[level-membership-for-allergy-and-immunology-category]
Chapter 25 Hypersensitivity (Type III)
• Immune complexes are formed when antibody meets antigen. They are removed by the mononuclear phagocyte system following complement activation. Persistence of antigen from chronic infection or in autoimmune disease can lead to immune complex disease.
• Immune complexes can trigger a variety of inflammatory processes. Fc–FcR interactions are the key mediators of inflammation. Most importantly, Fc regions within immune deposits within tissues engage Fc receptors on activated neutrophils, lymphocytes, and platelets to induce inflammation. During chronic inflammation B cells and macrophages are the predominant infiltrating cell type, and activation of endogenous cells within the organ participates in fibrosis and disease progression.
• Experimental models demonstrate the main immune complex diseases. Serum sickness can be induced with large injections of foreign antigen. Autoimmunity causes immune complex disease in the NZB/NZW mouse. Injection of antigen into the skin of presensitized animals produces the Arthus reaction.
• Immune complexes are normally removed by the mononuclear phagocyte system. Complement helps to disrupt antigen–antibody bonds and keeps immune complexes soluble. Primate erythrocytes bear a receptor for C3b and are important for transporting complement-containing immune complexes to the spleen for removal. Complement deficiencies lead to the formation of large, relatively insoluble complexes, which deposit in tissues.
• The size of immune complexes affects their deposition. Deposition of circulating, soluble immune complexes is limited by physical factors, such as the size and charge of the complexes. Small, positively charged complexes have the greatest propensity for deposition within vessels. Large immune complexes are rapidly removed in the liver and spleen.
• Immune complex deposition in the tissues results in tissue damage. Immune complexes can form both in the circulation, leading to systemic disease, and at local sites such as the lung. Charged cationic antigens have tissue-binding properties, particularly for the glomerulus, and help to localize complexes to the kidney. Factors that tend to increase blood vessel permeability enhance the deposition of immune complexes in tissues.
Immune complex diseases
Immune complex formation can result from:
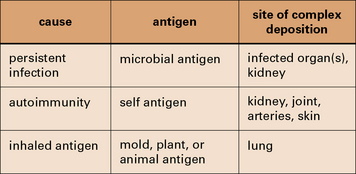
Fig. 25.1 Three categories of immune complex disease
This table indicates the source of the antigen and the organs most frequently affected.
Persistent infection with a weak antibody response can lead to immune complex disease
The combined effects of a low-grade persistent infection and a weak antibody response lead to chronic immune complex formation, and eventual deposition of complexes in the tissues (Fig. 25.2). Diseases with this etiology include:
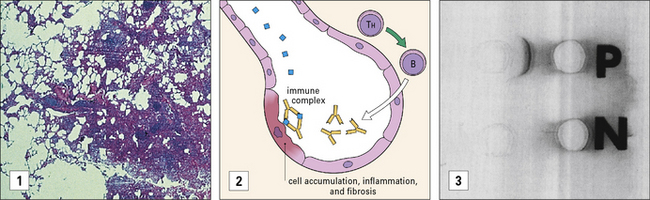
Immune complexes can be formed with inhaled antigens
Immune complexes may be formed at body surfaces following exposure to extrinsic antigens.
• farmer’s lung, where there are circulating antibodies to actinomycete fungi (found in moldy hay); and
• pigeon fancier’s lung, where there are circulating antibodies to pigeon antigens.
Both diseases are forms of extrinsic allergic alveolitis, and occur only after repeated exposure to the antigen. Note that the antibodies induced by these antigens are primarily IgG, rather than the IgE seen in type I hypersensitivity reactions. When antigen again enters the body by inhalation, local immune complexes are formed in the alveoli leading to inflammation and fibrosis (Fig. 25.3).
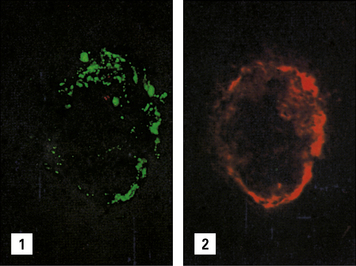
Immune complex disease occurs in autoimmune rheumatic disorders
Immune complex disease is common in autoimmune disease, where the continued production of autoantibody to a self antigen leads to prolonged immune complex formation. As the number of complexes in the blood increases, the systems responsible for the removal of complexes (mononuclear phagocyte, erythrocyte, and complement) become overloaded, and complexes are deposited in the tissues (see Fig. 25.16). Systemic lupus erythematosus (SLE) is the classic disease characterized by immune complex deposition and others include Henoch-Schönlein purpura and primary Sjögren’s syndrome.
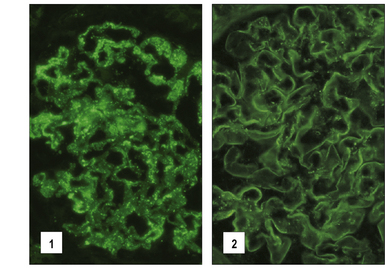
Fig. 25.16 Immunofluorescence study of immune complexes in autoimmune disease
(Courtesy of Dr S Thiru.)
