method of heating operating at 27.12 MHz. Int J Hyperthermia. 1989;5(4):451-466.
220 DeFord JA, et al. Effective estimation and computer control of minimum tumour temperature during conductive interstitial hyperthermia. Int J Hyperthermia. 1991;7(3):441-453.
221 Stauffer PR, Cetas TC, Jones RC. Magnetic induction heating of ferromagnetic implants for inducing localized hyperthermia in deep seated tumors. IEEE Trans Biomed Eng. 1984;31:235-251.
222 Brezovich IA, Atkinson WJ, Chakraborty DP. Temperature distributions in tumor models heated by self-regulating nickel-copper alloy thermoseeds. Med Phys. 1984;11:145-152.
223 Stauffer PR, et al. Practical induction heating coil designs for clinical hyperthermia with ferromagnetic implants. IEEE Trans Biomed Eng. 1994;41(1):17-28.
224 Cetas TC, Gross EJ, Contractor Y. A ferrite core/metallic sheath thermoseed for interstitial thermal therapies. IEEE Trans Biomed Eng. 1998;45(1):68-77.
225 Matsuki H, et al. High quality soft heating method utilizing temperature dependence of permeability and core loss of low curie temperature ferrite. IEEE Trans Magn. 1987;23:2440-2442.
226 Jordan A, Maier-Hauff K. Magnetic nanoparticles for intracranial thermotherapy. J Nanosci Nanotechnol. 2007;7(12):4604-4606.
227 Wust P, et al. Magnetic nanoparticles for interstitial thermotherapy—feasibility, tolerance and achieved temperatures. Int J Hyperthermia. 2006;22(8):673-685.
228 Stauffer PR, et al. Observations on the use of ferromagnetic implants for inducing hyperthermia. IEEE Trans Biomed Eng. 1984;31(1):76-90.
229 Master V, Shinohara K, Carroll P. Ferromagnetic thermal ablation of locally recurrent prostate cancer: prostate specific antigen results and immediate/intermediate morbidities. J Urelogy. 2004;172:2197-2202.
230 Thiesen B, Jordan A. Clinical applications of magnetic nanoparticles for hyperthermia. Int J Hyperthermia. 2008;24(6):467-474.
231 Diederich CJ. Ultrasound applicators with integrated catheter-cooling for interstitial hyperthermia: Theory and preliminary experiments. Int J Hyperthermia. 1996;12(2):279-297.
232 Diederich CJ, et al. Direct-coupled interstitial ultrasound applicators for simultaneous thermobrachytherapy: A feasibility study. Int J Hyperthermia. 1996;12(3):401-419.
233 Kinsey AM, et al. Transurethral ultrasound applicators with dynamic multi-sector control for prostate thermal therapy: In vivo evaluation under MR guidance. Med Phys. 2008;35(5):2081-2093.
234 Chen J, et al. Monitoring prostate thermal therapy with diffusion-weighted MRI. Magn Reson Med. 2008;59(6):1365-1372.
235 Diederich CJ. Thermal ablation and high-temperature thermal therapy: overview of technology and clinical implementation. Int J Hyperthermia. 2005;21(8):745-753.
236 Li DJ. Present status on treatment of esophageal cancer in China. Int J Radiat Oncol Biol Phys. 1990;18(2):477-484.
237 Sorbe B, Roos DI, Karlsson LG. The use of microwave-induced hyperthermia in conjunction with afterloading irradiation of vaginal carcinoma. A preliminary report. Acta Oncol. 1990;29(8):1029-1033.
238 Liu RL, et al. Heating pattern of helical microwave intracavitary oesophageal applicator. Int J Hyperthermia. 1991;7(4):577-586.
239 Fosmire H, et al. Feasibility and toxicity of transrectal ultrasound hyperthermia in the treatment of locally advanced adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys. 1993;26(2):253-259.
240 Smith NB, Buchanan MT, Hynynen K. Transrectal ultrasound applicator for prostate heating monitored using MRI thermometry. Int J Radiat Oncol Biol Phys. 1999;43(1):217-225.
241 Hurwitz MD, et al. Hyperthermia combined with radiation in treatment of locally advanced prostate cancer is associated with a favourable toxicity profile. Int J Hyperthermia. 2005;21(7):649-656.
242 Paglione R, et al. 27 MHz ridged waveguide applicators for localized hyperthermia treatment of deep-seated malignant tumors. Microw J. 1981;24(2):71-80.
243 Croghan MK, et al. A phase I study of the toxicity of regional hyperthermia with systemic warming. Am J Clin Oncol. 1993;16(4):354-358.
