Chapter 24 Hypersensitivity (Type II)
• Type II hypersensitivity is mediated by antibodies binding to specific cells. Type II hypersensitivity reactions are caused by IgG, IgA, or IgM antibodies against cell surface and extracellular matrix antigens. The antibodies damage cells and tissues by activating complement, and by binding and activating effector cells carrying Fc γ receptors.
• Red blood cells (blood groups) must be cross-matched for transfusion. Transfusion reactions to erythrocytes are produced by antibodies to blood group antigens, which may occur naturally or may have been induced by previous contact with incompatible tissue or blood following transplantation, transfusion, or during pregnancy.
• Hemolytic disease of the newborn occurs when maternal antibodies to fetal blood group antigens cross the placenta and destroy the fetal erythrocytes.
• Type II hypersensitivity reactions may target tissues. Damage to tissues may be produced by autoantibodies to extracellular matrix, cell surface molecules, or intracellular proteins. Examples of diseases caused by these mechanisms are myasthenia gravis, pemphigus, and Goodpasture’s syndrome.
• The role of autoantibodies in disease is not always clear. Antibodies to intracellular components are not necessarily pathogenic, but they may be diagnostically useful.
Mechanisms of tissue damage
• antibodies directed against cell surface antigens are usually pathogenic;
• antibodies directed against internal antigens are usually not pathogenic.
Type II reactions therefore differ from type III reactions, which involve antibodies directed against soluble antigens in the serum, leading to the formation of circulating antigen–antibody complexes. Damage occurs when the complexes are deposited non-specifically onto tissues and/or organs (see Chapter 25).
Effector cells engage their targets using Fc and C3 receptors
In type II hypersensitivity, antibody directed against cell surface or tissue antigens interacts with the Fc receptors (FcR) on a variety of effector cells and can activate complement to bring about damage to the target cells (Fig. 24.1).
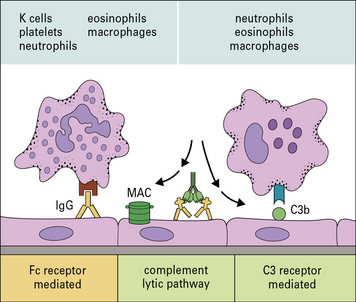
• complement fragments (C3a and C5a) generated by activation of complement attract macrophages and polymorphs to the site, and also stimulate mast cells and basophils to produce chemokines that attract and activate other effector cells;
• the classical complement pathway and activation loop lead to the deposition of C3b, C3bi, and C3d on the target cell membrane;
• the classical complement pathway and lytic pathway result in the production of the C5b–9 membrane attack complex (MAC) and insertion of the complex into the target cell membrane.
Effector cells – in this case macrophages, neutrophils, eosinophils, and NK cells – bind to either:
Cells damage targets by releasing their normal immune effector molecules
The mechanisms by which neutrophils and macrophages damage target cells in type II hypersensitivity reactions reflect their normal methods of dealing with infectious pathogens (Fig. 24.2).
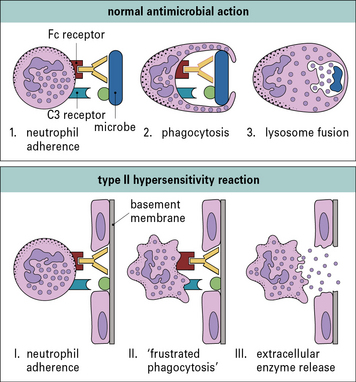
Normally pathogens would be internalized and then subjected to a barrage of microbicidal systems including defensins, reactive oxygen and nitrogen metabolites, hypohalites, enzymes, altered pH, and other agents that interfere with metabolism (see Chapters 7 and 14).
In some situations, such as the eosinophil reaction against schistosomes (see Chapter 15), exocytosis of granule contents is normal and beneficial. However, when the target is host tissue that has been sensitized by antibody, the result is damaging (Fig. 24.3).
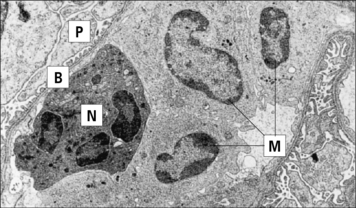
The resistance of a target cell to damage varies. Susceptibility depends on:
• the amount of antigen expressed on the target cell’s surface; and
• the inherent ability of different target cells to sustain damage.