Immune complexes and inflammation
Immune complexes are capable of triggering a wide variety of inflammatory processes:
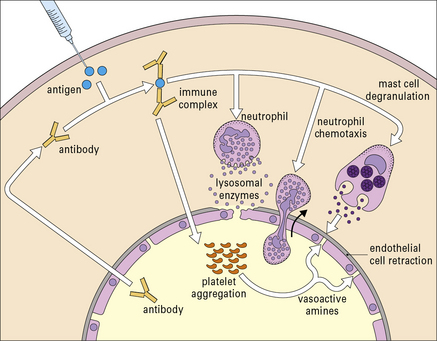
• they interact directly with basophils and platelets (via Fc receptors) to induce the release of vasoactive amines (Fig. 25.4);
• macrophages are stimulated to release cytokines, particularly tumor necrosis factor-α (TNFα) and interleukin-1 (IL-1), which have important roles in inflammation;
• they interact with the complement system to generate C3a and C5a, which stimulate the release of vasoactive amines (including histamine and 5-hydroxytryptamine) and chemotactic factors from mast cells and basophils; C5a is also chemotactic for basophils, eosinophils, and neutrophils.
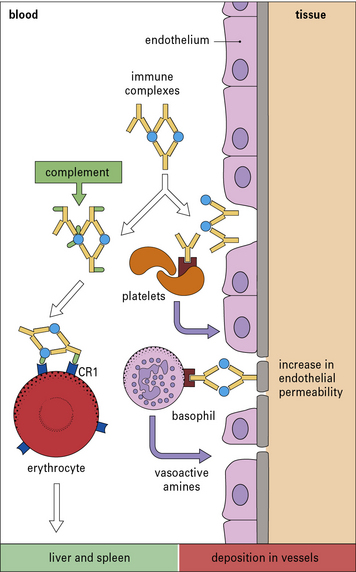
The vasoactive amines released by platelets, basophils, and mast cells cause endothelial cell retraction and thus increase vascular permeability, allowing the deposition of immune complexes on the blood vessel wall (Fig. 25.5). The deposited complexes continue to generate C3a and C5a.
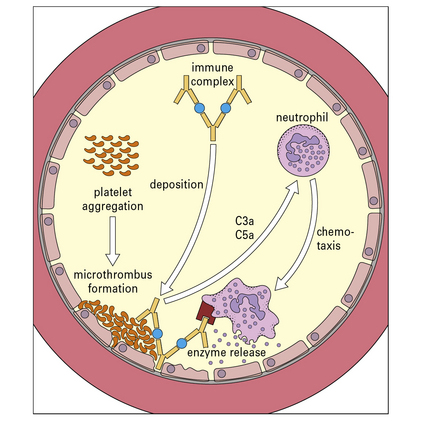
Polymorphs are chemotactically attracted to the site by C5a. They attempt to engulf the deposited immune complexes, but are unable to do so because the complexes are bound to the vessel wall. Therefore they exocytose their lysosomal enzymes onto the site of deposition (see Fig. 25.5). If simply released into the blood or tissue fluids these lysosomal enzymes are unlikely to cause much inflammation, because they are rapidly neutralized by serum enzyme inhibitors. But if the phagocyte applies itself closely to the tissue-trapped complexes through Fc binding, then serum inhibitors are excluded and the enzymes may damage the underlying tissue.
Complement is an important mediator of immune complex disease
• the kidney in various autoimmune glomerular diseases; and
• the skin in autoimmune diseases where cutaneous vasculitis is a feature such as SLE, Sjögren’s syndrome and Henoch–Schönlein purpura.
 be found in SLE.
be found in SLE.Experimental models of immune complex diseases
Experimental models are available for the main types of immune complex disease described above:
• serum sickness, induced by injections of foreign antigen, mimics the effect of a persistent infection;
• the NZB/NZW mouse demonstrates autoimmunity;
• the Arthus reaction is an example of local damage by extrinsic antigen.
Serum sickness can be induced with large injections of foreign antigen
Serum sickness is now commonly studied in rabbits by giving them an intravenous injection of a foreign soluble protein such as bovine serum albumin (BSA). After about 1 week antibodies are formed, which enter the circulation and complex with antigen. Because the reaction occurs in antigen excess, the immune complexes are small (Fig. 25.w1). These small complexes are removed only slowly by the mononuclear phagocyte system and therefore persist in the circulation.
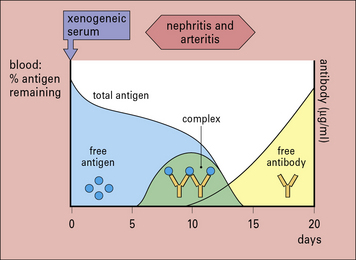
The formation of complexes is followed by an abrupt fall in total hemolytic complement.