244 De Leeuw AA, et al. Increasing the systemic temperature during regional hyperthermia: Effect of a cooling strategy on tumour temperatures and side-effects. Int J Hyperthermia. 2003;19(6):655-663.
245 Van Vulpen M, et al. Comparison of intra-luminal versus intra-tumoural temperature measurements in patients with locally advanced prostate cancer treated with the coaxial TEM system: report of a feasibility study. Int J Hyperthermia. 2003;19(5):481-497.
246 Kikuchi M, et al. Guide to the use of hyperthermic equipment. 1. Capacitively-coupled heating. Int J Hyperthermia. 1993;9(2):187-203.
247 Mochiki E, et al. Feasibility study of postoperative intraperitoneal hyperthermochemotherapy by radiofrequency capacitive heating system for advanced gastric cancer with peritoneal seeding. Int J Hyperthermia. 2007;23(6):493-500.
248 van Rhoon GC, et al. Radiofrequency capacitive heating of deep-seated tumours using pre-cooling of the subcutaneous tissues: results on thermometry in Dutch patients. Int J Hyperthermia. 1992;8(6):843-854.
249 Turner PF. Regional hyperthermia with an annular phased array. IEEE Trans Biomed Eng. 1984;31(1):106-114.
250 Fatehi D, van Rhoon GC. SAR characteristics of the Sigma-60-Ellipse applicator. Int J Hyperthermia. 2008;24(4):347-356.
251 Van Rhoon GC, et al. Characterization of the SAR-distribution of the Sigma-60 applicator for regional hyperthermia using a Schottky diode sheet. Int J Hyperthermia. 2003;19(6):642-654.
252 Albregts M, et al. A feasibility study in oesophageal carcinoma using deep loco-regional hyperthermia combined with concurrent chemotherapy followed by surgery. Int J Hyperthermia. 2004;20(6):647-659.
253 Gellermann J, et al. Clinical evaluation and verification of the hyperthermia treatment planning system hyperplan. Int J Radiat Oncol Biol Phys. 2000;47(4):1145-1156.
254 Kok HP, et al. Prospective treatment planning to improve locoregional hyperthermia for oesophageal cancer. Int J Hyperthermia. 2006;22(5):375-389.
255 Wust P, et al. Electric field distributions in a phased-array applicator with 12 channels: measurements and numerical simulations. Med Phys. 2000;27(11):2565-2579.
256 Carter D, et al. Magnetic resonance thermometry during hyperthermia for human high-grade sarcoma. Int J Radiat Oncol Biol Phys. 1998;40(4):815-822.
257 Gellermann J, et al. Noninvasive magnetic resonance thermography of soft tissue sarcomas during regional hyperthermia: correlation with response and direct thermometry. Cancer. 2006;107(6):1373-1382.
258 Li Z, et al. Towards the validation of a commercial hyperthermia treatment planning system (invited paper). Microw J. 2008;51(12):28-42.
259 Li Z, et al: Determining SAR/temperature distribution within an annular phased array using proton resonance frequency shift imaging with magnetic resonance imaging, Int J Hyperthermia (in press).
260 Stauffer P, et al. Clinical utility of magnetic resonance thermal imaging (MRTI) for realtime guidance of deep hyperthermia in SPIE. San Jose: SPIE Press; 2009.
261 Stakhursky V, et al: Real-time MRI guided hyperthermia treatment using a fast adaptive algorithm, Phys Med Biol (in press).
262 Hynynen K, et al. MR imaging-guided focused ultrasound surgery of fibroadenomas in the breast: A feasibility study. Radiology. 2001;219(1):176-185.
263 Jolesz FA, et al. MR imaging-controlled focused ultrasound ablation: a noninvasive image-guided surgery. Magn Reson Imaging Clin N Am. 2005;13(3):545-560.
264 Pettigrew RT, et al. Clinical effects of whole-body hyperthermia in advanced malignancy. Br Med J. 1974;4(5946):679-682.
265 Gerad H, et al. Doxorubicin, cyclophosphamide and whole body hyperthermia for treatment of advanced soft tissue sarcoma. Cancer. 1984;53(12):2585-2591.
266 Bull JM, et al. The importance of schedule in whole body thermochemotherapy. Int J Hyperthermia. 2008;24(2):171-181.
267 Repasky E, Issels R. Physiological consequences of hyperthermia: heat, heat shock proteins and the immune response. Int J Hyperthermia. 2002;18(6):486-489.
268 Gautherie M, editor. Methods of hyperthermia control. Clinical thermology: subseries thermotherapy. Berlin: Springer-Verlag, 1990.