Type II reactions against blood cells and platelets
• incompatible blood transfusions, where the recipient becomes sensitized to antigens on the surface of the donor’s erythrocytes;
• hemolytic disease of the newborn, where a pregnant woman has become sensitized to the fetal erythrocytes;
• autoimmune hemolytic anemias, where the patient becomes sensitized to his or her own erythrocytes.
Transfusion reactions occur when a recipient has antibodies against donor erythrocytes
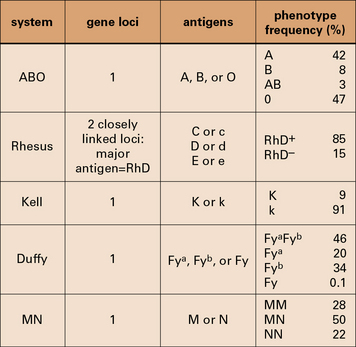
Some major human blood groups are listed in Figure 24.4.
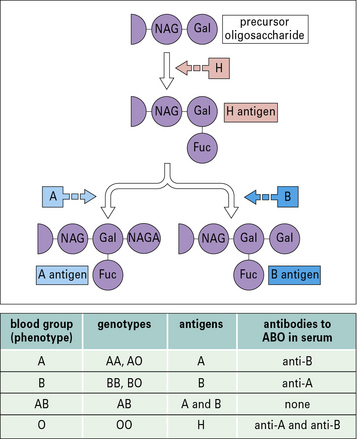
The ABO blood group system is of primary importance
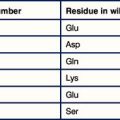
The epitopes of the ABO blood group system occur on many cell types in addition to erythrocytes and are located on the carbohydrate units of glycoproteins. The structure of these carbohydrates, and of those determining the related Lewis blood group system, is determined by genes coding for enzymes that transfer terminal sugars to a carbohydrate backbone (Fig. 24.5).
 RhD is the most important clinically due to its high immunogenicity, but in RhD– individuals the RhD locus is missing completely. The RhCcEe locus encodes a molecule that expresses the RhC/c and RhE/e epitopes.
RhD is the most important clinically due to its high immunogenicity, but in RhD– individuals the RhD locus is missing completely. The RhCcEe locus encodes a molecule that expresses the RhC/c and RhE/e epitopes.Transfusion reactions can be caused by minor blood groups
The related Ss system antigens are carried on glycophorin B.
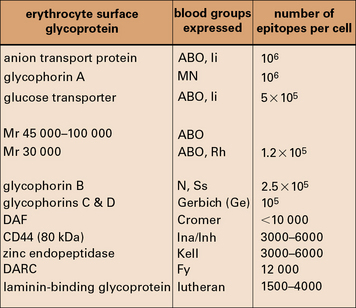
The relationship of the blood groups to erythrocyte surface proteins is listed in Figure 24.w1.
Cross-matching ensures that a recipient does not have antibodies against donor erythrocytes
• antibodies to ABO system antigens cause incompatible cells to agglutinate in a clearly visible reaction;
• minor blood group systems cause weaker reactions that may only be detectable by an indirect Coombs’ test (see Fig. 24.9).
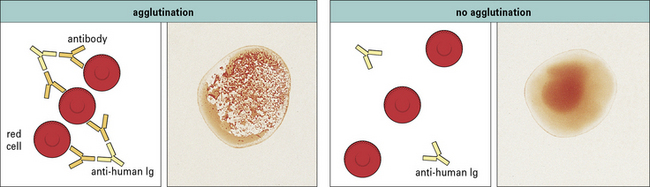
Transfusion reactions involve extensive destruction of donor blood cells
The severity of the reaction depends on the class and the amounts of antibodies involved:
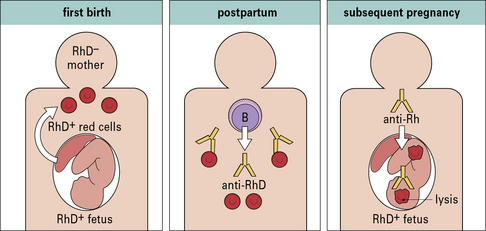
HDNB is due to maternal IgG reacting against the child’s erythrocytes in utero
Hemolytic disease of the newborn (HDNB) occurs when the mother has been sensitized to antigens on the infant’s erythrocytes and makes IgG antibodies to these antigens. These antibodies cross the placenta and react with the fetal erythrocytes, causing their destruction (Figs 24.6 and 24.7). Rhesus D (RhD) is the most commonly involved antigen.