Autoimmunity causes immune complex disease in the NZB/NZW mouse
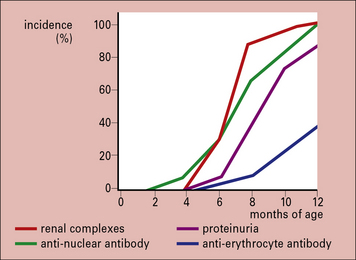
The F1 hybrid NZB/NZW mouse produces a range of autoantibodies (including anti-erythrocyte, anti-nuclear, anti-DNA, and anti-Sm) and suffers from an immune complex disease similar in many ways to SLE in humans. A NZB/NZW mouse is born clinically normal, but within 2–3 months shows sign of hemolytic anemia. Tests for anti-erythrocyte antibody (the Coombs’ test), anti-nuclear antibodies, lupus cells, and circulating immune complexes are all positive, and there are deposits in the glomeruli and choroid plexus of the brain. The disease is much more marked in the females, who die within a few months of developing symptoms (Fig. 25.w2).
Injection of antigen into the skin of pre-sensitized animals produces the Arthus reaction
An animal is immunized repeatedly until it has appreciable levels of serum antibody (mainly IgG). Following subcutaneous or intradermal injection of the antigen a reaction develops at the injection site, sometimes with marked edema and hemorrhage, depending on the amount of antigen injected. The reaction reaches a peak after 4–10 hours, then wanes and is usually minimal by 48 hours (Fig. 25.w3).
Immunofluorescence studies have shown that initial deposition of antigen, antibody, and complement in the vessel wall is followed by neutrophil infiltration and intravascular clumping of platelets (Fig. 25.w4). This platelet reaction can lead to vascular occlusion and necrosis in severe cases. After 24–48 hours the neutrophils are replaced by mononuclear cells, and eventually some plasma cells appear.
TNFα enhances cell-mediated immune responses in various ways (see Chapter 9). Treatment with antibodies to TNF can reduce severity in the Arthus reaction and, interestingly, anti-TNF therapy has become a highly effective disease modifying agent in treating rheumatoid arthritis.
Immune complexes clearance by the mononuclear phagocyte system
CR1 readily binds immune complexes that have fixed complement, as has been shown by experiments with animals lacking complement (Fig. 25.6).
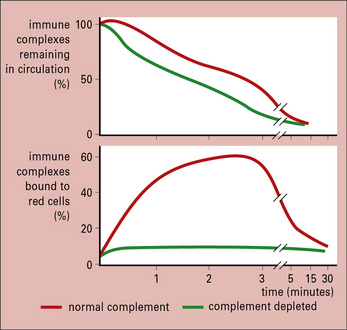
Fig. 25.6 Effects of complement depletion on handling of immune complexes
(Based on data from Waxman FJ, et al. J Clin Invest 1984;74:1329–1340.)
The complexes are transported to the liver and spleen, where they are removed by fixed tissue macrophages (Fig. 25.7). Most of the CR1 is also removed in the process, so in situations of continuous immune complex formation the number of active receptors falls rapidly, impairing the efficiency of immune complex handling.
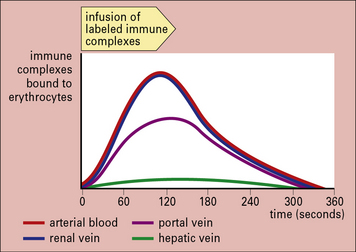
Fig. 25.7 Clearance of immune complexes in the liver
(Based on data from Cornacoff JB, et al. J Clin Invest 1983;71:236–247.)
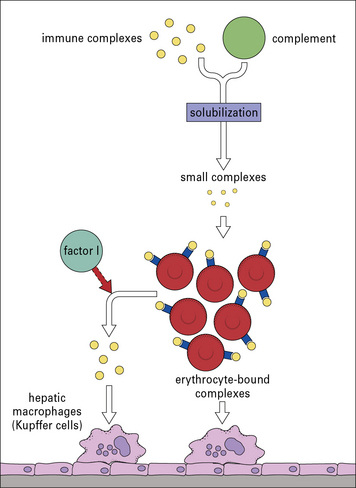
This action leaves a small fragment (C3dg) attached to the CR1 on the cell membrane. These soluble complexes are then removed by phagocytic cells, particularly those in the liver, bearing receptors for IgG Fc (Fig. 25.8).
Complement solubilization of immune complexes
Complement deficiency impairs clearance of complexes
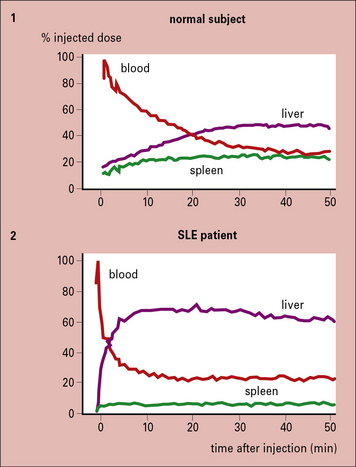
This might be expected to result in persistent immune complexes in the circulation, but in fact the reverse occurs, with the complexes disappearing rapidly from the circulation. These non-erythrocyte-bound complexes are taken up rapidly by the liver (but not the spleen) and are then released to be deposited in tissues such as skin, kidney, and muscle, where they can set up inflammatory reactions (Fig. 25.9).