269 Dickinson RG. Thermal conduction errors of manganin-constantan thermocouple arrays. Phys Med Biol. 1985;30:445-453.
270 Anhalt D, Hynynen K. Thermocouples—the Arizona experience with in-house manufactured probes. Med Phys. 1992;19(5):1325-1333.
271 Hoh LL, Waterman FM. Use of manganin-constantan thermocouples in thermometry units designed for copper-constantan thermocouples. Int J Hyperthermia. 1995;11(1):131-138.
272 Chan KW, Chou CK. Use of thermocouples in the intense fields of ferromagnetic implant hyperthermia. Int J Hyperthermia. 1993;9(6):831-848.
273 Chan KW, et al. Perturbations due to the use of catheters with non-perturbing probes. Int J Hyperthermia. 1988;4:699-702.
274 Sneed PK, et al. Survival benefit of hyperthermia in a prospective, randomized trial of brachytherapy boost ± hyperthermia for glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1998;40(2):287-295.
275 Tarczy-Hornoch P, et al. Automated mechanical thermometry probe mapping systems for hyperthermia. Int J Hyperthermia. 1992;8(4):543-554.
276 Dewhirst MW, et al. RTOG quality assurance guidelines for clinical trials using hyperthermia. Int J Radiat Oncol Biol PHys. 1990;18(5):1249-1259.
277 Emami B, et al. RTOG quality assurance guidelines for interstitial hyperthermia. Int J Radiat Oncol Biol Phys. 1991;20(5):1117-1124.
278 Waterman FM, Hoh LL. A recommended revision in the RTOG thermometry guidelines for hyperthermia administered by ultrasound. Int J Hyperthermia. 1995;11(1):121-130.
279 Sapozink MD, et al. RTOG quality assurance guidelines for clinical trials using hyperthermia for deep-seated malignancy. Int J Radiat Oncol Biol Phys. 1991;20(5):1109-1115.
280 Schneider CJ, et al. Quality assurance in various radiative hyperthermia systems applying a phantom with LED matrix. Int J Hyperthermia. 1994;10(5):733-747.
281 Tennant A, Conway J, Anderson AP. A robot-controlled microwave antenna system for uniform hyperthermia treatment of superficial tumours with arbitrary shape. Int J Hyperthermia. 1990;6(1):193-202.
282 Arunachalam K, et al. Performance evaluation of a conformal thermal monitoring sheet sensor array for measurement of surface temperature distributions during superficial hyperthermia treatments. Int J Hyperthermia. 2008;24(4):313-325.
283 Paulsen KD, et al. Initial in vivo experience with EIT as a thermal estimator during hyperthermia. Int J Hyperthermia. 1996;12(5):573-591.
284 Meaney PM, et al. Microwave thermal imaging: Initial in vivo experience with a single heating zone. Int J Hyperthermia. 2003;19(6):617-641.
285 Dubois L, et al. Temperature control and thermal dosimetry by microwave radiometry in hyperthermia. IEEE Trans Microw Theory Tech. 1996;44:1755-1761.
286 Arunachalam K, et al. Characterization of a digital microwave radiometry system for noninvasive thermometry using a temperature-controlled homogeneous test load. Phys Med Biol. 2008;53(14):3883-3901.
287 McDannold N, et al. Evaluation of referenceless thermometry in MRI-guided focused ultrasound surgery of uterine fibroids. J Magn Reson Imaging. 2008;28(4):1026-1032.
288 Gellermann J, et al. A practical approach to thermography in a hyperthermia/magnetic resonance hybrid system: validation in a heterogeneous phantom. Int J Radiat Oncol Biol Phys. 2005;61(1):267-277.
289 Cheng KS, et al. Fast temperature optimization of multi-source hyperthermia applicators with reduced-order modeling of ‘virtual sources’. Phys Med Biol. 2008;53(6):1619-1635.
290 Weihrauch M, et al. Adaptation of antenna profiles for control of MR guided hyperthermia (HT) in a hybrid MR-HT system. Med Phys. 2007;34(12):4717-4725.
291 Meyer JL, et al. Hyperthermic oncology: current biology, physics and clinical results. Pharmac Ther. 1989;42:251-288.
292 Perez CA, Emami B. Clinical trials with local (external and interstitial) irradiation and hyperthermia. Radiol Clin N Am. 1989;27(3):525-542.
293 Perez CA, et al. Randomized phase III study comparing irradiation and hyperthermia with irradiation alone in superficial measurable tumors. Final report by the Radiation Therapy Oncology Group. Am J Clin Oncol. 1991;14(2):133-141.