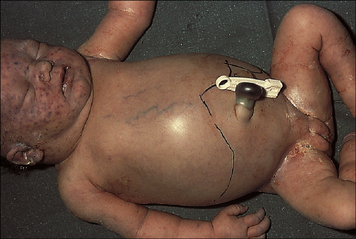
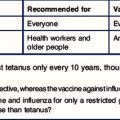
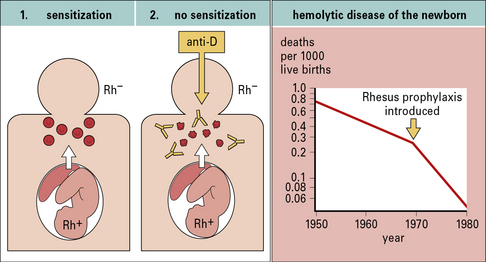
This notion led to the development of Rhesus prophylaxis – preformed anti-RhD antibodies are given to Rh– mothers immediately after delivery of Rh+ infants, with the aim of destroying fetal Rh+ erythrocytes before they can cause Rh− sensitization. This practice has successfully reduced the incidence of HDNB due to Rhesus incompatibility (Fig. 24.8). Although the number of cases of HDNB has fallen dramatically and progressively, the proportion of cases caused by other blood groups, including Kell and the ABO system, has increased.
Autoimmune hemolytic anemias arise spontaneously or may be induced by drugs
Autoimmune hemolytic anemia is suspected if a patient gives a positive result on a direct antiglobulin test (Fig. 24.9), which identifies antibodies present on the patient’s erythrocytes. These are usually antibodies directed towards erythrocyte antigens, or immune complexes adsorbed onto the erythrocyte surface.
• warm-reactive autoantibodies, which react with the antigen at 37 °C;
• cold-reactive autoantibodies, which can only react with antigen at below 37 °C;
Warm-reactive autoantibodies cause accelerated clearance of erythrocytes
Warm-reactive autoantibodies to other blood group antigens exist, but are relatively rare.
Cold-reactive autoantibodies cause erythrocyte lysis by complement fixation
However, some cases may follow infection with Mycoplasma pneumoniae, and these are acute-onset diseases of short duration with polyclonal autoantibodies. Such cases are thought to be due to cross-reacting antigens on the bacteria and the erythrocytes, producing a bypass of normal tolerance mechanisms (see Chapter 19).
Drug-induced reactions to blood components occur in three different ways
Drugs (or their metabolites) can provoke hypersensitivity reactions against blood cells, including erythrocytes and platelets. This can occur in three different ways (Fig. 24.10).
• The drug binds to the blood cells and antibodies are produced against the drug. In this case it is necessary for both the drug and the antibody to be present to produce the reaction. This phenomenon was first recorded by Ackroyd, who noted thrombocytopenic purpura (destruction of platelets leading to purpuric rash) following administration of the drug sedormid. Hemolytic anemias have been reported following the administration of a wide variety of drugs, including penicillin, quinine and sulfonamides. All of these conditions are rare.
• Drug–antibody immune complexes are adsorbed onto the erythrocyte cell membrane. When drug–antibody immune complexes are adsorbed on to the erythrocyte cell membrane damage occurs by complement-mediated lysis.
• The drug induces an allergic reaction. The drug induces an allergic reaction and autoantibodies are directed against the erythrocyte antigens themselves, as occurs in 0.3% of patients given α-methyldopa. The antibodies produced are similar to those in patients with warm-reactive antibody. However, the condition remits shortly after the cessation of drug treatment.