Failure to localize in the spleen not only results in immune complex disease, but may also have important implications for the development of appropriate immune responses. This is because the spleen plays a vital role in antigen processing and the induction of immune responses (see Chapter 2).
The size of immune complexes affects their deposition
In general, larger immune complexes are rapidly removed by the liver within a few minutes, whereas smaller complexes circulate for longer periods (Fig. 25.10). This is because larger complexes are:
• more effective at binding to Fc receptors and at fixing complement, so binding better to erythrocytes;
• released more slowly from the erythrocytes by the action of factor I.
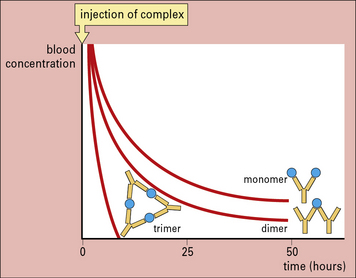
Anything that affects the size of complexes is therefore likely to influence clearance.
Immunoglobulin classes affect the rate of immune complex clearance
• IgG complexes are bound by erythrocytes and are gradually removed from the circulation;
• IgA complexes bind poorly to erythrocytes, but disappear rapidly from the circulation, with increased deposition in the kidney, lung, and brain.
Q. Provide an explanation for the different patterns of localization of immune complexes containing IgG and those containing IgA
A. IgG-containing immune complexes activate the complement classical pathway and can bind to CR1 on erythrocytes. IgA does not activate the classical pathway, but can bind to Fcα receptors on mononuclear phagocytes (see Fig. 3.15).
Immune complex deposition in tissues
Two questions are relevant to tissue deposition:
• Why are complexes deposited?
• Why do complexes show affinity for particular tissues in different diseases?
The most important trigger for immune complex deposition is probably an increase in vascular permeability
In studies of experimental immune complex disease in rabbits, long-term administration of vasoactive amine antagonists, such as chlorpheniramine and methysergide, has been shown to reduce immune complex deposition considerably (Fig. 25.11). More importantly young NZB/NZW mice which normally develop proteinuria by 9 months old, have less renal pathology, when treated with methysergide. Methysergide blocks the formation of the vasoactive amine 5-hydroxytryptamine (5-HT), and thus blocks a variety of inflammatory events (e.g. deposition of complexes, neutrophil infiltration of capillary walls, and endothelial proliferation), all of which produce the glomerular pathology.
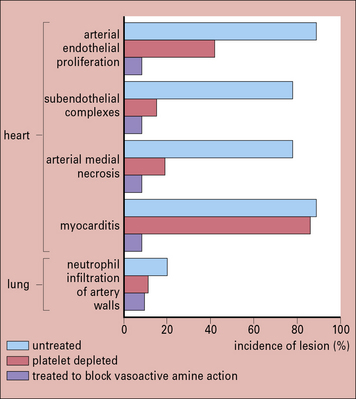
Immune complex deposition is most likely where there is high blood pressure and turbulence
Many macromolecules deposit in the glomerular capillaries, where the blood pressure is approximately four times that of most other capillaries (Fig. 25.12).
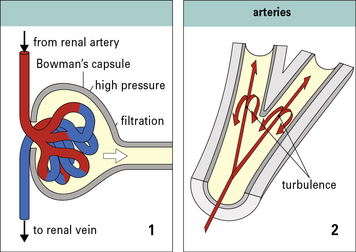
• at turns or bifurcations of arteries,
• in vascular filters such as the choroid plexus and the ciliary body of the eye.
Q. Why should the site of bifurcation in an artery be more susceptible to damage than other sites?
A. This site is subject to high pressure and more erratic shear forces, which may affect the integrity of the endothelium (see Fig. 25.12). In addition blood cells and platelets are not segregated from the vessel wall by laminar flow.
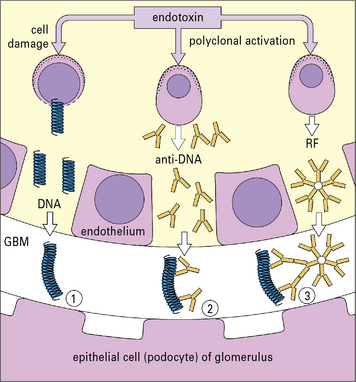
Affinity of antigens for specific tissues can direct complexes to particular sites
It is possible that the antigen in the complex provides the organ specificity, and a convincing model has been established to support this hypothesis. In the model, mice are given endotoxin causing cell damage and release of DNA, which then binds to healthy glomerular basement membrane. Anti-DNA is then produced by polyclonal activation of B cells, and is bound by the fixed DNA leading to local immune complex formation (Fig. 25.13). The production of rheumatoid factor (IgM anti-IgG) allows further immune complex formation to occur in situ.