294 Shrivastava PN, et al. Hyperthermia thermometry evaluation: Criteria and guidelines. Int J Radiat Oncol Biol Phys. 1988;14:327-335.
295 Valdagni R, Kapp DS, Valdagni C. N3 (TNM-UICC) metastatic neck nodes managed by combined radiation therapy and hyperthermia: Clinical results and analysis of treatment parameters. Int J Hyperthermia. 1986;2(2):189-200.
296 Perez CA, et al. Quality assurance problems in clinical hyperthermia and their impact on therapeutic outcome: a report by the Radiation Therapy Oncology Group. Int J Radiat Oncol Biol Phys. 1989;16:551-558.
297 Myerson RJ, et al. Tumor control in long-term survivors following superficial hyperthermia. Int J Radiat Oncol Biol Phys. 1990;18(5):1123-1129.
298 Myerson RJ, et al. Equilibrium temperature distributions in uniform phantoms for superficial microwave applicators: implications for temperature-based standards of applicator adequacy. Int J Hyperthermia. 1992;8(1):11-21.
299 Oleson JR, et al. Tumor temperature distributions predict hyperthermia effect. Int J Radiat Oncol Biol Phys. 1989;16:559-570.
300 Kapp DS, et al. Parameters predictive for complications of treatment with combined hyperthermia and radiation therapy. Int J Radiat Oncol Biol Phys. 1992;22:998-1008.
301 Dewey WC. Arrhenius relationships from the molecule and cell to the clinic. Int J Hyperthermia. 1994;10(4):457-483.
302 Myerson RJ, et al. Simultaneous superficial hyperthermia and external radiotherapy: report of thermal dosimetry and tolerance to treatment. Int J Hyperthermia. 1999;15(4):251-266.
303 Xu M, et al. Radiosensitization of heat resistant human tumour cells by 1 hour at 41.1 degrees C and its effect on DNA repair. Int J Hyperthermia. 2002;18(5):385-403.
304 Horsman MR, Overgaard J. Can mild hyperthermia improve tumour oxygenation? Int J Hyperthermia. 1997;13(2):141-147.
305 Iwata K, et al. Tumour pO2 can be increased markedly by mild hyperthermia. Br J Cancer. 1996;27(Suppl):S217-S221.
306 Myerson RJ, et al. Monitoring the effect of mild hyperthermia on tumour hypoxia by Cu-ATSM PET scanning. Int J Hyperthermia. 2006;22(2):93-115.
307 Kapp DS, et al. Two or six hyperthermia treatments as an adjunct to radiation therapy yield similar tumor responses: results of a randomized trial. Int J Radiat Oncol Biol Phys. 1990;19(6):1481-1495.
308 Emami B, et al. Combined hyperthermia and irradiation in the treatment of superficial tumors: Results of a prospective randomized trial of hyperthermia fractionation (1/wk vs 2/wk). Int J Radiat Oncol Biol Phys. 1992;24(1):145-152.
309 Leopold KA, et al. Preoperative hyperthermia and radiation for soft tissue sarcomas: Advantage of two vs. one hyperthermia treatments per week. Int J Radiat Oncol Biol Phys. 1989;16:107-115.
310 Arcangeli G, et al. One versus four heat treatments in combination with radiotherapy in metastatic mammary carcinoma. Int J Radiat Oncol Biol Phys. 1991;21(6):1569-1574.
311 Jones EL, et al. A randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol. 2005;23:3079-3085.
312 Myerson RJ, et al. Modelling heat-induced radiosensitization: clinical implications. Int J Hyperthermia. 2004;20(2):201-212.
313 Oleson JR, et al. Tumor temperature distributions predict hyperthermia effect. Int J Radiat Oncol Biol Phys. 1989;16:559-570.
314 Overgaard J. The design of clinical trials. In: Field SB, Franconi C, editors. Hyperthermic oncology: physics and technology of hyperthermia. Amsterdam: Martinus Nijhoff Publishers, 1982.
315 Arcangeli G, et al. Problem of sequence and fractionation in the clinical application of combined heat and radiation. Cancer Res. 1984;44(Suppl 10):4857s-4863s.
316 Bey P, et al. Potentialisation de la radiothérapie par hyperthermie locale. A propos de 90 tumeurs superficielles comparables (abstract). Bull Cancer. 1984;71:263.
317 Dunlop PRC, et al. An assessment of local hyperthermia in clinical practice. Int J Hyperthermia. 1986;2(1):39-50.
318 Hiraoka M, et al. Clinical results of radiofrequency hyperthermia combined with radiation in the treatment of radioresistant cancers. Cancer. 1984;54(12):2898-2904.