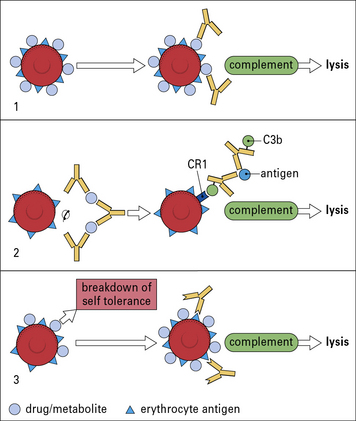
Autoantibodies to platelets may cause thrombocytopenia
 Thrombocytopenia may also be induced by drugs by similar mechanisms to those outlined in Figure 24.10.
Thrombocytopenia may also be induced by drugs by similar mechanisms to those outlined in Figure 24.10.
Reactions against neutrophils can occur in several autoimmune diseases
• antibody to proteinase-3 (PR3), a cytoplasmic antigen (c-ANCA), is associated with Wegener’s granulomatosis;
• antibodies to myeloperoxidase are seen more commonly in systemic lupus erythematosus (SLE), and are located in perinuclear granules (p-ANCA).
Other granule components may also act as antigens in SLE, but less commonly than myeloperoxidase. Such autoantigens are generally neutrophil specific and antibodies can be detected by immunofluorescent staining (Fig. 24.w2). (By contrast, antibodies to MHC antigens, which are also seen in SLE, are highly non-tissue specific.)
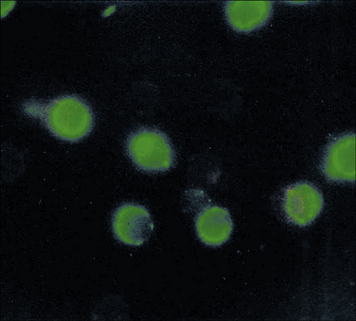
Type II hypersensitivity reactions in tissues
Pemphigus is caused by autoantibodies to an intercellular adhesion molecule
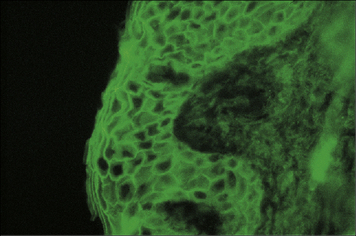
Pemphigus vulgaris is a serious blistering disease of the skin and mucous membranes. Patients have autoantibodies against desmoglein-1 and desmoglein-3, components of desmosomes, which form junctions between epidermal cells (Fig. 24.11). The antibodies disrupt cellular adhesion, leading to breakdown of the epidermis with separation of the superficial layers to form blisters.
Clinical disease profiles can be related to the specificity of the antibodies. For example:
• patients with only anti-desmoglein-3 tend to show mucosal disease;
• those with anti-desmoglein-1 and anti-desmoglein-3 have skin and mucosal involvement.
The disease profile is also partly dependent on the isotype of the antibodies produced; some patients show strong deposition of IgA (see Fig. 24.11) whereas other have particularly high titres of IgG4. Recently, antibodies to mitochondrial components have also been implicated in the pathology, by inducing apoptosis in keratinocytes.
In myasthenia gravis autoantibodies to acetylcholine receptors cause muscle weakness
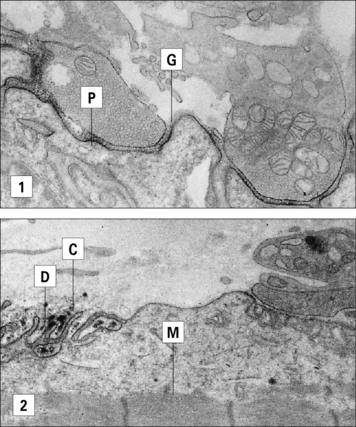
Analysis of the lesion in myasthenic muscles indicated that the disease was not due to an inability to synthesize acetylcholine, nor was there any problem in secreting it in response to a nerve impulse – the released acetylcholine was less effective at triggering depolarization of the muscle (Fig. 24.12).