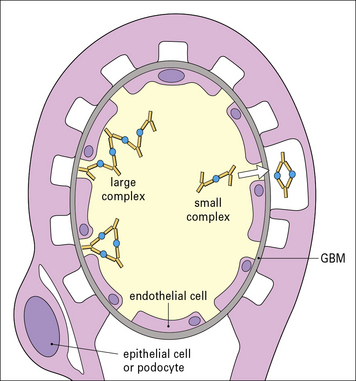
The site of immune complex deposition depends partly on the size of the complex
• small immune complexes can pass through the glomerular basement membrane, and end up on the epithelial side of the membrane;
• large complexes are unable to cross the membrane and generally accumulate between the endothelium and the basement membrane or the mesangium (Fig. 25.14).
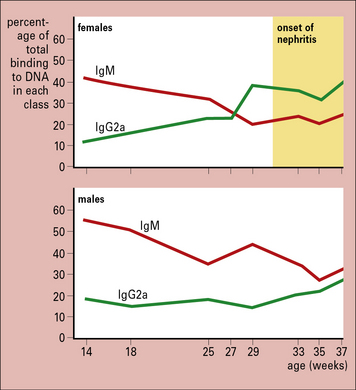
The class of immunoglobulin in an immune complex can influence deposition
Similarly, as NZB/NZW mice grow older there is a class switch, from predominantly IgM to IgG2a. This occurs earlier in females than in males and coincides with the onset of renal disease, indicating the importance of antibody class in the tissue deposition of complexes (Fig. 25.15).
Diagnosis of immune complex disease
The ideal place to look for immune complexes is in the affected organ (Figs. 25.2, 25.16).
• patients with the continuous, granular, subepithelial deposits of IgG found in membranous glomerulonephritis have a poor prognosis with prolonged heavy proteinuria;
• in contrast, those whose complexes are localized in the mesangium have a good prognosis and respond to immunosuppressive therapies.
Assays for immune complexes in serum are more readily performed than in-situ immunofluorescence, although the results have to be interpreted carefully (see Method box 25.1  ).
).
Method box 25.1 Assays for circulating immune complexes
Circulating complexes are found in two separate compartments:
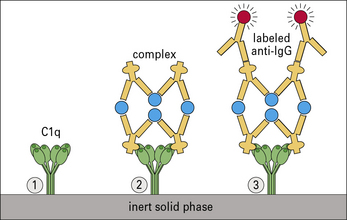
Circulating complexes are often identified by their affinity for complement C1q, using either radiolabeled C1q or solid-phase C1q. Complexes can also be detected by their low solubility in polyethylene glycol (Figs MB25.1.1 and MB25.1.2).
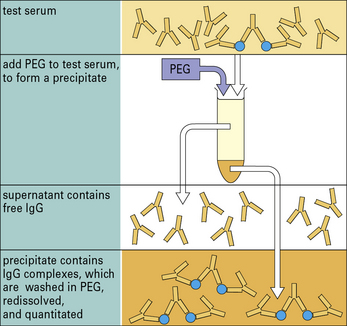
Polyethylene glycol (PEG) is added to the test serum containing IgG complexes and IgG monomer. When the concentration of PEG reaches 2%, complexes are selectively precipitated; the free antibody remains in solution. The test tube is then centrifuged and the complexes form a pellet at the bottom. The supernatant containing free antibody is removed. The precipitate is washed and redissolved so that the amount of complexed IgG can be measured (e.g. by single radial immunodiffusion, nephelometry, or radioimmunoassay (see Fig. MB25.1.2).
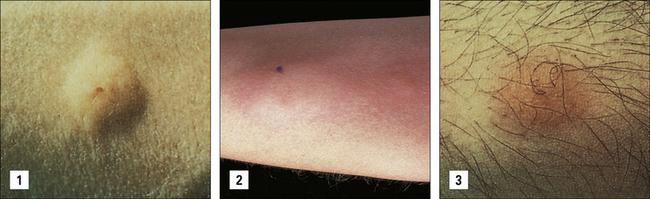
A 53-year-old woman presented with an inflammatory arthritis with a symmetrical pattern affecting her hands, wrists, knees and feet. She developed a pupuric rash on her lower limbs which was worse after exercise and especially on exposure to cold weather. This was followed by sensations of numbness and tingling in both legs below the knees (Fig. 25.w5).
Clinical examination showed a low grade inflammatory arthritis with synovitis affecting the metacarpophalangeal joints, wrists, knees and metatarsophalangeal joints. She had bilateral knee effusions. There was an extensive rash consisting of palpable purpura on her legs and there was a stocking distribution of sensory loss to the level of the knees with absent ankle reflexes (Fig. 25.w6). There was mild bilateral parotid enlargement and dry mouth and dry eyes were noted with a dry Schirmer’s tear test.

Critical thinking: Type III serum sickness after factor IX administration (see p. 444 for explanations)
An 8-year-old boy with factor IX deficiency has had repeated episodes of bleeding into his joints and skin, despite requiring administration of factor IX. Ten days after receiving a dose, he developed fever, swelling of multiple joints, and a skin rash. On physical exam, his temperature was 39 °C, he had a diffuse maculopapular skin rash involving his torso and extremities, and both elbows and knees were red, warm, and appeared inflamed. His mother thought the appearance and distribution were very different from the typical appearance after either minor trauma or bleeding into his joints, which he had sustained on multiple previous occasions. His pediatrician ordered the following tests (results shown in Table 25.1) and prescribed a short course of corticosteroids.