319 Howard GCW, et al. Hyperthermia and radiation in the treatment of superficial malignancy: an analysis of treatment parameters, response and toxicity. Int J Hyperthermia. 1987;3(1):1-8.
320 RY Li, et al. Effect of hyperthermia combined with radiation in the treatment of superficial malignant lesion in 90 patients. In: Overgaard J, editor. Hyperthermic oncology 1984, vol. 1. London: Taylor & Francis; 1984:395-397.
321 Lindholm CE, et al. Microwave-induced hyperthermia and radiotherapy in human superficial tumours: Clinical results with a comparative study of combined treatment versus radiotherapy alone. Int J Hyperthermia. 1987;3(5):393-411.
322 Scott RS, et al. Local hyperthermia in combination with definitive radiotherapy: increased tumor clearance, reduced recurrence rate in extended follow-up. Int J Radiat Oncol Biol Phys. 1984;10(11):2119-2123.
323 Steeves RA, et al. Matched-pair analysis of response to local hyperthermia and megavoltage electron therapy for superficial human tumors. Endocurietherapy/Hyperthermia Oncol. 1986;2(4):163-170.
324 Li GC, Hahn GM. A proposed operational model of thermotolerance based on effects of nutrients and the initial treatment temperature. Cancer Res. 1980;40:4501-4508.
325 Arcangeli G, et al. Tumor response to heat and radiation: prognostic variables in the treatment of neck node metastases from head and neck cancer. Int J Hyperthermia. 1985;1(3):207-217.
326 González González D, van Dijk JD, Blank LE. Chestwall recurrences of breast cancer: results of combined treatment with radiation and hyperthermia. Radiother Oncol. 1988;12(2):95-103.
327 Kjellén E, Lindholm CE, Nilsson P. Radiotherapy in combination with hyperthermia in recurrent or metastatic mammary carcinomas. In: Sugahara T, Saito M, editors. Hyperthermic oncology 1988, vol. 2. London: Taylor & Francis; 1989:426-429.
328 Van der Zee J, et al. Thermal enhancement of radiotherapy in breast carcinoma. In: Overgaard J, editor. Hyperthermic Oncology 1984, vol. 1. London: Taylor & Francis; 1984:345-348.
329 Arcangeli G, et al. Radiotherapy and hyperthermia: analysis of clinical results and identification of prognostic variables. Cancer. 1987;60(5):950-956.
330 González González D, et al. Combined treatment with radiation and hyperthermia in metastatic malignant melanoma. Radiother Oncol. 1986;6:105-113.
331 Kim JH, Hahn EW, Ahmed SA. Combination hyperthermia and radiation therapy for malignant melanoma. Cancer. 1982;50(3):478-482.
332 Overgaard J, Overgaard M. Hyperthermia as an adjuvant to radiotherapy in the treatment of malignant melanoma. Int J Hyperthermia. 1987;3(6):483-501.
333 Kapp DS, et al. Stanford University institutional report. Phase I evaluation of equipment for hyperthermia treatment of cancer. Int J Hyperthermia. 1988;4(1):75-115.
334 Dewhirst MW, et al. RTOG quality assurance guidelines for clinical trials using hyperthermia. Int J Radiat Oncol Biol Phys. 1990;18(5):1249-1259.
335 Egawa S, et al. A randomized clinical trial of hyperthermia and radiation versus radiation alone for superficially located cancers. J Jpn Soc Ther Radiol Oncol. 1989;1:135-140.
336 Overgaard J, et al. Thermoradiotherapy of malignant tumors. European randomized multicenter trials evaluating the effect of adjuvant hyperthermia in radiotherapy. In: Kogelnik HD, editor. Progress in Radio-Oncology V. Bologna: Monduzzi Editore; 1995:507-513.
337 Sherar M, et al. Relationship between thermal dose and outcome in thermoradiotherapy treatments for superficial recurrences of breast cancer: data from a phase III trial. Int J Radiat Oncol Biol Phys. 1997;39(2):371-380.
338 Hand JW, et al. Analysis of thermal parameters obtained during phase III trials of hyperthermia as an adjunct to radiotherapy in the treatment of breast carcinoma. Int J Hyperthermia. 1997;13(4):343-364.
339 Overgaard J, et al. Randomised trial of hyperthermia as adjuvant to radiotherapy for recurrent or metastatic malignant melanoma. Lancet. 1995;345:540-543.