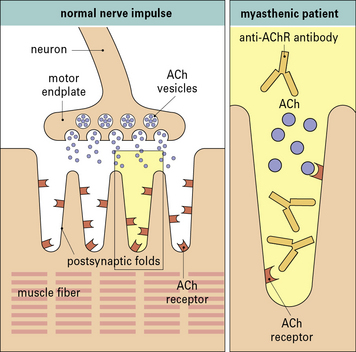
Examination of neuromuscular endplates by immunochemical techniques has demonstrated IgG and the complement proteins, C3 and C9, on the postsynaptic folds of the muscle (Fig. 24.13).
Autoantibodies and autoimmune disease
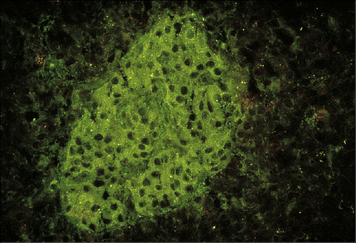
Although many autoantibodies react with tissue antigens, their significance in causing tissue damage and pathology in vivo is not always clear. For example, although autoantibodies to pancreatic islet cells can be detected in vitro using sera from some diabetic patients (Fig. 24.14), most of the immunopathological damage in autoimmune diabetes is thought to be caused by autoreactive T cells.
Finally, there are a group of conditions where autoantibodies actually stimulate the target cells. For example in some forms of autoimmune thyroid disease antibodies to the thyroid-stimulating hormone (TSH) receptor mimic TSH, thereby stimulating thyroid function (see Chapter 21).
Critical thinking: Blood groups and hemolytic disease of the newborn (see p. 443 for explanations)
• first child born 1968 – unaffected;
• second child born 1974 – mildly affected;
• third child born 1976 – seriously affected, required intrauterine blood transfusion;
1. From this information, what can you deduce about the blood group of the first child?
2. Why does HDNB usually become more serious with successive pregnancies?
3. What is the reason for giving anti-Rhesus D antibodies to the mother?
4. Why are the antibodies given postpartum and not earlier?
5. Give an explanation of why the Rhesus prophylaxis after the second delivery failed to prevent HDNB in the third child.
6. What explanation can be given to account for the fact that the fourth child is unaffected?
When the blood groups of the children are examined it is found that they are:
Alarçon-Segovia D., Ruiz-Argüelles A., Llorente L. Broken dogma: penetration of autoantibodies into living cells. Immunol Today. 1996;17:163–164.
Amagai M. Autoantibodies against desmosomal cadherins in pemphigus. J Dermatol Sci. 1999;20:92–102.
Anstee D.J. Blood group active substances of the human red blood cell. Vox Sang. 1990;58:1.
Black M., Mignogna M.D., Scully C. Pemphigus vulgaris. Oral Dis. 2005;11:119–130.
Dean F.G., Wilson G.R., Li M., Edgtton K.L., et al. Experimental autoimmune Goodpasture’s disease: a pathogenetic role for both effector cells and antibody injury. Kidney Int. 2005;67:566–575.
Engelfriet C.P., Reesink H.W., Judd W.J., et al. Current status of immunoprophylaxis with anti-D immunoglobulin. Vox Sang. 2003;85:328–337.
Lang B., Newsom-Davis J. Immunopathology of the Lambert–Eaton myasthenic syndrome. Springer Semin Immunopathol. 1995;17:3–15.
Mauro I., Colin Y., Chenif-Zahar B., et al. Molecular genetic basis of the human Rhesus blood group system. Nat Genet. 1993;5:62–65.
Payne A.S., Hanakawa Y., Amagai M., Stanley J.R. Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol. 2004;16:536–543.
Race R., Sanger R. Blood groups in man, 6th edn. Oxford: Blackwell Scientific Publications; 1975.
Russo D., Redman C., Lee S. Association of XK and Kell blood group proteins. J Biol Chem. 1998;273:13950–13956.
Schulz D.R., Tozman E.C. Anti-neutrophil cytoplasmic antibodies: major autoantigens, pathophysiology, and disease associations. Semin Arthritis Rheum. 1995;25:143–159.
Vincent A. Antibody-mediated disorders of neuro-muscular transmission. Clin Neurophysiol Suppl. 2004;57:147–158.
Yamamoto F.-I., Clausen H., White T., et al. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345:229.