1. What immunologic mechanisms are involved in this inflammatory reaction after the boy received the factor IX?
2. Why were corticosteroids prescribed?
3. What is the likelihood that this type of reaction will develop again?
4. What measures would you take to prevent this reaction from occurring again?
| Variable | Result (normal range) |
|---|---|
| C3 (mg/dL) | 38 (85–155) |
| C4 (mg/dL) | 4 (12–45) |
| anti-nuclear antibody | Negative |
| hemoglobin (g/dL) | 11.2 |
| white cell count (cells/mm3) | 11 000 |
| eosinophils (%) | 1 |
On physical exam, his blood pressure is elevated at 140/90 mmHg and his ankles are very edematous. His joints do not appear inflamed and the skin does not show either evidence of recent bleeding or inflammation. Results of tests are shown in Table 25.2.
5. What immunologic mechanisms are involved in this inflammatory reaction after the boy received the factor IX? How do they differ from the previous episode?
6. What is the likelihood that this type of reaction will develop again?
7. What measures would you take to prevent this reaction again?
| Variable | Result (normal range) |
|---|---|
| C3 (mg/dL) | 142 (85–155) |
| C4 (mg/dL) | 44 (12–45) |
| anti-nuclear antibody | Negative |
| hemoglobin (g/dL) | 11.6 |
| white cell count (cells/mm3) | 8600 |
| eosinophils (%) | < 1 |
| albumin (g/dL) | 2.5 (3.5–5.5) |
| urine protein (g/24 h) | 8 (< 0.2) |
Agnello V. Immune complex assays in rheumatic diseases. Hum Pathol. 1983;14:343–349.
Arthus M. Injections répétées de sérum de cheval chez le lapin. C R Seances Soc Biol Fil. 1903;55:817.
Birmingham D.J., Herbert L.A., Cosio F.G., et al. Immune complex erythrocyte complement receptor interactions in vivo during induction of glomerulonephritis in non-human primates. J Lab Clin Med. 1990;116:242–252.
Boackle S.A., Holer V.M., Karp D.R. CD21 augments antigen presentation in immune individuals. Eur J Immunol. 1997;27:122–129.
Boruchov A.M., Heller G., Veri M.C., et al. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest. 2005;115:2914–2923.
Bruhns P., Samuelsson A., Pollard J.W., Ravetch J.V. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity. 2003;18:573–581.
Clynes R., Maizes J.S., Guinamard R., et al. Modulation of immune complex-induced inflammation in vivo by the co-ordinate expression of activation and inhibitory Fc receptors. J Exp Med. 1999;189:179–185.
Cornacoff J.B., Hebert L.A., Smead W.L., et al. Primate erythrocyte immune complex clearing mechanism. J Clin Invest. 1983;71:236–247.
Czop J., Nussenzweig V. Studies on the mechanism of solubilization of immune precipitates by serum. J Exp Med. 1976;143:615–630.
Davies K.A., Hird V., Stewart S., et al. A study of in vivo immune complex formation and clearing in man. J Immunol. 1990;144:4613–4620.
Davies K.A., Peters A.M., Beynon H.L.C., Walport M.J. Immune complex processing in patients with systemic lupus erythematosus – in vivo imaging and clearance studies. J Clin Invest. 1992;90:2075–2083.
Davies K.A., Chapman P.T., Norsworthy P.J., et al. Clearance pathway of soluble immune complexes in the pig. Insights into the adaptive nature of antigen clearance in humans. J Immunol. 1995;155:5760–5768.
Davies K.A., Schifferli J.A., Walport M.J. Complement deficiency and immune complex diseases. Springer Semin Immunopathol. 1994;15:397–416.
Dixon F.J., Joseph D., Feldman J.D., et al. Experimental glomerulonephritis: the pathogenesis of a laboratory model resembling the spectrum of human glomerulonephritis. J Exp Med. 1961;113:899–919.
Dixon F.J., Vazquez J.J., Weigle W.O., et al. Pathogenesis of serum sickness. Arch Pathol. 1958;65:18–28.
Emlen W., Carl V., Burdick C.G. Mechanism of transfer of immune complexes from red blood cell CR1 to monocytes. Clin Exp Immunol. 1992;89:8–17.
Finbloom D.S., Magilvary D.B., Harford J.B., et al. Influence of antigen on immune complex behaviour in mice. J Clin Invest. 1981;68:214–224.
Fukuyama H., Nimmerjahn F., Ravetch J.V. The inhibitory Fc gamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G + anti-DNA plasma cells. Nat Immunol. 2005;6:99–106.
Heidelberger M. Quantitative chemical studies on complement or alexin. J Exp Med. 1941;73:681–709.
Inman R.D. Immune complexes in SLE. Clin Rheum Dis. 1982;8:49–62.
Johnston A., Auda G.R., Kerr M.A., et al. Dissociation of primary antigen–antibody bonds is essential for complement mediated solubilization of immune complexes. Mol Immunol. 1992;29:659–665.