340 Emami B, Perez CA. Combination of surgery, irradiation, and hyperthermia in treatment of recurrences of malignant tumors. Int J Radiat Oncol Biol Phys. 1987;13(4):611-613.
341 Kapp DS, et al. Thermoradiotherapy for residual microscopic cancer: elective or post-excisional hyperthermia and radiation therapy in the management of local-regional recurrent breast cancer [see comments]. Int J Radiat Oncol Biol Phys. 1992;24(2):261-277.
342 Sneed PK. Clinical interstitial hyperthermia: where have we been? Where are we going? In: Hagen U, et al, editors. Radiation Research 1895–1995, vol. 2. Würzburg: 10th ICRR Society; 1995:1009-1012.
343 Seegenschmiedt MH, Brady LW, Sauer R. Interstitial thermoradiotherapy: review on technical and clinical aspects. Am J Clin Oncol. 1990;13(4):352-363.
344 Seegenschmiedt MH, et al. Thermoradiotherapy for malignant brain tumors: review of biological and clinical studies. Endocurietherapy/Hyperthermia Oncol. 1995;11(4):201-221.
345 Sneed PK, et al. Thermoradiotherapy of recurrent malignant brain tumors. Int J Radiat Oncol Biol Phys. 1992;23(4):853-861.
346 Sneed PK, Larson DA, Gutin PH. Brachytherapy and hyperthermia for malignant astrocytoma. Semin Oncol. 1994;21(2):186-197.
347 Stea B, et al. Interstitial irradiation versus interstitial thermoradiotherapy for supratentorial malignant gliomas: a comparative survival analysis. Int J Radiat Oncol Biol Phys. 1994;30(3):591-600.
348 Moros EG, et al. Simultaneous delivery of electron beam therapy and ultrasound hyperthermia utilizing scanning reflectors: a feasibility study. Int J Radiat Oncol Biol Phys. 1995;31(4):893-904.
349 Armour EP, et al. Hyperthermic enhancement of high dose-rate irradiation in 9L gliosarcoma cells. Int J Radiat Oncol Biol Phys. 1994;28(1):171-177.
350 Feldmann HJ, et al. Deep regional hyperthermia: comparison between the annular phased array and the Sigma-60 applicator in the same patients. Int J Radiat Oncol Biol Phys. 1993;26(1):111-116.
351 Feldmann HJ, et al. Thermoradiotherapy in locally advanced deep seated tumours—thermal parameters and treatment results. Radiother Oncol. 1993;26(1):38-44.
352 Tilly W, et al. Temperature data and specific absorption rates in pelvic tumours: predictive factors and correlations. Int J Hyperthermia. 2001;17(2):172-188.
353 Fatehi D, et al. RF-power and temperature data analysis of 444 patients with primary cervical cancer: Deep hyperthermia using the Sigma-60 applicator is reproducible. Int J Hyperthermia. 2007;23(8):623-643.
354 Petrovich Z, et al. Regional hyperthermia for advanced tumors: a clinical study of 353 patients. Int J Radiat Oncol Biol Phys. 1989;16(3):601-607.
355 Myerson RJ, et al. A phase I/II study to evaluate radiation therapy and hyperthermia for deep-seated tumours: a report of RTOG 89–08. Int J Hyperthermia. 1996;12(4):449-459.
356 Karasawa K, et al. Thermoradiotherapy in the treatment of locally advanced nonsmall cell lung cancer. Int J Radiat Oncol Biol Phys. 1994;30(5):1171-1177.
357 Hiraoka M, et al. Regional hyperthermia combined with radiotherapy in the treatment of lung cancers. Int J Radiat Oncol Biol Phys. 1992;22(5):1009-1014.
358 Sullivan DM, Ben-Yosef R, Kapp DS. Stanford 3-D hyperthermia treatment planning system. Int J Hyperthermia. 1993;9(5):627-643.
359 Lagendijk JJW, et al. ESHO quality assurance guidelines for regional hyperthermia. Int J Hyperthermia. 1998;14(2):125-133.
360 Sapozink MD, et al. RTOG quality assurance guidelines for clinical trials using hyperthermia for deep-seated malignancy. Int J Radiat Oncol Biol Phys. 1991;20:1109-1115.
361 Van der Zee J, González González D. The Dutch Deep Hyperthermia Trial: results in cervical cancer. Int J Hyperthermia. 2002;18(1):1-12.
362 Harima Y, et al. A randomized clinical trial of radiation therapy versus thermoradiotherapy in stage IIIB cervical cancer. Int J Hyperthermia. 2001;17(2):97-105.