Kijlstrea H., Van Es L.A., Daha M.R. The role of complement in the binding and degradation of immunoglobulin aggregates by macrophages. J Immunol. 1979;123:2488–2493.
Lachmann P.J. Complement deficiency and the pathogenesis of autoimmune complex disease. Chem Immunol. 1980;49:245–263.
Lucisano Valim M., Lachmann P.J. The effects of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimaeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol. 1991;84:1–8.
McGaha T.L., Sorrentino B., Ravetch J.V. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307:590–593.
McKenzie S.E., Taylor S.M., Malladi P., et al. The role of the human Fc receptor FcγRIIA in the immune clearance of platelets: a transgene mouse model. J Immunol. 1999;162:4311–4318.
Miller G.W., Nussenzweig V. A new complement function: solubilization of antigen–antibody aggregates. Proc Natl Acad Sci USA. 1975;72:418–422.
Muñoz L.E., Lauber K., Schiller M., et al. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289.
Moll T., Nitschke L., Carroll M., et al. A critical role for Fc gamma RIIB in the induction of rheumatoid factors. J Immunol. 2004;173:4724–4728.
Olsson M., Bruhns P., Frazier W.A., et al. Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood. 2005;105:3577–3582.
Park S.Y., Ueda S., Ohno H., et al. Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238.
Qiao J.-H., Castellani L.W., Fishbein M.C., et al. Immune complex-mediated vasculitis increases coronary artery lipid accumulation in autoimmune-prone MRL mice. Arterioscler Thromb. 1993;13:932–943.
Ravetch J.V. Fc receptors. Curr Opin Immunol. 1997;9:121–125.
Ravetch J.V. A full complement of receptors in immune complex diseases. J Clin Invest. 2002;110:1759–1761.
Schifferli J.A., Ng Y.C., Peters D.K. The role of complement and its receptor in the elimination of immune complexes. N Engl J Med. 1986;315:488–495.
Sylvestre D.L., Ravetch J.V. A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity. 1996;5:387–390.
Takata Y., Tamura N., Fujita T. Interaction of C3 with antigen–antibody complexes in the process of solubilisation of immune precipitates. J Immunol. 1984;132:2531–2537.
Terino F.L., Powell M.S., McKenzie I.F., Hogarth P.M. Recombinant soluble human FcγRII: production, characterization, and inhibition of the Arthus reaction. J Exp Med. 1993;178:1617–1628.
Theofilopoulos A.N., Dixon F.J. The biology and detection of immune complexes. Adv Immunol. 1979;28:89–220.
Warren J.S., Yabroff K.R., Remick D.G., et al. Tumour necrosis factor participates in the pathogenesis of acute immune complex alveolitis in the rat. J Clin Invest. 1989;84:1873–1882.
Waxman F.J., Hebert L.E., Cornacoff J.B., et al. Complement depletion accelerates the clearance of immune complexes from the circulation of primates. J Clin Invest. 1984;74:1329–1340.
Whaley K. Complement and immune complex diseases. In: Whaley K., ed. Complement in health and disease. Lancaster: MTP Press Ltd, 1987.
Williams R.C. Immune complexes in clinical and experimental medicine. Massachusetts: Harvard University Press; 1980.
Technical report 606World Health Organization Scientific Group. The role of immune complexes in disease. Geneva: WHO, 1977.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 25 Hypersensitivity (Type III)
• Immune complexes are formed when antibody meets antigen. They are removed by the mononuclear phagocyte system following complement activation. Persistence of antigen from chronic infection or in autoimmune disease can lead to immune complex disease.
• Immune complexes can trigger a variety of inflammatory processes. Fc–FcR interactions are the key mediators of inflammation. Most importantly, Fc regions within immune deposits within tissues engage Fc receptors on activated neutrophils, lymphocytes, and platelets to induce inflammation. During chronic inflammation B cells and macrophages are the predominant infiltrating cell type, and activation of endogenous cells within the organ participates in fibrosis and disease progression.
• Experimental models demonstrate the main immune complex diseases. Serum sickness can be induced with large injections of foreign antigen. Autoimmunity causes immune complex disease in the NZB/NZW mouse. Injection of antigen into the skin of presensitized animals produces the Arthus reaction.
• Immune complexes are normally removed by the mononuclear phagocyte system. Complement helps to disrupt antigen–antibody bonds and keeps immune complexes soluble. Primate erythrocytes bear a receptor for C3b and are important for transporting complement-containing immune complexes to the spleen for removal. Complement deficiencies lead to the formation of large, relatively insoluble complexes, which deposit in tissues.
• The size of immune complexes affects their deposition. Deposition of circulating, soluble immune complexes is limited by physical factors, such as the size and charge of the complexes. Small, positively charged complexes have the greatest propensity for deposition within vessels. Large immune complexes are rapidly removed in the liver and spleen.
• Immune complex deposition in the tissues results in tissue damage. Immune complexes can form both in the circulation, leading to systemic disease, and at local sites such as the lung. Charged cationic antigens have tissue-binding properties, particularly for the glomerulus, and help to localize complexes to the kidney. Factors that tend to increase blood vessel permeability enhance the deposition of immune complexes in tissues.
Immune complex diseases
Immune complex formation can result from:
Fig. 25.1 Three categories of immune complex disease
This table indicates the source of the antigen and the organs most frequently affected.
Persistent infection with a weak antibody response can lead to immune complex disease
The combined effects of a low-grade persistent infection and a weak antibody response lead to chronic immune complex formation, and eventual deposition of complexes in the tissues (Fig. 25.2). Diseases with this etiology include:
Immune complexes can be formed with inhaled antigens
Immune complexes may be formed at body surfaces following exposure to extrinsic antigens.
• farmer’s lung, where there are circulating antibodies to actinomycete fungi (found in moldy hay); and
• pigeon fancier’s lung, where there are circulating antibodies to pigeon antigens.
Both diseases are forms of extrinsic allergic alveolitis, and occur only after repeated exposure to the antigen. Note that the antibodies induced by these antigens are primarily IgG, rather than the IgE seen in type I hypersensitivity reactions. When antigen again enters the body by inhalation, local immune complexes are formed in the alveoli leading to inflammation and fibrosis (Fig. 25.3).
Immune complex disease occurs in autoimmune rheumatic disorders
Immune complex disease is common in autoimmune disease, where the continued production of autoantibody to a self antigen leads to prolonged immune complex formation. As the number of complexes in the blood increases, the systems responsible for the removal of complexes (mononuclear phagocyte, erythrocyte, and complement) become overloaded, and complexes are deposited in the tissues (see Fig. 25.16). Systemic lupus erythematosus (SLE) is the classic disease characterized by immune complex deposition and others include Henoch-Schönlein purpura and primary Sjögren’s syndrome.
Fig. 25.16 Immunofluorescence study of immune complexes in autoimmune disease
(Courtesy of Dr S Thiru.)
Immune complexes and inflammation
Immune complexes are capable of triggering a wide variety of inflammatory processes:
• they interact directly with basophils and platelets (via Fc receptors) to induce the release of vasoactive amines (Fig. 25.4);
• macrophages are stimulated to release cytokines, particularly tumor necrosis factor-α (TNFα) and interleukin-1 (IL-1), which have important roles in inflammation;
• they interact with the complement system to generate C3a and C5a, which stimulate the release of vasoactive amines (including histamine and 5-hydroxytryptamine) and chemotactic factors from mast cells and basophils; C5a is also chemotactic for basophils, eosinophils, and neutrophils.
The vasoactive amines released by platelets, basophils, and mast cells cause endothelial cell retraction and thus increase vascular permeability, allowing the deposition of immune complexes on the blood vessel wall (Fig. 25.5). The deposited complexes continue to generate C3a and C5a.
Polymorphs are chemotactically attracted to the site by C5a. They attempt to engulf the deposited immune complexes, but are unable to do so because the complexes are bound to the vessel wall. Therefore they exocytose their lysosomal enzymes onto the site of deposition (see Fig. 25.5). If simply released into the blood or tissue fluids these lysosomal enzymes are unlikely to cause much inflammation, because they are rapidly neutralized by serum enzyme inhibitors. But if the phagocyte applies itself closely to the tissue-trapped complexes through Fc binding, then serum inhibitors are excluded and the enzymes may damage the underlying tissue.
Complement is an important mediator of immune complex disease
• the kidney in various autoimmune glomerular diseases; and
• the skin in autoimmune diseases where cutaneous vasculitis is a feature such as SLE, Sjögren’s syndrome and Henoch–Schönlein purpura.
Experimental models of immune complex diseases
Experimental models are available for the main types of immune complex disease described above:
• serum sickness, induced by injections of foreign antigen, mimics the effect of a persistent infection;
• the NZB/NZW mouse demonstrates autoimmunity;
• the Arthus reaction is an example of local damage by extrinsic antigen.
Serum sickness can be induced with large injections of foreign antigen
Serum sickness is now commonly studied in rabbits by giving them an intravenous injection of a foreign soluble protein such as bovine serum albumin (BSA). After about 1 week antibodies are formed, which enter the circulation and complex with antigen. Because the reaction occurs in antigen excess, the immune complexes are small (Fig. 25.w1). These small complexes are removed only slowly by the mononuclear phagocyte system and therefore persist in the circulation.
The formation of complexes is followed by an abrupt fall in total hemolytic complement.
Autoimmunity causes immune complex disease in the NZB/NZW mouse
The F1 hybrid NZB/NZW mouse produces a range of autoantibodies (including anti-erythrocyte, anti-nuclear, anti-DNA, and anti-Sm) and suffers from an immune complex disease similar in many ways to SLE in humans. A NZB/NZW mouse is born clinically normal, but within 2–3 months shows sign of hemolytic anemia. Tests for anti-erythrocyte antibody (the Coombs’ test), anti-nuclear antibodies, lupus cells, and circulating immune complexes are all positive, and there are deposits in the glomeruli and choroid plexus of the brain. The disease is much more marked in the females, who die within a few months of developing symptoms (Fig. 25.w2).