363 Vasanthan A, et al. Regional hyperthermia combined with radiotherapy for uterine cervical cancers: a multi-institutional prospective randomized trial of the international atomic energy agency.[see comment]. Int J Radiat Oncol Biol Phys. 2005;61(1):145-153.
364 Robins HI, Neville AJ, Cohen JD. Whole Body Hyperthermia: biological, physical and clinical aspects. Berlin: Springer-Verlag; 1992.
365 Kraybill WG, et al. A phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: Correlation with mouse models. Int J Hyperthermia. 2002;18(3):253-266.
366 Ostberg JR, Repasky EA. Emerging evidence indicates that physiologically relevant thermal stress regulates dendritic cell function. Cancer Immunol Immunother. 2006;55(3):292-298.
367 Vernon CC, et al. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: results from five randomized controlled trials. Int J Radiat Oncol Biol Phys. 1996;35(4):731-744.
368 Overgaard J, et al. Thermoradiotherapy of malignant tumors. European randomized multicenter trials evaluating the effect of adjuvant hyperthermia in radiotherapy. In: Kogelnik HD, editor. Progress in radio-oncology V. Bologna: Monduzzi Editore; 1995:507-513.
369 Van der Zee J, et al. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Lancet. 2000;355:1119-1125.
370 Sharma S, et al. Local thermo-radiotherapy in carcinoma cervix: improved local control versus increased incidence of distant metastases. Asia Oceania J Obstet Gynaecol. 1991;17(1):5-12.
371 Berdov BA, Menteshashvili GZ. Thermoradiotherapy of patients with locally advanced carcinoma of the rectum. Int J Hyperthermia. 1990;6(5):881-890.
372 Overgaard J: Personal communication. 1995.
373 Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia. 2001;17(1):1-18.
374 Bull JMC, et al. Chemotherapy resistant sarcoma treated with whole body hyperthermia (WBH) combined with 1-3-Bis(2-chloroethyl)-1-nitrosourea (BCNU). Int J Hyperthermia. 1992;8(3):297-304.
375 Engelhardt R, et al. Preliminary results in the treatment of oat cell carcinoma of the lung by combined application of chemotherapy (CT) and whole-body hyperthermia. Prog Clin Biol Res. 1982;107:761-765.
376 Engelhardt R, et al. Treatment of disseminated malignant melanoma with cisplatin in combination with whole-body hyperthermia and doxorubicin. Int J Hyperthermia. 1990;6(3):511-515.
377 Leopold KA, et al. Intraperitoneal cisplatin and regional hyperthermia for ovarian carcinoma. Int J Radiat Oncol Biol Phys. 1993;27(5):1245-1251.
378 Westermann AM, et al. A pilot study of whole body hyperthermia and carboplatin in platinum-resistant ovarian cancer. Eur J Cancer. 2001;37(9):1111-1117.
379 Sugimachi K, et al. Chemotherapy combined with or without hyperthermia for patients with oesophageal carcinoma: a prospective randomized trial. Int J Hyperthermia. 1994;10(4):485-493.
380 Issels RD, Schlemmer M. Current trials and new aspects in soft tissue sarcoma of adults. Cancer Chemother Pharmacol. 2002;49(Suppl 1):S4-S8.
381 Ghussen F, et al. Hyperthermic perfusion with chemotherapy for melanoma of the extremities. World J Surg. 1989;13(5):598-602.
382 Koops HS, et al. Prophylactic isolated limb perfusion for localized, high-risk limb melanoma: results of a multicenter randomized phase III trial. J Clin Oncol. 1998;16(9):2906-2912.
383 Park JW. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res. 2002;4(3):95-99.
384 Park JW, et al: Hyperthermia + doxil significantly enhances drug delivery and efficacy in metastatic breast cancer of the chest wall: A phase I/II study. In proceedings of the 37th annual meeting of American Society of Clinical Oncology, San Francisco, 2001.
385 Park JW, et al: Hyperthermia significantly enhances drug delivery and efficacy of pegylated liposomal doxorubicin in metastatic breast cancer of the chest wall: A phase I/II study. Paper presented at the forty-ninth annual meeting of the Radiation Research Society and the twentieth annual meeting of the North American Hyperthermia Society. Reno, Nevada, 2002.
386 Bull JMC, et al: A phase I-II clinical trial of fever-range whole-body hyperthermia combined with liposomal doxorubicin (Doxil), 5-fluorouracil (5-FU), & metronomic interferon-a (IFN-a): an interim report. Paper presented at the forty-ninth annual meeting of the Radiation Research Society and the twentieth annual meeting of the North American Hyperthermia Society. Reno, Nevada, 2002.
387 Kitamura K, et al. Prospective randomized study of hyperthermia combined with chemoradiotherapy for esophageal carcinoma. J Surg Oncol. 1995;60(1):55-58.
388 Kouloulias VE, et al. Liposomal doxorubicin in conjunction with reirradiation and local hyperthermia treatment in recurrent breast cancer: a phase I/II trial. Clin Cancer Res. 2002;8(2):374-382.
389 Colombo R, et al. Multicentric study comparing intravesical chemotherapy alone and with local microwave hyperthermia for prophylaxis of recurrence of superficial transitional cell carcinoma [see comment]. J Clin Oncol. 2003;21(23):4270-4276.
390 Janetzki S, et al. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96: A pilot study. Int J Cancer. 2000;88(2):232-238.
391 Amato RJ, et al: Active specific immunotherapy in patients with renal cell carcinoma (RCC) using autologous tumor derived heat shock protein-peptide complex-96 (HSPPC-96) vaccine (abstract). Paper presented at the thirty-fifth annual meeting of the American Society of Clinical Oncology, Atlanta, Georgia, 1999.
392 Eton O, et al: Autologous tumor-derived heat-shock protein peptide complex-96 (HSPPC-96) in patients (PTS) with metastatic melanoma, 2000, in American Association of Cancer Research.
393 Caudill MM, Li Z. HSPPC-96: A personalised cancer vaccine. Expert Opin Biol Ther. 2001;1(3):539-547.
394 Manjili MH, et al. Immunotherapy of cancer using heat shock proteins. Front Biosci. 2002;7:d43-d52.
395 Srivastava PK, et al. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity. 1998;8(6):657-665.
396 Blachere NE, Srivastava PK. Heat shock protein-based cancer vaccines and related thoughts on immunogenicity of human tumors. Semin Cancer Biol. 1995;6(6):349-355.
397 Nicchitta CV. Biochemical, cell biological and immunological issues surrounding the endoplasmic reticulum chaperone GRP94/gp96. Curr Opin Immunol. 1998;10(1):103-109.
398 Przepiorka D, Srivastava PK. Heat shock protein-peptide complexes as immunotherapy for human cancer. Mol Med Today. 1998;4(11):478-484.
399 Basu S, Srivastava PK. Heat shock proteins: the fountainhead of innate and adaptive immune responses. Cell Stress Chaperones. 2000;5(5):443-451.
400 Denman DL, et al. Therapeutic responses of spontaneous canine malignancies to combinations of radiotherapy and hyperthermia. Int J Radiat Oncol Biol Phys. 1991;21(2):415-422.
401 Gillette EL, et al. Response of canine oral carcinomas to heat and radiation. Int J Radiat Oncol Biol Phys. 1987;13(12):1861-1867.
402 Gillette SM, et al. Response of canine soft tissue sarcomas to radiation or radiation plus hyperthermia: a randomized phase II study. Int J Hyperthermia. 1992;8(3):309-320.
403 Frew D, et al. Response of cannice soft tissue sarcomas to radiation or radiation plus hyperthermia: results of a randomized phase III clinical study. Int J Hyperthermia. 1995;11:217.
404 Thrall DE, et al. Use of whole body hyperthermia as a method to heat inaccessible tumours uniformly: a phase III trial in canine brain masses. Int J Hyperthermia. 1999;15(5):383-398.
405 Thrall DE, et al. Radiation plus local hyperthermia versus radiation plus the combination of local and whole-body hyperthermia in canine sarcomas. Int J Radiat Oncol Biol Phys. 1996;34(5):1087-1096.
406 Novotney CA, et al. Phase I evaluation of doxorubicin and whole-body hyperthermia in dogs with lymphoma. J Vet Intern Med. 1992;6(4):245-249.
407 Page RL, et al. Phase III evaluation of doxorubicin and whole-body hyperthermia in dogs with lymphoma. Int J Hyperthermia. 1992;8(2):187-197.
408 Oleson JR. Hyperthermia from the clinic to the laboratory: a hypothesis. Int J Hyperthermia. 1995;11(3):315-322.
409 Raymond U, et al. Microwave-induced local hyperthermia in combination with radiotherapy of human malignant tumors. Cancer. 1980;45(4):638-646.
410 González González D, van Dijk JDP, Blank LECM. Chestwall recurrences of breast cancer: results of combined treatment with radiation and hyperthermia. Radiother Oncol. 1988;12(2):95-103.
411 González González D, et al. Combined treatment with radiation and hyperthermia in metastatic malignant melanoma. Radiother Oncol. 1986;6(2):105-113.